

SUMMARY
Anxiety disorders are associated with body weight changes in humans. However, mechanisms underlying anxiety-induced weight changes remain poorly understood. Using Emx1Cre/+ mice, we deleted the gene for brain-derived neurotrophic factor (BDNF) in the cortex, hippocampus, and some amygdalar subregions. Resulting mutant mice displayed impaired GABAergic transmission and elevated anxiety. They were leaner when fed either chow diet or high-fat diet, due to higher sympathetic activity, basal metabolic rate, brown adipocyte thermogenesis and beige adipocyte formation, compared to control mice. BDNF re-expression in the amygdala rescued the anxiety and metabolic phenotypes in mutant mice. Conversely, anxiety induced by amygdala-specific Bdnf deletion or administration of an inverse GABAA receptor agonist increased energy expenditure. These results reveal that increased activities in anxiogenic circuits can reduce body weight by promoting adaptive thermogenesis and basal metabolism via the sympathetic nervous system and suggest that amygdalar GABAergic neurons be a link between anxiety and metabolic dysfunction.
INTRODUCTION
Anxiety is a feeling of fear and apprehension about what is to come in the absence of immediate threat, accompanied by a state of high arousal and increased vigilance (Davis et al., 2010). Occasional anxiety is believed to aid survival by increasing awareness and enabling rapid responses to possible threats (Calhoon and Tye, 2015). However, persistent and disruptive anxiety that is disproportionate to actual threat is pathological. Anxiety disorders are often linked to body weight change in humans; however, the relationship between the disorders and body weight is complex. Anxiety disorders are reportedly associated with a higher body weight in children (Anderson et al., 2006; Rofey et al., 2009), whereas some anxiety patients have been known to complain about substantial weight loss.
Anxiety is characterized by activation of the sympathetic nervous system (SNS) and the neuroendocrine system, as revealed by physiological changes such as sweating, increased heart rate, and elevated levels of corticotropin releasing factor (CRF) and glucocorticoids (Calhoon and Tye, 2015; Kreibig, 2010). As sympathetic outflow is the main determinant of adaptive thermogenesis and lipolysis in adipose tissues (Bachman et al., 2002; Harms and Seale, 2013; Rothwell and Stock, 1984), frequent or persistent sympathetic activation associated with anxiety disorders could increase energy expenditure through heightened adaptive thermogenesis and thereby reduce the risk of developing obesity. On the contrary, high levels of glucocorticoids could lead to increased visceral adiposity, as displayed in patients with Cushing’s syndrome (Charmandari et al., 2005). To date, no studies have been reported to investigate how energy balance is altered in humans or mice with elevated anxiety.
Brain-derived neurotrophic factor (BDNF) is a growth factor that plays crucial roles in neuronal development and synaptic plasticity (Huang and Reichardt, 2001; Park and Poo, 2013). Its deficiency causes anxiety-like behaviors and obesity in mice and humans (Chen et al., 2006; Gray et al., 2006; Han et al., 2008; Rios et al., 2001; Soliman et al., 2010; Xu et al., 2003). Genetic and pharmacological studies indicate that BDNF expressed in the hypothalamus and brainstem regulates energy balance by suppressing food intake and promoting energy expenditure (Xu and Xie, 2016). It remains, however, poorly understood what are the neural substrate and mechanism through which BDNF regulates mood.
In this study, we used the Emx1Cre/+ driver (Gorski et al., 2002) to abolish Bdnf expression in the cortex, hippocampus and some parts of the amygdala. Resulting mutant mice displayed impaired GABAergic transmission and high levels of anxiety-like behaviors, sympathetic activity, CRF expression and circulating corticosterone. Remarkably, the mutant mice were lean and resistant to diet-induced obesity (DIO) due to an increase in basal metabolic rate and adaptive thermogenesis in both brown and white adipose tissues. Furthermore, we found that induction of anxiety with site-specific Bdnf deletion in the basolateral amygdala (BLA) and surrounding area also led to similar metabolic phenotypes. Importantly, viral expression of BDNF in the BLA and surrounding area normalized the abnormalities in mood and energy metabolism in Emx1Cre-mediated Bdnf mutant mice. We further showed that acute induction of anxiety with an inverse agonist of GABAA receptor, FG7142, significantly enhanced energy expenditure. These results indicate that increased activities in anxiogenic circuits enhance energy expenditure by stimulating adaptive thermogenesis of adipose tissues and basal metabolism through SNS activation, and consequently conveys resistance to DIO. The results also reveal a role for amygdalar BDNF in the control of mood through modulation of GABAergic transmission.
RESULTS
Bdnf deletion in the dorsal forebrain increases adaptive thermogenesis
To investigate potential roles of BDNF expressed outside the hypothalamus in energy balance, we sought to generate a mouse mutant, Bdnflox/lox;Emx1Cre/+, by crossing floxed Bdnf (Bdnflox/lox) mice (Rios et al., 2001) to Emx1Cre/+ mice (Gorski et al., 2002). Using Bdnfklox/+;Emx1Cre/+ mice in which a uniquely floxed Bdnf allele (Bdnfklox) expresses β-galactosidase in BDNF-expressing cells once it is recombined by Cre (Liao et al., 2012), we found that the Emx1Cre knock-in allele was able to abolish Bdnf gene expression in the cortex, hippocampus and some parts of the amygdala including the anterior part of the BLA (BLAa), posterior part of the BLA (BLAp) and posterior part of basomedial amygdala (BMAp) (Figures 1A and S1A). We further confirmed that Bdnf mRNA in the cortex and hippocampus was abolished in Bdnflox/lox;Emx1Cre/+ mice using in situ hybridization (Figure S1B). When fed chow diet, Bdnflox/lox;Emx1Cre/+ mice had significantly higher food intake than Bdnflox/lox control littermates (Figure 1B), while maintaining a body weight comparable to control mice (Figure 1C). However, the Bdnf deletion drastically reduced fat mass without affecting lean mass (Figures 1D and S1C). Furthermore, the Bdnf mutant mice showed an improvement in blood glucose management as revealed by both intraperitoneal glucose tolerance test (IPGTT) and insulin tolerance test (IPITT) (Figures S1D and S1E). Given that BDNF-TrkB signaling impairment in the brain or whole body leads to severe obesity (Rios et al., 2001; Xu et al., 2003), this is a surprising metabolic phenotype from a Bdnf mutant.
Figure 1. Bdnf deletion in Emx1-expressing neurons increased energy expenditure and induced browning in the inguinal white adipose tissue (iWAT).
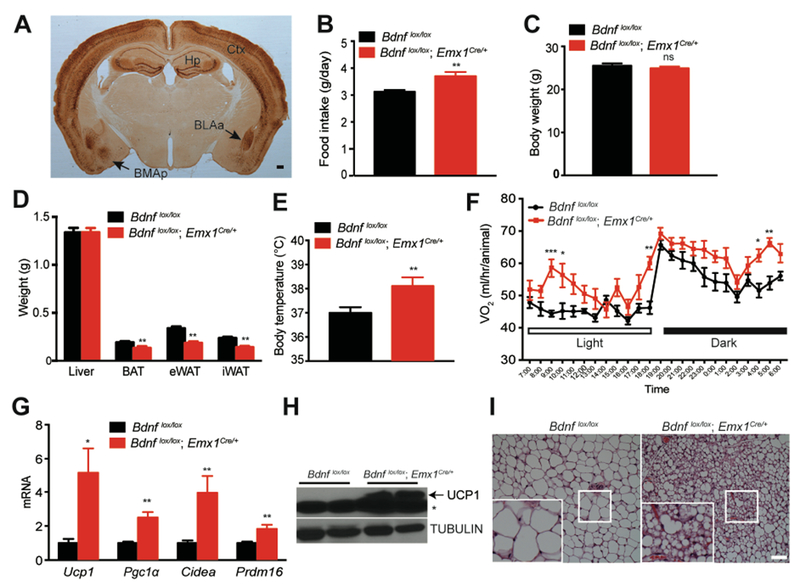
(A) Representative immunohistochemistry image showing expression of β-galactosidase in the brain of a Bdnfklox/+;Emx1Cre/+ mouse. Ctx, cortex; Hp, hippocampus; BLAa: anterior part of basolateral amygdala; BMAp, posterior part of basomedial amygdala. Scale bar, 200 μm.
(B) Daily intake of chow diet. N=8-9 male mice per group; **p<0.01 by Student’s t test.
(C) Body weight of 14-week-old male Bdnf mutants and control littermates raised on the chow diet. N=8-9 mice per group; ns, no significance.
(D) Weight of individual tissues including the liver, brown adipose tissue (BAT), epididymal white adipose tissue (eWAT) and inguinal white adipose tissue (iWAT) dissected from 14-week-old male mice raised on chow diet. N=7 mice per group; **p<0.01 by Student’s t test.
(E) Core body temperature of mice raised on chow diet, measured at around 4-5 pm. N=8-9 mice per group; **p<0.01 by Student’s t test.
(F) Oxygen consumption (VO2) of 10-week-old male mice raised on chow diet. N=7-9 mice per group; Two-way ANOVA followed by Bonferroni’s test: genotype, F(1,336)=99.31, p<0.0001; *p<0.05, **p<0.01 and ***p<0.001 when two genotypes were compared at individual time points.
(G) Gene expression analysis of mitochondrial proteins involved in iWAT thermogenesis. N=7-8 mice per group; *p<0.05 and **p<0.01 by Student’s t test.
(H) Western blotting analysis of UCP1 protein in iWAT dissected from 14-week-old male mice fed chow diet. * nonspecific band.
(I) Representative images from H&E staining of iWAT dissected from 14-week-old mice raised on chow diet. Scale bar, 20 μm.
All data are presented as mean ± SEM.
Increase in food intake and reduction in fat mass in Bdnflox/lox;Emx1Cre/+ mice could result from increased energy expenditure. Indeed, Bdnflox/lox;Emx1Cre/+ mice had significantly higher body temperature (Figure 1E) and oxygen consumption during both light and dark periods (Figures 1F and S1F). The increase in energy expenditure was not a result of changes in physical activity (Figure S1G). Furthermore, Bdnf deletion in the forebrain did not alter relative utilization of fat and carbohydrates as fuels, as there was no difference in respiratory exchange ratio (RER) between controls and Bdnf mutant mice (Figure S1H). Instead, our results indicate that the energy expenditure increase observed in mutant mice was in part attributable to development of brown-adipocyte-like beige cells in white adipose tissues, a process termed browning. We found that expression of genes important for adaptive thermogenesis, such as Ucp1, Pgc1α, Cidea and Prdm16, was significantly increased in the inguinal white adipose tissue (iWAT) of Bdnflox/lox;Emx1Cre/+ mice (Figures 1G and 1H). During thermogenesis in brown and beige adipocytes, uncoupling protein 1 (UCP1) uncouples proton gradients generated through β-oxidation of fatty acid from ATP synthesis in mitochondria and instead produces heat to maintain body temperature (Harms and Seale, 2013). Consequently, iWAT adipocytes in Bdnf mutant mice were smaller and contained multiple droplets (Figure 1I).
Collectively, these observations indicate that deletion of the Bdnf gene in the cortex, hippocampus and some parts of the amygdala induces browning of iWAT and increases energy expenditure, which leads to reduction in adiposity.
Bdnflox/lox;Emx1Cre/+ mice were resistant to diet-induced obesity
Because energy expenditure was increased in Bdnflox/lox;Emx1Cre/+ mice, we reasoned that these animals might be resistant to diet-induced obesity (DIO). To test this possibility, we fed male mice chow diet until 15 weeks of age and then high-fat diet (HFD, 60% calories from fat) for additional 12 weeks. Bdnf mutant mice and control mice displayed similar body weights by the age of 15 weeks; however, their body weights diverged shortly after the start of HFD feeding (Figure 2A). After 12 weeks of HFD feeding, mutant mice were much lower in body weight, fat mass and lean mass than control mice (Figures 2B, S2A and S2B), although they ingested more calories than control mice (Figure 2C). Furthermore, mutant mice showed better glucose tolerance and insulin sensitivity (Figures 2D and 2E) and lower levels of liver lipid (a sign for lack of hepatic steatosis; Figures S2C and S2D), compared with control mice. All of these measurements show that male Bdnflox/lox;Emx1Cre/+ mice are resistant to HFD-induced obesity. The resistance should result from elevated energy expenditure, as the Bdnf deletion in Emx1-expressing neurons led to a significant increase in O2 consumption (Figures 2F and S2E) and core body temperature (Figure 2G), but normal RER (Figure S2F). Note that female mutant mice also showed resistance to HFD-induced obesity, but to a lesser extent (Figures S2G-S2J).
