

Abstract
Currently speaking, it is noted that radiofrequency ablation (RFA) has been the most widely used treatment for hepatocellular carcinoma (HCC) occurring in patients. However, accumulating evidence has demonstrated that the incidence of insufficient RFA (IRFA) may result in the identified rapid progression of residual HCC in the patient, which can greatly hinder the effectiveness and patient reported benefits of utilizing this technique. Although many efforts have been proposed, the underlying mechanisms triggering the rapid progression of residual HCC after IRFA have not yet been fully clarified through current research literature reviews. It was shown in this study that cell proliferation, migration and invasion of residual HepG2 and SMMC7721 cells were significantly increased after the IRFA was simulated in vitro. In other words, it is noted that IRFA could do this by enhancing the image of autophagy of the residual HCC cell via the HIF-1α/BNIP3 pathway. Consequently, the down-regulation of BNIP3 may result in the inhibition of the residual HCC cell progression and autophagy after IRFA. Our present study results suggest that IRFA could promote residual HCC cell progression in vitro by enhancing autophagy via the HIF-1α/BNIP3 pathway. For this reason, it is noted that the targeting of the BNIP3 may be useful in preventing the rapid growth and metastasis of residual HCC after IRFA.
Keywords: Autophagy, Hepatocellular carcinoma, Radiofrequency ablation, Residual
INTRODUCTION
To begin with, it is known that hepatocellular carcinoma (HCC) worldwide ranks as the sixth most common cancer as occurring in patients, and is identified as the second leading cause of cancer mortality in all patients globally (1). In this relation, the radical treatment for HCC includes the utilization of a surgical resection, liver transplantation and local ablation therapy (2). However, surgical resection and liver transplantation are severely undermined and moderately used in these cases, owning to some limitations, such as for example, the shortage of donor organs, poor hepatic reservation, advanced neoplastic stage or multifocal distribution of tumor nodules (3). Currently, radiofrequency ablation (RFA) has been widely used for HCC in these cases, due to its definitive therapeutic effect, safety, cost-effectiveness, and minimal invasiveness as a surgical procedure for the patients (4, 5). However, insufficient RFA (IRFA) may result in the rapid progression of residual HCC, thus greatly hindering the effectiveness of this technique for the benefit of the patient. The underlying mechanisms triggering this phenomenon have not been fully clarified to date, although many efforts have been made in the past to identify the triggers to this condition to improve patient outcomes (6–10).
This brings us to understand that after liver tumor RFA, two zones will be produced in the ablation area. The central section identified in this region is the coagulating necrosis zone, which is characterized as without any living cells in it, while the surrounding tissue is referenced as a migration band, which may exist with residual tumor cells. All ablated foci will be in an absolutely or relatively found in an anoxic state after a long period of developed RFA (11). Compared with their parental cells, the residual HCC cells in this hypoxic microenvironment might exhibit changes including an enhanced invasive, metastatic, and chemoresistant abilities, as well as exhibiting mesenchymal characteristics (12). In this case, the hypoxia inducible factor-1 alpha (HIF-1α), only expressed highly in hypoxic microenvironment, has been found to correlate with lymph node metastasis and advanced pathological stage in HCC (13). We have demonstrated previously that HIF-1α could induce angiogenesis which plays an important role in the rapid growth of residual HCC after IRFA (14). In this context, it is noted that sorafenib in combination with RFA could improve the treatment of HCC for the reason that sorafenib suppresses cell proliferation and induces apoptosis in hepatoma cells by HIF-1α/VEGFA pathway (15, 16). In these terms, BNIP3 is a transcriptional target of HIF-1α that is upregulated in response to hypoxia (17). Additionally, BNIP3 was reported to be strongly associated with autophagy, which prevents tumor cell from surviving in a stressed environment (18, 19). The Bellot, et al. (20) study confirmed that when tumor cells were subjected to hypoxia (1% O2) or severe hypoxia (0.1% O2) treatment, the expression of BNIP3 and BNIP3 ligand (BNIP3L) mediated by HIF-1α contributed to the occurrence of mitochondrial autophagy, thereby preventing cell death as it relates to the patient.
As has been seen, autophagy is an evolutionarily conserved lysosome-dependent catabolic process which degrades a cell’s components in order to recycle substrates to exert optimally and adapt to conditions of starvation, hypoxia or infection (21). Recently, a study reported that IRFA can induce autophagy, leading to residual HCC cell proliferation, which could be inhibited by hydroxychloroquine, which characteristically acts as a kind of autophagy inhibitor (22). Based on these studies, we hypothesized that IRFA could induce autophagy through the HIF-1α/BNIP3 signaling pathway, which may contribute to the rapid progression of residual HCC.
