

Abstract
Background
We have previously shown that ATRAP (angiotensin II receptor–associated protein; Agtrap) interacts with AT1R (angiotensin II type 1 receptor) and promotes constitutive internalization of AT1R so as to inhibit hyperactivation of its downstream signaling. In response to angiotensin II, systemic ATRAP deficiency exacerbates angiotensin II–mediated hypertension via hyperactivation of renal tubular AT1R. Although ATRAP expression is abundant in renal proximal tubules, little is known about the actual function of renal proximal tubule ATRAP in angiotensin‐mediated hypertension.
Methods and Results
In this study, we examined the in vivo functional role of renal proximal tubule ATRAP in angiotensin‐dependent hypertension. We succeeded in generating proximal tubule–specific ATRAP knockout (PT‐KO) mice for the first time using the Cre/loxP system with Pepck‐Cre. Detailed analysis of renal ATRAP expression in PT‐KO mice estimated by immunohistochemical and laser‐capture microdissection analysis revealed that ATRAP mRNA expression decreased by ≈80% in proximal regions of the nephron in PT‐KO mice compared with wild‐type (WT) mice. We compared blood pressure of PT‐KO and WT mice using both tail‐cuff and radiotelemetric methods. Blood pressure of PT‐KO mice was comparable with that of WT mice at baseline. Moreover, no significant differences were noted in pressor response to angiotensin II (600 ng/kg per min or 1000 ng/kg per minute) infusion between PT‐KO and WT mice. In addition, angiotensin II–mediated cardiac hypertrophy was identical between PT‐KO and WT mice.
Conclusions
ATRAP deficiency in proximal tubules did not exacerbate angiotensin‐dependent hypertension in vivo. The results indicate that renal proximal tubule ATRAP has a minor role in angiotensin‐dependent hypertension in vivo.
Keywords: hypertension, kidney, renin–angiotensin system
Subject Categories: Hypertension, ACE/Angiotension Receptors/Renin Angiotensin System, Nephrology and Kidney
Clinical Perspective
What Is New?
Our previous investigation revealed that by regulating renal sodium reabsorption, ATRAP (angiotensin II receptor–associated protein; Agtrap) exerts negative regulatory effects as an endogenous compensatory system to suppress angiotensin‐mediated hypertension in vivo; however, little is known about the function of renal proximal tubule ATRAP in angiotensin‐mediated hypertension.
In this study, we investigated the in vivo functional role of renal proximal tubule ATRAP in angiotensin‐dependent hypertension.
ATRAP deficiency in proximal tubules does not exacerbate angiotensin‐dependent hypertension, which indicates that ATRAP in renal proximal tubules has a minor role in angiotensin‐dependent hypertension.
What Are the Clinical Implications?
The findings of this study and our previous studies suggest that the progression of hypertension could be ameliorated by therapeutic activation of ATRAP, especially in the renal distal nephron segments but not in proximal nephron segments.
Introduction
Accumulating evidence shows that activation of the tissue renin–angiotensin system through AT1R (angiotensin II type 1 receptor) plays a pivotal role in the pathogenesis of hypertension and associated end‐organ injury. In kidneys, AT1R signaling plays a key role in altered renal sodium handling: hyperactivation of renal AT1R signaling can evoke excessive sodium retention, resulting in hypertension.1, 2, 3 The carboxyl‐terminal domain of AT1R is involved in control of AT1R internalization independent of G‐protein coupling.4, 5 It plays an important role in linking receptor‐mediated signal transduction to the specific biological response to angiotensin II (Ang II). ATRAP (AT1R‐associated protein; Agtrap) has been identified as a specific binding protein of the carboxyl‐terminal domain of AT1R.6, 7 Previous studies have shown that ATRAP promotes constitutive internalization of AT1R and functions as an endogenous inhibitor that suppresses AT1R hyperactivation at local tissue sites.7, 8, 9, 10, 11, 12 Endogenous ATRAP is most abundantly expressed in the kidney, where it is highly expressed in tubular epithelial cells in proximal and distal tubules but only faintly expressed in glomeruli.13, 14 Our previous investigation using systemic ATRAP knockout (KO) mice revealed that ATRAP suppresses angiotensin‐dependent hypertension by regulating renal sodium reabsorption in vivo.15 In our previous study, systemic ATRAP KO mice exhibited exacerbation of Ang II‐induced hypertension compared with wild‐type (WT) mice, concomitant with increased sodium retention in an aldosterone‐independent manner. However, it is unknown which renal tubular ATRAP plays a major role in angiotensin‐dependent hypertension in vivo. Endogenous ATRAP is most abundantly expressed in proximal tubules in the kidney. Consequently, we examined whether ATRAP deficiency in proximal tubules exacerbates Ang II‐induced hypertension. To this end, we generated proximal tubule–specific ATRAP KO (PT‐KO) mice in this study. Furthermore, we compared blood pressure (BP) in response to Ang II treatment in WT mice and PT‐KO mice.
Materials and Methods
Data, analytical methods, and study materials will be made available by the corresponding author on reasonable request from other researchers for purposes of reproducing the results or replicating the procedure. This study was performed in accordance with the National Institutes of Health guidelines for the use of experimental animals. All animal studies were reviewed and approved by the animal studies committee of Yokohama City University.
Generation of PT‐KO Mice
The generation of floxed ATRAP (Agtrap fl/fl) mice has previously been described in detail previously.16 Agtrap fl/fl mice were intercrossed with Pepck‐Cre transgenic mice expressing Cre recombinase under control of the mouse PEPCK (phosphoenolpyruvate carboxykinase) promoter. The PEPCK promoter is reported to have high specificity for proximal tubules in the kidney.17 Pepck‐Cre transgenic mice were kindly provided by Dr Volker Haase at Vanderbilt University School of Medicine. The resulting Pepck‐Cre +/Agtrap fl/− mice were mated with Agtrap fl/fl mice to generate Pepck‐Cre +/Agtrap fl/fl mice as PT‐KO mice. Homozygous mice without Cre (Agtrap fl/fl) served as WT mice. All mice were housed under a 12/12‐hour day/night cycle at a temperature of 25°C. They were fed a standard diet (0.3% NaCl) throughout the current study.
Ang II Treatment
Ang II infusion was performed as described previously.12, 18 Briefly, Ang II (600 or 1000 ng/kg per minute) was infused subcutaneously in male WT and PT‐KO mice (9–12 weeks old) for 14 days using an osmotic minipump (model 2002; ALZET).
