

Abstract
An operationally simple protocol for the conversion of geranyl acetate to 8-hydroxygeraniol is reported. The convenient two-step procedure relies on an efficient, chemo-and regioselective SeO2-promoted oxidation, followed by straightforward deacetylation. This facile means to prepare 8-hydroxygeraniol is expected to enable biosynthetic studies pertaining to thousands of monoterpene indole alkaloids.
Graphical Abstract

Monoterpene indole alkaloids (MIAs) have provided chemists and biologists with the inspiration to pursue countless scientific endeavors.1–4 To date, over 3000 MIAs have been discovered, many of which possess striking biological activity. Select examples of MIAs are the notorious poison strychnine (1) and the life-changing anticancer drug vinblastine (2), both of which are shown in Figure 1. All MIAs are prepared by nature through a remarkable biosynthetic pathway, which has been under investigation for decades.1,5–11
Figure 1.
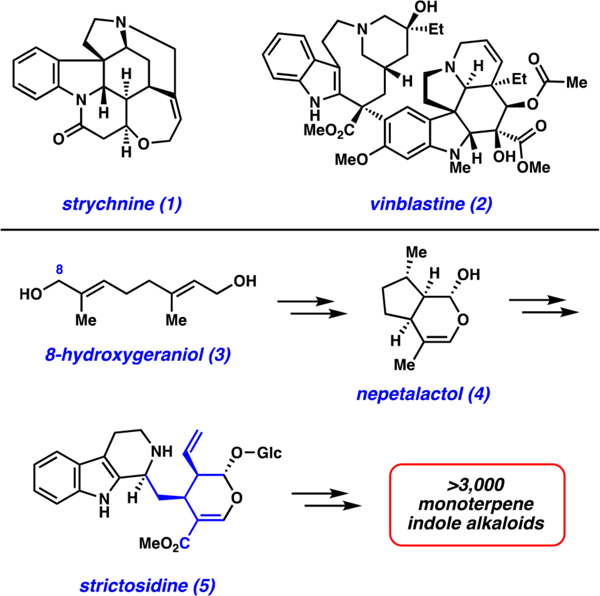
Role of 8-hydroxygeraniol (3) in the biosynthesis of all monoterpene indole alkaloids, including strychnine (1) and vinblastine (2).
This Note focuses on 8-hydroxygeraniol (3, Figure 1), an early biosynthetic precursor to MIAs. 8-Hydroxygeraniol (3) is made biosynthetically through a controlled enzymatic oxidation of geraniol6 before being elaborated to nepetalactol (4).12 Many further biosynthetic manipulations ultimately give rise to strictosidine (5), the last common biosynthetic precursor to all MIAs. Given the relative simplicity of 3 compared to its successors in the biosynthetic pathway (e.g., 5), 3 has been used to enable several biosynthetic studies, including the biosynthesis of nepetalactol (4)13,14 and the biosynthesis of strictosidine (5).15
Several synthetic approaches to 8-hydroxygeraniol (3) have been reported in the literature (Figure 2). The earliest reports appeared back-to-back in 1970, where multistep synthetic routes were developed beginning from either levulinaldehyde (6)16 or dehydrolinalool (7).17 An alternative strategy was reported by Williams and Lin, which involved photo-cycloaddition of 8 and 9, with subsequent elaboration using a thermolysis/Cope rearrangement strategy.18 Perhaps the most direct approach relies on the use of geranyl acetate (10) as the starting material. In this regard, Kobayashi has reported a procedure for the C8-oxidation of 10 using stoichiometric SeO2, which proceeds in low yields and with extensive overoxidation to the corresponding enal.19 Around the same time, Sharpless reported a similar protocol that relies on catalytic SeO2 and stoichiometric t-butyl hydroperoxide.20 This procedure, which has subsequently been repeated with similar results,21 leads to a significant recovery of the starting material20 with some minimization of the overoxidation byproduct (i.e., 45% yield of the desired alcohol and 19% enal21). A promising biocatalytic approach to 8-hydroxyger- aniol using a cytochrome P450 has also been described,21 although it has yet to be rendered practical for material throughput.