RESULTS
siRNA efficacy analysis in vitro
In this context, it is important that the assay can distinguish between siRNAs with different efficacies against a respective target. Likewise, to test for BNIP3 siRNA target-specific efficacy, a negative control siRNA (NC-siRNA) was analyzed to provide a benchmark for off-target effects on BNIP3 knockdown. In addition, we examined three BNIP3 siRNAs to target human BNIP3 as described in the referenced Supplemental Table. Our results showed that the BNIP3 mRNA was significantly inhibited in the siRNA1, 2 and 3 group respectively (P < 0.001, Supplemental Fig. 1). Additionally, the inhibitory effect of the siRNA 1 was the most obvious among the three different siRNA group (P < 0.001, Supplemental Fig. 1). In this repect, based on these results, we selected BNIP3 siRNA 1 as the most highly functional siRNA for further investigation in vitro.
Down-regulation of BNIP3 can reverse the proliferation, migration and invasion of HCC cells promoted by IRFA
In what follows, the first experiments aimed at looking at the morphological changes which occur in HCC cells treated by IRFA, following BNIP3 siRNA transfections. In this case, the HepG2 and SMMC7721 cells were randomly divided into three groups, namely, 37°C-si-NC (37°C-NC-siRNA) group, 47°C-si-NC (47°C-NC-siRNA) group, and 47°C-si-BNIP3 (47°CBNIP3-siRNA) group. Briefly, the HepG2 and SMMC7721 cells were submerged in a water bath at 37°C for 10 min. Next, after being exposed to the aforementioned heat treatment, the cells were transfected with NC-siRNA. The procedure of the other two groups were similar to the procedure highlighted and referenced above in this report. It is noted that the differences recorded were that the temperature of bath was 47°C as well as cells were transfected with NC-siRNA and BNIP3-siRNA respectively. It was found that the cells in group 37°C-si-NC had been adhered to the wall, the morphology was normal, the nucleus was intact, and the cell organelle was considered to be fine. As compared with group 37°C-si-NC, polymorphism of the cell morphology and increased vacuoles of the cells could be seen in the 47°C-si-NC group. Furthermore, the number of vacuoles of HepG2 cells was obviously reduced in the 47°C-si-BNIP3 group in comparison with the 47°C-si-NC group (Supplemental Fig. 2).
In the best effort to detect the heat treatment effect on tumor cell proliferation, we studied the cell viability at 0 h, 24 h, 48 h, and 72 h. It was found that compared with 37°C-si-NC group, the cell vitality in 47°C-si-NC group increased gradually. In addition, there was a statistically significant difference between the two groups at 72 h (1.00 ± 0.01 Vs. 1.12 ± 0.02, P < 0.01, Fig. 1). Notably, in contrast to 47°C-si-NC group, the cell viability decreased significantly in the 47°C-si-BNIP3 group at 72 h (P < 0.001, Fig. 1). It was therefore revealed that IRFA can promote the proliferation of HepG2 cells to a certain extent, which can be inhibited by suppressing the BNIP3 expression.
Fig. 1.
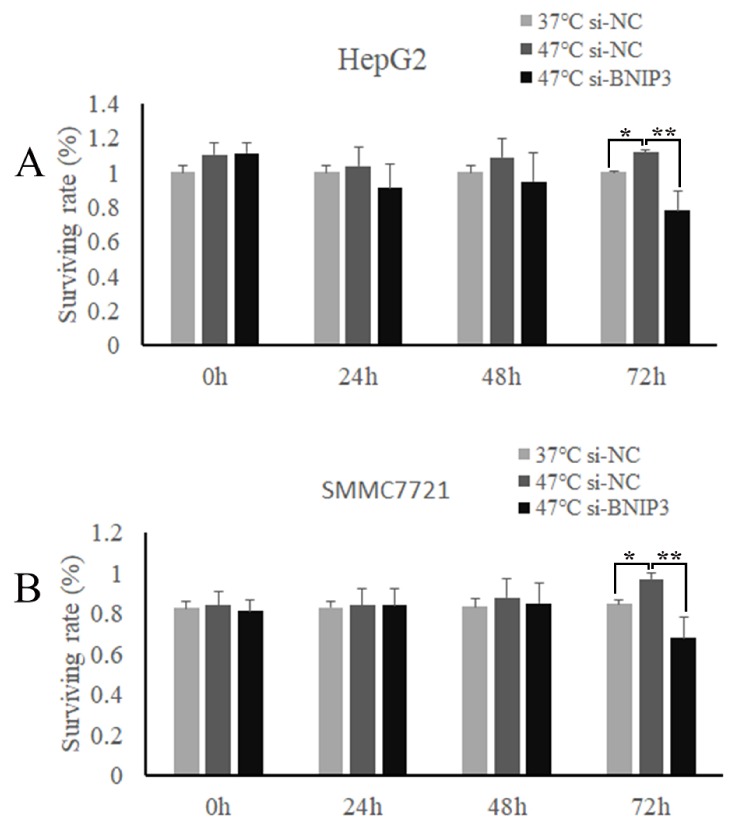
The HepG2 (A) and SMMC7721 (B) cell viability after heat treatment at 0 h, 24 h, 48 h, and 72 h. *P < 0.01, 47°C-si-NC group VS. 37°C-si-NC group. **P < 0.001, 47°C-si-BNIP3 group VS. 47°Csi-NC group.