BP and Heart Rate Measurements by a Tail‐Cuff Method
Systolic BP and heart rate were measured using a tail‐cuff method (BP monitor MK‐2000; Muromachi Kikai Co). The MK‐2000 monitor enabled measurement of BP without preheating the animals and using anesthesia. This procedure avoided this stressful condition, as described previously.11, 12, 19 All measurements were performed once between 10 am and 2 pm, and at least 8 values were taken for each measurement in a blinded manner.
BP Measurements by a Radiotelemetric Method
Direct BP measurement was performed by a radiotelemetric method using a BP transducer (model TA11PA‐C10; Data Science International), as described previously.12, 15, 18 Seven days after transplantation, when the circadian rhythm was restored, Ang II (1000 ng/kg per minute) was infused into the mice for 14 days. Systolic and diastolic BPs were recorded every 5 minutes using the software Ponemah 6.3 (Data Science International).
Metabolic Cage Analysis
Metabolic cage analysis was performed as described previously.15, 18 Briefly, the PT‐KO and WT mice were acclimated to metabolic cages (Techniplast) for 3 days. After an additional 2 days of baseline monitoring, Ang II (1000 ng/kg per minute) was continuously infused subcutaneously into the mice for 14 days. Body weight, food intake, and water intake were measured daily, and urine was collected. Mice were given free access to water and fed a normal diet (0.3% NaCl). Daily food intake was multiplied by sodium concentration in food, and the product was considered as daily sodium intake. Urine sodium concentration was assessed using an autoanalyzer (Hitachi 7170). Daily urine volume was multiplied by sodium concentration in urine, and the product was considered as daily sodium excretion. We calculated differences between daily sodium intake and excretion, and the amount of the differences during Ang II infusion was considered as cumulative sodium balance.
Real‐Time Quantitative Polymerase Chain Reaction Analysis
Total RNA was extracted from the kidney with ISOGEN (Nippon Gene), and cDNA was synthesized using the SuperScript III First‐Strand System (Invitrogen). Real‐time quantitative polymerase chain reaction was performed by incubating reverse transcription product with the TaqMan Universal PCR Master Mix and designed TaqMan probe (Applied Biosystems), as described previously.18, 19 The mRNA levels were normalized to 18S rRNA control.
Histological and Immunohistochemical Analysis
Histological analysis was performed as described previously.14, 15 Renal tissue from mice was fixed with 4% paraformaldehyde and subsequently embedded in paraffin. Sections (4‐μm thick) were stained with periodic acid‐Schiff and Masson's trichrome. Immunohistochemistry was performed as described previously.13, 14, 18 Sections of 4‐μm thickness were dewaxed and rehydrated, and antigen retrieval was performed by microwave heating. The sections were blocked for endogenous biotin activity using peroxidase blocking reagent (DAKO) and treated for 60 minutes with 10% normal goat serum in phosphate‐buffered saline. The sections were then incubated with one of the following: anti‐ATRAP antibody (diluted at 1:100), anti–calbindin D‐28K antibody (C9848 [Sigma–Aldrich]; diluted at 1:100), or anti–megalin antibody (NB110‐96417 [Novus Biologicals]; diluted at 1:100). The characterization and specificity of the anti–mouse ATRAP antibody was described previously in detail.14, 18, 20
Laser‐Capture Microdissection and Subsequent Reverse Transcription Quantitative Polymerase Chain Reaction Analysis
Laser‐capture microdissection (LMD) was performed using a Leica LMD system (LMD6000), as described previously.18 Briefly, formalin‐fixed paraffin‐embedded tissues were cut into 10‐μm‐thick sections, mounted on polyethylene terepthalate membrane slides, and stained with hematoxylin–eosin. Next, proximal or distal tubules in the renal cortex were microdissected using an LMD6000 microscope. The renal proximal tubule cells were identified via their unique brush border membranes.21 The other tubule cells in the renal cortex were recognized as distal tubule cells (Figure 1B). In total, 60 areas (≈500 000 μm2) of proximal or distal tubules were microdissected from the renal cortex per mouse in each group (6 mice in each group). Total RNA was extracted from microdissected tissue using the RNeasy FFPE Kit (Qiagen). The cDNA was synthesized using the SuperScript III First‐Strand System (Invitrogen) and applied to Taqman reverse transcription quantitative polymerase chain reaction analysis.
Figure 1.
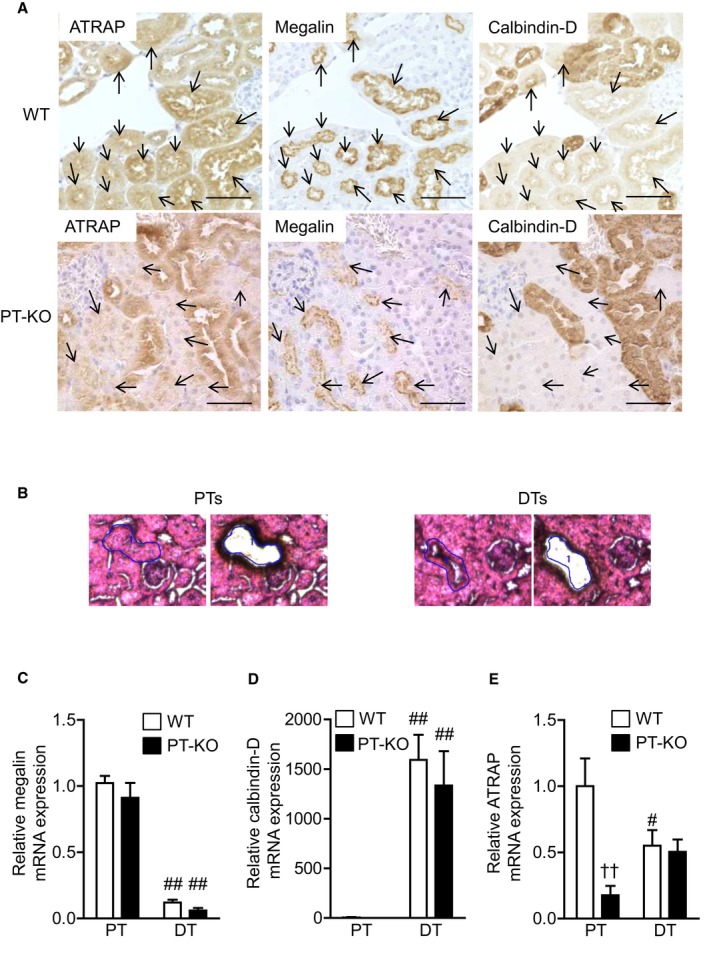
Expression and localization of ATRAP in PT‐KO and WT mice. A, Renal cortical sections showing ATRAP expression in renal tubules detected by anti‐ATRAP antibody (left panels). Consecutive sections were stained with a monoclonal antibody against megalin (middle panels), a specific marker of PTs, and a monoclonal antibody against calbindin‐D (right panels), a specific marker of distal convoluted tubule and connecting tubule. Arrows indicate PTs. Original magnification: ×400. B, Representative image of a hematoxylin–eosin‐stained section of Proximal tubules (PTs) and Distal tubules (DTs) in the renal cortex before and after laser‐capture microdissection (LMD). The renal PT cells were recognized by their distinct brush border membranes and circled (left). The other tubule cells were recognized as DT cells. Original magnification: ×400. Quantitative analysis of megalin (C), calbindin‐D (D), and ATRAP (E) mRNA expression in PTs and DTs of the renal cortex identified by the LMD method. Values are normalized relative to 18S rRNA control levels and expressed relative to mRNA levels in PTs of WT mice. Values are expressed as mean±SE (6 mice in each group). # P<0.05, ## P<0.01 vs PTs. †† P<0.01 vs WT mice. ATRAP indicates angiotensin II receptor–associated protein; DT, distal tubule; PT, proximal tubule; PT‐KO, proximal tubule–specific ATRAP knockout; WT, wild type.