Figure 2. Bdnflox/lox;Emx1Cre/+ mice were resistant to diet-induced obesity.
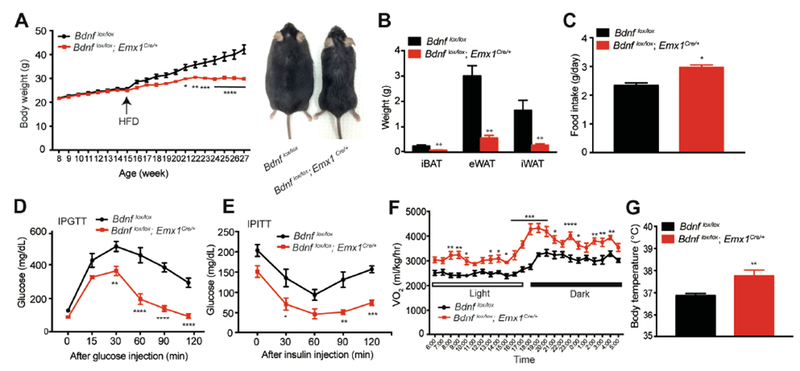
(A) Body weight (left) and representative picture (right) of control and mutant mice. Animals were fed chow diet until 15 weeks of age and then HFD for 12 weeks. N=6-9 mice per group; Two-way ANOVA followed by Bonferroni’s test: genotype, F(1,260)=139.9, p<0.0001; *p<0.05, **p<0.01, ***p<0.001 and ****p<0.0001 when comparisons were made at individual time points.
(B) Weight of individual fat tissues dissected from male mice after 12 weeks of HFD feeding. N=6-9 mice per group; **p<0.01 by Student’s t test.
(C) Daily intake of HFD. N=6-9 mice per group; **p<0.05 by Student’s t test.
(D) Intraperitoneal glucose tolerance test (IPGTT) of male mice fed HFD for 6-7 weeks. N=6-9 mice per group; Two-way ANOVA followed by Bonferroni’s test: genotype, F(1,48)=116.1, p<0.0001; **p<0.01 and ****p<0.0001 when comparisons were made at individual time points.
(E) Intraperitoneal insulin tolerance test (IPITT) of male mice fed HFD for 6-7 weeks. N=6-9 mice per group. Two-way ANOVA followed by Bonferroni’s test: genotype, F(1,40)=54.41, p<0.0001; *p<0.05, **p<0.01 and ***p<0.001 when comparisons were made at individual time points.
(F) Oxygen consumption (VO2) of male mice during the first week of HFD feeding. N=6-9 mice per group; Two-way ANOVA followed by Bonferroni’s test: genotype, F(1,312)=372.3, p<0.0001; *p<0.05, **p<0.01, ***p<0.001 and ****p<0.0001 when comparisons were made at individual time points.
(G) Core body temperature of male mice fed HFD, measured at around 4-5 pm. N=6-9 mice per group; **p<0.01 by Student’s t test.
All data are presented as mean ± SEM.
Lipid hydrolysis and adaptive thermogenesis were increased in Bdnflox/lox;Emx1Cre/+ mice
In consideration of thermogenesis in adipose tissues as a major component of energy expenditure in mice (Garland et al., 2011) and the observation that beige adipocytes were induced in the iWAT of Bdnflox/lox;Emx1Cre/+ mice fed chow diet (Figures 1G-I), we reasoned that increased thermogenesis in adipose tissues should significantly contribute to elevated energy expenditure in HFD-fed mutant mice. To this end, we examined morphology and gene expression in the iWAT, epididymal white adipose tissue (eWAT) and interscapular brown adipose tissue (iBAT) of HFD-fed male control and mutant mice.
In comparison with HFD-fed control mice, HFD-fed Bdnflox/lox;Emx1Cre/+ mice had smaller iWAT adipocytes, some of which resembled beige adipocytes with multiple lipid droplets (Figure 3A). In support of the presence of beige adipocytes, a marked increase in the protein level of UCP1 was observed in mutant iWAT (Figures 3A and 3C). Moreover, levels of mRNAs for Ucp1 and other thermogenic genes were significantly increased in mutant iWAT (Figure 3B). Importantly, tyrosine hydroxylase (TH), the rate-limiting enzyme in the synthesis of sympathetic neurotransmitter norepinephrine, was much more abundant in mutant iWAT than control iWAT (Figure 3C), suggesting enhanced sympathetic outflow into iWAT. Consistently, the expression of β3 adrenergic receptor (ADRB3), the main receptor for norepinephrine in adipocytes, was significantly increased in mutant iWAT (Figure 3D). In accordance with increased sympathetic activity and appearance of thermogenic beige adipocytes, active and phosphorylated hormone sensitive lipase (pHSL), which is activated by adrenergic receptors through phosphorylation and is a critical enzyme in lipid hydrolysis to produce fuel for adaptive thermogenesis (Harms and Seale, 2013), was markedly increased in its level in mutant iWAT (Figure 3C). Taken together, these results indicate that Bdnf deletion in Bdnflox/lox;Emx1Cre/+ mice increases the sympathetic nerve activity (SNA) and ADRB3 activation, thereby stimulating lipolysis and adaptive thermogenesis in iWAT.
Figure 3. Lipolysis and adaptive thermogenesis were elevated in adipose tissues of male Bdnflox/lox;Emx1Cre/+ mice.
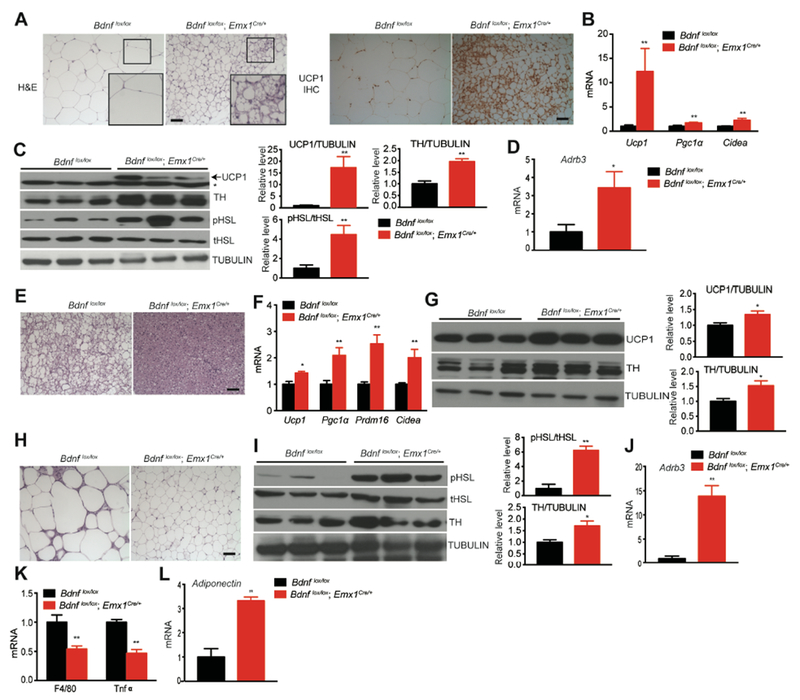
All analyses were done in fat tissues dissected from mice fed HFD for 12 weeks.
(A) Representative images showing H&E staining and UCP1 immunohistochemistry of iWAT. Scale bar, 20 μm.
(B) qRT-PCR analysis of mRNA for thermogenic genes in iWAT. N=6-9 mice per group; **p<0.01 by Student’s t test.
(C) Western blotting analysis and quantification of UCP1, tyrosine hydroxylase (TH), total hormone-sensitive lipase (tHSL) and phosphorylated hormone-sensitive lipase (pHSL) in iWAT. **p<0.01 by Student’s t test. * marks nonspecific bands.
(D) Relative levels of mRNA for β3 adrenergic receptor (Adrb3) in iWAT. N=6-9 mice per group; *p<0.05, by Student’s t test.
(E) Representative images showing H&E staining of iBAT. Scale bar, 20 μm.
(F) qRT-PCR analysis of mRNAs for genes involved in iBAT thermogenesis. N=6-9 mice per group; *p<0.05 and **p<0.01 by Student’s t test.
(G) Western blotting analysis and quantification of UCP1 and TH in iBAT.
(H) Representative images showing H&E staining of eWAT. Scale bar, 20 μm.
(I) Western blotting analysis and quantification of HSL and TH in eWAT. *p< 0.05 and **p< 0.01 by Student’s t test.
(J) Relative levels of Adrb3 mRNA in eWAT of male mice after 12 weeks of HFD feeding. **p<0.01 by Student’s t test.
(K) qRT-PCR analysis of mRNAs for proinflammatory markers in eWAT. N=6-9 mice per group.
(L) Levels of adiponectin mRNA in eWAT of male mice fed HFD for 12 weeks. **p<0.01 by Student’s t test.
All data were presented as mean ± SEM.
In iBAT (a key thermogenesis organ in mice (Kajimura et al., 2015)), lipid droplets were smaller in mutant mice than control mice after 12 weeks of HFD feeding, as revealed by H&E staining (Figure 3E). This morphological difference is indicative of a change in adaptive thermogenesis. In support of this view, the expression of several mitochondrial genes involved in adaptive thermogenesis, such as Ucp1, Pgc1α, Prdm16 and Cidea, was significantly increased in iBAT of Bdnflox/lox;Emx1Cre/+ mutant mice (Figure 3F). Levels of both UCP1 and TH proteins in iBAT were also significantly increased in mutant mice compared to control mice (Figure 3G). These results indicate that elevated SNA leads to enhanced adaptive thermogenesis in iBAT of Bdnflox/lox;Emx1Cre/+ mice.
Like iWAT, eWAT in HFD-fed Bdnf mutant mice had smaller adipocytes (Figure 3H) and higher levels of TH protein, pHSL protein (Figure 3I) and Adrb3 mRNA (Figure 3J), compared to HFD-fed control mice. These morphological and biochemical data indicate that sympathetic input and lipolysis are also elevated in eWAT of mutant mice. It is of interest that mutant mice showed a significant reduction in levels of mRNAs for F4/80 (a macrophage marker) and TNFα (an inflammatory cytokine) in eWAT, implying decreased macrophage infiltration and inflammatory response (Figure 3K). On the contrary, the level of mRNA for adiponectin was drastically elevated in mutant eWAT (Figure 3L). Given the beneficial effects of adiponectin in metabolism, this increase, together with reduced inflammation in eWAT, likely contribute to a substantial improvement in insulin sensitivity observed in Bdnflox/lox;Emx1Cre/+ mice.
Next, we asked if the metabolic phenotypes observed in Bdnf mutant mice could be partially attributable to a compensatory increase in BDNF-TrkB signaling in the hypothalamus or brainstem, the two brain regions important for the control of energy balance (Morton et al., 2014). There was no significant difference in BDNF levels and TrkB activation between control and mutant mice in either the hypothalamus (Figure S3A) or the brainstem (Figure S3B).
Taken together, our analyses of three fat tissues revealed that sympathetic outflow into fat tissues was enhanced in Bdnflox/lox;Emx1Cre/+ mice, which leads to an increase in adaptive thermogenesis of iWAT and iBAT and lipolysis, and thereby low mass of all fat tissues.
Bdnflox/lox;Emx1Cre/+ mice were resistant to DIO within the thermoneutral zone
Mice are normally housed around 22°C, a temperature that is below their thermoneutrality of 30°C. As a consequence, mice live under a chronic thermal stress so that they increase heat production to compensate for heat loss (Nedergaard and Cannon, 2014). As described above, Bdnflox/lox;Emx1Cre/+ mutant mice displayed leanness and increased thermogenesis in adipose tissues at 22°C when they were fed either chow diet or HFD. If elevated adaptive thermogenesis is fully responsible for leanness of Bdnflox/lox;Emx1Cre/+ mice, the resistance of these mutant mice to DIO should disappear within the thermoneutral zone. Otherwise, an increase in basal metabolic rate should contribute to the phenotype of leanness.
