

Abstract
We show that capillary zone electrophoresis-electrospray ionization-tandem mass spectrometry (CZE-ESI-MS/MS) generates very large numbers of peptide and protein identifications (IDs) by combining four technologies: a separation capillary coated to generate very low electroosmosis, an electrokinetically-pumped sheath-flow nanoelectrospray interface to produce high-sensitivity ionization, an Orbitrap Fusion Lumos Tribrid platform to provide high-speed analysis, and use of an advanced peak determination (APD) algorithm to take advantage of the mass spectrometer’s data acquisition speed. The use of the APD algorithm resulted in two-times more identification than the Standard Peak Algorithm. We also investigated the effect of the isolation window, injection time, and loading amount. Optimization of these parameters produced over 27,000 peptide identifications and nearly 4,400 protein group identifications from 220 ng of K562 cell digest in a single 120 min run, which is 2.7 times the number of IDs in the previous state of the art produced by CZE-ESI-MS/MS.
Graphical Abstract
In shotgun proteomics, a complex set of proteins is digested to create a much more complex set of peptides.1–3 These peptides undergo one or more stages of separation before analysis by tandem mass spectrometry. The vast majority of bottom-up proteomic studies use reversed-phase liquid chromatography (RPLC) for peptide separation. However, the performance of mass spectrometry is exceeding the capabilities of RPLC to provide peptides for analysis, and alternative separation technologies will be of value.4
Capillary zone electrophoresis (CZE) is such an alternative. CZE separations are complementary to RPLC, and the combination of the technologies provides deeper proteomic coverage than either alone.5 CZE is particularly useful for detection of hydrophilic peptides that are lost in the void volume of RPLC separations.6 CZE consistently produces more peptide identifications than RPLC when analyzing submicrogram amounts of a tryptic digest.5,7 CZE has very low operational costs, particularly if capillaries are coated in-house using straightforward chemistry.8 Finally, CZE separations are much easier to accurately model than RPLC; such models are very useful in detecting peptides that are misidentified by search engines.6
CZE-ESI-MS/MS analysis has undergone remarkably rapidly improvement in the number of peptide and protein identifications IDs. This improvement arises due to the development of a robust CZE-MS interface, the incremental improvement in capillary coatings with reduced electroosmotic flow, and the continual improvement in the performance of mass analyzers. 9–15 To date, the best single shot CZE-ESI-MS/MS bottom-up proteomic analyses of eukaryotic systems have identified approximately 10,000 peptides and 2,000 proteins.8, 10
In this paper, we more than double the number of IDs produced by the state-of-the-art in single-shot CZE–MS/MS for analysis of a complex proteome. This improvement results from the combination of a number of technologies. We employ a surface-confined aqueous reversible addition-fragmentation chain transfer polymerization for preparation of a coated capillary with very low electroosmotic flow.11 We also employ a third-generation electrokinetrically-pumped electrospray interface for high-sensitivity analysis. In addition, we use an advanced peak determination (APD) algorithm with an Orbitrap Fusion Lumos Tribrid platform.14 This mass spectrometer can generate up to 60 ion trap MS/MS scans per second. However, such high acquisition rates are significantly underutilized in most experiments because the instrument exhausts the number of detectable peptide determined by the standard peak determination (SPD) algorithm from a survey scan.14 The SPD algorithm only considers local maxima in a given mass-to-charge (m/z) region when annotating the most abundant peptide feature, and lower abundance features are not selected for data-dependent MS/MS. The APD algorithm uses an iterative approach to annotate densely populated m/z regions, and enables nearly full utilization of the sampling capacity of the mass spectrometer. This RPLC-MS/MS analysis generated ∼53,000 peptide IDs in a 90 min gradient from 1 μg of HeLa sample.14
MATERIALS AND METHODS
Chemicals and Materials
Formic acid (FA), acetic acid (HAc), acrylamide, trifluoroacetic acid (TFA), 4,4 -azobis(4-cyanovaleric acid), cyanomethyl [3-(trimethoxysilyl)propyl] trithiocarbonate, bovine pancreas TPCK-treated trypsin, tris(2-carboxyethyl)phosphine (TCEP), and iodoacetamide (IAA) were purchased from Sigma-Aldrich (St. Louis, USA). Methanol was purchased from Honeywell Burdick & Jackson (Wicklow, Ireland). Uncoated fused silica capillary (50 μm i.d. × 350 μm o.d.) was purchased from Polymicro Technologies (Phoenix, AZ). Water was deionized by a Nano Pure system from Thermo Scientific (Marietta, OH).