Figure 2.

Various approaches to 8-hydroxygeraniol (3).
One further point regarding 8-hydroxygeraniol (3) and its naming should be noted. Throughout the aforementioned literature and other sources, the compound is often referred to as “10-hydroxygeraniol” instead (Figure 3).6,7,12,13,16–19 We believe this is a simple nomenclature error that has propagated for many decades. Similarly, 8-oxogeranial (11), the biosynthetic successor to 8-hydroxygeraniol (3), has been referred to as “10-oxogeranial.”7,12,22 From a nomenclature standpoint, this is also incorrect. We suggest the chemical and biosynthetic community use the “8-” prefix going forward for 8-hydroxygeraniol to minimize confusion, as this is consistent with IUPAC standards where “8” should reflect the longest carbon chain in the molecule, with C8 being the trans substituent on the alkene.23,24 Additionally, the “8-” prefix is accepted according to various enzymology resources.25,26
Figure 3.
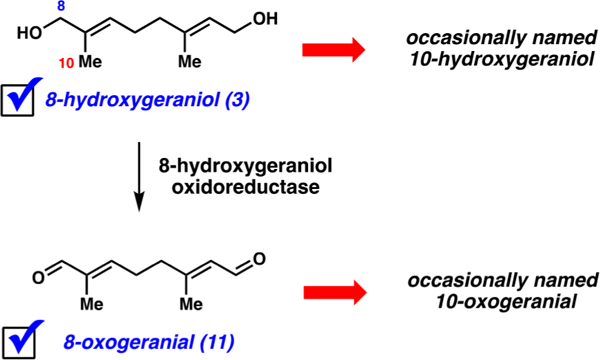
Confusion surrounding the naming of 8-hydroxygeraniol (3) and its biosynthetic successor 8-oxogeranial (11).
With the aim of developing a practical procedure for the preparation of 8-hydroxygeraniol (3) to enable our biosynthetic studies,14 we opted to pursue the chemo- and regioselective allylic oxidation of geranyl acetate (10), shown in Figure 2, as a starting point. Select results from our efforts to reproduce and optimize the catalytic SeO2 oxidation procedure are provided in Table 1. As shown in entries 1 and 2, the oxidation could be performed at 0 °C. After 5 h, significant amounts of unreacted geranyl acetate (10) remained (entry 1). However, longer reaction times at 0 °C showed promise for increasing the conversion (entry 2). For the sake of developing a more convenient protocol that would not require cooling for extended periods of time, we attempted the oxidation at 23 °C. After 30 min or 1 h, significant recovery of unreacted substrate 10 was observed (entries 3 and 4, respectively). When the reaction was performed for 1.5 h, a more desirable ratio was obtained (entry 5), with the desired product 12 being formed in 61% yield. At longer reaction times of 5 h, substrate 10 could be fully consumed, however, competitive overoxidation to enal 13 was observed (entry 6). Overall, entry 5 conditions were deemed ideal because of the convenience of the experimental protocol (23 °C, 1.5 h) and the optimal yield of 12 obtained.
Table 1.
Optimization of SeO2-Promoted Oxidation of 10a
 |
Ratios and yields were determined by 1H NMR analysis of the crude reaction mixtures (1,3,5-Trimethoxybenzene was used as an external standard).
With practical reaction conditions in hand for the efficient oxidation of geranyl acetate (10), we performed the preparation of 8-hydroxygeraniol (3) on a 3 mmol scale, as shown in Figure 4. SeO2-promoted oxidation of 10 proceeded smoothly under our optimized conditions in just 1.5 h at 23 °C. This gave the desired C8-hydroxylated product 12 in 64% isolated yield. Subsequent treatment of 12 with K2CO3 in methanol at 23 °C smoothly delivered 8-hydroxygeraniol (3) in 83% yield after flash column chromatography. This exceedingly simple protocol can be used to synthesize multimmol quantities of 3.