Bdnflox/lox;Emx1Cre/+ mice ingested similar amounts of calories on both chow diet and HFD to their control littermates under a 30°C housing condition (Figure 4A). This result suggests that elevated adaptive thermogenesis leads to the observed hyperphagia in mutant mice under the 22°C housing condition. To our surprise, mutant mice were leaner than control mice even on chow diet and showed robust resistance to DIO (Figures 4B and S4A-S4C). Remarkably, mutant mice gained little weight even after 12 weeks of HFD exposure, whereas control mice gained substantial weight and fat mass during the same period (Figures 4B and 4C). Consequently, compared with control mice, Bdnflox/lox;Emx1Cre/+ mice showed significantly improved insulin sensitivity and glycemic control (Figures 4D and 4E).
Figure 4. Male Bdnflox/lox;Emx1Cre/+ mice were resistant to DIO within the thermoneutral zone.
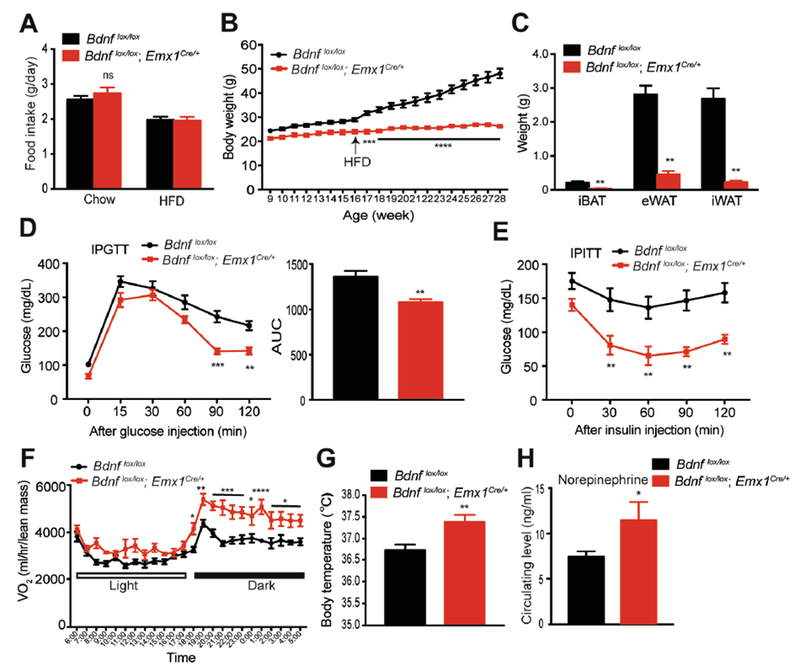
(A) Daily intake of chow diet and HFD at the thermoneutral temperature (30°C). N=6-8 mice per group; ns, no significance by Student’s t test.
(B) Body weight of male mice housed at thermoneutrality. Eight-week-old mice were switched to thermoneutrality from standard housing. They were maintained on chow diet until 16 weeks of age when they started receiving HFD for 12 weeks. N=6-8 mice per group; Two-way ANOVA followed by Bonferroni’s test: genotype, F(1,240)=689, p<0.0001; ***p<0.001 and ****p<0.0001 when comparisons were made at individual time points.
(C) Weight of individual fat tissues after HFD feeding for 12 weeks at thermoneutrality. N=6-8 mice per group; **p<0.01 by Student’s t test.
(D) IPGTT in mice fed HFD for 10-11 weeks at thermoneutrality. N=6-8 mice per group; Two-way ANOVA followed by Bonferroni’s test: genotype, F(1,72)=37.61, p<0.0001; **p<0.01 and ***p<0.001 when comparisons were made at individual time points.
(E) IPITT in mice fed HFD for 10-11 weeks at thermoneutrality. N=6-8 mice per group; Two-way ANOVA followed by Bonferroni’s test: genotype, F(1,51)=52.18, p<0.0001; **p<0.01 when comparisons were made at individual time points.
(F) O2 consumption of mice at thermoneutrality during the first week of HFD feeding. N=6-8 mice per group; Two-way ANOVA followed by Bonferroni’s test: genotype, F(1,288)=189.5, p<0.0001; *p<0.05, **p<0.01, ***p<0.001 and ****p<0.0001 when comparisons were made at individual time points.
(G) Body temperature of mice housed at thermoneutrality. N=6-8 mice per group; **p < 0.01 by Student’s t test.
(H) Concentration of norepinephrine in the plasma of HFD-fed mice. N=8-10 mice per group; *p<0.05 by Student’s t test.
All data were presented as mean ± SEM.
Adaptive thermogenesis is not required to maintain core body temperature under the thermoneutral condition. Indeed, there was no difference in the expression of thermogenic genes in iWAT between Bdnflox/lox;Emx1Cre/+ and control mice housed at 30°C (data not shown). However, mutant mice still consumed more O2 and had higher body temperature than control mice when they were fed HFD (Figures 4F, 4G and S4D). Although mutant mice showed a trend of higher ambulatory activities than control mice, the difference was not significant (Figure S4E). In addition, no correlation was found between ambulatory activities and weight gain on HFD in Bdnflox/lox;Emx1Cre/+ mice (Figure S4F). These results indicate that basal metabolic rate is also elevated in Bdnflox/lox;Emx1Cre/+ mutant mice. In support of this argument, circulating levels of norepinephrine, which is largely from the SNS, were increased in Bdnflox/lox;Emx1Cre/+ mice (Figure 4H). We next asked if sympathetic tone was also increased in some other organs than fat tissues of Bdnflox/lox;Emx1Cre/+ mice. TH expression in the liver and heart was comparable between control and Bdnf mutants (Figures S4G and S4H), while its expression in the kidney of Bdnf mutants was significantly increased compared to control mice (Figure S4I). Therefore, leanness and resistance to DIO in Bdnflox/lox;Emx1Cre/+ mice primarily result from increased sympathetic outflow to fat tissues, which leads to an increase in both adaptive thermogenesis and basal metabolic rate.
BDNF expressed in the motor cortex did not affect energy expenditure
As described earlier, Bdnf expression was abolished in the cortex, hippocampus and several parts of amygdala in Bdnflox/lox;Emx1Cre/+ mice. We investigated the possibility that some BDNF-expressing neurons in these brain areas regulate energy expenditure through polysynaptic connection to adipose tissues. We sought to identify BDNF neurons that are polysynaptically connected to iWAT using retrograde trans-neuronal tracing. Seven days after injection of GFP-expressing retrograde pseudorabies virus PRV152 into iWAT of BdnfLacZ/+ mice, GFP-expressing neurons were found in many brain regions. Among brain regions in which the Bdnf gene is deleted in Bdnflox/lox;Emx1Cre/+ mice, we detected many PRV152-infected neurons in layer 5 of the primary (M1) motor cortex (Figure 5A) and some infected neurons in the BLAp (Figure 5D). Colocalization analysis of GFP with β-galactosidase indicated that many PRV152-infected neurons in the M1 cortex and BLAp were BDNF-expressing neurons (Figures 5B, 5C, 5E and 5F). Thus, some BDNF neurons in the M1 layer 5 and BLAp are polysynaptically connected to iWAT.
Figure 5. Some BDNF-expressing neurons were polysynaptically linked to iWAT.
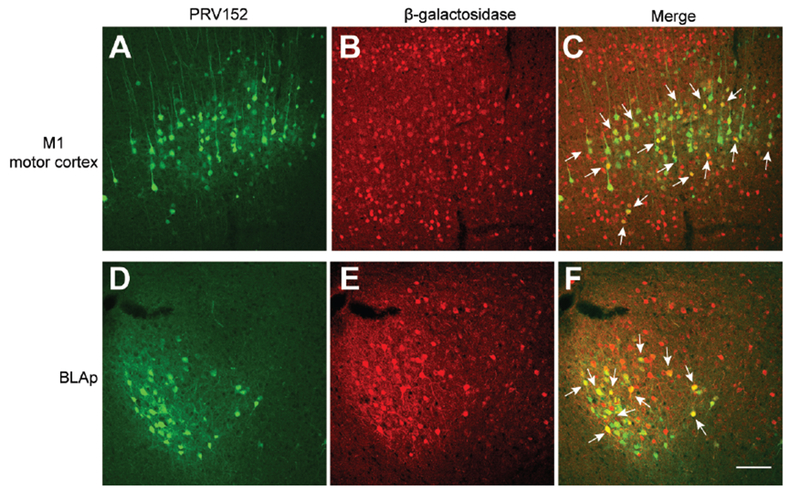
(A-C) Representative images showing infection of BDNF neurons in the M1 motor cortex of male BdnfLacZ/+ mice by GFP-expressing PRV152 injected into the iWAT. M1, primary motor cortex.
(D-F) Representative images showing infection of BDNF neurons in the BLAp of male BdnfLacZ/+ mice by GFP-expressing PRV152 injected into the iWAT. BLAp, posterior part of basolateral amygdala.
Immunohistochemistry against β-galactosidase revealed BDNF-expressing neurons in BdnfLacZ/+ mice. Arrows denote some neurons containing both PRV152 and β-galactosidase. Scale bar, 100 μm.
We next tested whether BDNF expressed in neurons of M1 layer 5 regulates iWAT activities and thereby accounts for iWAT browning and increased adaptive thermogenesis in Bdnflox/lox;Emx1Cre/+ mice. We stereotaxically injected AAV-Cre-GFP virus into M1 layer 5 of 8-week-old male Bdnflox/lox mice to delete the Bdnf gene (Figure S5A). AAV-GFP virus was used as control. Bdnf deletion in this brain region did not have any effect on food intake, energy expenditure and body weight even after 6 weeks of HFD feeding (Figures S5B-S5D). These data suggest that ablation of BDNF expression in neurons of the M1 cortex which are connected to adipose tissues is unlikely responsible for increased energy expenditure and DIO resistance observed in Bdnflox/lox;Emx1Cre/+ mice.
Bdnflox/lox,Emx1Cre/+ mice exhibited anxiety-like behaviors and deficits in GABAergic transmission
We next investigated the possibility that abnormal behaviors could indirectly alter energy balance in Bdnflox/lox;Emx1Cre/+ mice. BDNF modulates synaptic transmission and plasticity (Park and Poo, 2013), and its insufficiency has been linked to cognitive impairment (Bekinschtein et al., 2008; Gray et al., 2006), anxiety-like behaviors (Chen et al., 2006; Rios et al., 2001) and aggression (Ito et al., 2011; Lyons et al., 1999). Anxiety and aggression behaviors could increase energy expenditure because they may activate the autonomic nervous system for the flight or fight response for a short period of time. Deletion of the Bdnf gene in the hippocampal CA3 region was found to elevate aggression without affecting anxiety-like behaviors (Ito et al., 2011).
Bdnflox/lox;Emx1Cre/+ mice were aggressive, likely due to Bdnf deletion in the hippocampus. We had to singly house male mutant mice in order to avoid fighting-related injuries. The KA1-Cre mouse line has been used to delete the Bdnf gene primarily in the CA3 area, resulting in an aggressive mutant (Ito et al., 2011). We generated this mouse mutant and detected the aggression behavior. However, male Bdnflox/lox;KA1-Cre and control mice consumed comparable amounts of calories and gained comparable body weight after 6 weeks of HFD feeding (Figures S6A and S6B), suggesting that aggression is not the reason for low fat mass and resistance to DIO in Bdnflox/lox;Emx1Cre/+ mice.
We then examined whether Bdnflox/lox;Emx1Cre/+ mutant mice displayed anxiety-like behaviors by performing open field and light-dark box behavioral tests. Anxious mice tend to avoid the central zone in open field tests and the light chamber in light-dark box tests. In the first 5 minutes of open field tests, male mutant mice traveled less distance and spent less time in the central zone than male controls (Figures 6A and 6B). Male Bdnflox/lox;Emx1Cre/+ mice also significantly decreased their entries into the central zone compared with male control mice (Figure S6D), which was not a result of reduced locomotion in open field tests (Figure S6C). In light-dark box tests, male Bdnflox/lox;Emx1Cre/+ mice spent less time and moved shorter distance than male controls in the light chamber, and significantly reduced transition number between light and dark chambers (Figures 6C, 6D and S6E). Furthermore, female Bdnflox/lox;Emx1Cre/+ mice also displayed anxiety-like behaviors in these two tests (Figures S6F-S6I). These behavioral results indicate that anxiety is elevated in Bdnflox/lox;Emx1Cre/+ mice.