To evaluate the effect of IRFA on the migration and invasion of HepG2 cells, the cells of 3 groups (group 37°C-si-NC, group 47°C-si-NC, group 47°C-si-BNIP3) received heat treatment as described previously. It was found that the HepG2 cells in 47°C-si-NC group displayed enhanced migration and invasion abilities in comparison with the cells in the other two groups (P < 0.01, Fig. 2). However, it was also noted that the migration and invasion abilities of the HepG2 cells would decrease dramatically when their BNIP3 was specifically knocked down (Fig. 2). A review of these results further indicated that the heat treatment of simulating IRFA can promote the migration and invasion of HepG2 cells, while it can be inhibited by the down-regulation of BNIP3.
Fig. 2.

The migration and invasion results of HepG2 and SMMC7721 cells. (A, C) The migration results of HepG2 cells. (E, G) The migration results of SMMC7721 cells. (B, D) The invasion results of HepG2 cells. (F, H) The invasion results of SMMC7721 cells. *P < 0.01, 47°C-si-NC group VS. 37°C-si-NC group. **P < 0.001, 47°C-si-BNIP3 group VS. 47°C-si-NC group.
IRFA promotes residual HCC cell progression via autophagy, which can be inhibited by down-regulation of BNIP3
As is noted, compared with the cells of the 37°C-si-NC group, significantly more autophagic vacuoles were observed in the cells of the 47°C-si-NC group (Fig. 3A and 3F). Simply stated, the expression of LC3B-II, a marker of autophagy, in 47°C-si-NC group was significantly increased in comparison with that in 37°C-si-NC group, indicating that IRFA could induce autophagy in tumor cells. In comparison, the levels of HIF-1α/BNIP3 in the HepG2 cells of the 47°C-si-NC group were seen to have been markedly increased. However, when the BNIP3 was knocked down, the levels of HIF-1α as well as the LC3 were significantly decreased and the autophagy was exhibited as inhibited in that case (Fig. 3).
Fig. 3.
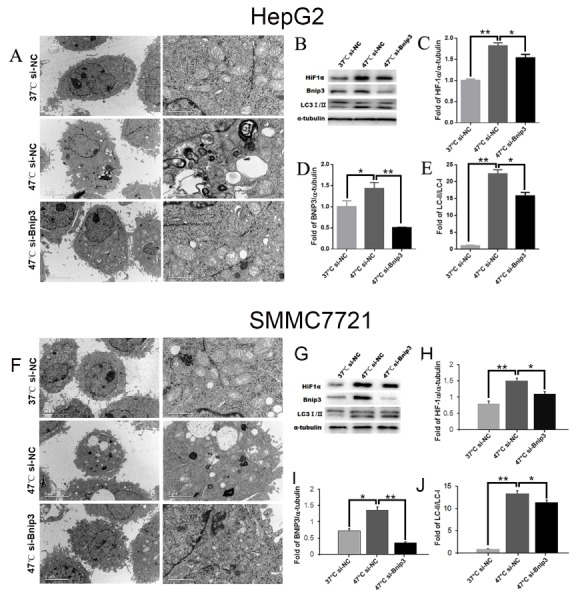
The results of the electron microscope and the western blotting. (A, F) The results of the electron microscope. (B–E, G–J) The expression of HIF-1α, BNIP3, LC3-I/LC3-II and the western blotting. *P < 0.01 and **P < 0.001.
To further confirm the influence of IRFA on autophagy and the changes of mitochondria, we applied the TOM20, a kind marker protein of chondriosome, and LC-antibody to Immunofluorescence assay. As we expected, the tumor cells in the 47°C-si-NC group, simulated of IRFA, had the further quantity of identified mitochondria as well as the higher expression of LC3 comparable to 37°C-si-NC group. Similar to the above referenced results, after we knocked down the BNIP3 of the cells, the number of mitochondria reduced significantly, and results were similar or the same as LC3 in the 47°C-si-BNIP3 group (Fig. 4).