Statistical Analysis
Statistical analysis was performed using GraphPad Prism software. All quantitative data are expressed as mean±SE. Differences were analyzed as follows. A 2‐way repeated measures ANOVA was used to test for differences over time (Figures 2A, 2C, 2E and 4A, 4C). A 2‐factor ANOVA followed by a Bonferroni multiple comparison test was used to test for differences between proximal and distal tubules within each genotype or vehicle‐infused group and Ang II–infused group within each genotype or differences between the WT and PT‐KO mice with the same treatment (Figures 1C through 1E, 2B, 2D, 2F, 3A through 3C, and 4B, 4D). An unpaired Student t test was used to test for differences between the WT and PT‐KO mice (Figure 4E). P<0.05 was considered statistically significant.
Figure 2.
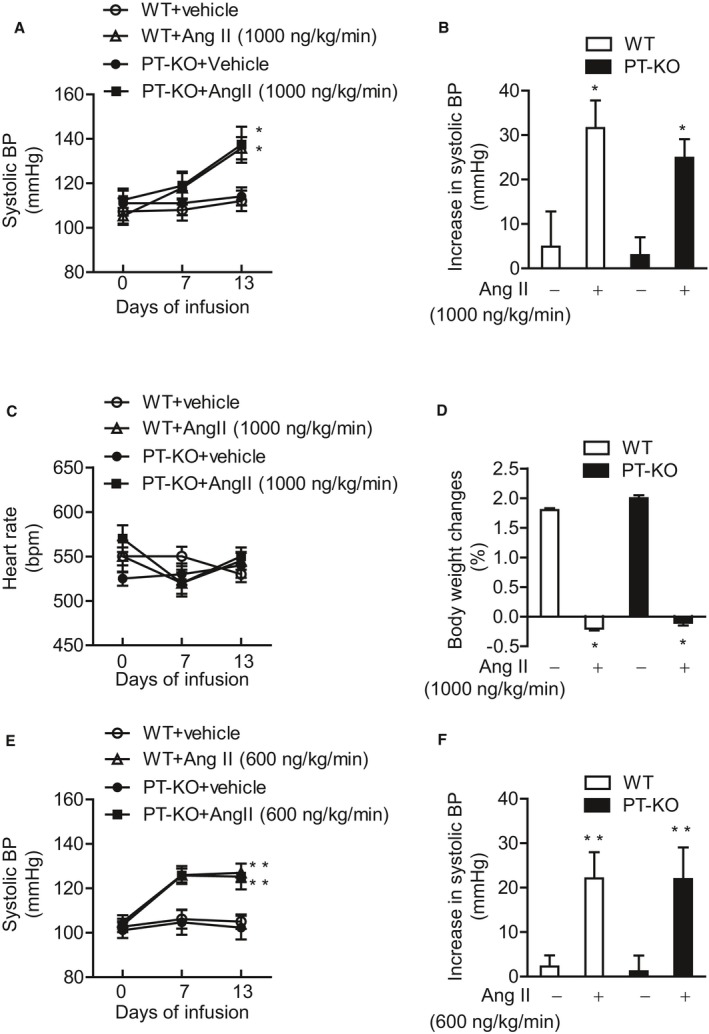
Effect of Ang II infusion (1000 ng/kg per minute) on indirect BP, heart rate, and body weight in WT and PT‐KO mice, and effect of a lower dose of Ang II infusion (600 ng/kg per minute) on indirect BP in WT and PT‐KO mice. A, Effect of Ang II (1000 ng/kg per minute) infusion on systolic BP in WT and PT‐KO mice using a tail‐cuff method. Values are expressed as mean±SE (7–8 mice in each group). B, Increase in systolic BP during 2 weeks of vehicle or Ang II (1000 ng/kg per minute) infusion in WT and PT‐KO mice. Values are expressed as mean±SE (7–8 mice in each group). C, Effect of Ang II (1000 ng/kg per minute) infusion on heart rate in WT and PT‐KO mice. Values are expressed as mean±SE (7–8 mice in each group). D, Body weight change during 2 weeks of vehicle or Ang II (1000 ng/kg per minute) infusion in WT and PT‐KO mice. Values are expressed as mean±SE (7–8 mice in each group). E, Effect of Ang II (600 ng/kg per minute) infusion on systolic BP in WT and PT‐KO mice using a tail‐cuff method. Values are expressed as mean±SE (6 mice in each group). F, Increase in systolic BP during 2 weeks of vehicle or Ang II (600 ng/kg per minute) infusion in WT and PT‐KO mice. Values are expressed as mean±SE (6 mice in each group). *P<0.05 vs vehicle‐infused group. ** P<0.01 vs vehicle‐infused group. Ang II indicates angiotensin II; BP, blood pressure; PT‐KO, proximal tubule–specific ATRAP (angiotensin II receptor–associated protein) knockout; WT, wild type.
Figure 4.