Sample preparation
Human K562 cells were homogenized in 6 M guanidine HCl by probe sonication, followed by boiling for 5 minutes. Methanol was added to 90% final concentration, samples were centrifuged at 10,000 × g for 5 min, and the supernatant was discarded. The protein pellet was dissolved in 8 M urea, 100 mM tris pH 8, 10 mM TCEP, and 40 mM chloroacetamide. Endoproteinase LysC was added to an estimated 50:1 (protein:enzyme) ratio and the digestion was incubated at ambient temperature for 2 hours. The digest was diluted to 2 M urea in 100 mM tris; trypsin was added at a 50:1 ratio followed by overnight incubation at ambient temperature. Samples were titrated with TFA to approximately pH 2, followed by centrifugation and solid phase extraction of the peptides in the supernatant, and dried down.
Preparation of linear poly(acrylamide) (LPA) coated capillaries by surface-confined aqueous reversible-addition fragmentation transfer polymerization
A published protocol was use for coating the capillary.8 Briefly, a piece of ∼110 cm long, 50-μm ID, 360 μm OD fused silica capillary was pretreated by flushing with 0.1 M NaOH for 2 h, flushing with water until the outflow reached pH ∼7.0, flushing with 0.1 M HCl for 12 h, flushing again with water until the outflow reached pH ∼7.0, and drying with a nitrogen stream overnight at room temperature. Then, the capillary was filled with 50% cyanomethyl [3-(trimethoxysilyl)propyl] trithiocarbonate solution in MeOH(v/v) and incubated in a water bath at 45 °C for 12 h. The capillary was then rinsed with MeOH to flush out the residual reagent and dried with a nitrogen stream. A polymerization mixture was prepared by mixing acrylamide (0.56 M) and 4,4’-azobis(4-cyanovaleric acid) (5.42 × 10−4 M) in an acetate buffer (pH 5.2, 0.27 M acetic acid, and 0.73 M sodium acetate) at room temperature and stirring for about 10 min under nitrogen to form a homogeneous solution. The mixture then was introduced into the pretreated capillary and incubated at 60 °C for 7 hours. The coated capillary was flushed with H2O and MeOH to remove residual reagents.
Capillary etching
The outer portion of a 5-mm length of the LPA coated separation capillary was etched with HF to a ∼80 μm diameter using a published protocol.10 The final length of the etched capillary was 105 cm.
Caution: use appropriate safety procedures while handling HF solutions.
CZE-ESI-MS/MS analysis
The CZE system consisted of two high-voltage power supplies (Spellman CZE 1000R) and an electrokinetically pumped nanospray interface,9–11 which coupled the CZE separation capillary to the mass spectrometer. The electrospray emitter was made from a borosilicate glass capillary (1.0 mm o.d. × 0.75 mm i.d., 10 cm long) pulled with a Sutter instrument P-1000 flaming/brown micropipette puller; the size of the emitter opening was 15-20 μm.8 As noted above, the distal tip of the capillary was etched to a ∼80-μm outer diameter. The electrospray sheath electrolyte was 10% (v/v) methanol with 0.5% FA. The background electrolyte was 1 M HAc in H2O. The sample was injected by nitrogen pressure. The separation voltage of 23.8 kV was applied at the injection end of the capillary. 1.8 kV was applied to the sheath flow reservoir for electrospray. Voltage programming was controlled by LabView software. The mass spectrometer’s operating parameters are described below.
Mass Spectrometer Operating Parameters
Peptides were analyzed on an Orbitrap Fusion Lumos Tribrid platform with Instrument Control Software version 3.0. Typical analyses used a 240 000 resolving power survey scan with an AGC of 106, followed by MS/MS of the most intense precursors for 1 s. The MS/MS analyses were performed by 0.4 m/z isolation with the quadrupole, normalized HCD collision energy of 25%, and analysis of fragment ions in the ion trap using the “Turbo” scan speed scanning from 200 to 1200 m/z. Dynamic exclusion was set to 20 s. Monoisotopic precursor selection (MIPS) was set to Peptide. For MS/MS analyses, the maximum injection time was set to 11 ms with an AGC target of 30 000, and charge states unknown, 1, or >5 were excluded. Advanced peak determination was either off or on for SPD and APD analyses, respectively.