Fig. 4.
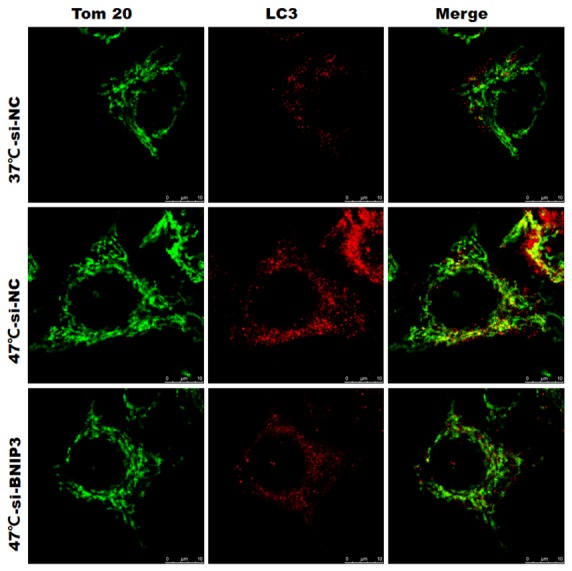
The results of the immunofluorescence.
DISCUSSION
Currently, RFA, as well as hepatectomy and liver transplantation, has been considered as one of the three major methods of curing HCC in patients diagnosed with this condition. However, the rapid progression of residual liver tumor after IRFA has been observed in many clinical centers (23–25), and has been a major impediment to the development of this technique. A recent study revealed that the identified epidermal growth factor receptor transactivation served as an important mechanism underlying IRFA induced HCC malignancy (6). Additionally, it was revealed that another study reported that the suboptimal RFA accelerated residual tumor proliferation by inducing the overexpression of ITGB3 which was mediated by FAK/PI3K/AKT/MMP2 signaling pathway (7). Consequently, our previous research has certified by a VX2 carcinoma rabbit model that residual hepatic VX2 carcinoma could facilitate its rapid progression and lung metastases after IRFA (10). In addition, we also found that epithelial-mesenchymal transition (EMT) involving the AKT/ERK signaling pathways play a vital role in the invasiveness and metastasis of the residual tumor cells after IRFA (26). We endeavor to note that it was demonstrated that the angiogenesis induced by HIF1a/VEGFA produced by altered cells after hyperthermia treatment may involve in the rapid growth of residual HCC (14). Although many efforts have been proposed, the mechanisms of rapid growth of the residual liver tumor after IRFA is not yet well understood to date. Our present study showed that the incidence of cell proliferation, migration and invasion of residual HepG2 cells was significantly increased after the IRFA simulated in vitro, which was consistent with the previous study (14, 26). What’s more, IRFA could promote residual HCC cell progression by enhancing autophagy via the HIF-1α/BNIP3 pathway.
To our knowledge, autophagy is an evolutionarily conserved catabolic process and cumulative studies have shed light on the importance of autophagy in cancer (27, 28). Interestingly, it noted that there is a double-edged sword of autophagy modulation in cancer which has been proposed as a viable theory in these cases (29). Autophagy may promote or prevent tumorigenesis, depending on the consequence and utilization of autophagy (30). In other words, autophagy defection has been found in human HCC (31). Another study reported that the activation of autophagy could promote metastasis and glycolysis in HCC cells (32). The Zhao et al. (22) study has reported that IRFA promotes proliferation of residual HCC via autophagy. However, as far as we know, there are fewer mechanisms researched and identified regarding this phenomenon until now. It was demonstrated in this study that the IRFA-induced residual HCC autophagy involves in the HIF-1α/BNIP3 pathway. For this reason, it is noted that HIF-1α is an important mediator of the hypoxia response and it regulates the expression of various chemokines involved in tumor growth, angiogenesis and metastasis. Notably, it is determined that BNIP3, a BH3-only Bcl-2 family member, can regulate the expression of autophagy-related genes including Beclin1 and Atg5 (33), and the upregulation of BNIP3 contributes to anoikis resistance of HCC cell which has shown to be associated with the metastasis of malignant cells (18). In this study, it was indicated that the residual HCC cell progression and autophagy after IRFA could be inhibited by the down-regulation of BNIP3.
Mitophagy, a selective form of autophagy, has been demonstrated that it can selectively remove damaged or unwanted mitochondria for both mitochondrial quantity and quality control in response to the presence of hypoxic conditions (34). It has been reported that IRFA can generate a chronically hypoxic tissue zone where the malignant phenotype could be cultivated, which also includes local invasion, metastatic progression, and chemoresistance (35). Many studies have demonstrated that HIF-1α/BNIP3 which can induce mitophagy under the condition of hypoxia (36–38). It is therefore revealed in this study that mitophagy can be induced by IRFA, as shown from the changes of LC3 and TOM20. In addition, the mitophagy as well as the expression of HIF-1α was inhibited when the BNIP3 was knocked down by using siRNA. However, further experiments will be needed in this area to confirm these and other similar results as noted.
