

Abstract
Read-across has become a primary approach to fill data gaps for chemical safety assessments. Chemical similarity based on structure, reactivity, and physic-chemical property information is a traditional approach applied for read-across toxicity studies. However, toxicity mechanisms are usually complicated in a biological system, so only using chemical similarity to perform the read-across for new compounds was not satisfactory for most toxicity endpoints, especially when the chemically similar compounds show dissimilar toxicities.
This study aims to develop an enhanced read-across method for chemical toxicity predictions. To this end, we used two large toxicity datasets for read-across purposes. One consists of 3,979 compounds with Ames mutagenicity data, and the other contains 7,332 compounds with rat acute oral toxicity data. First, biological data for all compounds in these two datasets were obtained by querying thousands of PubChem bioassays. The PubChem bioassays with at least five compounds from either of these two datasets showing active responses were selected to generate comprehensive bioprofiles. The read-across studies were performed by using chemical similarity search only and also by using a hybrid similarity search based on both chemical descriptors and bioprofiles. Compared to traditional read-across based on chemical similarity, the hybrid read-across approach showed improved accuracy of predictions for both Ames mutagenicity and acute oral toxicity. Furthermore, we could illustrate potential toxicity mechanisms by analyzing the bioprofiles used for this hybrid read-across study. The results of this study indicate that the new hybrid read-across approach could be an applicable computational tool for chemical toxicity predictions. In this way, the bottleneck of traditional read-across studies can be overcome by introducing public biological data into the traditional process. The incorporation of bioprofiles generated from the additional biological data for compounds can partially solve the “activity cliff” issue and reveal their potential toxicity mechanisms. This study leads to a promising direction to utilize data-driven approaches for computational toxicology studies in the big data era.
Keywords: computational toxicology, read-across, biosimilarity, hybrid approach, toxicity mechanisms, big data
1. Introduction
Numerous chemicals are used in our ordinary life, and over 100,000 chemicals have been put on the market (Johnson et al. 2017). However, only a small portion of these compounds have been tested for their toxicity potentials, and the toxicities of a great number of new chemicals wait to be evaluated. Traditional experimental toxicology protocols are usually based on animal tests, which are expensive and time-consuming (Hartung 2009). Moreover, these traditional protocols have raised ethical concerns regarding the well-being of animals (Balls 1994; Baumans 2004; Rollin 2003). This situation leads to an urgent need to develop alternatives for animal tests, so the regulatory agencies are developing pre-screening and prioritization programs to fill toxicity data gaps.
In 2007, the U.S. National Research Council recommended both high-throughput screening (HTS) and computational models as essential chemical toxicity evaluation tools in 21st-century toxicology (Gibb 2008). The HTS techniques have been widely applied in chemical screening with advantages of low expenses and faster turnaround time, which resulted in rich biological data accumulating in publically available databases (Zhu and Xia 2016). Motivated by these available data, computational toxicology has advanced to a big data era (Ciallella and Zhu 2019; Zhao and Zhu 2018; Zhu et al. 2014).
Quantitative structure-activity relationship (QSAR) approaches have been widely used in traditional computational toxicology modeling (Hansch et al. 1995; Schultz et al. 2003). QSAR models were based on the hypothesis that chemically similar compounds are likely to exhibit similar biological activities, including toxicities. Since all QSAR models were developed based on chemical structure information, the “activity cliff” issue (Maggiora 2006) (i.e., chemically similar compounds with distinctly different toxicity results) brings prediction errors to QSAR models, especially when using existing QSAR models to predict new compounds.
