

Abstract
Background
Gap junction channels made of Connexin37 (Cx37) are expressed by aortic endothelial and smooth muscle cells of hypertensive mice, as well as by the renin‐secreting cells of kidneys.
Methods and Results
To decipher whether Cx37 has any role in hypertension, angiotensin II (Ang II) was infused in normotensive wild‐type and Cx37‐deficient mice (Cx37−/−). After 2 to 4 weeks, the resulting increase in blood pressure was lower in Cx37−/− than in wild‐type mice, suggesting an alteration in the Ang II response. To investigate this possibility, mice were submitted to a 2‐kidney, 1‐clip procedure, a renin‐dependent model of hypertension. Two weeks after this clipping, Cx37−/− mice were less hypertensive than wild‐type mice and, 2 weeks later, their blood pressure had returned to control values, in spite of abnormally high plasma renin levels. In contrast, Cx37−/− and wild‐type mice that received N‐nitro‐l‐arginine‐methyl‐ester, a renin‐independent model of hypertension, featured a similar and sustained increase in blood pressure. The data indicate that loss of Cx37 selectively altered the Ang II‐dependent pathways. Consistent with this conclusion, aortas of Cx37−/− mice featured an increased basal expression of the Ang II type 2 receptors (AT2R), and increased transcripts levels of downstream signaling proteins, such as Cnksr1 and Ptpn6 (SHP‐1). Accordingly, the response of Cx37−/− mice aortas to an ex vivo AngII exposure was altered, since phosphorylation levels of several proteins of the AngII pathway (MLC2, ERK, and AKT) remained unchanged.
Conclusions
These findings provide evidence that Cx37 selectively influences Ang II signaling, mostly via a modulation of the expression of the Ang II type 2 receptor.
Keywords: angiotensin II, aorta, connexins, endothelial cells, hypertension, kidney, smooth muscle cells
Subject Categories: High Blood Pressure, Hypertension
Clinical Perspective
What Is New?
We submitted Connexin37 (Cx37)‐null mice to different models of angiotensin II (Ang II)‐dependent and ‐independent hypertension, and document that Cx37 selectively participates in Ang II‐mediated hypertension.
In the renin‐dependent model, Cx37−/− mice rapidly recover a normal blood pressure, in spite of increased plasma renin levels.
This response results from an altered expression of proteins implicated in the Ang II pathway, notably AT2R.
What Are the Clinical Implications?
Our data suggest the following scenario for the involvement of vascular Cx37 in the control of Ang II‐dependent hypertension: in the absence of Cx37, increased aortic levels of AT2R and of downstream targets of the AT2R‐dependent pathway associate to reduce the vasoconstrictive effects mediated by Ang II, eventually altering the development of hypertension.
Understanding how AT2R is upregulated in the absence of Cx37 may help in developing new therapeutic tools against hypertension.
Introduction
Vascular gap junctions formed of connexins (Cx)1 provide for communications between endothelial cells (EC), smooth muscle cells (SMC), and renin‐secreting cells, which are required to control blood pressure (BP).2, 3, 4 In the vasculature, EC predominantly express Cx37 and Cx40,5, 6, 7, 8, 9, 10 whereas SMC mostly express Cx43 and Cx45,6, 11, 12 and renin‐secreting cells express Cx40 and Cx37.2, 7, 9, 13, 14, 15 Various changes in the expression of these proteins have been documented in different models of vascular alterations, notably during chronic changes in BP.2, 3, 4 Specifically, Cx43 has been reported to allow for the rapid diffusion of calcium‐dependent contraction waves between SMC, which ultimately control vasomotor tone,16, 17 and we have documented a prominent role of Cx40 in renin‐dependent hypertension.13, 14, 18 This protein also interacts with endothelial nitric oxide synthase (eNOS) to increase the production of NO which, in turn, promotes the relaxation of SMC.5, 19 Both renin‐dependent and ‐independent models of chronic hypertension also induce the expression of Cx37 in vascular SMC2, 6, 9; still the protein appears dispensable for the physiological control of renin expression and function.7, 14, 15, 18 Thus, whether and how Cx37 contributes to control BP remains to be fully investigated.
To approach this question, we compared wild type (WT) and Cx37−/− mice after (1) an infusion of angiotensin II (Ang II) via osmotic mini‐pumps; (2) induction of a renin‐dependent hypertension, as provided by the 2‐kidney, 1‐clip (2K1C) surgical procedure; and (3) induction of a renin‐independent model of hypertension, as provided by a N‐nitro‐l‐arginine‐methyl‐ester (l‐NAME) treatment. We further compared the aortas of these animals ex vivo, under basal conditions and following an exposure to Ang II. Here, we show that loss of Cx37 partially protects mice against the development of Ang II‐dependent hypertension, but not against the hypertension induced by l‐NAME. We further document that the lower increase in BP of Cx37−/− mice in response to Ang II is associated with increased levels of Ang II type 2 receptors in their aortas, with selective changes in the transcript levels of proteins involved in the Ang II signaling, including SHP‐1 and CNKSR1, and with an altered phosphorylation of downstream targets of this signaling pathway (MLC2, ERK, and AKT). The data provide direct evidence that Cx37 plays a crucial, selective role in the control of Ang II‐dependent hypertension.
Methods
The data, analytic methods, and study materials will be available to other researchers, for purposes of reproducing the results or replicating the procedure, on request from the Department of Medicine, Lausanne, Switzerland.
Animals and Surgery
Experiments were performed using 2‐ to 3‐month‐old Cx37−/− males (a kind gift from Dr A. Simon),20 and WT littermate mice, all on a C57BL/6J genetic background. These mice were generated by breeding heterozygote Cx37+/− males and females. WT, heterozygous, and knockout animals were identified by polymerase chain reaction of genomic DNA, using the following primers: forward primer 5′‐TGCTAGACCAGGTCCAGGAAC‐3′ and reverse primer: 5′‐GATCTCTCGTGGGATCATTG‐3′ to detect the Cx37 knockout allele, and reverse primer: 5′‐GTCCCTTCGTGCCTTTATCTC‐3′ to detect the Cx37 WT allele. All mice were housed in individually ventilated cages, as per our institutional guidelines. Mouse care, surgery, and euthanasia procedures were approved by the Centre Hospitalier Universitaire Vaudois, and the Cantonal Veterinary Office (Service de la Consommation et des Affaires Vétérinaires SCAV‐EXPANIM, authorization numbers 2832.1 and 3102). All animal experimentation conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication Eighth Edition, 2011). The 2K1C procedure, which consists of placing a U‐shaped silver clip of 0.12‐mm internal diameter around the left renal artery,6, 9, 14 was performed in 8‐ to 10‐week‐old WT and Cx37−/− mice. Control mice were sham‐operated (no clipping of the renal artery). ALZET osmotic mini‐pumps were loaded with Ang II in 0.9% NaCl, to provide a daily output of either 0.25 or 1 mg hormone/kg body weight, and subcutaneously implanted as reported.21, 22 Control mice were similarly implanted with pumps containing only 0.9% NaCl. WT and Cx37−/− mice were also treated with Nω‐Nitro‐l‐arginine methyl ester hydrochloride (l‐NAME; Sigma‐Aldrich Chemie GmbH, Buchs, Switzerland), which was added at the concentration of 500 mg/L to the drinking water, for 4 weeks.6, 12, 23