There is a limitation to our study as will be described next in this report. Our study can be criticized for the lack of assessment of the outcomes in vivo. Within the limitation, our study does add to the current understanding of the underlying mechanisms related to the rapid progression of residual HCC after IRFA. We believed that the novel results presented in this manuscript would be of interest to clinical researchers reviewing and researching on this an ancillary topics. Our present results suggest that IRFA could promote residual HCC cell progression in vitro by enhancing autophagy via the HIF-1α/BNIP3 pathway. For this reason, the targeting of the BNIP3 may be useful in preventing the rapid growth and metastasis of residual HCC after IRFA.
MATERIALS AND METHODS
Cell culture
In this study, the HepG2 cells, a human hepatoma cell line, were brought from Shanghai Fu Xiang Biotechnology Co. Ltd. An established human HCC cell line, SMMC7721 was brought from the American Type Culture Collection (ATCC; Manassas, VA, USA). All of the cells were cultured in Dulbecco’s modified Eagle’s with high glucose supplement (DMEM, GIBCO, UK) containing 10% fetal bovine serum (FBS) in a humidified incubator at 37°C with an atmosphere of 5% CO2.
Small Interfering RNA (siRNA) and Transfection Assays
According to the human BNIP3 sequence, the siRNA of BNIP3 was designed, containing 3 interference fragments and 1 control fragments. To this end, the sequence information is shown in the Supplemental Table. Upon review, the HepG2 and SMMC7721 cells in 6-well plates were transfected with the siRNA containing the gene BNIP3 by using Lipofectamine 2000 (Invitrogen; Shanghai, China) according to the manufacturer’s instructions. The expression of BNIP3 was confirmed by Real-time PCR, using Glyceraldehydes 3-phosphate dehydrogenase (GAPDH) as the normalized control. The sequences of primers to determine the expression of the target gene were listed as follows: BNIP3 [5′-GTTCCAGCCTCGGTTTCTATT-3′ (forward); 5′-CCAATGCTATGGGTATCTGTTTC-3′ (reverse)] and GADPH [5′-GGAGCGAGATCCCTCCAAAAT-3′ (forward); 5′-GGCTGTTGTCATACTTCTCATGG -3′ (reverse)].
Heat treatment
IRFA was simulated in vitro using a previously described method (26). Briefly, HepG2 and SMMC7721 cells were seeded into 6 well plates (5 × 104 cells/well). After 24 h, the plates were sealed and submerged in a water bath at 47°C for 5 min. Subsequently, the cells were cultured in RPMI 1640 medium supplemented with 10% FBS in a humidified atmosphere of 5% CO2 at 37°C until residual populations reached 80% confluence. The surviving populations were propagated into the 6 well plates and exposed to the aforementioned heat treatment for 10 min. Then, it was noted that this process was repeated, and the cells were sequentially exposed to the above referenced heat treatment for 15 min, 20 min and 25 min. At that time, it was noted that the surviving cells were the residual tumor cells.
Cell proliferation
In this case, the proliferation was measured using a Cell Counting Kit-8 (CCK-8; Engreen (EC008)). Briefly, at that time the cell was cultured in 96-well plates at a concentration of 4 × 103 well, and each of them contained a culture medium of 100 μl. The cells were then cultured in the cell culture box, and the corresponding plasmid was transfected after the cells were adhered to the wall. After incubation for 0, 24, 48 or 72 h, 10 μl of CCK-8 reagent was added to each well. The absorbance was measured by the use of an enzyme-labeled instrument (PerkinElmer; USA) after 4 h incubation at 37°C.
Cell migration, invasion assay
The ability of the cell’s migration and invasion was assessed by transwell assay (Costar, USA). Briefly, 1 × 106 cells in serum-free DMEM were seeded into the upper chamber of each well of 24-well plates. DMEM containing 10% FBS was added to the lower chamber of each well. The cells were then allowed to migrate for 24 h at 37°C. At that time, the non-migrated cells were removed from the upper surface of the membrane by scraping with a cotton swab, and the migrating cells were fixed with methanol for 30 min, stained with crystal violet (Beyotime, Nantong, China) and photographed under an inverted fluorescence microscope (Olympus IX51) equipped with an Olympus Qcolor 3 digital camera (Olympus). The cell invasion assay was carried out similarly, except that 50 μl Matrigel (Corning, New York, USA) was added to each well 2 h before the cells were seeded on the membrane, the results were examined after 20 h.