Along with QSAR modeling studies in the past decade, the read-across strategy was developed to predict toxicity for new compounds using similar compounds with known toxicity results (Dimitrov and Mekenyan 2010; Modi et al. 2012; Raies and Bajic 2016; Schultz et al. 2015). Various software tools were developed to perform read-across studies in the toxicology field in recent years, such as ToxMatch and the OECD QSAR Toolbox. ToxMatch (Gallegos-Saliner et al. 2008; Van Ravenzwaay et al. 2016) is an open-source software application that encodes several chemical similarity calculation tools to facilitate the systematic development of chemical groupings and read-across. The OECD QSAR Toolbox (http://www.qsartoolbox.org/) (Dimitrov et al. 2016) is a software to systematically group chemicals into categories using chemical similarity read-across, trend analysis, or QSAR predictions. Similar to traditional QSAR models, these read-across tools are only based on the chemical structure information, which cannot deal with predictions of complex biological activities (e.g., animal toxicity). In order to solve the above issue, Low et al. (Low et al. 2013) proposed a hybrid approach, termed as chemical-biological read-across (CBRA), that relies not only on chemical descriptors but also on biological profiles generated from short-term experimental assays (i.e., biological descriptors). However, the CBRA approach was based on a small set of assays, which were manually selected. This method is applicable only when all experimental assay data are available for compounds in the training set and the target new compounds.
In this study, we developed a new hybrid read-across method to evaluate the chemical toxicity potentials. Unlike traditional read-across methods, the similarity between two compounds in this study was calculated by combining chemical similarity, which was based on chemical structures, and biosimilarity, which was based on publically available biological data. For biosimilarity searches, a large set of biological data was obtained and optimized from PubChem database using the in-house Chemical In Vitro-In Vivo Profiling (CIIPro) portal (Russo et al. 2017). This hybrid read-across method showed advantages compared with the traditional read-across strategy on modeling and predicting both Ames mutagenicity and acute oral toxicity datasets. It could be used as a universal strategy to deal with other complex toxicity endpoints when extra biological data are available.
2. Material and methods
2.1. Datasets
The two toxicity datasets used in this study were curated in-house or obtained from MultiCASE, Inc. (http://www.multicase.com/). They were selected because they are two of the largest toxicity datasets available, which contain thousands of diverse compounds. The first dataset contains 3,979 unique organic compounds with the Ames mutagenicity testing results collected from public sources (Hansen et al. 2009). These mutagenicity testing data were categorized as toxic (activity as 1) for 1,718 compounds and non-toxic (activity as 0) for 2,261 compounds. The second dataset (Zhu et al. 2009) contains 7,332 unique organic compounds with rat acute toxicity results. These acute toxicity results were previously collected and curated from ChemIDplus (https://chem.nlm.nih.gov/chemidplus/) and shown as the lethal dose (unit as moles per kilogram) that cause the death of 50% testing rats (LD50). In this study, the quantitative toxicity results were expressed as the negative logarithm values of LD50 (mol/kg) (− log10 LD50 ) ranging from −0.343 to 10.207.
2.2. Chemical similarity calculations
A total of 192 2-D chemical descriptors for each compound were generated using Molecular Operating Environment (MOE) software (version 2013) (http://www.chemcomp.com/), such as physical properties, atom and bond counts, and van der Waals surface area information. (Labute 2000). The descriptors were standardized and rescaled to range from 0 to 1. The set of MOE 2-D chemical descriptors for a compound could be treated as a 192-dimensional vector (a1, a2, …, a192). The pairwise chemical similarity Schem was calculated based on the Euclidean distance dEuc between two compounds, using Equation 1:
| (1) |
2.3. Biosimilarity calculations
Biological data of all compounds in these two datasets were obtained from the PubChem database (https://pubchem.ncbi.nlm.nih.gov/) using the CIIPro portal (http://ciipro.rutgers.edu/) (Russo et al. 2017). The biosimilarity Sbio between two compounds was calculated using Equation 2 (Ribay et al. 2016; Russo et al. 2017):
| (2) |
Here, Aa and Ba represent the active responses for compounds A and B in the same set of bioassays, respectively. And Ai and Bi represent the inactive responses. Our previous work (Kim et al. 2016) showed that the biosimilarity values rely on active data more than inactive data, since the active data indicates more significant information than inactive. The term w weights the inactive responses less than active in biosimilarity calculations. In this study, w was defined as the ratio for each compound pair and ranged from 0 to 1.
2.4. Read-across predictions and evaluations
The read-across prediction of a compound in the test set was made by the nearest neighbor compound in the training set. For traditional read-across, the prediction was made by the toxicity value of its chemical nearest neighbor, which was identified by chemical similarity calculations. Furthermore, the hybrid read-across prediction was made by the toxicity value of its chemical and biological nearest neighbor, which was identified by calculating biosimilarity between the test set compound and its chemical nearest neighbor in the training set.