Figure 4.

Preparation of 8-hydroxygeraniol (3) on a 3 mmol scale.
In summary, we have developed a simple and convenient procedure to synthesize 8-hydroxygeraniol (3). The procedure involves a regio- and chemoselective oxidation, followed by methanolysis. Both transformations are performed at ambient temperature and can be used to easily access multimmol quantities of 3. We expect this protocol will enable biosynthetic investigations pertaining to thousands of monoterpene indole alkaloids.
EXPERIMENTAL SECTION
Materials and Methods.
Unless stated otherwise, reactions were conducted in flame-dried glassware under an atmosphere of nitrogen using anhydrous solvents (either freshly distilled or passed through activated alumina columns). All commercially obtained reagents were used as received unless otherwise specified. Geranyl acetate (10) and potassium carbonate were purchased from Alfa Aesar. Selenium dioxide and tert-butyl hydroperoxide solution (~5.5 M in decane, over 4 Å molecular sieves) were obtained from Sigma-Aldrich. Reaction temperatures were controlled using an IKAmag temperature modulator, and unless stated otherwise, reactions were performed at 23°C. Thin-layer chromatography (TLC) was conducted with EMD gel 60 F254 precoated plates (0.25 mm) and visualized using anisaldehyde staining. Silicycle Siliaflash P60 (particle size 0.040–0.063 mm) was used for flash column chromatography. 1H NMR spectra were recorded on Bruker spectrometers (at 500 MHz) and are reported relative to the residual solvent signal. Data for 1H NMR spectra are reported as follows: chemical shift (δ ppm), multiplicity, coupling constant (Hz), and integration. 13C NMR spectra were recorded on Bruker spectrometers (at 125 MHz) and are reported relative to the residual solvent signal. Data for 13C NMR spectra are reported in terms of chemical shift. IR spectra were obtained on a PerkinElmer UATR Two FT-IR spectrometer and are reported in terms of frequency of absorption (cm–1). DART-MS spectra were collected on a Thermo Exactive Plus MSD (Thermo Scientific) equipped with an ID-CUBE ion source, a Vapur Interface (IonSense Inc.), and an Orbitrap mass analyzer. Both the source and MSD were controlled by Excalibur software v. 3.0. The analyte was spotted onto OpenSpot sampling cards (IonSense Inc.) using CH2Cl2 as the solvent. Ionization was accomplished using UHP He (Airgas Inc.) plasma with no additional ionization agents. The mass calibration was carried out using Pierce LTQ Velos ESI (+) and (–) ion calibration solutions (Thermo Fisher Scientific).
Representative Procedure for Optimization of Oxidation (Table 1, Entry 3 as an Example).
To a flame-dried 10 mL round-bottom flask equipped with a magnetic stir bar and selenium dioxide (23.4 mg, 0.20 mmol, 0.4 equiv) under N2 were added CH2Cl2 (2.5 mL, 0.20 M), tert-butyl hydroperoxide (5.5 M in decane, 0.29 mL, 1.58 mmol, 3.1 equiv), and geranyl acetate (10, 109 μL, 0.509 mmol, 1.0 equiv). After the mixture was stirred for 30 min at 23 °C, water (2 mL) and EtOAc (10 mL) were added, and the reaction was transferred to a separatory funnel. The layers were separated, and the organic layer was washed successively with deionized water (2 × 5 mL), saturated aqueous NaHCO3 (1 × 5 mL), deionized water (1 × 5 mL), and brine (1 × 5 mL). The organic layer was dried over MgSO4, filtered, and concentrated under reduced pressure. To the resulting crude product was added 1,3,5-trimethoxybenzene (28.3 mg, 0.33 equiv) as an external standard. The ratio and yields were determined by 1H NMR analysis.
8-Hydroxygeranyl Acetate (12).