Electron microscopy
After the designated treatments, the HepG2 and SMMC7721 cells were promptly fixed with 2.5% glutaraldehyde containing 0.1 mol/L sodium cacodylate and stored at 4°C. After fixation, the cells were dehydrated in a gradient of 50%–100% ethanol and embedded, and ultrathin (50–60 nm) sections were cut using an ultramicrotome (Leica Ultracut; Leica Microsystems, Wetzlar, Germany). The samples were then examined with an electron microscope (JEM-1400; JEOL, Tokyo, Japan) at 80 kV.
Western blotting
A Western blot was performed as described previously (39). Briefly, HepG2 and SMMC7721 cells were washed twice with phosphate-buffered saline (PBS) and lysed with lysis buffer [150 mM NaCl, 50 mM Tris–HCl (pH 8.0), 0.1% SDS, 1% Triton X-100]. Then, the cells were harvested using a cell scraper. Next, the cell lysates were centrifuged at 12,000 rpm for 5 min and the protein content was determined using a Bicinchoninic acid (BCA) protein assay kit. Following this procedure, the samples were then adjusted to contain 40 μg protein each; subsequently, they were subjected to SDS-PAGE and electrophoretically transferred to a polyvinylidene fluoride membrane. Subsequently, the membranes were blocked with 5% non-fat milk for 2 h and then incubated with respective primary antibody (HIF-1α, BNIP3, LC3-I/LC3-II, α-tubulin) overnight at 4°C followed by the incubation with the appropriate HRP-conjugated secondary antibody for 2 h at room temperature. Next, the blots were visualized with an ECL detection kit (Pierce, USA) and analyzed using Quantity One 1-D Analysis Software (Bio-Rad, Hercules, USA).
Immunofluorescence
The cells in this case were cultured on glass coverslips and then fixed in 4% paraformaldehyde for 15 min at room temperature and washed with PBS for 3 times. Then, the samples were permeabilized with 0.2% Triton X-100 for 10 min. For the detection of mitochondria, cells were incubated with the TOM20 antibody (1:200) for one night at RT. After being washed, the cells were incubated with anti-mouse secondary antibody for 10 min at room temperature. As to the referenced LC3, all of the procedures are similar to what has been stated above. The detection of the fluorescence was performed with a laser confocal fluorescence microscopy.
Statistical analysis
As indicated, Student’s t test or ANOVA test was used for comparison of the differences between groups using GraphPad Prism (GraphPad Software Inc, La Jolla, CA). P < 0.05 was considered statistically significant.
SUPPLEMENTARY INFORMATION
ACKNOWLEDGEMENTS
This work was partly supported by the National Natural Science Foundation of China (81572957, 81502650), the West Campus of Beijing Chaoyang Hospital Incubating Program (JXPY201605), and the Beijing Municipal Administration of Hospitals Incubating Program (PX2017014).
Footnotes
CONFLICTS OF INTEREST
The authors have no conflicting interests.
REFERENCES
- 1.Tang A, Hallouch O, Chernyak V, Kamaya A, Sirlin CB. Epidemiology of hepatocellular carcinoma: target population for surveillance and diagnosis. Abdom Radiol (NY) 2018;43:13–25. doi: 10.1007/s00261-017-1209-1. [DOI] [PubMed] [Google Scholar]
- 2.Daher S, Massarwa M, Benson AA, Khoury T. Current and Future Treatment of Hepatocellular Carcinoma: An Updated Comprehensive Review. J Clin Transl Hepatol. 2018;6:69–78. doi: 10.14218/JCTH.2017.00031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Feng K, Ma KS. Value of radiofrequency ablation in the treatment of hepatocellular carcinoma. World J Gastroenterol. 2014;20:5987–5998. doi: 10.3748/wjg.v20.i20.5987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nishikawa H, Kimura T, Kita R, Osaki Y. Radiofrequency ablation for hepatocellular carcinoma. Int J Hyperthermia. 2013;29:558–568. doi: 10.3109/02656736.2013.821528. [DOI] [PubMed] [Google Scholar]