Animal Monitoring
Systolic blood pressure (SBP) was measured on conscious mice by a noninvasive, computerized tail‐cuff method (BP‐2000, Visitech Systems). Mice were pretrained during 7 days, and thereafter pulse rate and BP were recorded by an automatic system of cuff inflation and deflation, over at least 15 days.7, 9 At the end of the experience, blood samples (300 μL) were collected from the tail vein into EDTA‐coated tubes. Plasma was separated by centrifugation at 4°C (8 minutes, 5720g), and stored at −80°C. Five microliters of plasma diluted in 100 μL 0.9% NaCl were incubated for 90 minutes at 37°C. Plasma from bilaterally nephrectomized male rats served as substrate for renin. Plasma renin concentration (PRC) was determined by measuring the capacity of plasma samples to generate AngI in the presence of an excess renin substrate. The generated angiotensin I (ng/mL·h−1) was determined by radioimmunoassay (Byk&DiaSorin Diagnostics, Dietzenbach, Germany).24
RNA and Protein Analysis
Mice were anesthetized with isoflurane, euthanized by cervical dislocation, and then depleted of blood by an intracardiac perfusion of 5 mL PBS. After PBS washing, aortas and kidneys were carefully removed, immersed into liquid nitrogen, reduced to powder, and stored at −80°C. For reverse transcription polymerase chain reaction analysis, the aortic and renal powders were homogenized in Tripure Isolation Reagent (Roche, Switzerland), and total RNA was extracted according to the manufacturer's instructions. RNA was analyzed by real‐time quantitative polymerase chain reaction, using the primers given in Table 1. For Western blots, aortic powder was homogenized by sonication in lysis buffer (62.5 mmol/L Tris HCl, 5% SDS, 10 mmol/L EDTA, pH=8). Protein content was measured using a detergent‐compatible DC protein assay kit (Bio‐Rad Laboratories, Reinach BL, Switzerland). Samples (30 μg) were loaded on a 7% or 10% polyacrylamide gel, separated by electrophoresis and transferred onto PVDF membranes (Immobilon‐P; Millipore, Volketswil, Switzerland). Membranes were incubated for 1 hour in PBS or TBS (Tris‐Buffered Saline) containing 5% milk and 0.1% Tween 20 (blocking buffer). The membranes were then incubated overnight at 4°C with 1 of the following primary antibodies: rabbit polyclonal antibodies against AT1R (Alomone labs, AAR‐011, 1:1000), AT2R (Alomone labs, AAR‐012, 1:1000), MLC2 (Cell Signaling, 3672, 1:1000), P‐MLC2 (Cell Signaling, 3674, 1:1000), ERK (Cell Signaling, 4695, 1:1000), P‐ERK (Cell Signaling, 9101, 1:1000), AKT (Cell Signaling, 9272S, 1:1000), Cx40 (Chemicon, AB1726; 1:250), Cx37 (Biotrend Chemikalien, Cx37A11‐A; 1:500), Cx43 (Cell Signaling, 3512S, 1:500) or Cx45 (Millipore, AB1745, 1:500); mouse monoclonal antibodies against eNOS (BD Biosciences, 610297, 1:500), PeNOS (BD Biosciences, 612392, 1:500), P‐AKT (Cell Signaling, 4051, 1:500), and α‐tubulin (Sigma‐Aldrich, T5168; 1:2500). The secondary antibodies were horseradish peroxidase–conjugated goat anti‐mouse immunoglobulins (Jackson Immuno research, 63343, diluted 1:20 000) or goat anti‐rabbit immunoglobulins (Thermo Scientific, 31460, diluted 1:5000), whichever was adequate. Bands were developed for enhanced chemiluminescence (Millipore, Immunobilon Western Chemiluminescent HRP substrate), and visualized using a supercooled CCD camera (Chemidoc XRS, Bio‐Rad Laboratories). Densitometric analysis was performed using ImageLab Software (3.0.1 Bio‐Rad Laboratories). For immunolabeling, freshly excised kidneys and aortas were rapidly frozen and processed for cryosectioning.5, 6, 7, 9 Cryostat sections were stained using 1 of the following antibodies: goat polyclonal antibodies against renin (R and D, AF4277), rabbit polyclonal against Cx40 (Chemicon, AB1726), Cx37 (Biotrend Chemikalien, Cx37A11‐A) or Cx43 (Cell Signaling, 3512S). All antibodies were diluted 1:100. Primary antibodies were detected using antibodies to rabbit immunoglobulins labeled with AlexaFluor 488 or to goat immunoglobulins labeled with AlexaFluor 350 (Invitrogen), all diluted 1:500.
Table 1.
Specific Primers for qPCR Analysis of Aortas and Kidneys
| Gene | Sense Primer (5′–3′) | Antisense Primer (5′–3′) |
|---|---|---|
| Renin | TCTCTGGGCACTCTTGTTGCTCTG | ATACGTCCCATTCAGCACTGAGCC |
| Agt | GTTGGCGCTGAAGGATACAC | GACCCAGGTCAAGATGCAGAA |
| Agtr1a | GTCGCACTCAAGCCTGTC | CCTGTCACTCCACCTCAG |
| Agtr1b | CGTGCACGGGTGCATTT | TAATTGTGCCTGCCAGCCTT |
| Agtr2 | CCAGAGATCTGGTGCAGTTACA | TCCCGCATGCACTCCTTAAA |
| Cnskr1 | ACACCCTGACAAGAGTCCCA | TACCTGCCTCAACACAGCAG |
| Ptpn6 | TACACCAACGTCTGGAAGGG | GGCTGTGGTCAAAGGGAAGA |
| Enpep | GGAAGGCAGAACATCACCCA | TGTGTAACCGAGCTCTGACG |
| Cma1 | TGCAGCAGCCCTGAGGA | ATCTCTCCAGCTTTGGTGCT |
| Rgs2 | GACTGCGTACCCATGGACAA | TCCTTTAAGAGTGTCCGCTTCA |
| Ace | TTACCATAGAGGGCAGCAAG | AGGTTCCAGGGGCATACAA |
| Ace2 | GAGGATAAGCCTAAAATCAGCTCTTG | TCGGAACAGGAACATTTCGTT |
| L27 | GATCCAAGATCAAGTCGTTTGTG | CTGGGTCTCTGAACACATCCT |
| Mas1 | CCTTTGGGAACCTGCATAAC | GGATACAGTGTTGCCGTTGC |
qPCR indicates quantitative polymerase chain reaction.
Statistical Analysis
Values are given as means+SEM in all bar graphs, and as means±SEM in Table 2. Mean values of different groups were compared by Student t test or 2‐way ANOVA, and the post hoc Tukey test. To compare blood pressure values over time, we calculated the areas under the systolic blood pressure curves (AUC), which reflect the cumulated blood pressure responses over the entire, 2‐week‐long observation period. The AUC values of different animal groups were then compared by 2‐way ANOVAs, followed by the post hoc Tukey test. P<0.05 were considered as significant. GraphPad Prism 7 was used for statistical analyses.
Table 2.
Characteristics of the Mice Subjected to the 2K1C Procedure for 4 Weeks
| Mice | Groups | n | SBP (mm Hg) | Body Weight (g) | Cwi (mg/g) | LKi (mg/g) | RKi (mg/g) |
|---|---|---|---|---|---|---|---|
| WT | Sham | 16 | 96.5±1.1 | 26.2±0.5 | 5.2±0.1 | 6.5±0.1 | 6.9±0.1 |
| 2K1C | 19 | 115.3±1.5‡ | 25.5±0.5 | 5.8±0.2* | 5.4±0.3† | 7.7±0.2* | |
| Cx37−/− | Sham | 14 | 92.3±0.8 | 27.3±0.6 | 5.1±0.1 | 6.1±0.2 | 6.5±0.2 |
| 2K1C | 17 | 97.2±1.4∥ | 27.9±0.6§ | 5.5±0.2 | 5.7±0.2* | 7.6±0.2† |
Values are mean±SEM. Cwi indicates cardiac weight index (mg heart weight/g body weight); Cx37, Connexin37; 2K1C, 2 kidney, 1 clip; LKi, left kidney index (mg left kidney weight/g body weight); n, number of mice; RKi, right kidney index (mg right kidney weight/g body weight); SBP, systolic blood pressure; WT, wild type.
*P≤0.05, † P≤0.01, ‡ P≤0.001 vs sham mice.
§ P≤0.05, ∥ P≤0.001 vs WT mice given by 2‐way ANOVA.
Results
Loss of Cx37 Attenuates the Increase in Blood Pressure Induced by Ang II
Basal SBP was similar in WT and Cx37−/− mice (Figure 1 and Table 2). A systemic, 2‐week‐long infusion of NaCl did not alter the SBP of control WT and Cx37−/− mice (Figure 1A). In contrast, the daily infusion of 1 mg Ang II/kg body weight markedly increased (P<0.001) SBP in both types of animals (Figure 1A), even though this increase was significantly lower (P<0.001) in Cx37−/− than WT mice (Figure 1A). Infusion of a lower dose of Ang II (0.25 mg/kg body weight) revealed a comparable difference between the 2 types of mice, in spite of a modest increase in SBP (Figure 1B).
Figure 1.