Figure 6. Elevated anxiety levels and reduced GABAergic transmission in Bdnflox/lox;Emx1Cre/+ mice.
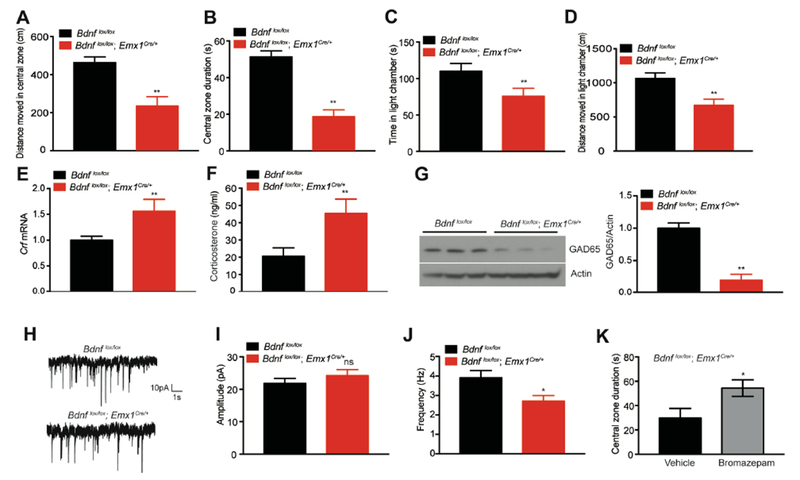
(A and B) Distance moved in central zone and time spent in central zone (central zone duration) during the first 5 minutes of open field tests. N=8-13 mice per group; **p<0.01 by Student’s t test.
(C and D) Time spent and distance moved in the light chamber during light-dark box tests. N=8-13 mice per group; **p<0.01 by Student’s t test.
(E) Gene expression of corticotropin-releasing factor (Crf) in the hypothalamus, determined by qRT-PCR. Hypothalami were dissected from mice fed HFD for 12 weeks. N=6-8 mice per group. **p<0.01 by Student’s t test.
(F) Corticosterone levels in the plasma of HFD-fed male Bdnflox/lox;Emx1Cre/+ and Bdnflox/lox mice. N=8-10 mice per group; **p<0.01 by Student’s t test.
(G) Western blotting analysis and quantification of GAD65 in the cortex. ** p<0.01 by Student’s t test.
(H) Representative traces of mIPSCs in layer 5 pyramidal neurons of the mPFC.
(I) Amplitudes of mIPSCs. N=12-14 neurons from 3 mice per group; ns, not significant by Student’s t test.
(J) Frequency of mIPSCs. N=12-14 neurons from 3 mice per group; * p<0.05 by Student’s t test.
(K) Time spent in central zone during the first 5 minutes of open field tests. Male Bdnflox/lox;Emx1Cre/+ mice were treated with vehicle or bromazepam (50 μg/kg). N=7 mice per group; *p<0.05 by Student’s t test.
All data were presented as mean ± SEM.
Given a well-established connection between hypothalamic corticotropin-releasing factor (CRF) and anxiety-like behavior (Binder and Nemeroff, 2010), we examined CRF expression and production of CRF-regulated corticosterone in Bdnflox/lox;Emx1Cre/+ mice. Mutant mice had significantly higher levels of hypothalamic Crf mRNA and circulating corticosterone (Figures 6E and 6F). Hypothalamic CRF has been shown to be the central player in fibroblast growth factor 21 (FGF21)-mediated increase in the SNA and adaptive thermogenesis in adipose tissues (Owen et al., 2014).
Impairment in BDNF-TrkB signaling reduces levels of the 65-kDa glutamic acid decarboxylase (GAD65), the presynaptic GABA synthesizing enzyme (Kaneko et al., 2012; Sanchez-Huertas and Rico, 2011), and GABAergic deficits are associated with anxiety disorders (Mohler, 2012). We therefore reasoned that Bdnf deletion in the forebrain could impair GABAergic transmission, which then leads to the anxiety phenotype. To investigate this possibility, we first examined the expression of GAD65 and miniature inhibitory postsynaptic currents (mIPSCs) in the cortex. GAD65 levels in the cortex were greatly reduced in Bdnflox/lox;Emx1Cre/+ mice (Figure 6G). Whole-cell recordings in layer 5 pyramidal neurons of the medial prefrontal cortex revealed that Bdnf mutant mice had a lower frequency, but normal amplitude of mIPSCs, compared to control mice (Figures 6H–6J). These results indicate that BDNF deficiency reduces GABA synthesis and consequently probability of GABA release in Bdnflox/lox;Emx1Cre/+ mice. To determine if this GABAergic transmission deficit is linked to elevated anxiety in Bdnf mutants, we administered bromazepam, an enhancer of the GABAA receptor activity, to Bdnflox/lox;Emx1Cre/+ mice. The treatment reduced anxiety levels of these mice, as indicated by an increase in the duration the mice spent in the central zone in open field tests (Figure 6K). Taken together, these results indicate that Bdnf deletion in the forebrain causes anxiety, which is largely due to impaired GABAergic transmission.
Bdnf deletion in the amygdala elevated anxiety and reduced HFD-induced weight gain
The aforementioned results indicate a strong correlation of anxious behaviors and metabolic phenotypes in Bdnflox/lox;Emx1Cre/+ mice. The results, however, do not rule out the possibility that the metabolic phenotypes result from an abnormal behavior we did not detect, because Bdnf deletion occurred in a large part of the brain in these mice and could lead to many abnormal behaviors. To address this issue, we decided to elevate anxiety by deleting Bdnf in a small brain region and determine whether resulting mutant mice still display resistance to DIO. We deleted the Bdnf gene in 8-week-old male Bdnflox/lox mice by stereotaxically injecting AAV-Cre-GFP into the BLA (both BLAa and BLAp), which is critical for the control of anxiety-related behaviors (Calhoon and Tye, 2015; Tovote et al., 2015). Among the mice in which the Cre was expressed in the BLA, we also detected AAV infection in the central nucleus of the amygdala (CeA) and to a lesser extent, the medial amygdala and basomedial amygdala (Figure 7A). We did not notice an increase in fighting between mice injected with AAV-Cre-GFP, indicating that this site-specific Bdnf deletion does not lead to aggression. These mutant mice displayed a reduced probability of entering open arms but traveled similar distance in open arms in elevated plus maze tests (Figures 7B, S7A and S7B) and decreased time spent in the light chamber in light-dark box tests (Figure 7C), compared with control mice injected with AAV-GFP. However, mice injected with AAV-Cre-GFP did not show abnormal behaviors in open field tests (data not shown). These behavioral observations indicate that Bdnf deletion in the BLA and surrounding area increases anxiety levels in mice, but not to the extent as observed in Bdnflox/lox;Emx1Cre/+ mice.
Figure 7. BDNF deficiency in the amygdala causes anxiety and associated metabolic changes.
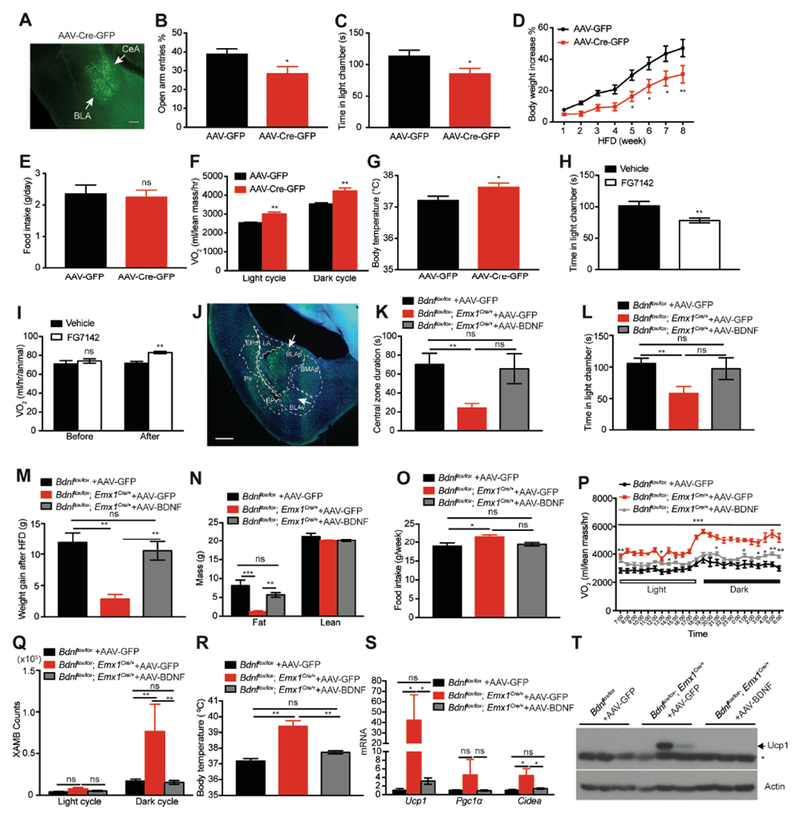
(A) A GFP expression image showing AAV-Cre-GFP infection in the BLA and CeA. Scale bar, 200 μm.
(B) Probability of entering open arms in elevated plus maze tests. N=12-13 mice per group. *p<0.05 by Student’s t test.
(C) Time spent in the light chamber during light-dark box tests. N=12-13 mice per group. *p<0.05 by Student’s t test.
(D) Body weight increase of AAV-injected male Bdnflox/lox mice during 8 weeks of HFD feeding. N=12-13 mice per group. Two-way ANOVA followed by Bonferroni’s test: F(1,168)=43.48, p<0.0001; *p<0.05 and **p<0.01 when comparisons were made at individual time points.
(E) Daily HFD intake of AAV-injected male Bdnflox/lox mice. N=12-13 mice per group. ns, no significance by Student’s t test.
(F) Oxygen consumption (VO2) of AAV-injected male Bdnflox/lox mice during HFD feeding. N=7-8 mice per group; **p<0.01 by Student’s t test.
(G) Core body temperature of AAV-injected male Bdnflox/lox mice during HFD feeding. N=12-13 mice per group; *p<0.05 by Student’s t test.
(H) Time spent in light chamber during light-dark exploration tests after FG7142 treatment (5 mg/kg). N=10 male WT mice per group. **p<0.01 by Student’s t test.
(I) O2 consumption of male C57BL6 WT mice during 1-h period before and 1h after vehicle or FG7142 treatment. N=8 mice per group. ns, no significance; **p<0.01 by Student’s t test.
(J) A representative image showing AAV-BDNF infection in the BLA and surround area. BLAv, ventral part of basolateral amygdala; LA, lateral amygdalar nucleus; EPd, endopiriform nucleus, dorsal part; EPv, endopiriform nucleus, ventral part; Pir, piriform area. Scale bar, 500 μm.
(K) Time spent in the central zone during the first 5 minutes of open field tests. N=7-16 mice per group; **p<0.01 by one-way ANOVA.
(L) Time spent in the light chamber during light-dark box tests. N=7-16 mice per group; **p<0.01 by one-way ANOVA.
(M) Weight gain of AAV-injected male Bdnflox/lox and Bdnflox/lox;Emx1Cre/+ mice after 7 weeks of HFD. N=7-16 mice per group. ns, no significance; **p<0.01 by one-way ANOVA.
(N) Body composition of AAV-injected male Bdnflox/lox and Bdnflox/lox;Emx1Cre/+ mice fed HFD for 4 weeks. N=7-16 mice per group. ns, no significance; **p<0.01 and ***p<0.001 by one-way ANOVA.
(O) Weekly HFD intake of AAV-injected male Bdnflox/lox and Bdnflox/lox;Emx1Cre/+ mice. N=7-16 mice per group. *p<0.05 by one-way ANOVA.
(P) Oxygen consumption (VO2) of AAV-injected male Bdnflox/lox and Bdnflox/lox;Emx1Cre/+ mice during HFD feeding. N=7-16 mice per group; **p<0.01 by two-way ANOVA.