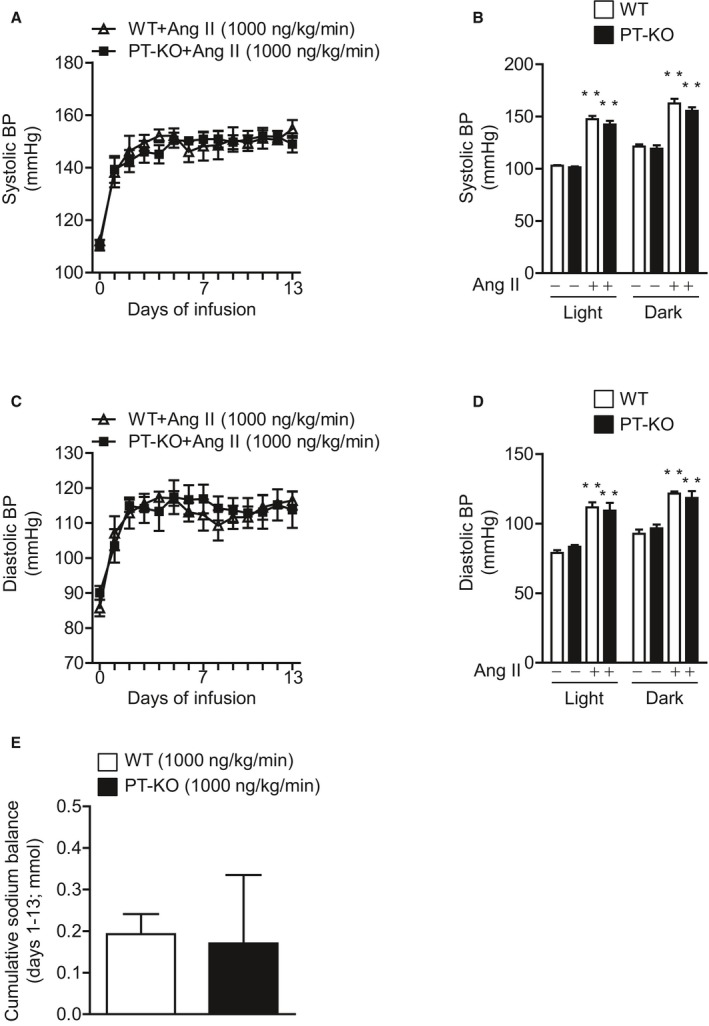
Effect of Ang II on direct BP and cumulative sodium balance in WT and PT‐KO mice. The 24‐hour mean systolic BP (A) and diastolic BP (C) during 2 weeks of Ang II (1000 ng/kg per minute) infusion using a radiotelemetric method are shown. Values are expressed as mean±SE (6 mice in each group). Mean systolic BP (B) and diastolic BP (D) before and after 2 weeks of Ang II (1000 ng/kg per minute) infusion in the light and dark periods. Values are expressed as mean±SE (6 mice in each group). **P<0.01, vs before Ang II infusion. E, The cumulative positive sodium balance during 2 weeks of Ang II (1000 ng/kg per minute) infusion. Values are expressed as mean±SE (6 mice in each group). Ang II indicates angiotensin II; BP, blood pressure; PT‐KO, proximal tubule–specific ATRAP (angiotensin II receptor–associated protein) knockout; WT, wild type.
Figure 3.

Effect of Ang II infusion on cardiac hypertrophy, kidney weight/body weight ratio, renal KIM1 (kidney injury molecule 1) mRNA, and renal histology in WT and PT‐KO mice. A, Effect of 2 weeks of Ang II (1000 ng/kg per minute) infusion on heart weight/body weight ratio in WT and PT‐KO mice. Values are expressed as mean±SE (7–8 mice in each group). **P<0.01 vs vehicle‐infused group. B, Effect of 2 weeks of Ang II (1000 ng/kg per minute) infusion on kidney weight/body weight ratio in WT and PT‐KO mice. Values are expressed as mean±SE (7–8 mice in each group). C, Quantitative analysis of KIM1 mRNA expression in kidneys. Values are normalized relative to 18S rRNA control levels and expressed relative to KIM1 mRNA in vehicle‐infused WT mice. Values are expressed as mean±SE (7–8 mice in each group). D, Representative images of sections of kidneys from WT and PT‐KO mice stained with periodic acid‐Schiff (top; original magnification, ×400; bar, 50 μm) and Masson's trichrome (bottom; original magnification, ×200; bar, 100 μm). Ang II indicates angiotensin II; PT‐KO, proximal tubule–specific ATRAP (angiotensin II receptor–associated protein) knockout; WT, wild type.
Results
Generation of PT‐KO Mice
To investigate the functional role of renal proximal tubule ATRAP in angiotensin‐dependent hypertension, we generated PT‐KO mice using the Cre/loxP system and Pepck‐Cre. To determine expression and distribution of ATRAP in the kidneys of PT‐KO mice, we performed immunohistochemical examination using anti‐ATRAP antibody and antibodies against specific nephron markers. Specifically, we used a monoclonal antibody against calbindin‐D, a calcium‐binding protein expressed primarily in distal tubules, and a monoclonal antibody against megalin, which is specifically expressed in the apical side of proximal tubules. Consecutive sections stained with specific antibodies showed specifically reduced ATRAP immunostaining in renal proximal tubules of PT‐KO mice compared with WT mice (Figure 1A).
To more strictly evaluate proximal tubule–specific reduction of ATRAP expression in PT‐KO mice, we compared ATRAP expression in proximal and distal tubules between PT‐KO and WT mice using LMD. We distinguished between proximal and distal tubules based on morphological features (eg, brush border) and then microdissected them from the kidneys of PT‐KO and WT mice (Figure 1B). Megalin mRNA expression was predominantly observed in a sample from proximal tubules but not distal tubules (Figure 1C). Calbindin‐D mRNA expression was almost all observed in a sample from distal tubules but not proximal tubules (Figure 1D). These results show correct sampling by LMD. ATRAP mRNA expression was decreased by ≈80% in proximal tubules of PT‐KO mice compared with WT mice (2‐factor ANOVA followed by Bonferroni multiple comparison test, P<0.01; Figure 1E). In contrast, ATRAP mRNA expression in distal tubules was comparable between PT‐KO and WT mice.
Effect of Ang II on Indirect BP, Heart Rate, and Body Weight in PT‐KO and WT Mice
To investigate the functional role of renal proximal tubule ATRAP in angiotensin‐dependent hypertension, we compared BP in response to Ang II treatment in PT‐KO and WT mice by indirect BP measurement using a tail‐cuff method. Age‐matched male PT‐KO and WT mice were divided into 4 groups: (1) vehicle‐infused PT‐KO mice, (2) Ang II–infused (1000 ng/kg per minute) PT‐KO mice, (3) vehicle‐infused WT mice, and (4) Ang II–infused (1000 ng/kg per minute) WT mice. Systolic BP of PT‐KO mice was comparable with WT mice at baseline (vehicle‐infused PT‐KO mice versus vehicle‐infused WT mice, 2‐way repeated measures ANOVA, F=0.2744, P=0.6092; Figure 2A). Ang II infusion for 2 weeks significantly and similarly increased systolic BP in both PT‐KO and WT mice (vehicle‐infused PT‐KO mice versus Ang II–infused PT‐KO mice, 2‐way repeated measures ANOVA, F=4.845, P=0.0450; vehicle‐infused WT mice versus Ang II–infused WT mice, 2‐way repeated measures ANOVA, F=5.945, P=0.0299; Ang II–infused PT‐KO versus Ang II–infused WT mice, 2‐way repeated measures ANOVA, F=0.1014, P=0.7548; Figure 2A). Systolic BP elevation in response to Ang II infusion was also comparable between PT‐KO and WT mice (Figure 2B). In contrast, there were no differences in heart rate among the 4 groups (Figure 2C). Ang II infusion for 2 weeks significantly inhibited body weight gain during infusion to the same extent in PT‐KO and WT mice (Figure 2D).