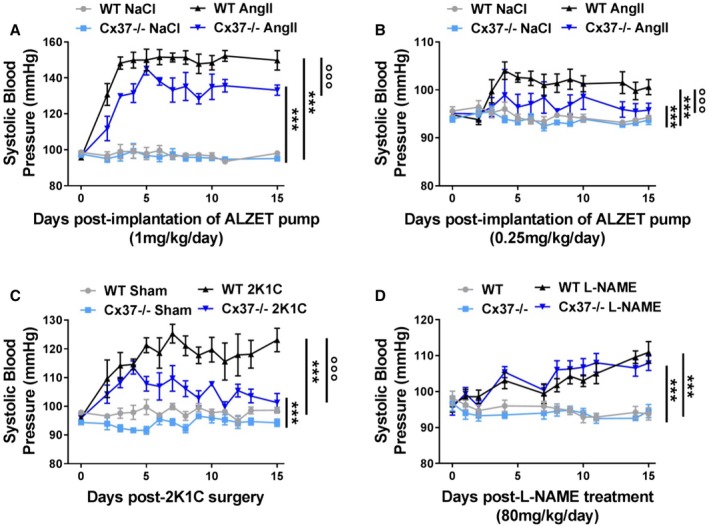
Loss of Cx37−/− alters the increase in blood pressure in models of renin‐dependent hypertension. A, WT mice receiving daily 1 mg Ang II/kg body weight displayed a 50% increase in systolic blood pressure (SBP). Under the same conditions, Cx37−/− mice displayed a significantly lower increase of SBP. WT NaCl N=5; Cx37−/− NaCl, WT Ang II (angiotensin II), Cx37−/− Ang II N=6. B, Similar observations were made in mice receiving daily 0.25 mg Ang II/kg body weight WT NaCl, Cx37−/− NaCl N=7; WT Ang II, Cx37−/− Ang II N=8. C, Two weeks after the 2K1C (2‐kidney, 1‐clip) procedure, Cx37−/− mice also showed a lower increase in SBP than WT controls. WT sham (sham‐operated controls) WT 2K1C, N=5; Cx37−/− sham, Cx37−/− 2K1C N=6. D, In contrast, both types of mice became similarly hypertensive after a treatment with l‐NAME (N‐nitro‐l‐arginine‐methyl‐ester); N=6. ***P≤0.001 vs corresponding controls (NaCl‐infused or sham‐operated animals); °°° P≤0.001 vs WT mice, as given by 2‐way ANOVA of AUC (areas under the SBP curve). Ang II indicates angiotensin II; Cx37, connexin37; WT, wild type.
During the first 2 weeks following the 2K1C surgery, known to induce a renin‐dependent hypertension, SBP was also increased in both WT and Cx37−/− mice and, again, this increase was lower (P<0.001) in the latter than in the former mice (Figure 1C). In contrast, such a difference was not observed after an l‐NAME treatment, known to induce a renin‐independent hypertension (Figure 1D). The data suggest a selective role of Cx37 in the control of Ang II‐dependent pathways.
Sustained Clipping of the Renal Artery Did Not Alter SBP of Cx37−/− Mice
Four weeks after the 2K1C surgery, SBP remained significantly higher than control values in WT mice (Table 2 and Figure 2A). In contrast, SBP of Cx37−/− mice had returned to the levels observed in sham‐operated controls (Table 2 and Figure 2A), documenting that the hypertension observed immediately after the 2K1C procedure (Figure 1C) was transient. A 4‐week‐long, daily infusion of 0.25 mg Ang II/kg body weight also increased SBP significantly less (P<0.001) in Cx37−/− than in WT mice (Figure 2B), whereas an l‐NAME treatment of the same duration had a similar hypertensive effect (P<0.001) in both types of mice (Figure 2C).
Figure 2.
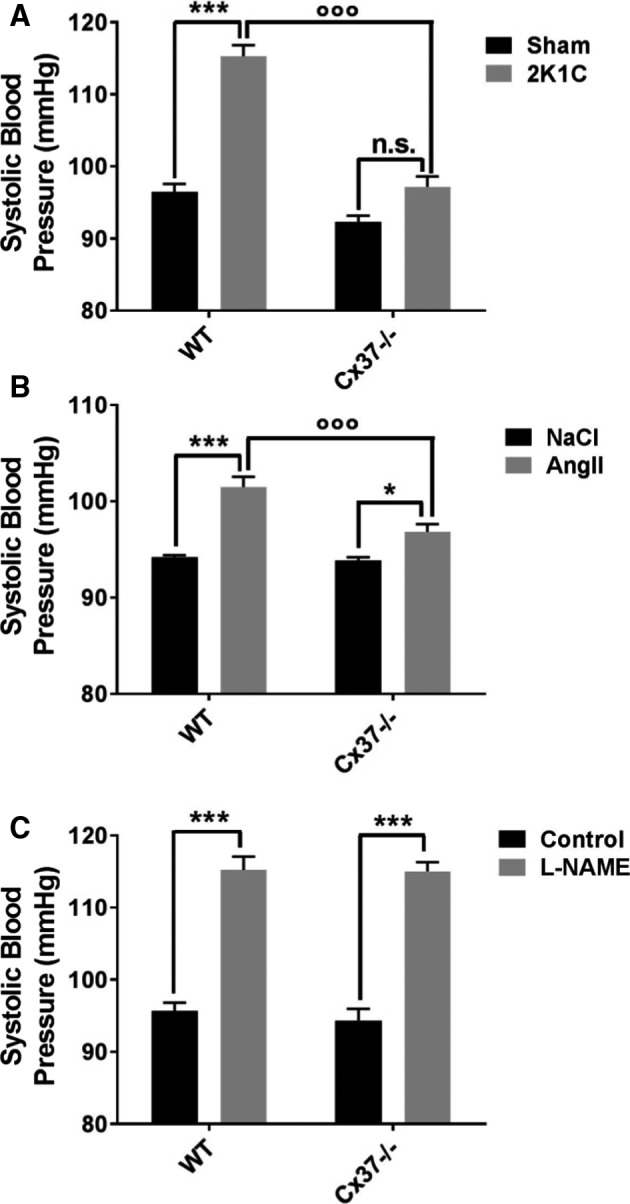
Cx37−/− mice become rapidly normotensive after the 2K1C procedure. A, One month after the 2K1C surgery, WT mice were significantly hypertensive, compared with their sham‐operated controls, whereas Cx37−/− mice featured a control SBP (systolic blood pressure). WT sham (sham‐operated controls) N=15; Cx37−/− sham N=14; WT 2K1C (2‐kidney, 1‐clip) N=19; Cx37−/− 2K1C N=17. B, Compared with controls infused with NaCl, both WT and Cx37−/− mice showed a significant increase in SBP after a daily infusion of 0.25 mg Ang II/kg body weight during 1 month. However, this increase was significantly lower in Cx37−/− than in WT mice. WT NaCl, Cx37−/− NaCl N=7; WT Ang II (angiotensin II); Cx37−/− Ang II N=8. C, In contrast, the mice of these 2 genotypes showed a similar increase in blood pressure, after a daily treatment with l‐NAME (N‐nitro‐l‐arginine‐methyl‐ester, 500 mg/L in the drinking water) for 1 month. N=6. Results are means+SEM. *P≤0.05 and ***P≤0.001 vs respective control mice; °°° P≤0.001 vs WT mice, as given by 2‐way ANOVA. Ang II indicates angiotensin II; Cx37, connexin37; n.s., not significant; WT, wild type.
Evaluation of the organs obtained 4 weeks after the 2K1C surgery revealed the anticipated decrease in the relative weight index of the clipped kidney, and the associated increase in the relative weight of the unclipped kidney, in both WT and Cx37−/− mice, demonstrating that the 2K1C procedure similarly affected these organs in both types of animals (Table 2). However, the heart weight increased in the hypertensive 2K1C WT mice, but was not affected in Cx37−/− mice (Table 2). The data document that, in the absence of Cx37, the modulation of hypertension because of the 2K1C procedure is transient, in spite of persistent renal effects of the surgery.
Accordingly, the levels of renin transcripts were similarly increased over control levels, in the clipped kidneys of both WT and Cx37−/− mice (Figure 3A). Parallel immunostaining of renin showed that the expression of this hormone increased in the hypoperfused kidneys of both WT and Cx37−/− 2K1C mice, and decreased in the unclipped, contralateral kidneys of the very same animals (Figure 3C), still was higher in the kidneys of Cx37−/− than of WT mice. Consistent with these observations, the concentration of circulating renin was significantly higher in the plasma of Cx37−/− than in WT mice, in both the sham‐operated mice and in the mice that were subjected to the whole 2K1C procedure (Figure 3B). The data document that, while the transcriptional expression of renin was similarly modulated by the 2K1C procedure in the kidneys of WT and Cx37−/− mice, the latter animals featured a higher renal content and plasma secretion of the hormone.