Since the read-across procedure was performed by using the above two datasets with different types of toxicity values, universal statistical metrics were needed to evaluate the performance of the models developed individually. The same parameters were used to evaluate the computational models in our previous studies (Kim et al. 2014; Solimeo et al. 2012; Wang et al. 2015). The results were harmonized by using 1) sensitivity (percentage of compounds predicted correctly within the toxic class, Equation 3), specificity (percentage of compounds predicted correctly within the nontoxic class, Equation 4), and CCR (correct classification rate or balanced accuracy, Equation 5) for the Ames mutagenicity dataset; and 2) coefficient of determination (R02, Equation 6) and mean absolute error (MAE, Equation 7) for the acute oral toxicity dataset.
| (3) |
| (4) |
| (5) |
| (6) |
| (7) |
3. Results
3.1. Overview of the workflow
Figure 1 summarized the workflow of the hybrid read-across procedure used in this study. For all compounds in the test set, 192 MOE 2-D chemical descriptors were used to calculate the chemical similarity for identifying their chemical nearest neighbors in the training set. Then, our in-house profiling tool CIIPro was used to extract all relevant biological data and to generate bioprofiles for these compounds. Biosimilarity was calculated to determine, for a target compound, whether its chemical nearest neighbor is also biosimilar. A read-across toxicity prediction was made when the chemical nearest neighbor was also identified to be biosimilar.
Fig. 1.

The hybrid read-across workflow
3.2. Bioprofile generation
The bioprofile was generated by extracting all relevant biological data from PubChem database using the CIIPro portal for all the compounds in these two datasets. Over 50,000 PubChem bioassays with at least one compound in the datasets showing an active response were extracted as the original bioprofile. This original bioprofile contains over ten million data points for all the compounds in these two datasets. However, PubChem assays containing very few data points in the original bioprofile would be useless for read-across. Thus, to optimize this original bioprofile, the bioassays with less than five active responses within either of the two databases were removed. This effort resulted in 1,716 bioassays in the bioprofile for 2,025 compounds in the Ames mutagenicity dataset, with a ratio of active data (’1’) to inactive data (‘−1’) of 8.38%, and 1,091 bioassays in the bioprofile for 2,208 compounds in the acute toxicity dataset, with an active/inactive data ratio of 7.51%.
The optimized bioprofiles could reveal rich biological information for compounds in these two datasets. For example, 4′-Chlorodiazepam (CID 1688), which is a mutagen in Ames dataset, contains 69 PubChem bioassays testing results in the bioprofile and 21 of them were active responses. Most of these 21 PubChem bioassays are related to toxicity testing, such as cytotoxicity assay (AID 449705) and hepatotoxicity related assays (AID 678712, 678713 and etc.). Another compound, 4-Dihydroxypyrimidine (CID 1174) from acute oral toxicity dataset, which has LD50 value of 0.00034 mol/kg, contains 651 PubChem bioassays testing results in the bioprofile and 159 of them were active responses. Not surprisingly, most of these assays are also related to toxicity testing, including some assays from Tox21 program related to identify antagonists of cell signaling pathways (AID 1224838, 1259244 and etc.).
3.3. Similarity calculation
Using chemical descriptors and bioprofiles generated above, pairwise similarity was calculated for all compounds in these two dataset, respectively. For each target compound, its nearest neighbor was defined as the most similar compound, which should be the compound with the largest Schem and/or Sbio in the dataset. The hypothesis of traditional QSAR models and read-across studies is that chemically similar compounds have similar bioactivities. For this reason, it is worth to compare the two types of similarity indices based on chemical descriptors and bioprofiles. Figure 2 shows the distribution of compounds with at least one chemical nearest neighbor (Schem > 0.80) for these two datasets. These compounds and their chemical nearest neighbors were also classified as biosimilar (Sbio > 0.80) and biodissimilar (Sbio < 0.80).
Fig. 2.