Effect of a Lower Dose of Ang II on BP in WT and PT‐KO Mice
To further investigate the functional role of renal proximal tubule ATRAP expression in angiotensin‐dependent hypertension, we used a lower dose of Ang II (600 ng/kg per minute) treatment in additional experiments. We compared BP in response to Ang II infusion (600 ng/kg per minute) in PT‐KO and WT mice by indirect BP measurement using the tail‐cuff method. Age‐matched male PT‐KO and WT mice were divided into 4 groups: (1) vehicle‐infused PT‐KO mice; (2) Ang II–infused (600 ng/kg per minute) PT‐KO mice; (3) vehicle‐infused WT mice; and (4) Ang II–infused (600 ng/kg per minute) WT mice. Ang II infusion (600 ng/kg per minute) for 2 weeks significantly and similarly increased systolic BP in both PT‐KO and WT mice (vehicle‐infused PT‐KO mice versus Ang II–infused PT‐KO mice, 2‐way repeated measures ANOVA, F=94.87, P<0.01; vehicle‐infused WT mice versus Ang II–infused WT mice, 2‐way repeated measures ANOVA, F=176.9, P<0.01; Ang II–infused PT‐KO versus Ang II–infused WT mice, 2‐way repeated measures ANOVA, F=0.8077, P=0.3899; Figure 2E). Systolic BP elevation in response to Ang II infusion (600 ng/kg per minute) was also comparable between PT‐KO and WT mice (Figure 2F). These results are consistent with the earlier results showing the comparable effects of the 1000‐ng/kg‐per‐minute dose of Ang II on BP in PT‐KO and WT mice.
Effect of Ang II on Cardiac Hypertrophy in PT‐KO and WT Mice
Because cardiac hypertrophy is closely associated with BP elevation, we further examined heart weight/body weight ratio in PT‐KO and WT mice. Ang II infusion (1000 ng/kg per minute) for 2 weeks significantly increased heart weight/body weight ratio to the same extent in both PT‐KO and WT mice (Figure 3A). These results are consistent with those showing comparably increased BP in response to Ang II infusion in PT‐KO and WT mice.
Effect of Ang II on Kidney Injury in PT‐KO and WT Mice
We next examined the effect of Ang II infusion on kidney injury in PT‐KO and WT mice. No significant differences in kidney weight/body weight ratio and KIM1 (renal kidney injury molecule 1) mRNA levels were noted among the 4 groups (Figure 3B and 3C). The results of histological analysis also showed that Ang II infusion (1000 ng/kg per minute) for 2 weeks did not obviously cause glomerular sclerosis or tubulointerstitial fibrosis in either PT‐KO or WT mice (Figure 3D).
Effect of Ang II on Direct BP and Sodium Balance in PT‐KO and WT Mice
In addition, we strictly compared the effects of Ang II infusion on BP in PT‐KO and WT mice by direct BP measurement using a radiotelemetric method. The 24‐hour mean systolic BP was significantly and similarly increased in response to 2 weeks of Ang II infusion (1000 ng/kg per minute) in both PT‐KO and WT mice (Ang II–infused PT‐KO versus Ang II–infused WT mice, 2‐way repeated measures ANOVA, F=0.04280, P=0.8407; Figure 4A). Ang II infusion (1000 ng/kg per minute) for 2 weeks also significantly increased 24‐hour mean systolic BP, in not only in the light period but also in the dark period and to the same extent in both types of mice (Figure 4B). Similarly, 24‐hour mean diastolic BP was significantly and similarly increased in response to 2 weeks of Ang II infusion (1000 ng/kg per minute) in both PT‐KO and WT mice (Ang II–infused PT‐KO versus Ang II–infused WT mice, 2‐way repeated measures ANOVA, F=0.02541, P=0.8769; Figure 4C). Ang II infusion (1000 ng/kg per minute) for 2 weeks also significantly increased 24‐hour mean diastolic BP, in not only in the light period but also in the dark period and to the same extent in both types of mice (Figure 4D). Furthermore, we compared the sodium balance of PT‐KO and WT mice during Ang II (1000 ng/kg per minute) treatment. Cumulative sodium balance during 2 weeks of Ang II treatment was comparable for PT‐KO and WT mice (P=0.9075, unpaired t test; Figure 4E).
Discussion
In this study, we generated genetically modified mice in which, for the first time, ATRAP expression was deleted specifically in renal proximal tubules. Detailed analysis of renal ATRAP expression revealed that ATRAP mRNA expression was decreased by ≈80% in proximal regions of the nephron in PT‐KO mice compared with WT mice. Furthermore, using these mice, we investigated the in vivo functional role of proximal tubule ATRAP in angiotensin‐dependent hypertension. Our most important finding is that ATRAP deficiency in proximal nephrons does not obviously enhance the pressor response to Ang II.
The intrarenal renin–angiotensin system regulates a diversity of renal hemodynamic and transport processes, which contribute to both sodium balance and BP homeostasis.1, 2, 3 Ang II is the most potent renin–angiotensin system component and exerts pleiotropic effects on renal microvasculature, tubular networks, and interstitial tissue. Through its effects on AT1R, Ang II regulates the vascular tone of afferent and efferent arterioles and the glomerular filtration coefficient. It also modulates sensitivity of the tubuloglomerular feedback mechanism and regulates medullary microvasculature by directly constricting pericytes in the vasa recta.22 More important, Ang II exerts a direct influence on the renal tubule transport function, including NHE3 (sodium‐proton antiporter 3) in proximal tubules and epithelial sodium channel and Na+–Cl− cotransporter in distal nephron segments.23, 24, 25 These multiple effects of Ang II act in a concerted manner to increase kidney conservation of sodium and enable maintenance of BP under conditions of sodium depletion or loss of extracellular fluid volume. Moreover, these effects also evoke excessive sodium retention, resulting in hypertension when inappropriately stimulated.