Figure 3.
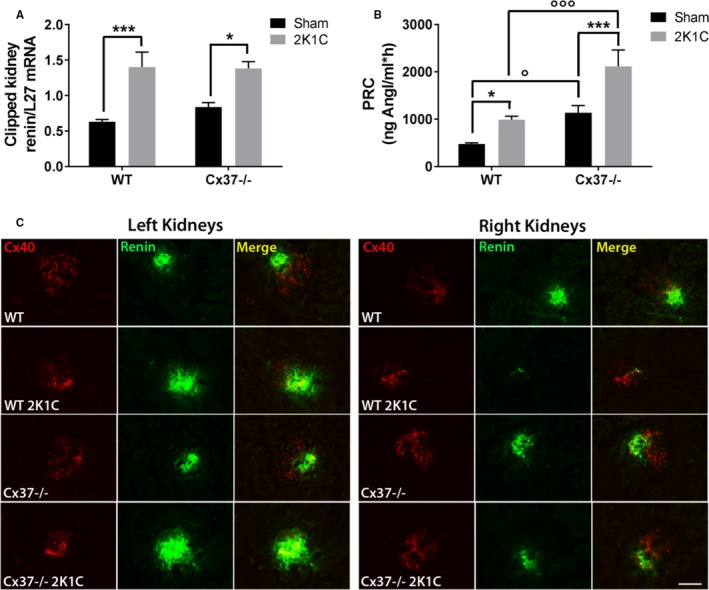
Renin expression and secretion are increased by the 2K1C surgery in both WT and Cx37−/− mice. A, Four weeks after surgery, the basal levels of the renin transcript increased in the clipped, hypoperfused kidney of both WT and Cx37−/− mice. WT sham (sham‐operated controls) N=14; Cx37−/− sham N=13; WT 2K1C N=16; Cx37−/− 2K1C N=12. B, The plasmatic concentration of renin (PRC) was also significantly increased in the 2 types of mice. However, Cx37−/− mice had basal and 2K1C‐induced levels of PRC significantly higher than WT animals. WT sham N=8; Cx37−/− sham N=6; WT 2K1C (2‐kidney, 1‐clip) N=9; Cx37−/− 2K1C N=6. Results are means+SEM. *P≤0.05, ***P≤0.001 vs respective control mice; ° P≤0.05, °°° P≤0.001 vs WT mice, as given by 2‐way ANOVA. C, Renin immunostaining (green) increased in the hypoperfused left kidney of both WT and Cx37−/− mice, while decreasing in their contralateral right kidney, and was higher in Cx37−/− than in WT mice. Cx40 (Connexin40). Immunostaining (red) localizes the renin‐secreting cells of the afferent arterioles. Bar=50 μm. AngI indicates angiotensin I; WT, wild type.
Cx37 Participates in Controling Ang II Signaling in Aortas
To investigate the mechanism associated with the reduced effect of Ang II in Cx37−/− mice, we studied the expression of transcripts coding for proteins involved in the renin‐angiotensin system. The renal levels of the transcripts coding for the angiotensin‐converting enzymes (ACE and ACE2) were similar in WT and Cx37−/− mice, under both basal conditions and following the 2K1C surgery (Figure 4). The transcript levels coding for angiotensinogen and the angiotensin receptors Agtr1a and Agtr1b were also similarly expressed in the aortas of WT and Cx37−/− mice (Figure 5), and this was also the case for the cognate AT1R protein (Figure 6A, 6B, and 6D). In contrast, the aortic levels of Agtr2 transcripts and of the cognate AT2R protein were higher in Cx37−/− than of WT mice (Figures 5 and 6A, 6C, and 6E). The expression of both ATR1 and ATR2 proteins was not significantly altered in the aortas of Cx37−/− mice, after either the 2K1C surgery or the infusion of Ang II (Figure 6).
Figure 4.
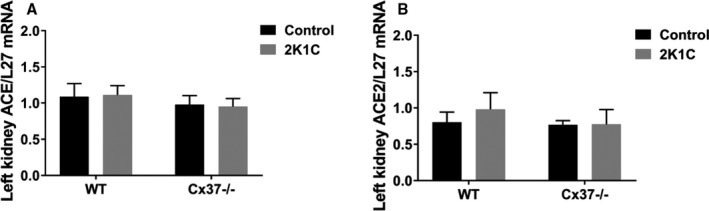
The ACEs were similarly expressed in the kidneys of WT and Cx37−/− mice. The levels of the ACE (A) and the ACE2 (B) transcripts were comparable in the hypoperfused kidneys after the 2K1C (2‐kidney, 1‐clip) surgery and in the sham‐operated controls of both WT and Cx37−/− mice. WT sham N=8; Cx37−/− sham N=7; WT 2K1C N=8; Cx37−/− 2K1C N=10. Results are means+SEM. Statistical analysis was performed using 2‐way ANOVA. ACE and ACE2 indicates angiotensin‐converting enzymes; WT, wild type.
Figure 5.
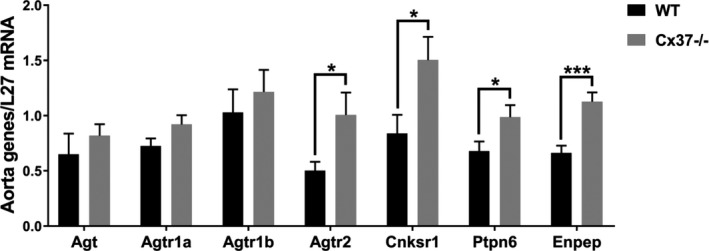
Loss of Cx37 selectively alters the expression of some Ang II‐stimulated signals. The levels of transcripts coding for Angiotensinogen (Agt), Angiotensin receptor 1a (Agtr1a), and Angiotensin receptor 1b (Agtr1b) were similar in the aortas of WT and Cx37−/− mice. In contrast, the transcripts coding for Angiotensin receptor 2 (Agtr2), Connector enhancer of kinase suppressor of Ras 1 (Cnksr1), SH2 domain‐containing tyrosine phosphatase 2 (Ptpn6), and Glutamyl aminopeptidase A (Enpep), which all contribute to control the Ang II pathway, were increased in the aortas of Cx37−/− mice. N=9. Results are means+SEM. *P≤0.05, ***P≤0.001 vs WT mice, as given by t test. Ang II indicates angiotensin II; Cx37, Connexin37; WT, wild type.
Figure 6.
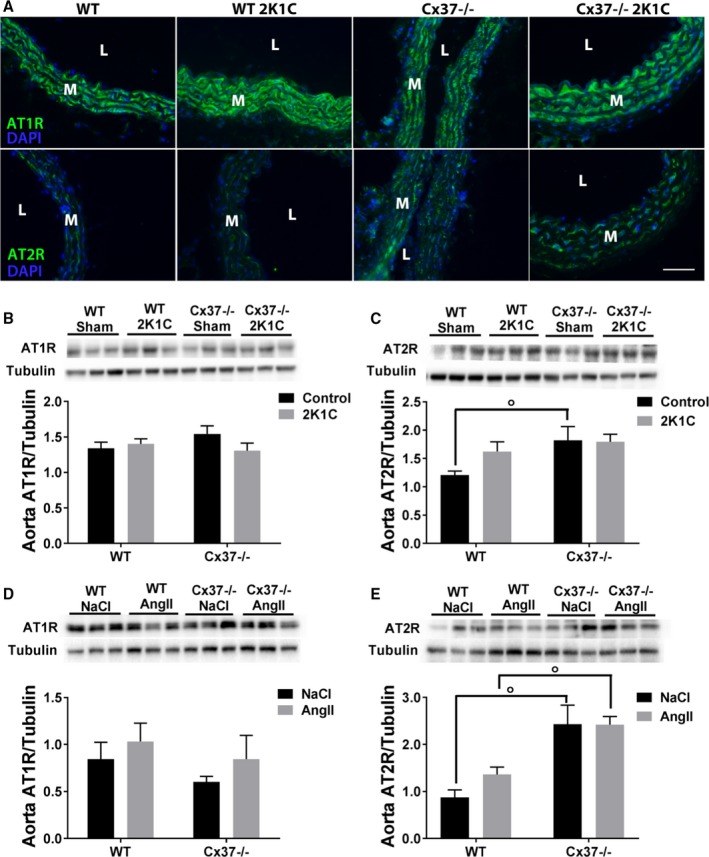
Loss of Cx37 selectively alters the expression of the AT2 receptors in the aortas of Cx37−/− mice. A, Immunofluorescence showed a similar staining for AT1R (upper panels) in the aortas of all WT and Cx37−/− mice. In contrast, the staining for AT2R was more intense in the aortas of Cx37−/− than in WT mice. Bar=20 μm. L indicates lumen; M, media. B, Western blot showed that the aortas of WT and Cx37−/− mice expressed similar levels of the AT1R protein. C, In contrast, the basal levels of the AT2R protein were higher in Cx37−/− than in WT mice, and were not increased after the 2K1C surgery. WT sham N=21; Cx37−/− sham N=17; WT 2K1C N=19; Cx37−/− 2K1C N=17. D and E, Comparable observations were made for both AT1R (D) and AT2R (E) in mice infused daily with 1 mg Ang II/kg body weight WT NaCl N=4; Cx37−/− NaCl, Cx37−/− Ang II N=3, WT Ang II N=6. Results are means+SEM. ° P≤0.05 vs WT mice, as given by 2‐way ANOVA. 2K1C indicates 2‐kidney, 1‐clip; AngII, angiotensin II; AT1R, Ang II type 1 receptors; AT2R, Ang II type 2 receptors; Cx37, Connexin37; DAPI, 4′,6‐diamidino‐2‐phenylindole; Sham, sham‐operated controls; WT, wild type.