Database Searching
Database searching of the raw files was performed in Proteome Discoverer 2.2 (Thermo) with Sequest HT and MS Amanda 2.0 database search engines.17, 18 The SwissProt database with taxonomy as Homo sapiens was downloaded from Uniprot and for the K562 cell line proteome digest. The database searching parameters included full tryptic digestion and allowed up to two missed cleavages, the precursor mass tolerance was set at 10 ppm, fragment mass tolerance was 0.4 Da. Carbamidomethylation of cysteines (+57.0215 Da) was set as a fixed modification, and variable modifications of methionine oxidation (+15.9949 Da), Acetyl (N-terminus) (+42.011 Da) and deamination (Asparagine or Glutamine) (+0.984 Da) were allowed. The false discovery rate (FDR) was determined by using a target-decoy search strategy. The decoy sequence database contains each sequence in reverse orientations, enabling FDR estimation. On the peptide level, the corresponding FDR on peptide level was less than 1%. On the protein level, protein grouping was enabled, and a Protein FDR Validator was used with the target FDR (strict) set at 1%.
For peak widths analysis, raw files were also analyzed by MaxQuant (version 1.61.0.). MS/MS spectra were searched by the Andromeda search engine against the Uniprot human protein database using the default parameters.19 The distribution of the peak widths was generated from the “evidence” file.
RESULTS AND DISCUSSION
We investigated the effect of a number of experimental parameters on the numbers of peptide and protein IDs, Table 1. We first compared the APD and the SPD algorithms for single shot CZE-ESI-MS/MS analysis of whole cell K562 lysates. Under the same separation conditions, the APD algorithm generated nearly twice the number of peptide IDs as the SPD algorithm. The fractional increase in the number of peptide IDs produced by the APD algorithm for CZE-ESI-MS/MS is over twice the increase produced by this algorithm for LC-ESI-MS/MS analysis.14 CZE-ESI-MS/MS benefits more from the APD algorithm than LC-ESI-MS/MS presumably due to co-migration of peptides with similar m/z in CZE experiments.
Table 1–
Numbers of peptide and protein IDs for CZE-ESI-MS/MS
| Algorithm | Isolation window | Injection time | Loading amount ng | Peptide IDs | Protein IDs |
|---|---|---|---|---|---|
| Effect of algorithm | |||||
| SPD | 0.4 m/z | 11 ms | 770 ng | 14,144 | 2,963 |
| APD | 0.4 m/z | 11 ms | 770 ng | 27,678 | 4,387 |
| Effect of isolation window width | |||||
| APD | 0.7 m/z | 18 ms | 386 ng | 20,355 | 3,454 |
| APD | 0.4 m/z | 18 ms | 386 ng | 22,433 | 3,720 |
| Effect of injection time | |||||
| APD | 0.4 m/z | 60 ms | 220 ng | 25,553 | 3,828 |
| APD | 0.4 m/z | 11 ms | 220 ng | 27,113 | 4,397 |
| Effect of loading amount | |||||
| APD | 0.4 m/z | 11 ms | 48 ng | 19,874 | 3,566 |
| APD | 0.4 m/z | 11 ms | 95 ng | 20,707 | 3,618 |
| APD | 0.4 m/z | 11 ms | 220 ng | 27,113 | 4,397 |
| APD | 0.4 m/z | 11 ms | 770 ng | 27,678 | 4,387 |
We also investigated the effect of the isolation window on the number of IDs. We observed a ∼15% increase in the number of peptide IDs for a 0.4 m/z isolation window compared with a 0.7 m/z isolation window. Decreasing the isolation window reduces co-fragmentation of the precursors and improves the quality of the MS/MS spectra, which leads to an increase in the numbers of IDs. Therefore, a 0.4 m/z isolation window was used in subsequent studies.
The mass spectrometer’s injection time had a limited effect on the number of IDs. Use of an 11 ms injection time generated 6% more peptide IDs compared to a 60 ms injection time. While there was not a significant difference in the number of identified peptides, there was a large decrease in the MS/MS scan rate when the injection time was increased from 11 ms to 60 ms. 323,303 tandem mass spectra were collected for the 11 ms injection time, but number of tandem mass spectra decreased to 131,524 when the injection time was increased to 60 ms. The injection time was kept at 11 ms in subsequent studies.