- 5.Bruix J, Gores GJ, Mazzaferro V. Hepatocellular carcinoma: clinical frontiers and perspectives. Gut. 2014;63:844–855. doi: 10.1136/gutjnl-2013-306627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dai H, Jia G, Wang H, Yang J, Jiang H, Chu M. Epidermal growth factor receptor transactivation is involved in the induction of human hepatoma SMMC7721 cell proliferation by insufficient radiofrequency ablation. Oncol Lett. 2017;14:2463–2467. doi: 10.3892/ol.2017.6463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang N, Ma D, Wang L, et al. Insufficient Radiofrequency Ablation Treated Hepatocellular Carcinoma Cells Promote Metastasis by Up-Regulation ITGB3. J Cancer. 2017;8:3742–3754. doi: 10.7150/jca.20816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dong S, Kong J, Kong F, et al. Sorafenib suppresses the epithelial-mesenchymal transition of hepatocellular carcinoma cells after insufficient radiofrequency ablation. BMC Cancer. 2015;15:939. doi: 10.1186/s12885-015-1949-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kong J, Kong L, Kong J, et al. After insufficient radiofrequency ablation, tumor-associated endothelial cells exhibit enhanced angiogenesis and promote invasiveness of residual hepatocellular carcinoma. J Transl Med. 2012;10:230. doi: 10.1186/1479-5876-10-230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ke S, Ding XM, Kong J, et al. Low temperature of radiofrequency ablation at the target sites can facilitate rapid progression of residual hepatic VX2 carcinoma. J Transl Med. 2010;8:73. doi: 10.1186/1479-5876-8-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Onishi H, Matsushita M, Murakami T, et al. MR appearances of radiofrequency thermal ablation region: histopathologic correlation with dog liver models and an autopsy case. Acad Radiol. 2004;11:1180–1189. doi: 10.1016/j.acra.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 12.Lee DH, Lee JM, Yoon JH, Kim YJ, Han JK. Thermal Injury-induced Hepatic Parenchymal Hypoperfusion: Risk of Hepatocellular Carcinoma Recurrence after Radiofrequency Ablation. Radiology. 2017;282:880–891. doi: 10.1148/radiol.2016152322. [DOI] [PubMed] [Google Scholar]
- 13.Xiang ZL, Zeng ZC, Fan J, Tang ZY, Zeng HY, Gao DM. Gene expression profiling of fixed tissues identified hypoxia-inducible factor-1alpha, VEGF, and matrix metalloproteinase-2 as biomarkers of lymph node metastasis in hepatocellular carcinoma. Clin Cancer Res. 2011;17:5463–5472. doi: 10.1158/1078-0432.CCR-10-3096. [DOI] [PubMed] [Google Scholar]
- 14.Kong J, Kong J, Pan B, et al. Insufficient radiofrequency ablation promotes angiogenesis of residual hepatocellular carcinoma via HIF-1alpha/VEGFA. PLoS One. 2012;7:e37266. doi: 10.1371/journal.pone.0037266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu M, Zheng YL, Xie XY, et al. Sorafenib blocks the HIF-1alpha/VEGFA pathway, inhibits tumor invasion, and induces apoptosis in hepatoma cells. DNA Cell Biol. 2014;33:275–281. doi: 10.1089/dna.2013.2184. [DOI] [PubMed] [Google Scholar]
- 16.Gong Q, Qin Z, Hou F. Improved treatment of early small hepatocellular carcinoma using sorafenib in combination with radiofrequency ablation. Oncol Lett. 2017;14:7045–7048. doi: 10.3892/ol.2017.7174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chourasia AH, Tracy K, Frankenberger C, et al. Mitophagy defects arising from BNip3 loss promote mammary tumor progression to metastasis. EMBO Rep. 2015;16:1145–1163. doi: 10.15252/embr.201540759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sun L, Li T, Wei Q, et al. Upregulation of BNIP3 mediated by ERK/HIF-1alpha pathway induces autophagy and contributes to anoikis resistance of hepatocellular carcinoma cells. Future Oncol. 2014;10:1387–1398. doi: 10.2217/fon.14.70. [DOI] [PubMed] [Google Scholar]
- 19.Awan MU, Hasan M, Iqbal J, et al. Neuroprotective role of BNIP3 under oxidative stress through autophagy in neuroblastoma cells. Mol Biol Rep. 2014;41:5729–5734. doi: 10.1007/s11033-014-3444-7. [DOI] [PubMed] [Google Scholar]
- 20.Bellot G, Garcia-Medina R, Gounon P, et al. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol. 2009;29:2570–2581. doi: 10.1128/MCB.00166-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu L, Liao JZ, He XX, Li PY. The role of autophagy in hepatocellular carcinoma: friend or foe. Oncotarget. 2017;8:57707–57722. doi: 10.18632/oncotarget.17202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao Z, Wu J, Liu X, et al. Insufficient radiofrequency ablation promotes proliferation of residual hepatocellular carcinoma via autophagy. Cancer Lett. 2018;421:73–81. doi: 10.1016/j.canlet.2018.02.024. [DOI] [PubMed] [Google Scholar]