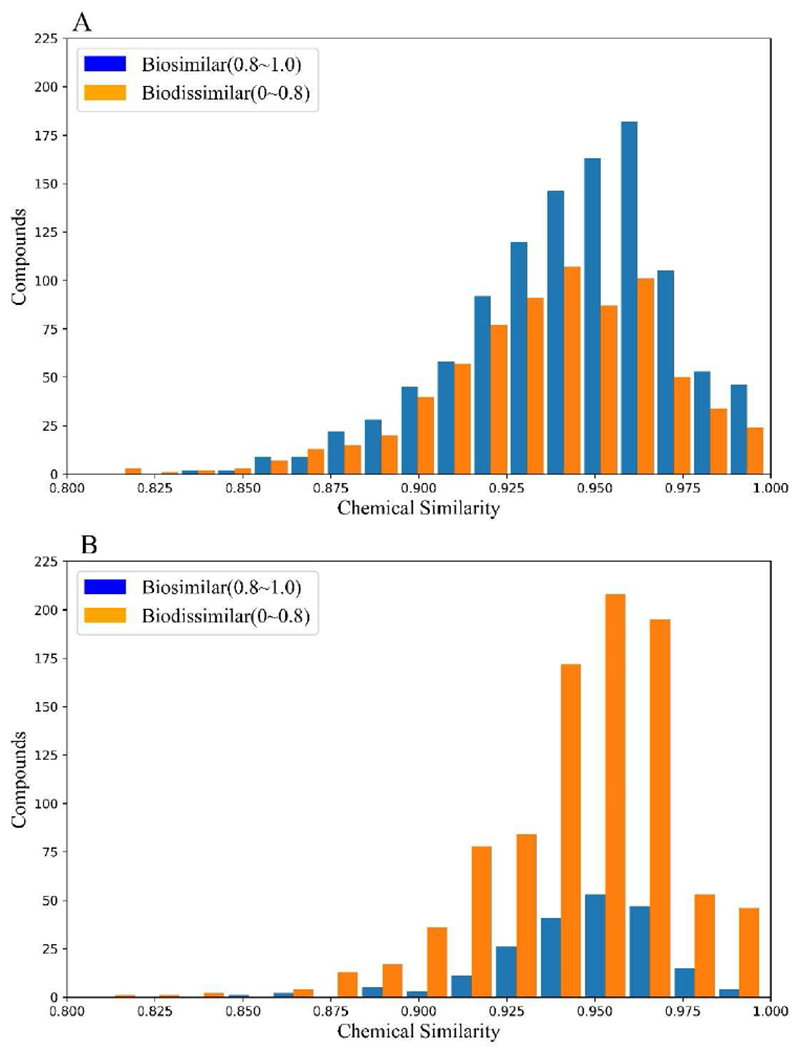
The comparison of biosimilarity results of the compounds with their chemical nearest neighbors for (A) Ames mutagenicity; (B) Rat acute oral toxicity. A biosimilarity threshold (0.80) was set to evaluate whether a target compound and its chemical nearest neighbor are biologically similar or not. The blue columns represent the numbers of compounds which are also biosimilarity to their chemcial nearest neighbors (Sbio>0.80); the red columns represent the numbers of compounds which are biodissimilar to their chemical nearest neighbors (Sbio<0.80).
The similarity distribution in Ames mutagenicity dataset fulfilled the hypothesis of traditional read-across. As shown in Figure 2A, when two compounds are chemically similar, they are more likely to be biosimilar (represented by blue bars) than biodissimilar (represented by orange bars). However, in oral acute toxicity dataset, two chemically similar compounds are likely to have dissimilar bioprofiles, as shown in Figure 2B. This result showed an opposite condition to the above hypothesis. These results indicated the reason that much better modeling results (i.e. higher predictivity) could be obtained previously from QSAR studies of Ames mutagenicity (Bakhtyari et al. 2013; Hillebrecht et al. 2011; Votano et al. 2004; Xu et al. 2012) than those of acute oral toxicity dataset (Devillers and Devillers 2009; Lagunin et al. 2011). In this study, it was also expected that read-across based on only chemical structures would likely to cause significant prediction errors for the acute oral toxicity.