Figure 1 (top) presents a representative base-peak electropherogram of the CZE-ESI-MS/MS analysis of a K562 cell proteome digest. The separation window is approximately 100 min wide, which is two times wider than a previous report using a linear polyacrylamide coated capillary prepared by free radical polymerization.18 Figure 1 (bottom) presents the number of peptide IDs per second. The system generated a peak of 20 peptide IDs/sec, stayed above 10 IDs/sec for much of the time between 25 and 70 minutes, and dropped below 5 peptide IDs/sec at 85 min.
Figure 1.
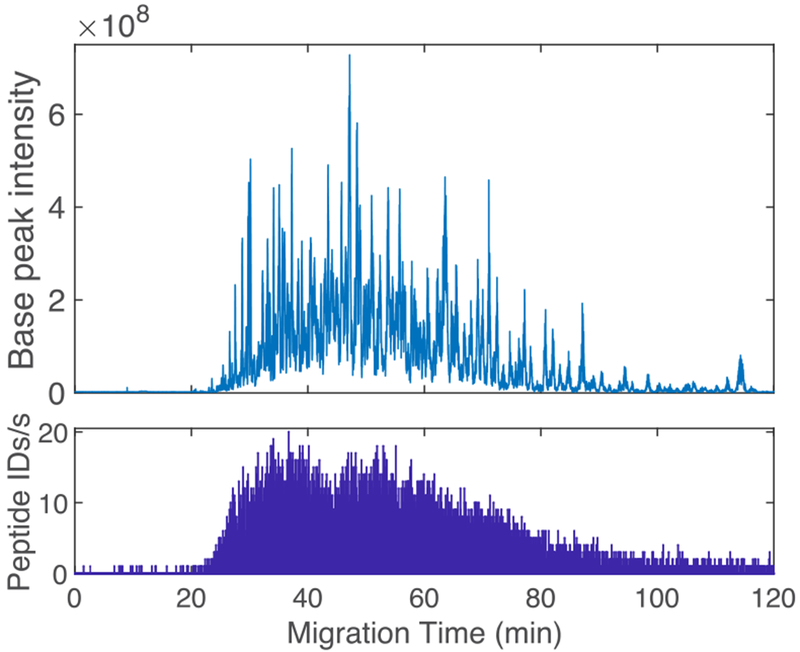
Single-shot CZE-ESI-MS/MS analysis of K562 cell lysate digest. Approximately 220 ng of the digest was loaded for CZE analysis. Top-base peak electropherogram. Bottom-unique peptide IDs per second.
The effect of the sample loading amount on the number of IDs is listed in Table 1. The numbers of protein groups and peptides IDs increased when the sample loading amount increased from 48 ng to 220 ng, and then plateaued for higher loading amounts. We determined the peak widths for different sample loading amounts (S-Figure 1). It seems that the peak broadening for larger injections is not a serious problem when the sample loading amount increased from 220 ng to 772 ng and when employing the pH junction for injection.20–24 The plateau in IDs for loading amounts greater then 220 ng may be because sample overloading leads to co-migration of the peptides and compromises the quality of the MS/MS spectra used for identification. Nevertheless, these results suggest that the CZE is particularly useful for comprehensive proteomics analysis using small amounts of sample.25,26
The previous state-of-the-art for single-shot CZE-ESI-MS/MS was 10,274 peptide IDs, which was generated from 440 ng of a HeLa digest using a commercial linear polyacrylamide-coated capillary and an Orbitrap Fusion mass spectrometer operating with the SPD algorithm.10 The number of identifications presented in this paper is 2.7 times larger than the previous record. This improvement results from the wider separation window produced by the improved capillary coating, the faster MS/MS spectra collection rate of the Orbitrap Fusion Lumos Tribrid platform, and the use of the APD algorithm. We summarize recent CZE-MS/MS results for analysis of complex proteome digests in S-Table 1 in the supporting information.