(Q) Ambulatory activity at x axis (XAMB) of AAV-injected male Bdnflox/lox and Bdnflox/lox;Emx1Cre/+ mice during HFD feeding. N=7-16 mice per group. ns, no significance; **p<0.01 by one-way ANOVA.
(R) Core body temperature of AAV-injected male Bdnflox/lox and Bdnflox/lox;Emx1Cre/+ mice during HFD feeding. N=7-16 mice per group. ns, no significance; **p<0.01 by one-way ANOVA.
(S) Gene expression of thermogenic genes in iWAT of AAV-injected male Bdnflox/lox and Bdnflox/lox;Emx1Cre/+ mic. N=7-16 mice per group. ns, no significance; *p<0.05 by one-way ANOVA.
(T) Western blotting analysis of UCP1 in iWAT of AAV-injected male Bdnflox/lox and Bdnflox/lox;Emx1Cre/+ mice after 7 weeks of HFD. * marks nonspecific bands.
All data were presented as mean ± SEM.
Mice injected with AAV-Cre-GFP gained smaller weight than control mice when fed HFD (Figure 7D). This was a result of increased energy expenditure, as these mice ingested normal amounts of food (Figure 7E) but had higher O2 consumption (Figures 7F and S7C) and core body temperature (Figure 7G) compared with control mice. However, the Bdnf deletion in the amygdala did not significantly alter physical activity (Figure S7D), expression of thermogenic genes Ucp1 and Pgc1a in iWAT (Figure S7E), and expression of the Adrb3 gene in iWAT (Figure S7F). Therefore, the Bdnf deletion in the amygdala did not alter adaptive thermogenesis in fat tissues and gave mice some resistance to DIO by increasing basal metabolic rate.
Pharmacologic induction of anxiety acutely enhanced energy expenditure
To further establish the link between anxiety and energy metabolism, we asked if pharmacologically induced anxiety could enhance energy expenditure. We injected FG7142, an inverse agonist of the GABAA receptor and a potent anxiogenic drug (Arrant et al., 2013), into male wild-type (WT) mice to induce anxiety. The treatment induced anxiety (Figures 7H and S7G) and enhanced energy expenditure (Figures 7I and S7H), suggesting that activation of anxiogenic circuits increases energy expenditure. FG7142 treatment, however, had a significant effect on food intake, which makes it difficult to evaluate the effect of chronic FG7142 treatment on DIO.
As described above, our results indicate that impairment in GABAergic transmission contributes to elevated anxiety observed in Bdnflox/lox;Emx1Cre/+ mice (Figures 6G-K). We reasoned that enhancing GABAergic transmission would reduce energy expenditure in Bdnflox/lox;Emx1Cre/+ mice by ameliorating anxiety. Indeed, administration of bromazepam acutely reduced O2 consumption in Bdnf mutant mice (Figures S7I and S7J). We did not examine the effect of chronic bromazepam administration on energy expenditure and body weight of Bdnflox/lox;Emx1Cre/+ mice because we noticed that bromazepam treatment suppressed food intake. Thus, our results suggest a cascade of BDNF deficiency → impaired GABAergic transmission → increased anxiogenic circuit activity → increased energy expenditure.
Viral BDNF expression in the amygdala of Bdnflox/lox,Emx1Cre/+ mice attenuated anxiety and increased HFD-induced weight gain
We described earlier that site-specific Bdnf deletion in the amygdala caused anxiety and some resistance to DIO. This observation suggests that amygdala BDNF is necessary for mood control. To determine if amygdala BDNF is sufficient to regulate mood and normal energy metabolism, we sought to rescue the anxiety and metabolic phenotypes in Bdnflox/lox;Emx1Cre/+ mice by stereotaxically injecting BDNF-expressing AAV into the amygdala. As does the Bdnf gene (An et al., 2008), AAV-BDNF virus expresses Bdnf mRNA variants with either a short or a long 3’ untranslated region (UTR). The virus was delivered to the BLA and surrounding area such as BMAp and endopiriform nucleus (Figure 7J). In agreement with earlier observations (Figures 6A-D), AAV-GFP-injected Bdnflox/lox;Emx1Cre/+ mice displayed higher anxiety levels than AAV-GFP-injected Bdnflox/lox control mice in both open field and light-dark box tests, as indicated by reduced time in the central zone and in the light chamber than control mice (Figures 7K and 7L). Remarkably, AAV-BDNF-injected Bdnflox/lox;Emx1Cre/+ mice showed similar levels of anxiety to AAV-GFP-injected Bdnflox/lox mice in the two behavioral tests (Figures 7K and 7L). These results indicate that viral BDNF expression in the BLA and surrounding area ameliorates anxiety-like behaviors in Bdnflox/lox;Emx1Cre/+ mice.
When fed HFD, AAV-BDNF-injected Bdnflox/lox;Emx1Cre/+ mice drastically gained more weight and had higher fat mass than AAV-GFP-injected Bdnf mutant mice (Figures 7M and 7N, S7K and S7L). Furthermore, viral BDNF expression reduced food intake of mutant mice to a level similar to that of AAV-GFP-injected Bdnflox/lox mice (Figure 7O). These results suggest that AAV-BDNF-injected Bdnf mutant mice have lower energy expenditure than AAV-GFP-injected mutant mice. Indeed, viral BDNF expression significantly reduced energy expenditure at both light and dark cycles in Bdnf mutant mice (Figures 7P and S7M). This batch of Bdnflox/lox;Emx1Cre/+ mice displayed higher locomotion activities than control Bdnflox/lox mice (Figure 7Q). Surprisingly, viral BDNF expression completely rescued the hyper-activity (Figure 7Q). In addition, viral BDNF expression decreased body temperature (Figure 7R) and the expression of thermogenic genes in iWAT (Figures 7S and 7T). The change in energy expenditure in AAV-BDNF-injected Bdnflox/lox;Emx1Cre/+ mice was not due to reduced locomotion activities because there was no correlation between ambulatory activities and weight gain in these mice (Figure S7N).
Taken together, these viral rescue experiments show that BDNF overexpression in the BLA and surrounding area is sufficient to reduce anxiety levels and normalize energy expenditure and HFD-induced weight gain in Bdnflox/lox;Emx1Cre/+ mice. These results not only further show a causal relationship between enhanced activities in anxiogenic circuits and increased energy expenditure, but also indicate a critical role for amygdalar BDNF in mood control.
DISCUSSION
This study reveals that activation of anxiogenic circuits elevate basal metabolic rate of the body and promote adaptive thermogenesis of both brown and white adipose tissues by stimulating sympathetic outflow, leading to increased energy expenditure and leanness. In addition to this main discovery linking anxiogenic circuits to energy metabolism, our results also indicate that BDNF expressed in the amygdala regulates mood through GABAergic transmission.
A causal link between enhanced anxiogenic circuit activity and leanness
Deletion of the Bdnf gene in the whole brain or the hypothalamus or disruption of BDNF-TrkB signaling in the whole body leads to marked hyperphagia and severe obesity in mice (An et al., 2015; Rios et al., 2001; Xu et al., 2003). It is surprising to find that Bdnf deletion in the cortex, hippocampus and some parts of the amygdala led to leanness, high core body temperature and resistance to DIO. In agreement with a previous report that Bdnf deletion in the hippocampal CA3 regions increased aggression (Ito et al., 2011), male Bdnflox/lox;Emx1Cre/+ mice were abnormally aggressive and constantly attacked other males when they were in the same cage. In addition to aggression, we found that Bdnflox/lox;Emx1Cre/+ mice displayed anxiety-like behaviors, indicating the importance of BDNF expressed in Emx1 neurons in the control of moods. We generated CA3-specific Bdnf knockout mice as previously reported (Ito et al., 2011), and male mutant mice were aggressive but did not show leanness. Our physiological and biochemical analyses revealed that the lean phenotype in Bdnflox/lox;Emx1Cre/+ mice resulted from increased sympathetic activity, which elevates basal metabolic rate and adaptive thermogenesis in adipose tissues. As anxiety is known to increase sympathetic activity (Calhoon and Tye, 2015; Kreibig, 2010), these results suggest that BDNF deficiency elevates the activity of anxiogenic circuits, which leads to the lean phenotype in Bdnflox/lox;Emx1Cre/+ mice.
The α2 adrenergic receptor antagonist yohimbine was found to be anxiogenic in mice and rats (Figlewicz et al., 2014; Funk et al., 2008) and increase adaptive thermogenesis and lipolysis during fasting in dogs (Galitzky et al., 1991). However, these pharmacological studies did not establish a link between anxiogenic circuits and energy metabolism; yohimbine could stimulate adaptive thermogenesis and lipolysis by acting on peripheral tissues or central neural circuits that do not control mood, because α2 adrenergic receptor is widely expressed in the nervous system and non-neuronal tissues (Lorenz et al., 1990; MacDonald and Scheinin, 1995).
We employed both genetic and pharmacologic approaches to obtain four lines of evidence in support of a causal link between enhanced activities in anxiogenic circuits and leanness in Bdnflox/lox;Emx1Cre/+ mice. First, we found that site-specific Bdnf deletion in the BLA and surrounding area induced anxiety and conveyed resistance to DIO by boosting basal metabolism and energy expenditure. Second, administration of anxiolytic bromazepam acutely reduced energy expenditure in Bdnflox/lox;Emx1Cre/+ mice. Third, viral BDNF re-expression in the BLA and surrounding area lowered anxiety levels, reduced energy expenditure, and consequently promoted HFD-induced adiposity in Bdnflox/lox;Emx1Cre/+ mice. Lastly, administration of anxiogenic FG7142 acutely induced anxiety and increased energy expenditure in WT mice. This FG7142 result also suggests that our finding about the link between anxiogenic circuits and energy metabolism could be applied to anxiety disorders caused by factors other than BDNF deficiency.
Anxiety induced by site-specific Bdnf deletion in the BLA and surrounding area was not as severe as the one observed in Bdnflox/lox;Emx1Cre/+ mice. This could result from incompleteness of AAV-mediated ablation of BDNF expression in the neural circuits of anxiety. We found, however, that viral BDNF expression in the BLA and surrounding area completely reversed the anxiety phenotype in Bdnflox/lox;Emx1Cre/+ mice. The discrepancy between the results from the deletion and rescue experiments could be attributable to a difference in BDNF expression levels. The AAV-BDNF vector expresses BDNF at a much higher level than the Bdnf gene so that viral transduction of a fraction of neurons at the injection site would produce a sufficient amount of BDNF to ameliorate anxiety. One study reported that Bdnf deletion in Emx1 neurons did not elevate anxiety (Gorski et al., 2003), which could be a result of utilization of a hypomorphic floxed Bdnf allele (termed Bdnfklox) that does not express the long 3’ UTR Bdnf mRNA (An et al., 2008). Mice homozygous for this Bdnf conditional allele (Bdnfklox/klox) display deficits in synaptic plasticity and severe obesity without any Cre recombinase (An et al., 2008; Liao et al., 2012). Because Bdnfklox/klox mice have an established phenotype, it would be difficult to interpret the effect of an additional deletion. We found that both basal metabolic rate and thermogenesis were elevated in Bdnflox/lox;Emx1Cre/+ mice, whereas Bdnf deletion in the BLA and surrounding area only increased basal metabolic rate. This observation suggests that the threshold for the activity of anxiogenic circuits is higher to stimulate thermogenesis than to increase basal metabolic rate.
How do our findings reconcile with reports that anxiety is associated with increased body mass index in children? Whether anxiety leads to leanness or obesity could be dependent on the cause of the disorder. Several brain structures, such as bed nucleus of the stria terminalis and paraventricular hypothalamus (PVH), are involved in the control of mood and food intake (Betley et al., 2013; Calhoon and Tye, 2015; Xu and Xie, 2016). Dysfunction in some neuronal populations could lead to anxiety disorders as well as hyperphagic obesity.