The present study is not designed to reveal the identical pressor response to Ang II between PT‐KO and WT mice. We examined whether PT‐KO exacerbates angiotensin‐induced hypertension compared with WT mice and found no significant differences in the pressor response to Ang II infusion between PT‐KO and WT mice. With respect to the functional role of ATRAP in the kidney, systemic ATRAP KO mice exhibit exacerbation of angiotensin‐dependent hypertension, concomitant with an increase in sodium retention.15 In contrast to systemic ATRAP KO mice, ATRAP transgenic mice dominantly overexpressing ATRAP in renal tubules exhibit suppression of angiotensin‐dependent hypertension, concomitant with a decrease in sodium retention.18 As a key mechanism, Ang II–induced upregulation of the epithelial sodium channel, a major sodium transporter in distal tubules, was significantly enhanced in systemic ATRAP KO mice compared with WT mice.15 In contrast, Ang II–induced Na+–Cl− cotransporter activation and epithelial sodium channel induction were suppressed in renal tubule‐dominant ATRAP transgenic mice compared with WT mice.18 However, there were no differences in renal expression of NHE3, a major sodium transporter in proximal tubules, between systemic ATRAP KO mice and WT mice or renal tubule–dominant ATRAP transgenic mice and WT mice.15, 18 Given the results of the present study together with previous knowledge, it appears that proximal tubular ATRAP plays a minor role in angiotensin‐dependent hypertension in vivo. Alternatively, the inhibitory effect of renal ATRAP on angiotensin‐dependent hypertension appears to act mainly through a distal tubule ATRAP‐mediated mechanism.
Regarding the regulatory role of renal proximal tubule AT1R in physiological and pathophysiological BP regulation, Gurley et al reported that proximal tubule–specific AT1R deficiency reduces baseline BP and further suppresses BP elevation in response to chronic Ang II infusion. In their study, they used proximal tubule‐specific AT1R‐deficient mice that were generated by the Cre/loxP system using PEPCK‐Cre.17 In contrast, using another line of proximal tubule‐specific AT1R‐deleted mice, Li et al showed that proximal tubule–specific AT1R deficiency reduces baseline BP but does not affect the pressor response to chronic Ang II infusion.26 These controversial results indicate that pathophysiology of BP elevation in response to chronic Ang II infusion is not exclusively mediated through the mechanism of AT1R activation in proximal nephrons.
In addition, a previous study reported that chronic Ang II stimulation does not affect proximal tubule fluid reabsorption or sodium delivery to distal nephron segments but rather that sodium reabsorption was increased in distal nephron segments by dual distal nephron blockade with amiloride and bendroflumethiazide in Ang II–infused mice.27 Hashimoto et al also observed that disruption of tissue angiotensin‐converting enzyme did not alter proximal tubule fluid reabsorption, as assessed by a micropuncture technique in a tissue‐specific angiotensin‐converting enzyme KO model.28 These results suggest that distal, but not proximal, nephron segments play a critical role in AT1R signal‐mediated renal sodium reabsorption and BP regulation, at least under in vivo conditions of chronic Ang II stimulation. This may be one of the reasons why ATRAP deficiency in proximal nephrons does not obviously affect the pressor response to Ang II.
With respect to hypertension‐related organ injury, chronic Ang II infusion did not cause histological renal injury in either PT‐KO or WT mice in the present study. In addition, renal mRNA levels of KIM1, a marker for renal proximal tubular damage,29 were not increased by 2 weeks of Ang II infusion in either PT‐KO or WT mice. Chronic Ang II stimulation reportedly induces renal injury such as tubulointerstitial fibrosis and glomerular sclerosis in rats or 129/Sv strain mice, whereas C57BL/6 strain mice are known to be genetically resistant to Ang II–induced kidney injury.30, 31, 32 PT‐KO and WT mice used in the present study were generated on a C57BL/6 background. Therefore, Ang II infusion of a duration >2 weeks would be required to cause renal injury.33 In contrast, 2 weeks of Ang II infusion (600 or 1000 ng/kg per minute) significantly increased the heart weight/body weight ratio in both types of mice. In the development of cardiac hypertrophy, BP‐independent effects of Ang II have been reported. Notably, we previously demonstrated that cardiomyocyte‐specific enhancement of ATRAP suppresses Ang II–induced cardiac hypertrophy independent of BP, concomitant with a suppression of p38 mitogen‐activated protein kinase.12 However, the PEPCK promoter used to generate PT‐KO mice in the current study does not drive Cre expression in the heart.17 Consequently, the similar extent of Ang II–induced cardiac hypertrophy in PT‐KO and WT mice suggests comparable BP elevation in response to Ang II between the genotypes.
Endogenous ATRAP is highly expressed in proximal tubules, with one question being the in vivo functional role of proximal tubule ATRAP. In this regard, we have recently shown that ATRAP plays an important role in inhibiting kidney aging, possibly through a SIRT1 (sirtuin 1)–mediated mechanism independent of blocking Ang II–AT1R signaling, which further protects normal lifespan.34 Compared with WT mice, systemic ATRAP KO mice show more advanced age–associated mitochondrial abnormalities and subsequently increased reactive oxygen species production in proximal tubules of the kidney, as well as exacerbated age‐associated tubulointerstitial fibrosis. Consequently, further studies are needed to investigate the functional role of proximal tubule ATRAP in kidney aging using PT‐KO mice.
This study has some limitations. First, we could not exclude the possibility of functional compensation in the mechanism due to embryonic silencing of the gene. Some gene deletions show no phenotypic changes because of functional compensation by closely related or duplicate genes. Functional compensation is also attributed to alternative metabolic pathways or regulatory networks.35 Further studies are needed to exclude the possibility of functional compensation for the deletion of ATRAP in proximal tubules. Second, because of the statistical design, the results of this study do not unequivocally prove that ATRAP deficiency in the proximal nephron does not affect angiotensin‐dependent hypertension. However, considering the results of our previous studies using systemic ATRAP KO mice and renal tubule–dominant ATRAP transgenic mice,15, 18 ATRAP in the proximal tubules is not likely to play a major role in angiotensin‐dependent hypertension. In conclusion, ATRAP deficiency in the proximal nephron does not obviously enhance angiotensin‐mediated hypertension in vivo.