There are a few limitations in the CZE experiment. We noticed that the slowly migrating components undergo significant longitudinal diffusion, leading to the repeated acquisition of data from the same precursor and waste of a significant amount of mass spectrometer time. Optimization of the CZE conditions and MS parameters will be required to improve the identification results.7 In addition, the first 20 minutes of the CZE separation generated very few IDs, and the number of IDs drops significantly after 85 minutes. The identification rate will be improved by using a multi-segment sequential injection method that has been recently reported.27,28
We note that state-of-art RPLC-MS/MS analysis has generated approximately 53,000 peptide IDs in a 90 min gradient from 1 μg of HeLa sample with the APD algorithm using the same mass spectrometer that was used in this paper.14 Additional optimization will be required for CZE to match the performance of RPLC for bottom-up proteomics.
Supplementary Material
Acknowledgements
We thank Drs. William Boggess and Mathew Champion of the University of Notre Dame Mass Spectrometry and Proteomic Facility for their assistance. This work was supported by grants from the National Institute of Health (R01GM096767, R01HD084399, R35 GM 118110, and P41GM108538).
Footnotes
Supporting Information
Summary of CZE-ESI-MS/MS data for bottom-up proteomics and distribution of peak widths. An Excel spreadsheet with information on peptide and protein identifications. The Supporting Information is available free of charge on the ACS Publications website.
References
- 1.Pandey A; Mann M Proteomics to study genes and genomes. Nature 2000, 405, 837–846. [DOI] [PubMed] [Google Scholar]
- 2.Yates JR III, The Revolution and Evolution of Shotgun Proteomics for Large-Scale Proteome Analysis. J. Am. Chem. Soc 2013, 135, 1629–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Malmstrom J; Beck M; Schmidt A ; Lange V ; Deutsch EW; Aebersold R, Proteome-wide cellular protein concentrations of the human pathogen Leptospira interrogans. Nature 2009, 460, 762–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shishkova E; Hebert AS; Coon J Now, More Than Ever, Proteomics Needs Better Chromatography. J. Cell Systems 2016, 3, 321–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhu G; Sun L; Yan X; Dovichi NJ Single-Shot Proteomics Using Capillary Zone Electrophoresis-Electrospray Ionization-Tandem Mass Spectrometry with Production of More than 1 250 Escherichia coli Peptide Identifications in a 50 min Separation. Anal. Chem 2013, 85, 2569–2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Krokhin OV; Anderson G; Spicer V; Sun L; Dovichi NJ Predicting Electrophoretic Mobility of Tryptic Peptides for High-Throughput CZE-MS Analysis. Anal. Chem 2017, 89, 2000–2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang Z; Dovichi NJ Optimization of mass spectrometric parameters improve the identification performance of capillary zone electrophoresis for single-shot bottom-up proteomics analysis. Anal. Chim. Acta 2018, 1001, 93–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang Z; Peuchen EH; Dovichi NJ Surface-Confined Aqueous Reversible Addition-Fragmentation Chain Transfer (SCARAFT) Polymerization Method for Preparation of Coated Capillary Leads to over 10 000 Peptides Identified from 25 ng HeLa Digest by Using Capillary Zone Electrophoresis-Tandem Mass Spectrometry. Anal. Chem 2017, 89, 6774–6780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wojcik R; Dada OO; Sadilek M; Dovichi NJ Simplified capillary electrophoresis nanospray sheath-flow interface for high efficiency and sensitive peptide analysis, Rapid Commun. Mass Spectrom 2010, 24, 2554–2560. [DOI] [PubMed] [Google Scholar]