A critical role for amygdala BDNF in mood control
We found that deletion of the Bdnf gene in the amygdala increased anxiety levels whereas viral expression of BDNF in the amygdala of Bdnflox/lox;Emx1Cre/+ mice lessened anxiety-like behaviors. In these two gene expression manipulations the BLA was most heavily transduced by AAV. Furthermore, the Bdnf gene in the BLA is also deleted in Bdnflox/lox;Emx1Cre/+ mice. These results strongly indicate that BDNF-expressed in the amygdala, especially the BLA, plays an essential role in the control of mood. However, interference of BDNF expression with antisense oligodeoxynucleotides in the CeA and medial amygdala, but not the BLA, was found to provoke anxiety-like behaviors in rats (Pandey et al., 2006). This discrepancy could result from a difference in species used in the studies and off-target effects of antisense oligodeoxynucleotides. Nevertheless, both of the studies implicate deficiency of amygdalar BDNF in anxiety. Further studies are warranted to define the brain structures where BDNF is produced to modulate mood.
Impaired GABAergic transmission is associated with anxiety disorders (Mohler, 2012). Our results suggest that deficits in GABAergic transmission lead to elevated anxiety in Bdnflox/lox;Emx1Cre/+ mice. We found that the Bdnf mutant mice had much lower GAD65 levels and fewer mIPSC events in the cortex than control mice. Furthermore, enhancing GABAergic transmission with bromazepam reduced anxiety levels in Bdnflox/lox;Emx1Cre/+ mice. Future studies on identification of GABAergic neurons that are regulated by amygdalar BDNF would help delineate the neural circuits of anxiety.
Neural circuits linking anxiety to energy expenditure
Acute psychological stress transiently activates BAT thermogenesis and increases core body temperature (Oka, 2015), which involves the canonical thermoregulation neural circuit from neurons in the dorsomedial hypothalamus to the SNS via sympathetic premotor neurons in the rostral medullary raphe region (rMR) (Kataoka et al., 2014). In addition to activating BAT thermogenesis, we found that activation of anxiogenic circuits also increased thermogenesis in iWAT by stimulating development of beige adipocytes in Bdnflox/lox;Emx1Cre/+ mice. This is somewhat similar to persistent psychological stress induced by severe burn injury, which leads to iWAT browning in both mice and humans (Porter et al., 2015; Sidossis et al., 2015). These findings suggest that anxiety-related worrying in the absence of real threats and trauma induced by severe stressors have the same impact on the SNS and adipose tissue thermogenesis. In addition, our results show that activation of anxiogenic circuits by Bdnf deletion also increased basal metabolic rate. Therefore, neural circuits that link anxiety and psychological stress to energy metabolism are likely to be overlapping but distinct.
Neurons that link anxiety and energy metabolism should be at the interface of mood-controlling circuit and metabolism-controlling circuit. Using retrograde pseudorabies virus, we identified two groups of neurons that are polysynaptically connected to iWAT and in which BDNF expression is abolished in Bdnflox/lox;Emx1Cre/+ mice: M1 layer 5 neurons and BLA neurons. The polysynaptic connection from M1 cortex to iWAT has been observed before (Bartness et al., 2014; Stanley et al., 2010; Zeng et al., 2015). As no evidence implicates the M1 motor cortex in the control of mood (Calhoon and Tye, 2015; Tovote et al., 2015), M1 layer 5 neurons do not satisfy the requisite as neurons linking anxiety and energy metabolism. Indeed, we found that deleting Bdnf in the M1 motor cortex had no effect on energy expenditure. Although it will be important to better understand the function of the connection between the motor cortex and iWAT in future studies, our results do not support dysfunction of this connection as the cause for the lean phenotype observed in Bdnflox/lox;Emx1Cre/+ mice. Conversely, BDNF-expressing neurons in the BLA appear to be good candidate neurons that link anxiety to metabolism. These neurons have been shown to be part of the neural circuitry of anxiety (Calhoon and Tye, 2015). Furthermore, our viral experiments suggest that altering BDNF expression in the BLA changes anxiety levels and energy metabolism.
It is very likely that the canonical thermoregulation pathway is a critical part of the neural circuit linking anxiety and adaptive thermogenesis. We found that Crf expression in the hypothalamus is significantly increased in Bdnflox/lox;Emx1Cre/+ mice. Neurons in the PVH release CRF to promote production of corticotropin in the anterior pituitary, which in turns stimulates synthesis and release of glucocorticoids in the adrenal gland. Additionally, CRF acts on neurons expressing CRF receptors throughout the CNS (Perrin and Vale, 1999). Interestingly, CRF has been found to mediate the stimulatory effect of FGF21 on adaptive thermogenesis (Bookout et al., 2013; Owen et al., 2014). Therefore, the action of CRF in the brain could contribute to the increase in basic metabolic rate and adaptive thermogenesis as a result of increased activities in anxiogenic circuits. We notice an interesting phenomenon in Bdnflox/lox;Emx1Cre/+ mice: the enhancement in adaptive thermogenesis is accompanied with compensatory food intake increase, while the increase in basal metabolic rate does not alter food intake. This suggests that increasing basal metabolic rate could be more effective than augmenting adaptive thermogenesis in treating obesity. Furthermore, basal metabolic rate accounts for ~70% of total daily energy expenditure in sedentary individual (Ravussin and Bogardus, 1992). Thus, elucidation of the neural circuit linking anxiety to basal metabolic rate could uncover a novel strategy to develop effective therapeutic interventions of obesity.
Limitations of study
Our Bdnf deletion and re-expression experiments strongly indicate that the BLA is a brain region that produces BDNF to regulate mood and to link anxiety with high energy expenditure. However, our study does not rule out the possibility that the two functions are carried out by BDNF expressed in a nearby brain region, because AAV injected into the BLA spread to surrounding structures in our experiments. Definitive identification of the amygdalar nucleus in future studies is necessary for elucidation of neural circuits that control mood and energy expenditure. The neural circuits of anxiety are complex and consist of neurons in multiple brain regions (Calhoon and Tye, 2015; Tovote et al., 2015). Anxiety disorders as a result of neuronal dysfunction at different nodes of the circuits may not have the same impact on energy expenditure and body weight.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Baoji Xu (bxu@scripps.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
All mice were on the C57BL/6 background. Bdnflox/+ (Stock No: 004339), Emx1Cre/+ (Stock No: 005628) and KA1-Cre (Stock No: 006474) mouse strains (Gorski et al., 2002; Nakazawa et al., 2002; Rios et al., 2001) were obtained from the Jackson Laboratory (Bar harbor, ME). Bdnfklox/+ and BdnfLacZ/+ mouse strains were previously described (An et al., 2008; Liao et al., 2012). Unless specified otherwise, mice were maintained at 22°C with a 12/12 hr light/dark cycle and had free access to water and food. For thermoneutral experiments, mice were housed at 30°C with a 12/12 hr light/dark cycle and had free access to water and food. The chow diet (2920×, Teklad Diets) and HFD (D12492, Research Diets) contain 16% and 60% of calories from fat, respectively. Male Bdnflox/lox;Emx1Cre/+ mice, male Bdnflox/lox;KA1-Cre mice and their male Bdnflox/lox littermate controls were singly housed after weaning due to aggressive behaviors of mutant mice. All other mice were housed in group. All animal procedures were approved by the Animal Care and Use Committee at Scripps Florida.
METHOD DETAILS
Construction of BDNF-overexpressing AAV vector
To generate pAAV-CMV-BDNF-Myc plasmid, the CMV-BDNF-Myc fragment was cut out of pBK-CMV-BDNF-Myc construct (Liao et al., 2012) and then inserted into pAAV vector at NotI sites. The resultant pAAV-CMV-BDNF-Myc was used to produce AAV-BDNF (serotype 2) by Vigene Biosciences (Rockville, MD). An antibody against Myc was used in immunohistochemistry to detect BDNF overexpression at injected sites.
Administration of anxiogenic and anxiolytic drugs
β-Carboline-3-carboxylic acid N-methylamide (FG7142) and 7-bromo-5-(2-pyridyl)-3H-1,4-benzodiazepin-2(1H)-one (bromazepam) were purchased from Sigma-Aldrich (St. Louis, MO). FG7142 was dissolved in saline with 0.3% Tween 80 (Chaki et al., 2003) and bromazepam was dissolved in saline (Uriguen et al., 2004). Both FG7142 (5 mg/kg) and bromazepam (5 μg/kg) were intraperitoneally injected into mice 20 min before behavioral tests.
Immunoblotting analysis
As previously described (Mansuy-Aubert et al., 2013), animal tissues were harvested and homogenized in lysis buffer containing 1% Triton X-100, 50 mM Hepes (pH 7.4), 137 mM NaCl and proteinase inhibitor cocktail (Roche). In general, 20-30 μg of total protein was resolved in SDS/PAGE and electro-transferred to PVDF membranes. Primary antibodies were incubated with membranes at 4°C overnight. Secondary antibodies (HRP) were incubated with membranes at room temperature for 1 hr. An ECL detection system was applied to develop signals.
Physiological measurements
Measurement of food intake, body weight, core body temperature, and fat pads was carried out as described previously (An et al., 2015; Liao et al., 2012). Body composition was assessed using a Minispec LF-50/mq 7.5 NMR Analyzer (Brucker Optics). VO2 and locomotor activity were monitored with a comprehensive lab animal monitoring system (CLAMS, Columbus Instruments). Circulating norepinephrine and corticosterone levels were measured using a Norepinephrine ELISA kit (Abnova, # KA1891) and a Corticosterone ELISA kit (Enzo Life Sciences, # ADI-900097), respectively.
Histology and Immunohistochemistry
Adipose tissues were fixed with 4% paraformaldehyde and embedded in paraffin. H&E staining was performed on paraffin-embedded tissue sections by the Histology Core at Scripps Florida. Liver samples were flash-frozen, and sections were prepared for both H&E and Oil Red O staining by the Histology Core at Scripps Florida. For brain immunohistochemistry, animals were anesthetized and perfused with 4% paraformaldehyde. Coronal brain sections were prepared at 40-μm thickness using a microtome. Immunohistochemistry was performed as previously described (Liao et al., 2012). The following primary antibodies were used for immunohistochemistry: rabbit polyclonal antibody against β-galactosidase (#55976; Cappel) and rabbit polyclonal antibody against UCP1 (ab10983, Abcam).
qRT-PCR
qRT-PCR was performed as previously described (Mansuy-Aubert et al., 2013). Briefly, tissues were homogenized in TRIzol (Invitrogen), and total RNA was extracted, reverse-transcribed to cDNA with random primers, and quantified with real-time PCR.
IPGTT and IPITT
Intraperitoneal glucose tolerance test (IPGTT) and intraperitoneal insulin tolerance test (IPITT) were performed as described (Mansuy-Aubert et al., 2013). For IPGTT, animals were fasted overnight (16 h) and received an intraperitoneal injection of glucose (1.0 g/kg body weight). For IPITT, animals were fasted for 6 h and injected intraperitoneally with insulin (1.5 U/kg body weight). Blood was collected from the tail vain at different time points after glucose or insulin injection. A portable glucometer (OneTouch Ultra) was used to measure glucose concentrations.
Stereotaxic injection of AAV
AAV was stereotaxically injected into specific brain sites as described previously (An et al., 2015). In brief, mice were anesthetized with isoflurane before surgery. AAV-GFP, AAV-Cre-GFP (serotype 2, UNC Vector Core) or AAV-BDNF viruses were injected bilaterally into the M1 motor cortex or BLA of at least 8-week-old male Bdnflox/lox or Bdnflox/lox;Emx1Cre/+ mice using a Hamilton syringe with a 33-gauge needle. The coordinate for the M1 motor cortex was AP = −0.5 mm, ML = ± 1.25 mm and DV = −1.30 mm. The coordinate for the BLA was AP = −1.5 mm, ML = ± 3.07 mm and DV = −5.15 mm. Following injections, mice received Metacam (1 mg / kg) for analgesia and were returned to their home cages.
Retrograde trans-neuronal tracing.
Recombinant GFP-expressing PRV-Bartha PRV152 was used in the trans-neuronal tracing study as previously described (Stanley et al., 2010). Ten-week-old male BdnfLacZ/+ mice were anesthetized with isoflurane, and 5 μl of PRV152 was injected into the right inguinal white fat pad with a Hamilton syringe. The needle was left in the fat pad for 1-2 additional minutes. Seven days after PRV152 injection, mice were perfused with 4% PFA. Coronal brain sections at 40-μm thickness were stained with an antibody against β-galactosidase.
In situ Hybridization
Radioactive in situ hybridization of brain sections was performed using 35S-labeled riboprobes as described before (Xu et al., 2003).