The levels of transcripts coding for downstream proteins contributing to the AT2R‐dependent pathway, including the AT2R‐associated protein CNKSR1 (Cnksr1),25 the AT2R downstream tyrosine phosphatase SHP‐1 (Ptpn6),26 and the glutamyl aminopeptidase A (Enpep) converting Ang II to Ang III,27 were also significantly higher in the aortas of Cx37−/− than of WT mice (Figure 5). In contrast, the mRNA expression of several other signaling proteins of the Ang II pathway, including MAS1 (Mas1) the receptor for Ang‐(1–7), which is known to promote vasodilation,28, 29 the chymase (Cma1), which catalyzes the Ang I‐cleavaged to Ang II,30 RGS2 (Rgs2), which negatively regulates the signaling by the G protein subunits of AT1R,31 and DUSP1 (Dusp1),32 an AT2R‐downstream phosphatase involved in the AT2R‐dependent ERK 1/2 dephosphorylation, was unaltered (Figure 7).
Figure 7.
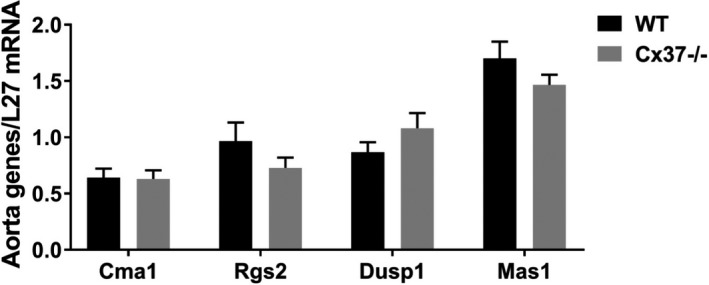
Loss of Cx37 does not alter the expression of several signals involved in the Ang II effects. The levels of transcripts coding for the chymase (Cma1), which converts Ang I to Ang II, the Regulator of G‐protein signaling 2 (Rgs2), which negatively regulates AT1R function by modulating its Gαq subunit activity, the Dual specificity protein phosphatase 1 (Dusp1), an AT2R‐associated phosphatase that contributes to AT2R‐mediated inactivation of the ERK cascade, and Mas1, the receptor for Ang‐(1–7) that promotes vasodilatation, were similar in the aortas of WT and Cx37−/− mice. WT N=9; Cx37−/− N=8. Results are means+SEM. Statistical analysis was performed using t test. Ang indicates angiotensin; AT1R, Ang II type 1 receptors; AT2R, Ang II type 2 receptors; Cx37, Connexin37; WT, wild type.
To decipher whether these modulations altered the Ang II‐mediated response, freshly isolated WT and Cx37−/− mice aortas were exposed to 40 μmol/L Ang II during 2 hours. This exposure increased the phosphorylation levels of the ERK (Figure 8A), MLC2 (Figure 8B), and AKT proteins (Figure 8C) in aortas of WT, but not in those of Cx37−/− mice. Collectively, these data document that the regulation of the Ang II pathway is selectively altered in mice lacking Cx37.
Figure 8.
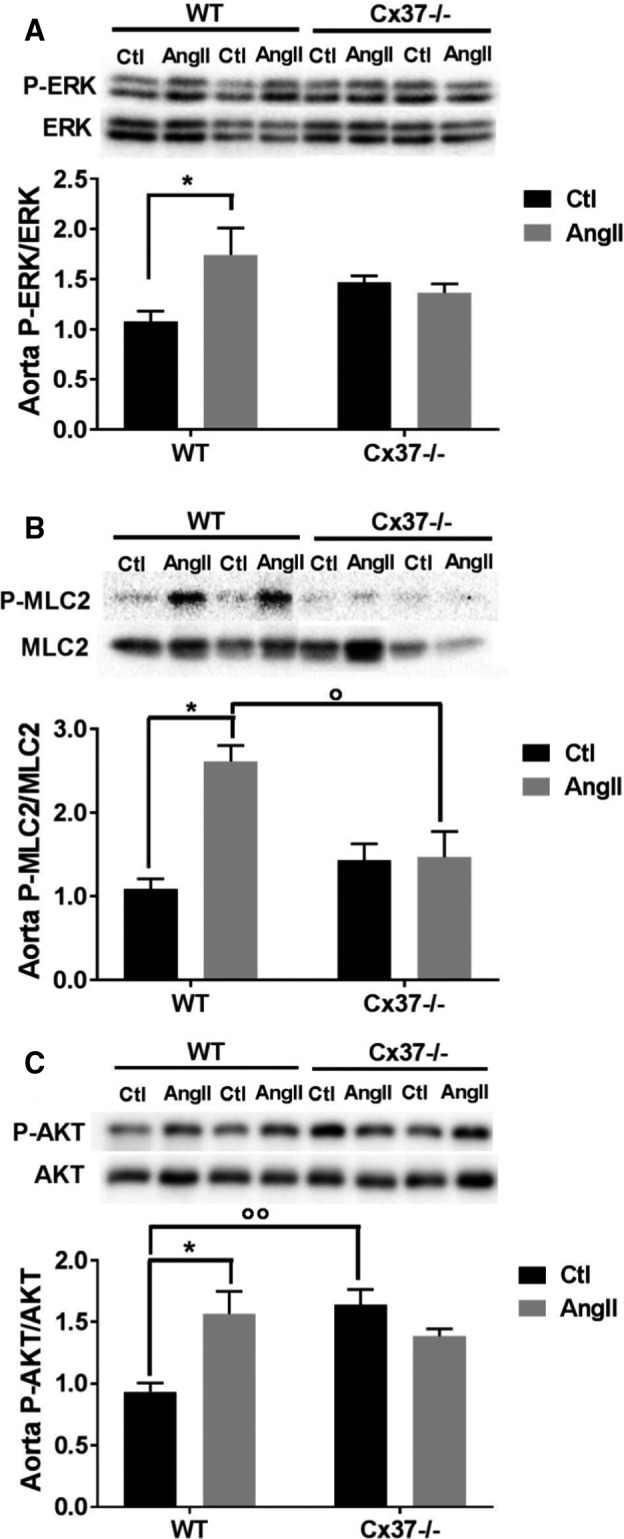
Loss of Cx37−/− alters the activation of downstream key signaling proteins to the Ang II signaling pathway. A through C, After a 2‐hour exposure to 40 μmol/L Ang II, the levels of phosphorylated ERK (A), MLC2 (B), and AKT (C) increased in the aortas of WT but not of Cx37−/− mice. WT Ctl, WT Ang II N=3; Cx37−/− Ctl, Cx37−/− Ang II N=5. Results are means+SEM. *P≤0.05 vs respective control mice and ° P≤0.05, °° P≤0.01 vs WT mice, as given by 2‐way ANOVA. Ang II indicates angiotensin II; Ctl, control; Cx37, Connexin37; MLC2, myosin light chain 2; WT, wild type.