To a flame-dried 100 mL round-bottom flask equipped with a magnetic stir bar and selenium dioxide (226 mg, 2.04 mmol, 0.4 equiv) under N2 were added CH2Cl2 (25 mL, 0.20 M), tert-butyl hydroperoxide (5.5 M in decane, 2.9 mL, 15.8 mmol, 3.1 equiv), and geranyl acetate (10, 1.09 mL, 5.09 mmol, 1.0 equiv). After the mixture was stirred for 1.5 h at 23 °C, the reaction mixture was concentrated under reduced pressure. The crude oil was transferred to a separatory funnel with EtOAc (50 mL). The organic layer was washed successively with deionized water (2 × 20 mL), saturated aqueous NaHCO3 (1 × 20 mL), deionized water (1 × 10 mL), and brine (1 × 10 mL). The combined aqueous layers were back-extracted with EtOAc (1 × 80 mL). The combined organic layers were dried over Na2SO4, filtered, and concentrated under reduced pressure. The resulting crude oil was purified via flash chromatography (6:1→2:1 hexanes/EtOAc) to afford 8-hydroxygeranyl acetate (12, 688 mg, 64% yield) as a colorless oil. 8-Hydroxygeranyl acetate (12): Rf 0.43 (2:1 hexanes/EtOAc); 1H NMR (500 MHz, CDCl3) δ 5.39–5.31 (m, 2H), 4.58 (d, J = 7.1, 2H), 3.99 (s, 2H), 2.17 (dt, J = 7.4, 7.4, 2H), 2.11–2.07 (m, 2H), 2.05 (s, 3H), 1.71 (s, 3H), 1.66 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 171.4, 141.9, 135.4, 125.4, 118.8, 69.0, 61.6, 39.2, 25.8, 21.2, 16.5, 13.8; IR (film) 3424, 2919, 2860, 1736, 1671, 1229 cm–1; HRMS-APCI (m/z) [M + H]+ calcd for C12H21O3+ 213.1485, found 213.1478. Spectral data match those previously reported.14
8-Hydroxygeraniol (3).
A flame-dried 50 mL round-bottom flask equipped with a magnetic stir bar was charged with 8-hydroxygeranyl acetate (12, 633 mg, 2.98 mmol, 1 equiv) and methanol (19 mL, 0.16 M). Potassium carbonate (495 mg, 3.58 mmol, 1.2 equiv) was added in one portion. After the mixture was stirred at 23 °C for 2.5 h, the solvent was removed under reduced pressure, and the reaction mixture was transferred to a separatory funnel with deionized water (10 mL). The aqueous layer was extracted with diethyl ether (3 × 20 mL). The combined organic layers were washed successively with 0.5 M aqueous HCl (1 × 10 mL), saturated aqueous NaHCO3 (1 × 10 mL), brine (1 × 10 mL), and deionized water (1 × 10 mL). Next, the organic layers were dried over MgSO4, filtered, and concentrated under reduced pressure. The resulting crude oil was purified via flash chromatography (1:1 hexanes/EtOAc) to afford 8-hydroxygeraniol (3, 490 mg, 83% yield) as a light yellow oil. 8-Hydroxygeraniol (3): Rf 0.18 (1:1 hexanes/EtOAc); 1H NMR (500 MHz, CDCl3) δ 5.41–5.34 (m, 2H), 4.14 (d, J = 6.9, 2H), 3.98 (s, 2H), 2.17 (dt, J = 7.5, 7.1, 2H), 2.09–2.04 (m, 2H), 1.67 (s, 3H), 1.65 (s, 3H), 1.44 (br s, 2H); 13C NMR (125 MHz, CDCl3) δ 139.1, 135.3, 125.6, 123.9, 68.9, 59.4, 39.1, 25.8, 16.3, 13.8; IR (film) 3307, 2916, 2859, 1669, 999 cm–1; HRMS-APCI (m/z) [M + H]+ calcd for C10H19O2+ 171.1380, found 171.1375. Spectral data match those previously reported.14
Supplementary Material
ACKNOWLEDGMENTS
The authors are grateful to the University of California, Los Angeles, the National Science Foundation (CBET1605877 to Y.T.), the Packard Foundation (Y.T.), the Foote Family (F.M.I.), and the Chemistry–Biology Interface training program (J.S.B., USPHS National Research Service Award 5T32GM008496–20) for financial support. These studies were supported by shared instrumentation grants from the NSF (CHE-1048804) and the NIH NCRR (S10RR025631).