Anxiety-like behavioral tests
All behavioral tests were performed during the active phase as previously described (Chen et al., 2006; Muller et al., 2003). Prior to behavioral tests, mice were handled for at least 3 consecutive days to minimize stress during experiments. For light-dark box tests, a mouse was put into the light chamber first and its behavior was recorded for 5 min using a sensitive video recorder that was connected to a computer running EthoVision software. We quantified the time animals spent in each chamber, total distance mice moved in the light chamber, as well as the total number of entries into each chamber. In open field tests, a mouse was placed at the center of the open arena and its subsequent behavior was recorded using a sensitive video recorder that was connected to a computer running EthoVision software for a total of 30 min. To measure anxiety levels, we quantified the total distance moved in the central zone, time spent in the central zone, and total number of entries into the central zone during the first 5 min. In elevated plus-maze tests, a mouse was put at the center of the plus maze and its total behavior was recorded using a sensitive video recorder that was connected to a computer running EthoVision software for 5 minutes. We quantified the total distance moved in the open arms, percentage of time spent in the open arms, and percentage of entries into open arms.
Electrophysiology
Mice at P28-P35 were used for electrophysiological recordings. Mice were transcardially perfused under isoflurane anesthesia with oxygen-saturated ice-cold cutting solution containing (mM): 124 choline chloride, 26 NaHCO3, 2.5 KCl, 3.3 MgCl2, 1.2 NaH2PO4, 0.5 CaCl2 and 1 D-glucose. 300-μm medial prefrontal cortical (mPFC) coronal slices were obtained in cold cutting solution using a vibratome (Leica VT 1200S, Germany) and then transferred to artificial cerebrospinal fluid (aCSF) composed of (mM): 124 NaCl, 3 KCl, 26 NaHCO3, 1.25 NaH2PO4, 1 MgSO4, 2 CaCl2 and 10 D-glucose, equilibrated with 95% O2 and 5% CO2 at 32 °C for recovery.
Slices were incubated in oxygenated aCSF at 32 °C for at least 1 hour, and then maintained at room temperature (22-25°C) for another 30 min before recording. The mPFC slices were gently transferred to a recording chamber (RC-27, Warner Instruments, Hamden, CT) at room temperature. The chamber was perfused with circulated oxygenated aCSF at a flow rate of 2-3 ml/min. Prelimbic layer 5 pyramidal neurons were visually identified in the slices using infrared-differential interference contrast microscope (Scientifica, UK). Whole-cell patch-clamp recordings were made through borosilicate glass pipettes (ID: 0.68 mm, OD: 1.2 mm, WPI, Sarasota, FL) of 3-5 MΩ pulled with a micropipette puller (P-1000; Sutter Instrument, Novato, CA). For mIPSCs, the recording pipettes were filled with internal solution containing (in mM): 100 CsCl, 35 CsMeSO3, 10 Hepes, 0.5 EGTA, 4 MgATP, 0.3 Na3GTP, 5 QX-314, and 10 Na2-Phosphocreatine (pH 7.3 with CsOH, osmolarity 285 mM). mIPSCs were recorded by holding the neurons at −70 mV in voltage-clamp mode without series resistance and liquid junction compensation. Only neurons with Ra < 25 MΩ were recorded. In order to isolate mIPSCs, we used 1 μM tetrodotoxin (TTX), 20 μM CNQX and 50 μM APV.
Experimenters were blind to genotypes. Signals were acquired with Multiclamp 700B and Digidata 1550A (Molecular Devices, San Jose, CA). Data were low-pass filtered at 2.9 KHz and sampled at 10 kHz. Analysis of miniature events were performed using Clampfit 10.6 software. Template search method was used using a template match threshold of 2 and manually selecting responses. First 200 responses, which are above 6 pA, were used to calculate average event amplitude and frequency for each recording (Ozkan et al., 2014).
QUANTIFICATION AND STATISTICAL ANALYSIS
All data were presented as mean ± SEM. Statistical significance was analyzed using unpaired Student’s t test (Excel, 2011) or one or two-way ANOVA (GraphPad Prism 7). p < 0.05 was considered statistically significant. All the data were exported into Adobe Illustrator CS6 for preparation of figures.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-UCP1 | Abcam | Cat# ab10983 RRID:AB_2241462 |
| Mouse monoclonal anti-GAD65 | Abcam | Cat# ab26113, RRID:AB_448989 |
| Rabbit polyclonal anti-UCP1 | ThermoFisher Scientific | Cat# PA1-24894, RRID:AB_2241459 |
| Rabbit polyclonal anti-HSL | Cell Signaling Technology | Cat# 4107 RRID:AB_2296900 |
| Rabbit polyclonal anti-phospho-HSL (Ser563) | Cell Signaling Technology | Cat# 4139, RRID:AB_2135495 |
| Mouse monoclonal anti-Myc-Tag (9B11) | Cell Signaling Technology | Cat# 2276, RRID:AB_331783 |
| Mouse monoclonal anti-β-Actin | Sigma-Aldrich | Cat# A5441, RRID:AB_476744 |
| Mouse monoclonal anti-α-Tubulin | Sigma-Aldrich | Cat# T8203, RRID:AB_1841230 |
| Rabbit polyclonal anti-Tyrosine Hydroxylase | Sigma-Aldrich | Cat# AB152, RRID:AB_390204 |
| Rabbit polyclonal anti-β-galactosidase | Cappel/ICN Pharmaceuticals | Cat# 55976, RRID:AB_2313707 |
| Goat anti-Rabbit IgG (H+L) Secondary Antibody, HRP | ThermoFisher Scientific | Cat# A16110, RRID:AB_2534782 |
| Goat anti-Mouse IgG (H+L) Secondary Antibody, HRP | ThermoFisher Scientific | Cat# 31430, RRID:AB_228307 |
| Alexa Fluor® 594 AffiniPure Goat anti-Rabbit IgG, F(ab’)2 fragment specific | Jackson ImmunoResearch Labs | Cat# 111-585-006, RRID:AB_2338060 |
| Biotinylated Goat anti-Rabbit IgG Antibody | Vector Laboratories | Cat# BA-1000, RRID:AB_2313606 |
| Bacterial and Virus Strains | ||
| AAV-GFP (AAV2-CMV-GFP) | University of North Carolina’s Vector Core | N/A |
| AAV-Cre-GFP (AAV2-CMV-Cre-GFP) | University of North Carolina’s Vector Core | N/A |
| AAV-BDNF (AAV2-CMV-BDNF-Myc) | This paper | N/A |
| PRV152 | NIH Center for Neuroanatomy with Neurotropic Viruses | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| β-Carboline-3-carboxylic acid N-methylamide (FG7142) | Sigma-Aldrich | Cat# E006-100MG |
| 7-bromo-5-(2-pyridyl)-3H-1,4-benzodiazepin-2(1H)-one (bromazepam) | Sigma-Aldrich | Cat# B4144-100MG |
| Insulin (Humulin) (IPITT) | Eli Lilly | Cat# VI7510 |
| CNQX | Tocris | Cat# 0190 |
| DL-APV | Sigma-Aldrich | Cat# A5282-10MG |
| Tetrodotoxin (TTX) | American Radiolabeled Chemicals | Cat# 50-753-2807 |
| Tween 80 | Sigma-Aldrich | Cat# P1754-25ML |
| Trizo | ThermoFisher Scientific | Cat# 15596018 |
| D-(+)-Glucose (IPGTT) | Sigma-Aldrich | Cat# G8270-1kg |
| FastStart Universal SYBR Green Master (Rox) | Roche | Cat# 4913914001 |
| Critical Commercial Assays | ||
| Norepinephrine ELISA Kit | Abnova | Cat# KA1891 |
| Corticosterone ELISA kit | Enzo Life Sciences | Cat# ADI-900-097 |
| Deposited Data | ||
| Raw and analyzed data | This paper | |
| Experimental Models: Organisms/Strains | ||
| C57BL/6J WT mice | The Jackson Laboratory | JAX: 000664 |
| Bdnflox/+ mice | The Jackson Laboratory | JAX: 004339 |
| Emx1Cre/+ mice | The Jackson Laboratory | JAX: 005628 |
| KA1-Cre mice | The Jackson Laboratory | JAX: 006474 |
| Bdnfklox/+ mice | The Jackson Laboratory | JAX: 021055 |
| BdnfLacZ/+ mice | Liao et al., 2012 | N/A |
| Oligonucleotides | ||
| A full list of Primers is in Table S1 | This paper | N/A |
| Software and Algorithms | ||
| Clampex 10.6 | Molecular devices | https://www.moleculardevices.com |
| Clampfix 10.6 | Molecular devices | https://www.moleculardevices.com |
| GraphPad Prism7 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| ImageJ bundled with Java 1.8.0_172 | NIH ImageJ | https://imagej.nih.gov/ij/ |
| EthoVision XT 11.5 | Noldus | https://www.noldus.com/animal-behavior-research/products/ethovision-xt |
| Adobe CS6 | Adobe | https://www.adobe.com |
| Excel (2011) | Microsoft | https://www.microsoft.com/enus/download/office.aspx |
| Stepone software | Applied Biosystems | https://www.thermofisher.com/us/en/home/technical-resources/softwaredownloads/StepOne-and-StepOnePlus-Real-Time-PCR-System.html |
| NIS-Elements software | Nikon | https://www.nikoninstruments.com/Products/Software |
| Other | ||
| Regular Chow Diet | Harlan Teklad | Cat# 2920× |
| High Fat Diet | Research Diets | Cat# D12492 |
ACKNOWLEDGEMENTS
We thank Tingting Xia for assistance in tissue collection, Clint E. Kinney for help in mouse surgery, and Shaw-wen Wu for critical comments on the manuscript. This work was supported by the National Institutes of Health grants to BX (R01 DK103335 and R01 DK105954): and JH (F32 NS106810). HX and ZXX were partially supported by a Training Grant in Alzheimer’s Drug Discovery from the Lottie French Lewis Fund of the Community Foundation for Palm Beach and Martin Counties. PRV152 was provided by the Center for Neuroanatomy with Neurotropic Viruses, which was supported by an NIH grant (P40 RR018604).
Footnotes
DECLARATION OF INTERESTS
The authors declare that they have no competing financial interests.