Loss of Cx37 Alters the Basal Expression of Aortic Cx43 and the Expression of Vascular Connexins After the 2K1C Surgery
To determine whether the loss of Cx37 altered the expression of the other vascular connexins, we analyzed aortic extracts by Western blots. We found that the loss of Cx37 did not affect the basal, control levels of Cx40 and Cx45, but significantly increased those of Cx43 (Figure 9). Four weeks after the 2K1C surgery, the levels of Cx37, Cx40, and Cx43 were significantly increased in the aortas of WT mice (Figure 9A through 9C and 9E), whereas those of Cx45 were unchanged (Figure 9D). Under the same conditions, the aortas of Cx37−/− mice shown unchanged levels of Cx40, and decreased levels of Cx43 and Cx45 (Figure 9B through 9D and 9E). The data document that loss of Cx37 specifically affects aortic Cx43, and altered its modulation under conditions of Ang II‐dependent hypertension.
Figure 9.
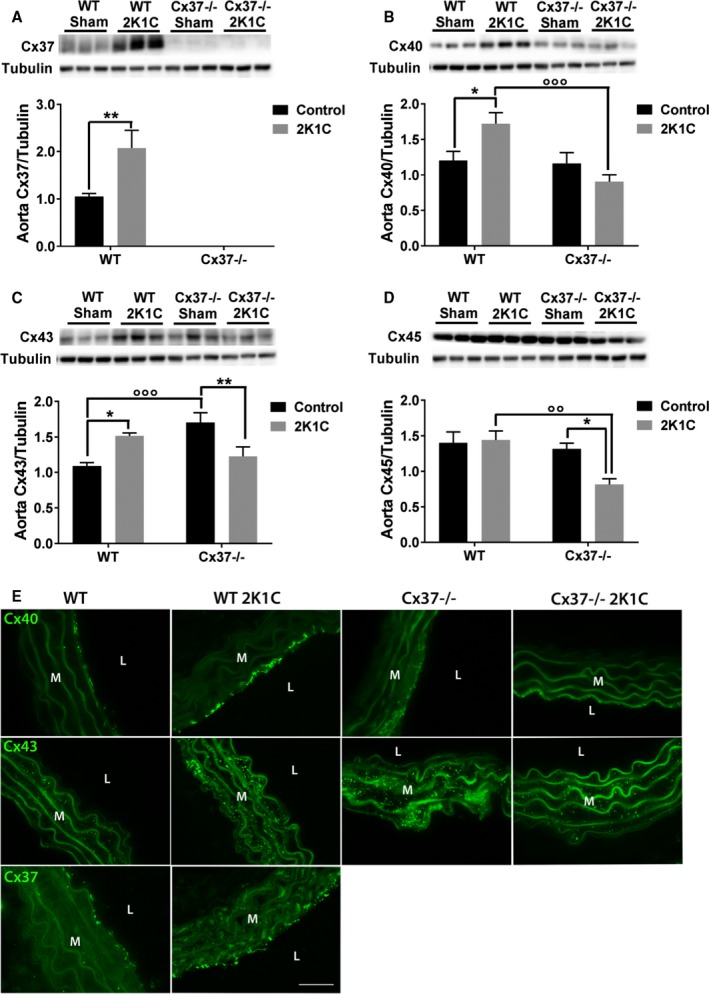
Loss of Cx37 alters the basal expression of aortic Cx43, and protects against the overexpression of Cx40 and Cx45 after the 2K1C procedure. A, Four weeks after the 2K1C procedure, Western blots showed a marked increase of Cx37 levels in the aortas of the WT mice that had become hypertensive. WT sham N=9; Cx37−/− sham N=7; WT 2K1C N=9; Cx37−/− 2K1C N=11. B, The levels of Cx40 were also increased in the aortas of WT made hypertensive, but not in Cx37−/− mice. WT sham N=18; Cx37−/− sham N=15; WT 2K1C N=16; Cx37−/− 2K1C N=14. C, The levels of Cx43 were enhanced in response to a renin‐dependent model of hypertension (WT 2K1C). Cx37‐deficient mice also displayed elevated Cx43 protein levels, which decreased after the 2K1C procedure. WT sham N=17; Cx37−/− sham N=13; WT 2K1C N=14; Cx37−/− 2K1C N=17. D, Comparable levels of Cx45 were evaluated in the aortas of control WT and Cx37−/− mice. After the 2K1C surgery, these levels did not change in the aortas of WT mice, but decreased in those of Cx37−/− mice. WT sham N=9; Cx37−/− sham N=7; WT 2K1C N=9; Cx37−/− 2K1C N=11. Results are means+SEM. *P≤0.05, **P≤0.01 vs respective control mice; °° P≤0.01, °°° P≤0.001 vs WT mice, as given by 2‐way ANOVA. E, After the 2K1C surgery, the immunostaining of Cx40 (upper panels) was increased between the aortic EC of WT, but not of Cx37−/−mice. Analogous changes were seen with regard to the immunostaining of Cx43 (middle panels) between the SMC of the media. The immunostaining of Cx37 (lower panels) increased in the aortic media of WT mice after the 2K1C surgery. L indicates lumen; M, media; Bar=20 μm. 2K1C indicates 2‐kidney, 1‐clip; Cx37, Connexin37; Cx40, Connexin40; Cx43, Connexin43; Cx45, Connexin45; EC, endothelial cells; Sham, sham‐operated controls; SMC, smooth muscle cells; WT, wild type.
Loss of Cx37 Does Not Affect the Expression and Activation of Aortic eNOS
Since Cx37 directly interacts with eNOS,19 we evaluated the aortic levels of this enzyme. Western blots showed that these levels were similar in WT and Cx37−/− mice, under both basal conditions, and 4 weeks after the 2K1C procedure (Figure 10A). Similar observations were made with regard to the active, phosphorylated form of eNOS (Figure 10B). The data do not support a participation of the eNOS pathway in the altered regulation of blood pressure in Cx37−/− 2K1C mice.
Figure 10.
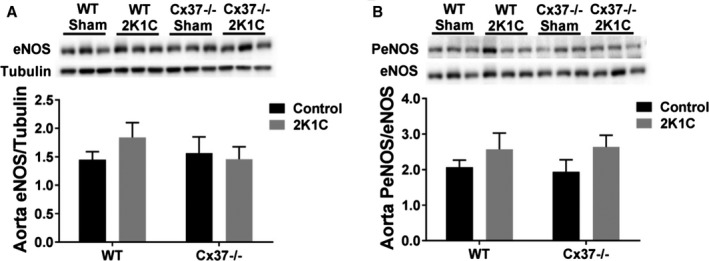
Loss of Cx37 did not alter the expression and activation of aortic eNOS. Western blots showed similar levels of the eNOS protein (A) and of its activated, phosphorylated form (B) in the aortas of all groups of mice. WT sham N=21; Cx37−/− sham N=15; WT 2K1C N=17; Cx37−/− 2K1C N=15. Results are means+SEM. Statistical analysis was performed using 2‐way ANOVA. Cx37 indicates Connexin37; 2K1C, 2‐kidney, 1‐clip; eNOS, endothelial nitric oxide synthase; Sham, sham‐operated controls; WT, wild type.
Discussion
Previous studies have documented the participation of Cx43, Cx40, and Cx37 in different models of hypertension.3, 5, 6, 7, 9 While the role of the 2 former connexins has been understood by multiple studies of different research groups,2, 6, 7, 9, 13, 14, 33 the involvement of the latter has been the topic of fewer studies,6, 7, 8, 15, 34 and remains to be fully validated. Here, we provide direct evidence that Cx37 is selectively involved in the control of renin‐dependent hypertension, via a mechanism that implicates its control of several proteins key to the Ang II‐dependent pathway. Thus, loss of Cx37 did not alter the hypertension induced by a treatment with l‐NAME, but reduced the hypertension induced by either an infusion of Ang II or a 2K1C surgery, 2 conditions that cause a marked renin‐dependent hypertension in control mice.
Our data document that this reduction did not result from a defective renin or eNOS production in the animals lacking Cx37, but involved the specific increased expression of AT2 receptors. In this setting, we observed selective alterations in the signaling pathway elicited by Ang II. Thus, loss of Cx37 did not modify the expression of AT1 receptors, but increased that of AT2 receptors. Previous studies have shown that AT1 receptors are responsible for the vasoconstrictive effect of Ang II, which markedly contributes to hypertension,35, 36, 37, 38, 39 whereas AT2 receptors antagonize this effect by promoting a vasodilation that counteracts hypertension.40, 41, 42, 43, 44 Indeed, it was demonstrated that loss of AT2 receptors in mice resulted in a hypertension that associated with increased vasopressor response to Ang II.45 Accordingly, a SMC‐specific overexpression of AT2R in transgenic mice abolished the vasoconstrictive effect elicited by Ang II.46 These data document for the first time that the upregulated expression of AT2 receptors observed in Cx37−/− mice at least partially accounts for the mitigated hypertension that these mice developed when exposed to either an infusion of Ang II or to the 2K1C surgery.