3.4. Read-across for toxicity prediction
In traditional read-across studies, prediction of a new compound was obtained from the experimental toxicity value of its nearest neighbor identified using chemical similarity. However, since biological systems are complex and two chemically similar compounds could show opposite toxic effect in biological test, prediction errors could always occur using the traditional read-across strategy. This issue is known as an “activity cliff’ (Cruz-Monteagudo et al. 2014; Maggiora 2006; Tropsha 2010; Zhu et al. 2009). In order to solve this problem, a hybrid read-across study was performed based on the combination of chemical similarity and biosimilarity calculation.
Figure 3 showed the distribution of read-across prediction for all target compounds on Ames mutagenicity dataset obtained from five-fold cross validation procedure. Traditionally the toxicity prediction of a target compound was made if there was a chemical nearest neigbor that could be identified from training set (i.e. Schem > 0.90). The predictivity of the traditional read-across was indicated as CCR of 0.80, sensitivity of 0.84 and specificity of 0.77 (Table 1). In this study, we further applied biosimilarity results into read-across prediction. To this end, the biosimilairty value of a compound with its chemical nearest neighbor was also calculated. Based on the correlation between chemical similarity and biosimilarity results, as shown in Figure 3, compound pairs (the target compound with its nearest neighbor) can be classified as: 1) both chemically similar (Schem > 0.90) and biosimilar (Sbio > 0.80) (area A); 2) chemically similar (Schem > 0.90) and biodissimilar (Sbio ≤ 0.80) (area B); 3) chemically dissimilar (Schem ≤ 0.90) and biosimilar (Sbio > 0.80) (area C); or 4) chemically dissimilar (Schem ≤ 0.90) and biodissimilar (Sbio ≤ 0.80) (area D). When the hybrid read-across was performed, a compound was predicted if its chemical nearest neighbor was also biosimilar (as area A in Fig. 3.). The predicitvity was moderately increased and CCR increased from 0.80 to 0.82 (Table 1).
Fig. 3.
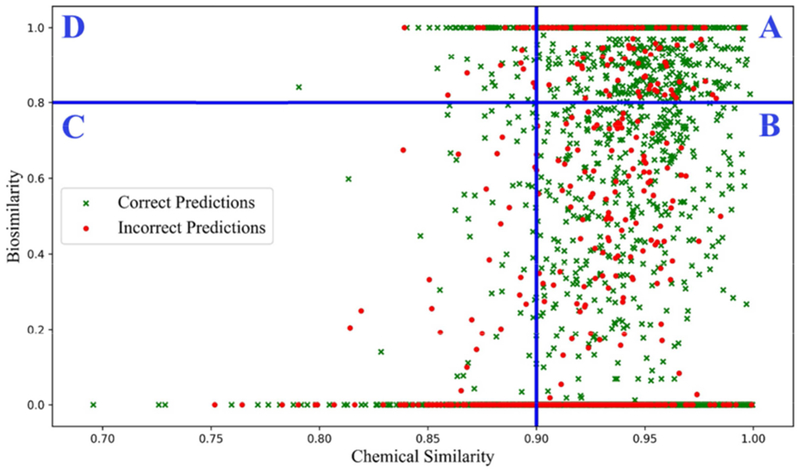
The distribution of read-across predictions for compounds in Ames mutagenicity. The green crosses are correct predictions and the red round dots are incorrect predictions. The read-across predictions were divided into four areas by using two threshold values (Chemical similarity = 0.90 and Biosimilarity = 0.80): The area A includes compound pairs with high chemical similarity and high biosimilarity; the area B includes compound pairs with high chemical similarity and low biosimilarity; the area C includes compound pairs with low chemical similarity and low biosimilarity; and the area D includes compound pairs with low chemical similarity and high biosimilarity.
Table 1.