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.joc.8b01544.
1H NMR and 13C NMR spectra for compounds 12 and 3 (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).O’Connor SE; Maresh JJ Chemistry and Biology of Monoterpene Indole Alkaloid Biosynthesis. Nat. Prod. Rep. 2006, 23, 532–547. [DOI] [PubMed] [Google Scholar]
- (2).Pickens LB; Tang Y; Chooi Y-H Metabolic Engineering for the Production of Natural Products. Annu. Rev. Chem. Biomol. Eng. 2011, 2, 211–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).O’Connor SE Strategies for Engineering Plant Natural Products: The Iridoid-Derived Monoterpene Indole Alkaloids of Catharanthus roseus In Methods in Enzymology; Hopwood DA, Ed.; Academic Press: 2012; Vol. 515, pp 189–206. [DOI] [PubMed] [Google Scholar]
- (4).Pritchett BP; Stoltz BM Enantioselective Palladium- Catalyzed Allylic Alkylation Reactions in the Synthesis of Aspidosperma and Structurally Related Monoterpene Indole Alkaloids. Nat. Prod. Rep. 2018, 35, 559–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Pan Q; Mustafa NR; Tang K; Choi YH; Verpoorte R Monoterpenoid Indole Alkaloids Biosynthesis and its Regulation in Catharanthus roseus: A Literature Review. Phytochem. Rev. 2016, 15, 221–250. [Google Scholar]
- (6).Madyastha KM; Meehan TD; Coscia CJ Characterization of a Cytochrome P-450 Dependent Monoterpene Hydroxylase from the Higher Plant. Biochemistry 1976, 15, 1097–1102. [DOI] [PubMed] [Google Scholar]
- (7).Ikeda H; Esaki N; Nakai S; Hashimoto K; Uesato S; Soda K; Fujita T Acyclic Monoterpene Primary Alcohol: NADP+ Oxidoreductase of Rauwolfia serpentina Cells: The Key Enzyme in Biosynthesis of Monoterpene Alcohols. J. Biochem. 1991, 109, 341–347. [PubMed] [Google Scholar]
- (8).Madyastha KM; Guarnaccia R; Baxter C; Coscia CJ S-Adenosyl-L-methionine: Loganic Acid Methyltransferase: A Carboxyl- Alkylating Enzyme from Vinca rosea. J. Biol. Chem. 1973, 248, 2497–2501. [PubMed] [Google Scholar]
- (9).Irmler S; Schroder G; St-Pierre B; Crouch NP; Hotze M; Schmidt J; Strack D; Matern U; Schroder J Indole Alkaloid Biosynthesis in Catharanthus roseus: New Enzyme Activities and Identification of Cytochrome P450 CYP72A1 as Secologanin Synthase. Plant J. 2000, 24, 797–804. [DOI] [PubMed] [Google Scholar]
- (10).Stockigt J; Zenk MH Isovincoside (Strictosidine), the Key Intermediate in the Enzymatic Formation of Indole Alkaloids. FEBS Lett. 1977, 79, 233–237. [Google Scholar]
- (11).Caputi L; Franke J; Farrow SC; Chung K; Payne RME; Nguyen T-D; Dang T-TT; Carqueijeiro IST; Koudounas K; de Bernonville TD; Ameyaw B; Jones DM; Vieira IJC; Courdavault V; O’Connor SE Missing Enzymes in the Biosynthesis of the Anticancer Drug Vinblastine in Madagascar periwinkle. Science 2018, 360, 1235–1239. [DOI] [PubMed] [Google Scholar]