The molecular mechanism whereby increased AT2R expression may prevent an overt and chronic hypertension remains to be fully elucidated. Although the bradykinin/NO/cGMP vasodilator cascade is the main downstream pathway responsible for the AT2R‐mediated vasodepressor effect, we did not observe altered aortic levels of eNOS and P‐eNOS in Cx37−/− mice. In contrast, we found that Cx37−/− mice displayed an increased expression of the transcripts coding for SHP‐1 (Ptpn6), an important protein tyrosine phosphatase downstream of AT2R and known to induce an SLK‐mediated RhoA phosphorylation, which inhibits RhoA‐dependent arterial contraction and promotes vasodilation.26 Accordingly, we also found that the phosphorylation‐dependent activation of the myosin light chain 2 (MLC2), which is responsible for SMC contraction in arteries, was not induced by Ang II in the aortas of Cx37−/− mice. Consistent with these results, we further document that, after an Ang II stimulation, loss of Cx37 altered the phosphorylation of ERK and AKT, 2 other main downstream targets of AT1 receptors. Other proteins may be involved in the dysregulation of the Ang II pathway, inasmuch as we also found that loss of Cx37 increased the mRNA expression of CNSKR1, the connector enhancer of kinase suppressor of RAS 1, and of glutamyl aminopeptidase A. Full validation of this conclusion now calls for further experiments (eg, in the presence of antagonists and agonists of the AT2 receptors). Similar experiments could similarly target AT1 receptors to exclude a plausible alteration in their sensitivity.
Eventually, our data also revealed that loss of Cx37 is associated with a significant, selective increase in the expression of Cx43 in SMC. This connexin allows for the rapid diffusion of calcium‐dependent contraction waves between SMCs, modulating the vasomotor tone16, 17 which, in turn, is key for the development and maintenance of renin‐ and Ang II‐dependent hypertension.6, 7, 9, 14, 23, 33 The specific role that may be played by this connexin in the context of Cx37−/− mice exposed to hypertensive conditions remains to be elucidated in future studies. Even though the expression of Cx37, Cx43, and Cx405, 6, 8, 9, 12, 47 is usually coordinated, a contribution of the latter connexin appears unlikely in this context, inasmuch as Cx40 is not expressed in the SMCs of either normotensive or hypertensive mice,3, 6, 7, 9, 10, 14 and its vascular levels were not altered after loss of Cx37−/−.8
In summary, our findings document that loss of Cx37 partly protects against the development of Ang II‐dependent hypertension, because of a selective increase in the expression of AT2R and of some of the downstream effectors involved in the signaling elicited by this form of Ang II receptors, notably SHP‐1. Even though the molecular mechanism whereby the loss of Cx37 induces these alterations remains to be fully unraveled, these findings highlight a so far unsuspected, specific role of Cx37 in the control of the Ang II signaling pathway, setting the first, obligatory in vivo basis to now envisage further analyses to fine‐tune the mechanistic scenario drafted here.
Source of Funding
This work was supported by grants from the Swiss National Science Foundation (31003A‐175452/1), the Swiss Cancer League (KFS‐3796‐02‐2016), the Octav and the Marcella Botnar Foundation, and the Novartis Foundation to Haefliger.
Disclosures
None.
Acknowledgments
We acknowledge the Mouse Pathology Facility, as well as the CAF/EMIF platform of Lausanne University for their assistance in blood pressure measurements and the 2K1C surgery procedure.
(J Am Heart Assoc. 2019;8:e010823 DOI: 10.1161/JAHA.118.010823.)
References
- 1. Bosco D, Haefliger JA, Meda P. Connexins: key mediators of endocrine function. Physiol Rev. 2011;91:1393–1445. [DOI] [PubMed] [Google Scholar]
- 2. Kurtz A. Connexins, renin cell displacement and hypertension. Curr Opin Pharmacol. 2015;21:1–6. [DOI] [PubMed] [Google Scholar]
- 3. Meda P, Haefliger JA. Connexins and pannexins: from biology towards clinical targets. Swiss Med Wkly. 2016;146:w14365. [DOI] [PubMed] [Google Scholar]
- 4. Meda P. Gap junction proteins are key drivers of endocrine function. Biochim Biophys Acta. 2018;1860:124–140. [DOI] [PubMed] [Google Scholar]
- 5. Alonso F, Boittin FX, Beny JL, Haefliger JA. Loss of connexin40 is associated with decreased endothelium‐dependent relaxations and eNOS levels in the mouse aorta. Am J Physiol Heart Circ Physiol. 2010;299:H1365–H1373. [DOI] [PubMed] [Google Scholar]
- 6. Alonso F, Krattinger N, Mazzolai L, Simon A, Waeber G, Meda P, Haefliger JA. An angiotensin II‐ and NF‐kappaB‐dependent mechanism increases connexin 43 in murine arteries targeted by renin‐dependent hypertension. Cardiovasc Res. 2010;87:166–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Le Gal L, Alonso F, Wagner C, Germain S, Haefliger DN, Meda P, Haefliger JA. Restoration of connexin 40 (Cx40) in renin‐producing cells reduces the hypertension of Cx40 null mice. Hypertension. 2014;63:1198–1204. [DOI] [PubMed] [Google Scholar]
- 8. Meens MJ, Alonso F, Le Gal L, Kwak BR, Haefliger JA. Endothelial connexin37 and connexin40 participate in basal but not agonist‐induced NO release. Cell Commun Signal. 2015;13:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Le Gal L, Alonso F, Mazzolai L, Meda P, Haefliger JA. Interplay between connexin40 and nitric oxide signaling during hypertension. Hypertension. 2015;65:910–915. [DOI] [PubMed] [Google Scholar]
- 10. Haefliger JA, Allagnat F, Hamard L, Le Gal L, Meda P, Nardelli‐Haefliger D, Genot E, Alonso F. Targeting Cx40 (connexin40) expression or function reduces angiogenesis in the developing mouse retina. Arterioscler Thromb Vasc Biol. 2017;37:2136–2146. [DOI] [PubMed] [Google Scholar]
- 11. Haefliger JA, Nicod P, Meda P. Contribution of connexins to the function of the vascular wall. Cardiovasc Res. 2004;62:345–356. [DOI] [PubMed] [Google Scholar]
- 12. Allagnat F, Dubuis C, Lambelet M, Le Gal L, Alonso F, Corpataux JM, Deglise S, Haefliger JA. Connexin37 reduces smooth muscle cell proliferation and intimal hyperplasia in a mouse model of carotid artery ligation. Cardiovasc Res. 2017;113:805–816. [DOI] [PubMed] [Google Scholar]
- 13. Wagner C, de Wit C, Kurtz L, Grunberger C, Kurtz A, Schweda F. Connexin40 is essential for the pressure control of renin synthesis and secretion. Circ Res. 2007;100:556–563. [DOI] [PubMed] [Google Scholar]
- 14. Krattinger N, Capponi A, Mazzolai L, Aubert JF, Caille D, Nicod P, Waeber G, Meda P, Haefliger JA. Connexin40 regulates renin production and blood pressure. Kidney Int. 2007;72:814–822. [DOI] [PubMed] [Google Scholar]
- 15. Wagner C, Kurtz L, Schweda F, Simon AM, Kurtz A. Connexin 37 is dispensable for the control of the renin system and for positioning of renin‐producing cells in the kidney. Pflugers Arch. 2009;459:151–158. [DOI] [PubMed] [Google Scholar]