Comparisons of traditional read-across and hybrid read-across prediction results.
| Parameters | Traditional read-across | Hybrid read-across |
|---|---|---|
| Ames mutagenicity | ||
| Sensitivity | 0.84 | 0.90 |
| Specificity | 0.77 | 0.74 |
| CCR | 0.80 | 0.82 |
| Acute oral toxicity | ||
| R02 | 0.36 | 0.68 |
| MAE | 0.55 | 0.44 |
For acute oral toxicity dataset, traditional read-across strategy resulted in low prediction accuracy (R02 = 0.36, MAE = 0.55) (Table 1). Furthermore, we also integrated biosimilarity result into the traditional read-across prediction. Based on the correlation between chemical similarity and biosimilarity results, as shown in Figure 4, pairs of a target compound with its chemical nearest neighbor can be classified as: 1) both chemically similar (Schem > 0.90) and biosimilar (Sbio > 0.80) (red dots); 2) chemically dissimilar (Schem ≤ 0.90) and/or biosimilar (Sbio ≤ 0.80) (black dots). By applying hybrid read-across approach, a compound was predicted by its chemical nearest neighbor if they are also biosimilar (as red dots in Fig. 4.). Through this way, the prediction accuracy was increased significantly (R02 = 0.68, MAE = 0.44) (Table 1).
Fig. 4.
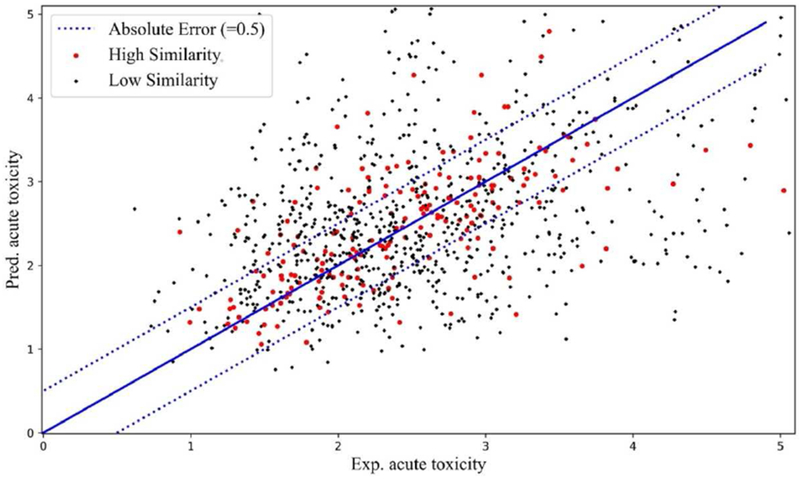
The correlation between experimental and predicted acute toxicity values for compounds in acute oral toxicity dataset (Values shown as −log10 LD50). The red dots represent compound pairs with high chemical similarity and high biosimilarity; the black dots represent pairs in other cases (i.e. either chemically disimiar or biodisimiar). The dots between two dashed lines represent accurate predictions (absolute errors less than 0. 50).
4. Discussion
The hybrid read-across approach used in this study increased predictivity for compounds in both datasets. The slight decrease of specificity of Ames dataset fits to the results obtained from our previous study (Ribay et al. 2016). The biosimilarity, which relies mostly on active data, is more meaningful for the predictions of toxicants instead of non-toxicants (Russo et al. 2019). With additional similarity calculations based on bioprofiles, read-across prediction can be strengthened by comparing the bioprofiles of chemical nearest neighbors. Several examples of the nearest neighbors (both chemically similar and biosimilar) identified by hybrid read-across, were listed in the Supplemental Table S1 and S2.
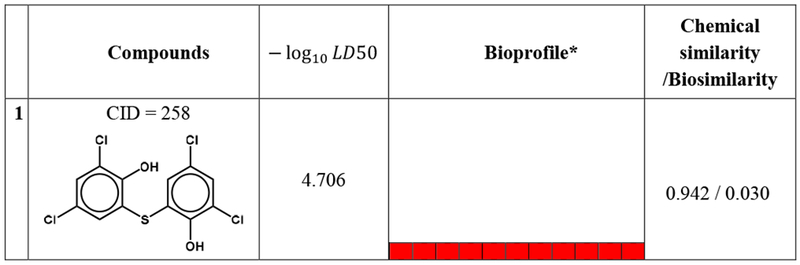
By analyzing the bioprofiles, it is also feasible to find the “activity cliffs” existing in these two datasets. Table 2 and 3 show five representative activity cliffs in these two datasets. Some of these nearest neighbor compounds are chemically similar but have opposite toxicity results. For example, masoprocol (CID 1593), which is a lipoxygenase inhibitor (Gowri et al. 2000), is shown as a mutagen in Ames dataset. However, its chemical nearest neighbor diphenolic acid (CID 2265) is a non-mutagen in Ames dataset. The only difference in the structures of these two compounds is the radical group between the two benzene rings (Table 2). If one of these two compounds is in the training set and the other is in the test set, a prediction error will occur. However, when comparing their bioprofiles, which are shown in Table 2, a significant difference can be noticed. Moreover, the biosimilarity value between these two compounds is only 0.189, indicating the biodissimilarity of these two compounds. A similar condition can also be seen in acute oral toxicity dataset (Table 3). For example, Blasticidin S (CID 258) is an antibiotic isolated from Streptomyces griseochromogenes (Takeuchi et al. 1958) with a −log10 LD50 value of 4.706. Its chemical nearest neighbor AC1L1K32 (CID 5317), however, has a −log 10 LD50 value of 1.913. The only difference in the structures of these two compounds is the substituent on the para-position of the benzene ring, which causes Blasticidin S to be acutely toxic. The biosimilarity between these two compounds is 0.030. These two compounds can also potentially induce the “activity cliff’ issue.