REFERENCES
- An JJ, Gharami K, Liao GY, Woo NH, Lau AG, Vanevski F, Torre ER, Jones KR, Feng Y, Lu B, et al. (2008). Distinct role of long 3’ UTR BDNF mRNA in spine morphology and synaptic plasticity in hippocampal neurons. Cell 134, 175–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An JJ, Liao GY, Kinney CE, Sahibzada N, and Xu B (2015). Discrete BDNF Neurons in the Paraventricular Hypothalamus Control Feeding and Energy Expenditure. Cell Metab 22, 175–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson SE, Cohen P, Naumova EN, and Must A (2006). Association of depression and anxiety disorders with weight change in a prospective community-based study of children followed up into adulthood. Arch Pediatr Adolesc Med 160, 285–291. [DOI] [PubMed] [Google Scholar]
- Arrant AE, Schramm-Sapyta NL, and Kuhn CM (2013). Use of the light/dark test for anxiety in adult and adolescent male rats. Behav Brain Res 256, 119–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachman ES, Dhillon H, Zhang CY, Cinti S, Bianco AC, Kobilka BK, and Lowell BB (2002). betaAR signaling required for diet-induced thermogenesis and obesity resistance. Science 297, 843–845. [DOI] [PubMed] [Google Scholar]
- Bartness TJ, Liu Y, Shrestha YB, and Ryu V (2014). Neural innervation of white adipose tissue and the control of lipolysis. Frontiers in neuroendocrinology 35, 473–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekinschtein P, Cammarota M, Katche C, Slipczuk L, Rossato JI, Goldin A, Izquierdo I, and Medina JH (2008). BDNF is essential to promote persistence of long-term memory storage. Proc Natl Acad Sci U S A 105, 2711–2716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betley JN, Cao ZF, Ritola KD, and Sternson SM (2013). Parallel, redundant circuit organization for homeostatic control of feeding behavior. Cell 155, 1337–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder EB, and Nemeroff CB (2010). The CRF system, stress, depression and anxiety-insights from human genetic studies. Mol Psychiatry 15, 574–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bookout AL, de Groot MH, Owen BM, Lee S, Gautron L, Lawrence HL, Ding X, Elmquist JK, Takahashi JS, Mangelsdorf DJ, et al. (2013). FGF21 regulates metabolism and circadian behavior by acting on the nervous system. Nat Med 19, 1147–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calhoon GG, and Tye KM (2015). Resolving the neural circuits of anxiety. Nat Neurosci 18, 1394–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaki S, Kawashima N, Suzuki Y, Shimazaki T, and Okuyama S (2003). Cocaine- and amphetamine-regulated transcript peptide produces anxiety-like behavior in rodents. Eur J Pharmacol 464, 49–54. [DOI] [PubMed] [Google Scholar]
- Charmandari E, Tsigos C, and Chrousos G (2005). Endocrinology of the stress response. Annual review of physiology 67, 259–284. [DOI] [PubMed] [Google Scholar]
- Chen ZY, Jing D, Bath KG, leraci A, Khan T, Siao CJ, Herrera DG, Toth M, Yang C, McEwen BS, et al. (2006). Genetic variant BDNF (Val66Met) polymorphism alters anxiety-related behavior. Science 314, 140–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis M, Walker DL, Miles L, and Grillon C (2010). Phasic vs sustained fear in rats and humans: role of the extended amygdala in fear vs anxiety. Neuropsychopharmacology 35, 105–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figlewicz DP, Hill SR, Jay JL, West CH, Zavosh AS, and Sipols AJ (2014). Effect of recurrent yohimbine on immediate and post-hoc behaviors, stress hormones, and energy homeostatic parameters. Physiol Behav 129, 186–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funk D, Li Z, Coen K, and Le AD (2008). Effects of pharmacological stressors on c-fos and CRF mRNA in mouse brain: relationship to alcohol seeking. Neurosci Lett 444, 254–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galitzky J, Vermorel M, Lafontan M, Montastruc P, and Berlan M (1991). Thermogenic and lipolytic effect of yohimbine in the dog. Br J Pharmacol 104, 514–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garland T Jr., Schutz H, Chappell MA, Keeney BK, Meek TH, Copes LE, Acosta W, Drenowatz C, Maciel RC, van Dijk G, et al. (2011). The biological control of voluntary exercise, spontaneous physical activity and daily energy expenditure in relation to obesity: human and rodent perspectives. The Journal of experimental biology 214, 206–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorski JA, Balogh SA, Wehner JM, and Jones KR (2003). Learning deficits in forebrain-restricted brain-derived neurotrophic factor mutant mice. Neuroscience 121, 341–354. [DOI] [PubMed] [Google Scholar]
- Gorski JA, Talley T, Qiu M, Puelles L, Rubenstein JL, and Jones KR (2002). Cortical excitatory neurons and glia, but not GABAergic neurons, are produced in the Emx1-expressing lineage. J Neurosci 22, 6309–6314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray J, Yeo GS, Cox JJ, Morton J, Adlam AL, Keogh JM, Yanovski JA, El Gharbawy A, Han JC, Tung YC, et al. (2006). Hyperphagia, severe obesity, impaired cognitive function, and hyperactivity associated with functional loss of one copy of the brain-derived neurotrophic factor (BDNF) gene. Diabetes 55, 3366–3371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han JC, Liu QR, Jones M, Levinn RL, Menzie CM, Jefferson-George KS, Adler-Wailes DC, Sanford EL, Lacbawan FL, Uhl GR, et al. (2008). Brain-derived neurotrophic factor and obesity in the WAGR syndrome. N Engl J Med 359, 918–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harms M, and Seale P (2013). Brown and beige fat: development, function and therapeutic potential. Nat Med 19, 1252–1263. [DOI] [PubMed] [Google Scholar]
- Huang EJ, and Reichardt LF (2001). Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci 24, 677–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito W, Chehab M, Thakur S, Li J, and Morozov A (2011). BDNF-restricted knockout mice as an animal model for aggression. Genes Brain Behav 10, 365–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajimura S, Spiegelman BM, and Seale P (2015). Brown and Beige Fat: Physiological Roles beyond Heat Generation. Cell Metab 22, 546–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko M, Xie Y, An JJ, Stryker MP, and Xu B (2012). Dendritic BDNF synthesis is required for late-phase spine maturation and recovery of cortical responses following sensory deprivation. The Journal of neuroscience : the official journal of the Society for Neuroscience 32, 4790–4802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kataoka N, Hioki H, Kaneko T, and Nakamura K (2014). Psychological stress activates a dorsomedial hypothalamus-medullary raphe circuit driving brown adipose tissue thermogenesis and hyperthermia. Cell Metab 20, 346–358. [DOI] [PubMed] [Google Scholar]
- Kreibig SD (2010). Autonomic nervous system activity in emotion: a review. Biol Psychol 84, 394–421. [DOI] [PubMed] [Google Scholar]
- Liao GY, An JJ, Gharami K, Waterhouse EG, Vanevski F, Jones KR, and Xu B (2012). Dendritically targeted Bdnf mRNA is essential for energy balance and response to leptin. Nat Med 18, 564–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz W, Lomasney JW, Collins S, Regan JW, Caron MG, and Lefkowitz RJ (1990). Expression of three alpha 2-adrenergic receptor subtypes in rat tissues: implications for alpha 2 receptor classification. Mol Pharmacol 38, 599–603. [PubMed] [Google Scholar]
- Lyons WE, Mamounas LA, Ricaurte GA, Coppola V, Reid SW, Bora SH, Wihler C, Koliatsos VE, and Tessarollo L (1999). Brain-derived neurotrophic factor-deficient mice develop aggressiveness and hyperphagia in conjunction with brain serotonergic abnormalities. Proc Natl Acad Sci U S A 96, 15239–15244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald E, and Scheinin M (1995). Distribution and pharmacology of alpha 2-adrenoceptors in the central nervous system. J Physiol Pharmacol 46, 241–258. [PubMed] [Google Scholar]
- Mansuy-Aubert V, Zhou QL, Xie X, Gong Z, Huang JY, Khan AR, Aubert G, Candelaria K, Thomas S, Shin DJ, et al. (2013). Imbalance between neutrophil elastase and its inhibitor alpha1-antitrypsin in obesity alters insulin sensitivity, inflammation, and energy expenditure. Cell Metab 17, 534–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohler H (2012). The GABA system in anxiety and depression and its therapeutic potential. Neuropharmacology 62, 42–53. [DOI] [PubMed] [Google Scholar]
- Morton GJ, Meek TH, and Schwartz MW (2014). Neurobiology of food intake in health and disease. Nat Rev Neurosci 15, 367–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller MB, Zimmermann S, Sillaber I, Hagemeyer TP, Deussing JM, Timpl P, Kormann MS, Droste SK, Kuhn R, Reul JM, et al. (2003). Limbic corticotropin-releasing hormone receptor 1 mediates anxiety-related behavior and hormonal adaptation to stress. Nat Neurosci 6, 1100–1107. [DOI] [PubMed] [Google Scholar]
- Nakazawa K, Quirk MC, Chitwood RA, Watanabe M, Yeckel MF, Sun LD, Kato A, Carr CA, Johnston D, Wilson MA, et al. (2002). Requirement for hippocampal CA3 NMDA receptors in associative memory recall. Science 297, 211–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedergaard J, and Cannon B (2014). The browning of white adipose tissue: some burning issues. Cell Metab 20, 396–407. [DOI] [PubMed] [Google Scholar]
- Oka T (2015). Psychogenic fever: how psychological stress affects body temperature in the clinical population. Temperature (Austin) 2, 368–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen BM, Ding X, Morgan DA, Coate KC, Bookout AL, Rahmouni K, Kliewer SA, and Mangelsdorf DJ (2014). FGF21 acts centrally to induce sympathetic nerve activity, energy expenditure, and weight loss. Cell Metab 20, 670–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozkan ED, Creson TK, Kramar EA, Rojas C, Seese RR, Babyan AH, Shi Y, Lucero R, Xu X, Noebels JL, et al. (2014). Reduced cognition in Syngap1 mutants is caused by isolated damage within developing forebrain excitatory neurons. Neuron 82, 1317–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey SC, Zhang H, Roy A, and Misra K (2006). Central and medial amygdaloid brain-derived neurotrophic factor signaling plays a critical role in alcohol-drinking and anxiety-like behaviors. J Neurosci 26, 8320–8331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H, and Poo MM (2013). Neurotrophin regulation of neural circuit development and function. Nat Rev Neurosci 14, 7–23. [DOI] [PubMed] [Google Scholar]
- Perrin MH, and Vale WW (1999). Corticotropin releasing factor receptors and their ligand family. Ann N Y Acad Sci 885, 312–328. [DOI] [PubMed] [Google Scholar]
- Porter C, Herndon DN, Bhattarai N, Ogunbileje JO, Szczesny B, Szabo C, Toliver-Kinsky T, and Sidossis LS (2015). Severe Burn Injury Induces Thermogenically Functional Mitochondria in Murine White Adipose Tissue. Shock 44, 258–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravussin E, and Bogardus C (1992). A brief overview of human energy metabolism and its relationship to essential obesity. Am J Clin Nutr 55, 242S–245S. [DOI] [PubMed] [Google Scholar]
- Rios M, Fan G, Fekete C, Kelly J, Bates B, Kuehn R, Lechan RM, and Jaenisch R (2001). Conditional deletion of brain-derived neurotrophic factor in the postnatal brain leads to obesity and hyperactivity. Mol Endocrinol 15, 1748–1757. [DOI] [PubMed] [Google Scholar]
- Rofey DL, Kolko RP, Iosif AM, Silk JS, Bost JE, Feng W, Szigethy EM, Noll RB, Ryan ND, and Dahl RE (2009). A longitudinal study of childhood depression and anxiety in relation to weight gain. Child Psychiatry Hum Dev 40, 517–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothwell NJ, and Stock MJ (1984). Effects of denervating brown adipose tissue on the responses to cold, hyperphagia and noradrenaline treatment in the rat. The Journal of physiology 355, 457–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Huertas C, and Rico B (2011). CREB-Dependent Regulation of GAD65 Transcription by BDNF/TrkB in Cortical Interneurons. Cereb Cortex 21, 777–788. [DOI] [PubMed] [Google Scholar]
- Sidossis LS, Porter C, Saraf MK, Borsheim E, Radhakrishnan RS, Chao T, Ali A, Chondronikola M, Mlcak R, Finnerty CC, et al. (2015). Browning of Subcutaneous White Adipose Tissue in Humans after Severe Adrenergic Stress. Cell Metab 22, 219–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soliman F, Glatt CE, Bath KG, Levita L, Jones RM, Pattwell SS, Jing D, Tottenham N, Amso D, Somerville LH, et al. (2010). A genetic variant BDNF polymorphism alters extinction learning in both mouse and human. Science 327, 863–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley S, Pinto S, Segal J, Perez CA, Viale A, DeFalco J, Cai X, Heisler LK, and Friedman JM (2010). Identification of neuronal subpopulations that project from hypothalamus to both liver and adipose tissue polysynaptically. Proc Natl Acad Sci U S A 107, 7024–7029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovote P, Fadok JP, and Luthi A (2015). Neuronal circuits for fear and anxiety. Nat Rev Neurosci 16, 317–331. [DOI] [PubMed] [Google Scholar]
- Uriguen L, Perez-Rial S, Ledent C, Palomo T, and Manzanares J (2004). Impaired action of anxiolytic drugs in mice deficient in cannabinoid CB1 receptors. Neuropharmacology 46, 966–973. [DOI] [PubMed] [Google Scholar]
- Xu B, Goulding EH, Zang K, Cepoi D, Cone RD, Jones KR, Tecott LH, and Reichardt LF (2003). Brain-derived neurotrophic factor regulates energy balance downstream of melanocortin-4 receptor. Nat Neurosci 6, 736–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu B, and Xie X (2016). Neurotrophic factor control of satiety and body weight. Nat Rev Neurosci 17, 282–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng W, Pirzgalska RM, Pereira MM, Kubasova N, Barateiro A, Seixas E, Lu YH, Kozlova A, Voss H, Martins GG, et al. (2015). Sympathetic neuro-adipose connections mediate leptin-driven lipolysis. Cell 163, 84–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.