- 10.Sun L; Hebert AS; Yan X; Zhao Y; Westphall MS; Rush MJ; Zhu G; Champion MM; Coon JJ; Dovichi NJ, Over 10,000 peptide identifications from the HeLa proteome by using single-shot capillary zone electrophoresis combined with tandem mass spectrometry. Angew. Chem. Int. Ed. Engl 2014, 53, 13931–13933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sun L; Zhu G; Zhang Z; Mou S; Dovichi NJ, Third-Generation Electrokinetically Pumped Sheath-Flow Nanospray Interface with Improved Stability and Sensitivity for Automated Capillary Zone Electrophoresis-Mass Spectrometry Analysis of Complex Proteome Digests. J. Proteome Res 2015; 14, 2312–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Y; Fonslow BR; Wong CCL ; Nakorchevsky A; Yates JR III, Improving the Comprehensiveness and Sensitivity of Sheath less Capillary Electrophoresis-Tandem Mass Spectrometry for Proteomic Analysis. Anal. Chem 2012, 84, 8505–8513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yan X; Essaka DC; Sun L; Zhu G; Dovichi NJ Bottom-up proteome analysis of E. coli using capillary zone electrophoresis-tandem mass spectrometry with an electrokinetic sheath-flow electrospray interface. Proteomics 2013, 13, 2546–2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hebert AS ; Thöing C; Riley NM; Kwiecien NW; Shiskova E; Huguet R; Cardasis HL; Kuehn A; Eliuk S; Zabrouskov V; Westphall MS; McAlister GC; Coon JJ, Improved Precursor Characterization for Data-Dependent Mass Spectrometry. Anal. Chem 2018, 90, 2333–2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen D; Shen X; Sun L, Strong cation exchange-reversed phase liquid chromatography-capillary zone electrophoresis-tandem mass spectrometry platform with high peak capacity for deep bottom-up proteomics. Anal. Chim. Acta 2018, 1012, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eng JK; McCormack AL; Yates JR, An approach to correlate tandem mass-spectral data of peptides with amino-acid-sequences in a protein database J. Am. Soc. Mass Spectrom 1994, 5, 976–989. [DOI] [PubMed] [Google Scholar]
- 18.Dorfer V; Pichler P; Stranzl T; Stadlmann J; Taus T; Winkler S; Mechtler K, MS Amanda, a Universal Identification Algorithm Optimized for High Accuracy Tandem Mass Spectra. J. Proteome Res 2014, 13, 3679–3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cox J; Mann M, MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotech 2008, 26, 1367–1372. [DOI] [PubMed] [Google Scholar]
- 20.Britz-McKibbin P; Bebault GM; Chen DD Velocity-difference induced focusing of nucleotides in capillary electrophoresis with a dynamic pH junction. Anal. Chem 2000, 72, 1729–1735. [DOI] [PubMed] [Google Scholar]
- 21.Zhu G; Sun L; Yan X; Dovichi NJ, Bottom-Up Proteomics of Escherichia coli Using Dynamic pH Junction Preconcentration and Capillary Zone Electrophoresis-Electrospray Ionization-Tandem Mass Spectrometry. Anal. Chem 2014, 86, 6331–6336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Z; Sun L; Zhu G; Cox OF; Huber PW, Dovichi NJ Nearly 1000 Protein Identifications from 50 ng of Xenopus laevis Zygote Homogenate Using Online Sample Preparation on a Strong Cation Exchange Monolith Based Microreactor Coupled with Capillary Zone Electrophoresis. Anal. Chem 2016, 88, 877–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang Z; Yan X; Sun L; Zhu G; Dovichi NJ, Detachable Strong Cation Exchange Monolith, Integrated with Capillary Zone Electrophoresis and Coupled with pH Gradient Elution, Produces Improved Sensitivity and Numbers of Peptide Identifications during Bottom-up Analysis of Complex Proteomes. Anal. Chem 2015, 87, 4572–4577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Z; Sun L; Zhu G; Yan X; Dovichi NJ, Integrated strong cation-exchange hybrid monolith coupled with capillary zone electrophoresis and simultaneous dynamic pH junction for large-volume proteomic analysis by mass spectrometry. Talanta 2015, 138, 117–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Altelaar AFM; Maarten AF; Heck A, Trends in ultrasensitive proteomics. Curr. Opin. Chem. Bio 2012, 16, 206–213. [DOI] [PubMed] [Google Scholar]
- 26.Lombard-Banek C; Moody SA; Nemes P, Single-Cell Mass Spectrometry for Discovery Proteomics: Quantifying Translational Cell Heterogeneity in the 16-Cell Frog (Xenopus) Embryo. Angew. Chem. Int. Ed 2016, 55, 2454–2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boley DA; Zhang Z; Dovichi NJ, Multisegment injections improve peptide identification rates in capillary zone electrophoresis-based bottom-up proteomics. J. Chromatogr A 2017, 1523, 123–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Faserl K; Sarg B; Sola L; Lindner HH, Enhancing Proteomic Throughput in Capillary Electrophoresis-Mass Spectrometry by Sequential Sample Injection. Proteomics 2017, 17, 1700310. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.