Table 2.
The five representative compounds and their chemical nearest neighbors in Ames mutagenicity dataset.
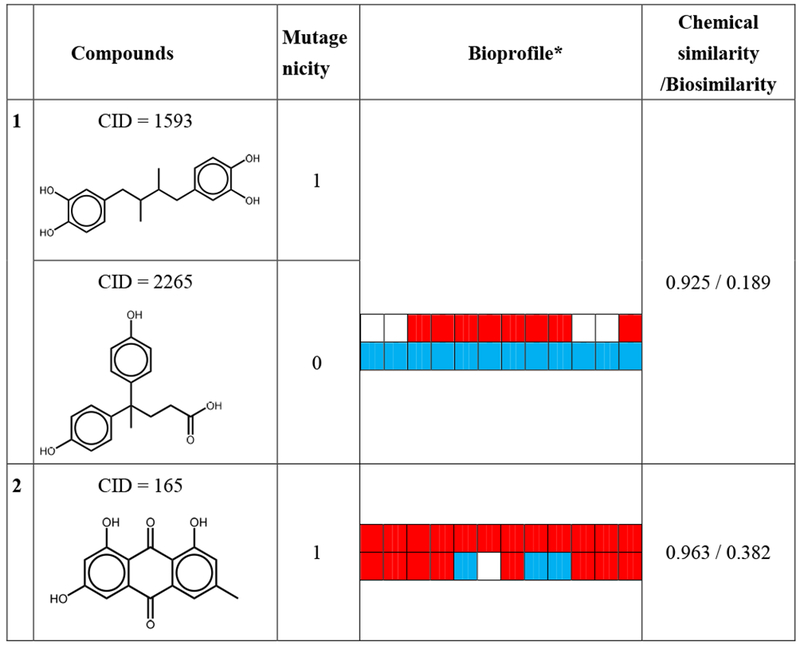 |
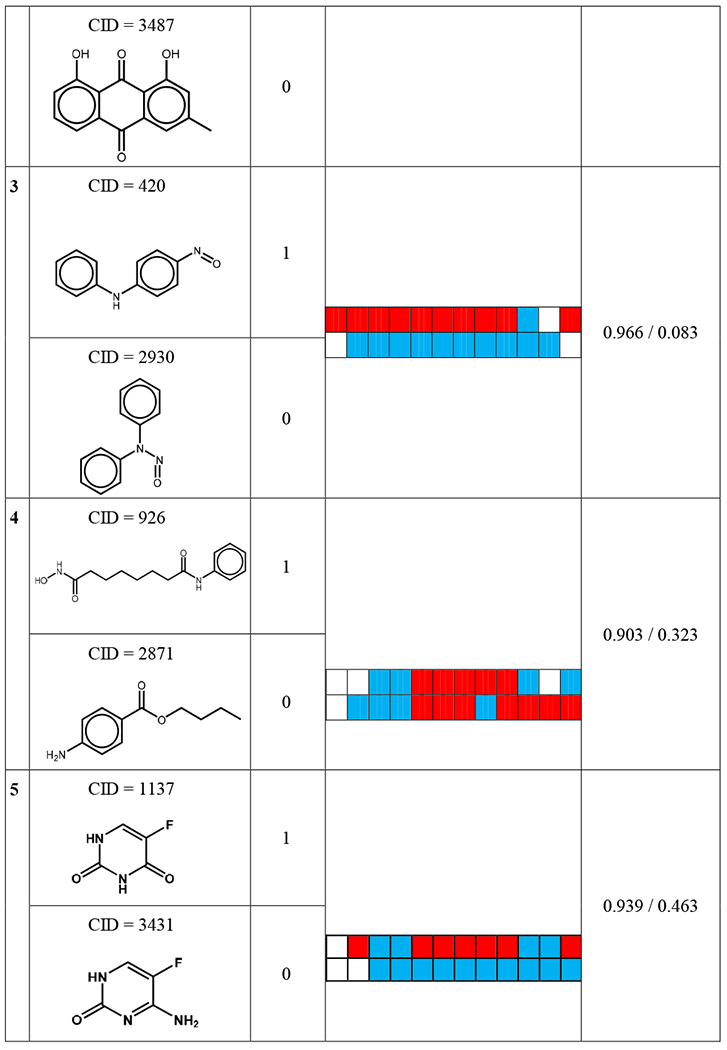 |
Bioprofiles AIDs: 651741, 651838, 720635, 720637, 743012, 743014, 743015, 743064, 743065, 743122, 1224892, 1259243.
For bioprofiles, the red color indicates an active response, the blue color indicates an inactive response and white color indicates no data available.
Table 3.
The five representative compounds and their chemical nearest neighbor in acute oral toxicity dataset.
 |
 |
 |
Bioprofile AID: 720635, 720637, 743012, 743014, 743015, 743064, 743065, 1159529, 1224871, 1224874, 1259243.
For bioprofiles, the red color indicates active response, blue color indicates inactive response and white color indicates no data available.
Some compounds were considered to be chemical nearest neighbors based on calculation results, but they are not actual similar in structure. This issue is due to the limitation of chemical descriptors, which cannot distinguish their structural diversity. A potential solution is to various chemical descriptors in the modeling process, such as reported in our previous studies (Solimeo et al. 2012; Zhao et al. 2017). For example, as shown in Table 2, compound with CID 926 is a dinucleotide and related to nicotinamide adenine dinucleotide (NAD) (Belenky et al. 2007), a cofactor in cells. Its chemical nearest neighbor Coumaphos (CID 2871) is a organothiophosphorus cholinesterase inhibitor that acts as an anthelmintic, insecticide, and as a nematocide (Gregorc et al. 2018). Their chemical similarity Schem was 0.903 but their structures actually differ significantly. This issue is due to the limitation of chemical descriptors, which cannot distinguish their structural diversity. The biosimilarity calculation result (Sbio = 0.323) indicated their difference and can avoid this prediction error in read-across process.