Sources of Funding
This work was supported by grants from the Yokohama Foundation for Advancement of Medical Science, a Uehara Memorial Foundation grant, Grants‐in‐Aid for Scientific Research from the Japan Society for the Promotion of Science, grants from Senshin Medical Research, the Banyu Life Science Foundation International, and the Salt Science Research Foundation (18C4), and a grant‐in‐aid from the Cardiovascular Research Fund, Tokyo, Japan. This research was also supported by the grant of Strategic Research Project of Yokohama City University, Japan Agency for Medical Research and Development (AMED), Yokohama City University research grant “KAMOME Project” and by TR‐SPRINT (Translational Research program; Strategic PRomotion for practical application of INnovative Medical Technology) from AMED.
Disclosures
None.
Acknowledgments
We would like to thank E. Maeda (Yokohama City University) for help with the experiments.
(J Am Heart Assoc. 2019;8:e012395 DOI: 10.1161/JAHA.119.012395.)
Contributor Information
Hiromichi Wakui, Email: hiro1234@yokohama-cu.ac.jp.
Kengo Azushima, Email: azushima@yokohama-cu.ac.jp.
Kouichi Tamura, Email: tamukou@med.yokohama-cu.ac.jp.
References
- 1. Coffman TM. The inextricable role of the kidney in hypertension. J Clin Invest. 2014;124:2341–2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kobori H, Nangaku M, Navar LG, Nishiyama A. The intrarenal Renin‐Angiotensin system: from physiology to the pathobiology of hypertension and kidney disease. Pharmacol Rev. 2007;59:251–287. [DOI] [PubMed] [Google Scholar]
- 3. Navar LG, Kobori H, Prieto MC, Gonzalez‐Villalobos RA. Intratubular renin‐angiotensin system in hypertension. Hypertension. 2011;57:355–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hein L, Meinel L, Pratt RE, Dzau VJ, Kobilka BK. Intracellular trafficking of angiotensin II and its AT1 and AT2 receptors: evidence for selective sorting of receptor and ligand. Mol Endocrinol. 1997;11:1266–1277. [DOI] [PubMed] [Google Scholar]
- 5. Miura S, Saku K, Karnik SS. Molecular analysis of the structure and function of the angiotensin II type 1 receptor. Hypertens Res. 2003;26:937–943. [DOI] [PubMed] [Google Scholar]
- 6. Daviet L, Lehtonen JY, Tamura K, Griese DP, Horiuchi M, Dzau VJ. Cloning and characterization of ATRAP, a novel protein that interacts with the angiotensin II type 1 receptor. J Biol Chem. 1999;274:17058–17062. [DOI] [PubMed] [Google Scholar]
- 7. Lopez‐Ilasaca M, Liu X, Tamura K, Dzau VJ. The angiotensin II type I receptor‐associated protein, ATRAP, is a transmembrane protein and a modulator of angiotensin II signaling. Mol Biol Cell. 2003;14:5038–5050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Azuma K, Tamura K, Shigenaga A, Wakui H, Masuda S, Tsurumi‐Ikeya Y, Tanaka Y, Sakai M, Matsuda M, Hashimoto T, Ishigami T, Lopez‐Ilasaca M, Umemura S. Novel regulatory effect of angiotensin II type 1 receptor‐interacting molecule on vascular smooth muscle cells. Hypertension. 2007;50:926–932. [DOI] [PubMed] [Google Scholar]
- 9. Azushima K, Ohki K, Wakui H, Uneda K, Haku S, Kobayashi R, Haruhara K, Kinguchi S, Matsuda M, Maeda A, Toya Y, Yamashita A, Umemura S, Tamura K. Adipocyte‐specific enhancement of angiotensin II type 1 receptor‐associated protein ameliorates diet‐induced visceral obesity and insulin resistance. J Am Heart Assoc. 2017;6:e004488 DOI: 10.1161/JAHA.116.004488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tanaka Y, Tamura K, Koide Y, Sakai M, Tsurumi Y, Noda Y, Umemura M, Ishigami T, Uchino K, Kimura K, Horiuchi M, Umemura S. The novel angiotensin II type 1 receptor (AT1R)‐associated protein ATRAP downregulates AT1R and ameliorates cardiomyocyte hypertrophy. FEBS Lett. 2005;579:1579–1586. [DOI] [PubMed] [Google Scholar]
- 11. Wakui H, Dejima T, Tamura K, Uneda K, Azuma K, Maeda A, Ohsawa M, Kanaoka T, Azushima K, Kobayashi R, Matsuda M, Yamashita A, Umemura S. Activation of angiotensin II type 1 receptor‐associated protein exerts an inhibitory effect on vascular hypertrophy and oxidative stress in angiotensin II‐mediated hypertension. Cardiovasc Res. 2013;100:511–519. [DOI] [PubMed] [Google Scholar]
- 12. Wakui H, Tamura K, Tanaka Y, Matsuda M, Bai Y, Dejima T, Masuda S, Shigenaga A, Maeda A, Mogi M, Ichihara N, Kobayashi Y, Hirawa N, Ishigami T, Toya Y, Yabana M, Horiuchi M, Minamisawa S, Umemura S. Cardiac‐specific activation of angiotensin II type 1 receptor‐associated protein completely suppresses cardiac hypertrophy in chronic angiotensin II‐infused mice. Hypertension. 2010;55:1157–1164. [DOI] [PubMed] [Google Scholar]
- 13. Masuda S, Tamura K, Wakui H, Maeda A, Dejima T, Hirose T, Toyoda M, Azuma K, Ohsawa M, Kanaoka T, Yanagi M, Yoshida S, Mitsuhashi H, Matsuda M, Ishigami T, Toya Y, Suzuki D, Nagashima Y, Umemura S. Expression of angiotensin II type 1 receptor‐interacting molecule in normal human kidney and IgA nephropathy. Am J Physiol Renal Physiol. 2010;299:F720–F731. [DOI] [PubMed] [Google Scholar]
- 14. Tsurumi Y, Tamura K, Tanaka Y, Koide Y, Sakai M, Yabana M, Noda Y, Hashimoto T, Kihara M, Hirawa N, Toya Y, Kiuchi Y, Iwai M, Horiuchi M, Umemura S. Interacting molecule of AT1 receptor, ATRAP, is colocalized with AT1 receptor in the mouse renal tubules. Kidney Int. 2006;69:488–494. [DOI] [PubMed] [Google Scholar]