- (12).Krithika R; Srivastava PL; Rani B; Kolet SP; Chopade M; Soniya M; Thulasiram HV Characterization of 10-Hydroxygeraniol Dehydrogenase from Catharanthus roseus Reveals Cascaded Enzymatic Activity in Iridoid Biosynthesis. Sci. Rep. 2015, 5, 8258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Campbell A; Bauchart P; Gold ND; Zhu Y; De Luca V; Martin VJJ Engineering of a Nepetalactol-Producing Platform Strain of Saccharomyces cerevisiae for the Production of Plant Seco- Iridoids. ACS Synth. Biol. 2016, 5, 405–414. [DOI] [PubMed] [Google Scholar]
- (14).Billingsley JM; DeNicola AB; Barber JS; Tang M-C; Horecka J; Chu A; Garg NK; Tang Y Engineering the Biocatalytic Selectivity of Iridoid Production in Saccharomyces cerevisiae. Metab. Eng. 2017, 44, 117–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Brown S; Clastre M; Courdavault V; O’Connor SE De novo Production of the Plant-derived Alkaloid Strictosidine in Yeast. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 3205–3210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Escher S; Loew P; Arigoni D The Role of Hydroxygeraniol and Hydroxynerol in the Biosynthesis of Loganin and Indole Alkaloids. J. Chem. Soc. D 1970, 823–825. [Google Scholar]
- (17).Battersby AR; Brown SH; Payne TG Preparation and Isolation of Deoxyloganin: Its Role as Precursor of Loganin and the Indole Alkaloids. J. Chem. Soc. D 1970, 827–828. [Google Scholar]
- (18).Williams JR; Lin C Photocycloaddition of 2,5- Dihydrothiophen SS-Dioxides to α,β-Unsaturated Cyclic Anhydrides. Synthesis of 10-Hydroxygeraniol. J. Chem. Soc., Chem. Commun. 1981, 752–753. [Google Scholar]
- (19).Inouye H; Ueda S-I; Uesato S-I; Kobayashi K Studies on Monoterpene Glucosides and Related Natural Products. XXXVII. Biosynthesis of the Iridoid Glucosides in Lamium amplexicaule, Deutzia crenata and Galium spurium var. echinospermon. Chem. Pharm. Bull. 1978, 26, 3384–3394. [Google Scholar]
- (20).Umbreit MA; Sharpless KB Allylic Oxidation of Olefins by Catalytic and Stoichiometric Selenium Dioxide with tert-Butyl Hydroperoxide. J. Am. Chem. Soc. 1977, 99, 5526–5528. [Google Scholar]
- (21).Bogazkaya AM; von Buhler CJ; Kriening S; Busch A; Seifert A; Pleiss J; Laschat S; Urlacher VB Selective Allylic Hydroxylation of Acyclic Terpenoids by CYP154E1 from Thermobi- fidafusca YX. Beilstein J. Org. Chem. 2014, 10, 1347–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Geu-Flores F; Sherden NH; Courdavault V; Burlat V; Glenn WS; Wu C; Nims E; Cui Y; O’Connor SE An Alternative Route to Cyclic Terpenes by Reductive Cyclization in Iridoid Biosynthesis. Nature 2012, 492, 138–142. [DOI] [PubMed] [Google Scholar]
- (23).IUPAC. Nomenclature of Organic Chemistry, Sections A, B, C, D, E, F, and H. Pergamon Press: Oxford, 1979. [Google Scholar]
- (24).Acyclic Terpenes. In System of Nomenclature for Terpene Hydrocarbons; Advances in Chemistry Series; American Chemical Society: Washington, DC, 1955; Vol. 14, pp 12–14. [Google Scholar]
- (25).Swiss Institute of Bioinformatics. ExPASy. ENZYME entry: EC https://enzyme.expasy.org/EC71.14.14.83 (accessed June 18, 2018).
- (26).Kyoto University Bioinformatics Center. KEGG. Enzyme 1.14.14.83. https://www.genome.jp/dbget-bin/www_bget?ec:1.14.14.83 (accessed June 18, 2018).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.