- 23.Shiozawa K, Watanabe M, Takahashi M, Wakui N, Iida K, Sumino Y. Analysis of patients with rapid aggressive tumor progression of hepatocellular carcinoma after percutaneous radiofrequency ablation. Hepatogastroenterology. 2009;56:1689–1695. [PubMed] [Google Scholar]
- 24.Seki T, Tamai T, Ikeda K, et al. Rapid progression of hepatocellular carcinoma after transcatheter arterial chemoembolization and percutaneous radiofrequency ablation in the primary tumour region. Eur J Gastroenterol Hepatol. 2001;13:291–294. doi: 10.1097/00042737-200103000-00014. [DOI] [PubMed] [Google Scholar]
- 25.Pua U. Rapid intra-hepatic dissemination of hepatocellular carcinoma with pulmonary metastases following combined loco-regional therapy. Korean J Radiol. 2013;14:640–642. doi: 10.3348/kjr.2013.14.4.640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dong S, Kong J, Kong F, et al. Insufficient radiofrequency ablation promotes epithelial-mesenchymal transition of hepatocellular carcinoma cells through Akt and ERK signaling pathways. J Transl Med. 2013;11:273. doi: 10.1186/1479-5876-11-273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kondo Y, Kanzawa T, Sawaya R, Kondo S. The role of autophagy in cancer development and response to therapy. Nat Rev Cancer. 2005;5:726–734. doi: 10.1038/nrc1692. [DOI] [PubMed] [Google Scholar]
- 28.Kimmelman AC. The dynamic nature of autophagy in cancer. Genes Dev. 2011;25:1999–2010. doi: 10.1101/gad.17558811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.White E, DiPaola RS. The double-edged sword of autophagy modulation in cancer. Clin Cancer Res. 2009;15:5308–5316. doi: 10.1158/1078-0432.CCR-07-5023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou S, Zhao L, Kuang M, et al. Autophagy in tumorigenesis and cancer therapy: Dr. Jekyll or Mr. Hyde? Cancer Lett. 2012;323:115–127. doi: 10.1016/j.canlet.2012.02.017. [DOI] [PubMed] [Google Scholar]
- 31.Bao L, Chandra PK, Moroz K, et al. Impaired autophagy response in human hepatocellular carcinoma. Exp Mol Pathol. 2014;96:149–154. doi: 10.1016/j.yexmp.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fan Q, Yang L, Zhang X, et al. Autophagy promotes metastasis and glycolysis by upregulating MCT1 expression and Wnt/beta-catenin signaling pathway activation in hepatocellular carcinoma cells. J Exp Clin Cancer Res. 2018;37:9. doi: 10.1186/s13046-018-0673-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang H, Bosch-Marce M, Shimoda LA, et al. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem. 2008;283:10892–10903. doi: 10.1074/jbc.M800102200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 34.Wu H, Chen Q. Hypoxia activation of mitophagy and its role in disease pathogenesis. Antioxid Redox Signal. 2015;22:1032–1046. doi: 10.1089/ars.2014.6204. [DOI] [PubMed] [Google Scholar]
- 35.Chang Q, Jurisica I, Do T, Hedley DW. Hypoxia predicts aggressive growth and spontaneous metastasis formation from orthotopically grown primary xenografts of human pancreatic cancer. Cancer Res. 2011;71:3110–3120. doi: 10.1158/0008-5472.CAN-10-4049. [DOI] [PubMed] [Google Scholar]
- 36.Gong LL, Yang S, Zhang W, et al. Akebia saponin D alleviates hepatic steatosis through BNip3 induced mitophagy. J Pharmacol Sci. 2018;136:189–195. doi: 10.1016/j.jphs.2017.11.007. [DOI] [PubMed] [Google Scholar]
- 37.Chen SJ, Hoffman NE, Shanmughapriya S, et al. A splice variant of the human ion channel TRPM2 modulates neuroblastoma tumor growth through hypoxia-inducible factor (HIF)-1/2alpha. J Biol Chem. 2014;289:36284–36302. doi: 10.1074/jbc.M114.620922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Belibi F, Zafar I, Ravichandran K, et al. Hypoxia-inducible factor-1alpha (HIF-1alpha) and autophagy in polycystic kidney disease (PKD) Am J Physiol Renal Physiol. 2011;300:F1235–1243. doi: 10.1152/ajprenal.00348.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu Z, Dai H, Jia G, Li Y, Liu X, Ren W. Insufficient radiofrequency ablation promotes human hepatoma SMMC7721 cell proliferation by stimulating vascular endothelial growth factor overexpression. Oncol Lett. 2015;9:1893–1896. doi: 10.3892/ol.2015.2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.