- 15. Ohsawa M, Tamura K, Wakui H, Maeda A, Dejima T, Kanaoka T, Azushima K, Uneda K, Tsurumi‐Ikeya Y, Kobayashi R, Matsuda M, Uchida S, Toya Y, Kobori H, Nishiyama A, Yamashita A, Ishikawa Y, Umemura S. Deletion of the angiotensin II type 1 receptor‐associated protein enhances renal sodium reabsorption and exacerbates angiotensin II‐mediated hypertension. Kidney Int. 2014;86:570–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ohki K, Wakui H, Azushima K, Uneda K, Haku S, Kobayashi R, Haruhara K, Kinguchi S, Matsuda M, Ohsawa M, Maeda A, Minegishi S, Ishigami T, Toya Y, Yamashita A, Umemura S, Tamura K. ATRAP expression in brown adipose tissue does not influence the development of diet‐induced metabolic disorders in mice. Int J Mol Sci. 2017;18:E676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gurley SB, Riquier‐Brison AD, Schnermann J, Sparks MA, Allen AM, Haase VH, Snouwaert JN, Le TH, McDonough AA, Koller BH, Coffman TM. AT1A angiotensin receptors in the renal proximal tubule regulate blood pressure. Cell Metab. 2011;13:469–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wakui H, Tamura K, Masuda S, Tsurumi‐Ikeya Y, Fujita M, Maeda A, Ohsawa M, Azushima K, Uneda K, Matsuda M, Kitamura K, Uchida S, Toya Y, Kobori H, Nagahama K, Yamashita A, Umemura S. Enhanced angiotensin receptor‐associated protein in renal tubule suppresses angiotensin‐dependent hypertension. Hypertension. 2013;61:1203–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kobayashi R, Wakui H, Azushima K, Uneda K, Haku S, Ohki K, Haruhara K, Kinguchi S, Matsuda M, Ohsawa M, Toya Y, Nishiyama A, Yamashita A, Tanabe K, Maeshima Y, Umemura S, Tamura K. An angiotensin II type 1 receptor binding molecule has a critical role in hypertension in a chronic kidney disease model. Kidney Int. 2017;91:1115–1125. [DOI] [PubMed] [Google Scholar]
- 20. Wakui H, Tamura K, Matsuda M, Bai Y, Dejima T, Shigenaga A, Masuda S, Azuma K, Maeda A, Hirose T, Ishigami T, Toya Y, Yabana M, Minamisawa S, Umemura S. Intrarenal suppression of angiotensin II type 1 receptor binding molecule in angiotensin II‐infused mice. Am J Physiol Renal Physiol. 2010;299:F991–F1003. [DOI] [PubMed] [Google Scholar]
- 21. Yee JY, Limenta LM, Rogers K, Rogers SM, Tay VS, Lee EJ. Ensuring good quality RNA for quantitative real‐time PCR isolated from renal proximal tubular cells using laser capture microdissection. BMC Res Notes. 2014;7:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Navar LG. Intrarenal renin‐angiotensin system in regulation of glomerular function. Curr Opin Nephrol Hypertens. 2014;23:38–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Castaneda‐Bueno M, Cervantes‐Perez LG, Vazquez N, Uribe N, Kantesaria S, Morla L, Bobadilla NA, Doucet A, Alessi DR, Gamba G. Activation of the renal Na+:Cl‐ cotransporter by angiotensin II is a WNK4‐dependent process. Proc Natl Acad Sci USA. 2012;109:7929–7934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. He P, Klein J, Yun CC. Activation of Na+/H+ exchanger NHE3 by angiotensin II is mediated by inositol 1,4,5‐triphosphate (IP3) receptor‐binding protein released with IP3 (IRBIT) and Ca2+/calmodulin‐dependent protein kinase II. J Biol Chem. 2010;285:27869–27878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mamenko M, Zaika O, Prieto MC, Jensen VB, Doris PA, Navar LG, Pochynyuk O. Chronic angiotensin II infusion drives extensive aldosterone‐independent epithelial Na+ channel activation. Hypertension. 2013;62:1111–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li H, Weatherford ET, Davis DR, Keen HL, Grobe JL, Daugherty A, Cassis LA, Allen AM, Sigmund CD. Renal proximal tubule angiotensin AT1A receptors regulate blood pressure. Am J Physiol Regul Integr Comp Physiol. 2011;301:R1067–R1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhao D, Seth DM, Navar LG. Enhanced distal nephron sodium reabsorption in chronic angiotensin II‐infused mice. Hypertension. 2009;54:120–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hashimoto S, Adams JW, Bernstein KE, Schnermann J. Micropuncture determination of nephron function in mice without tissue angiotensin‐converting enzyme. Am J Physiol Renal Physiol. 2005;288:F445–F452. [DOI] [PubMed] [Google Scholar]
- 29. Waanders F, van Timmeren MM, Stegeman CA, Bakker SJ, van Goor H. Kidney injury molecule‐1 in renal disease. J Pathol. 2010;220:7–16. [DOI] [PubMed] [Google Scholar]
- 30. Kitayama H, Maeshima Y, Takazawa Y, Yamamoto Y, Wu Y, Ichinose K, Hirokoshi K, Sugiyama H, Yamasaki Y, Makino H. Regulation of angiogenic factors in angiotensin II infusion model in association with tubulointerstitial injuries. Am J Hypertens. 2006;19:718–727. [DOI] [PubMed] [Google Scholar]
- 31. Yang S, Yao B, Zhou Y, Yin H, Zhang MZ, Harris RC. Intrarenal dopamine modulates progressive angiotensin II‐mediated renal injury. Am J Physiol Renal Physiol. 2012;302:F742–F749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wesseling S, Ishola DA Jr, Joles JA, Bluyssen HA, Koomans HA, Braam B. Resistance to oxidative stress by chronic infusion of angiotensin II in mouse kidney is not mediated by the AT2 receptor. Am J Physiol Renal Physiol. 2005;288:F1191–F1200. [DOI] [PubMed] [Google Scholar]
- 33. Ichikawa D, Kamijo‐Ikemori A, Sugaya T, Yasuda T, Hoshino S, Igarashi‐Migitaka J, Hirata K, Kimura K. Renal liver‐type fatty acid binding protein attenuates angiotensin II‐induced renal injury. Hypertension. 2012;60:973–980. [DOI] [PubMed] [Google Scholar]
- 34. Uneda K, Wakui H, Maeda A, Azushima K, Kobayashi R, Haku S, Ohki K, Haruhara K, Kinguchi S, Matsuda M, Ohsawa M, Minegishi S, Ishigami T, Toya Y, Atobe Y, Yamashita A, Umemura S, Tamura K. Angiotensin II Type 1 receptor‐associated protein regulates kidney aging and lifespan independent of angiotensin. J Am Heart Assoc. 2017;6:e006120. doi: 10.1161/JAHA.117.006120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gu Z, Steinmetz LM, Gu X, Scharfe C, Davis RW, Li WH. Role of duplicate genes in genetic robustness against null mutations. Nature. 2003;421:63–66. [DOI] [PubMed] [Google Scholar]