- 16. Halidi N, Alonso F, Burt JM, Beny JL, Haefliger JA, Meister JJ. Intercellular calcium waves in primary cultured rat mesenteric smooth muscle cells are mediated by connexin43. Cell Commun Adhes. 2012;19:25–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Quijano JC, Raynaud F, Nguyen D, Piacentini N, Meister JJ. Intercellular ultrafast Ca(2+) wave in vascular smooth muscle cells: numerical and experimental study. Sci Rep. 2016;6:31271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kurtz L, Janssen‐Bienhold U, Kurtz A, Wagner C. Connexin expression in renin‐producing cells. J Am Soc Nephrol. 2009;20:506–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pfenniger A, Derouette JP, Verma V, Lin X, Foglia B, Coombs W, Roth I, Satta N, Dunoyer‐Geindre S, Sorgen P, Taffet S, Kwak BR, Delmar M. Gap junction protein Cx37 interacts with endothelial nitric oxide synthase in endothelial cells. Arterioscler Thromb Vasc Biol. 2010;30:827–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Simon AM, McWhorter AR. Decreased intercellular dye‐transfer and downregulation of non‐ablated connexins in aortic endothelium deficient in connexin37 or connexin40. J Cell Sci. 2003;116:2223–2236. [DOI] [PubMed] [Google Scholar]
- 21. Shen M, Lee J, Basu R, Sakamuri SS, Wang X, Fan D, Kassiri Z. Divergent roles of matrix metalloproteinase 2 in pathogenesis of thoracic aortic aneurysm. Arterioscler Thromb Vasc Biol. 2015;35:888–898. [DOI] [PubMed] [Google Scholar]
- 22. Shen M, Morton J, Davidge ST, Kassiri Z. Loss of smooth muscle cell disintegrin and metalloproteinase 17 transiently suppresses angiotensin II‐induced hypertension and end‐organ damage. J Mol Cell Cardiol. 2017;103:11–21. [DOI] [PubMed] [Google Scholar]
- 23. Allagnat F, Haefliger JA, Lambelet M, Longchamp A, Berard X, Mazzolai L, Corpataux JM, Deglise S. Nitric oxide deficit drives intimal hyperplasia in mouse models of hypertension. Eur J Vasc Endovasc Surg. 2016;51:733–742. [DOI] [PubMed] [Google Scholar]
- 24. Gerl M, Vockl J, Kurt B, van Veen TA, Kurtz A, Wagner C. Inducible deletion of connexin 40 in adult mice causes hypertension and disrupts pressure control of renin secretion. Kidney Int. 2015;87:557–563. [DOI] [PubMed] [Google Scholar]
- 25. Castrop H. Angiotensin receptor‐associated proteins: local modulators of the renin‐angiotensin system. Pflugers Arch. 2013;465:111–119. [DOI] [PubMed] [Google Scholar]
- 26. Guilluy C, Rolli‐Derkinderen M, Loufrani L, Bourge A, Henrion D, Sabourin L, Loirand G, Pacaud P. Ste20‐related kinase SLK phosphorylates Ser188 of RhoA to induce vasodilation in response to angiotensin II Type 2 receptor activation. Circ Res. 2008;102:1265–1274. [DOI] [PubMed] [Google Scholar]
- 27. Yang Y, Liu C, Lin YL, Li F. Structural insights into central hypertension regulation by human aminopeptidase A. J Biol Chem. 2013;288:25638–25645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fraga‐Silva RA, Ferreira AJ, Dos Santos RA. Opportunities for targeting the angiotensin‐converting enzyme 2/angiotensin‐(1‐7)/Mas receptor pathway in hypertension. Curr Hypertens Rep. 2013;15:31–38. [DOI] [PubMed] [Google Scholar]
- 29. Santos RAS, Sampaio WO, Alzamora AC, Motta‐Santos D, Alenina N, Bader M, Campagnole‐Santos MJ. The ACE2/angiotensin‐(1‐7)/MAS axis of the renin‐angiotensin system: focus on angiotensin‐(1‐7). Physiol Rev. 2018;98:505–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ju H, Gros R, You X, Tsang S, Husain M, Rabinovitch M. Conditional and targeted overexpression of vascular chymase causes hypertension in transgenic mice. Proc Natl Acad Sci USA. 2001;98:7469–7474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hercule HC, Tank J, Plehm R, Wellner M, da Costa Goncalves AC, Gollasch M, Diedrich A, Jordan J, Luft FC, Gross V. Regulator of G protein signalling 2 ameliorates angiotensin II‐induced hypertension in mice. Exp Physiol. 2007;92:1014–1022. [DOI] [PubMed] [Google Scholar]
- 32. Calo LA, Schiavo S, Davis PA, Pagnin E, Mormino P, D'Angelo A, Pessina AC. Angiotensin II signaling via type 2 receptors in a human model of vascular hyporeactivity: implications for hypertension. J Hypertens. 2010;28:111–118. [DOI] [PubMed] [Google Scholar]
- 33. Haefliger JA, Krattinger N, Martin D, Pedrazzini T, Capponi A, Doring B, Plum A, Charollais A, Willecke K, Meda P. Connexin43‐dependent mechanism modulates renin secretion and hypertension. J Clin Invest. 2006;116:405–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Boittin FX, Alonso F, Le Gal L, Allagnat F, Beny JL, Haefliger JA. Connexins and M3 muscarinic receptors contribute to heterogeneous Ca(2+) signaling in mouse aortic endothelium. Cell Physiol Biochem. 2013;31:166–178. [DOI] [PubMed] [Google Scholar]
- 35. Berry C, Touyz R, Dominiczak AF, Webb RC, Johns DG. Angiotensin receptors: signaling, vascular pathophysiology, and interactions with ceramide. Am J Physiol Heart Circ Physiol. 2001;281:H2337–H2365. [DOI] [PubMed] [Google Scholar]
- 36. Crowley SD, Tharaux PL, Audoly LP, Coffman TM. Exploring type I angiotensin (AT1) receptor functions through gene targeting. Acta Physiol Scand. 2004;181:561–570. [DOI] [PubMed] [Google Scholar]
- 37. Oliverio MI, Coffman TM. Angiotensin‐II‐receptors: new targets for antihypertensive therapy. Clin Cardiol. 1997;20:3–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cervenka L, Horacek V, Vaneckova I, Hubacek JA, Oliverio MI, Coffman TM, Navar LG. Essential role of AT1A receptor in the development of 2K1C hypertension. Hypertension. 2002;40:735–741. [DOI] [PubMed] [Google Scholar]
- 39. Griendling KK, Ushio‐Fukai M, Lassegue B, Alexander RW. Angiotensin II signaling in vascular smooth muscle. New concepts. Hypertension. 1997;29:366–373. [DOI] [PubMed] [Google Scholar]
- 40. Chen D, Coffman TM. AT1 angiotensin receptors—vascular and renal epithelial pathways for blood pressure regulation. Curr Opin Pharmacol. 2015;21:122–126. [DOI] [PubMed] [Google Scholar]
- 41. Siragy HM, Xue C, Abadir P, Carey RM. Angiotensin subtype‐2 receptors inhibit renin biosynthesis and angiotensin II formation. Hypertension. 2005;45:133–137. [DOI] [PubMed] [Google Scholar]
- 42. Carey RM. Angiotensin type‐2 receptors and cardiovascular function: are angiotensin type‐2 receptors protective? Curr Opin Cardiol. 2005;20:264–269. [DOI] [PubMed] [Google Scholar]
- 43. Batenburg WW, Tom B, Schuijt MP, Danser AH. Angiotensin II type 2 receptor‐mediated vasodilation. Focus on bradykinin, NO and endothelium‐derived hyperpolarizing factor(s). Vascul Pharmacol. 2005;42:109–118. [DOI] [PubMed] [Google Scholar]
- 44. Duke LM, Evans RG, Widdop RE. AT2 receptors contribute to acute blood pressure‐lowering and vasodilator effects of AT1 receptor antagonism in conscious normotensive but not hypertensive rats. Am J Physiol Heart Circ Physiol. 2005;288:H2289–H2297. [DOI] [PubMed] [Google Scholar]
- 45. Ichiki T, Labosky PA, Shiota C, Okuyama S, Imagawa Y, Fogo A, Niimura F, Ichikawa I, Hogan BL, Inagami T. Effects on blood pressure and exploratory behaviour of mice lacking angiotensin II type‐2 receptor. Nature. 1995;377:748–750. [DOI] [PubMed] [Google Scholar]
- 46. Tsutsumi Y, Matsubara H, Masaki H, Kurihara H, Murasawa S, Takai S, Miyazaki M, Nozawa Y, Ozono R, Nakagawa K, Miwa T, Kawada N, Mori Y, Shibasaki Y, Tanaka Y, Fujiyama S, Koyama Y, Fujiyama A, Takahashi H, Iwasaka T. Angiotensin II type 2 receptor overexpression activates the vascular kinin system and causes vasodilation. J Clin Invest. 1999;104:925–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fang JS, Angelov SN, Simon AM, Burt JM. Cx40 is required for, and Cx37 limits, postischemic hindlimb perfusion, survival and recovery. J Vasc Res. 2012;49:2–12. [DOI] [PMC free article] [PubMed] [Google Scholar]