Previous QSAR models were usually questioned as “black box” (Fraczkiewicz et al. 2009; Polishchuk et al. 2013) by providing predictions without explaining the mechanisms of the toxicity. By examining the bioassays included in the bioprofiles, the hybrid read-across in this study could reveal the potential toxicity mechanisms. For example, the bioprofiles in Table 2 listed totally 12 PubChem bioassays (AIDs 651741, 651838, 720635, 720637, 743012, 743014, 743015, 743064, 743065, 743122, 1224892, 1259243). Among them, there were five assays related to cytotoxicity (AIDs 651838, 743012, 743014, 743015, 743064), two assays related to mitochondria membrane potential testing (AIDs 720635, 720637), and five assays related to antagonists of signaling pathways (AIDs 651741, 743065, 743122, 1224892, 1259243). These bioassays could be used for investigating the mechanism of compounds in Ames dataset for their mutagenicity. Similar analysis could also be done for acute toxicity dataset, all the information for bioassays list in Table 2 and 3 could be found in details from PubChem through their AID. Thus, using the hybrid read-across strategy demonstrated in this study, these prediction results could be further analyzed through investigating the bioprofiles. This strategy could be applied in future study studies for other toxicity endpoint predictions.
5. Conclusion
Traditional read-across was based on the use of chemical structure information and induce prediction errors in many toxicity studies. The availability of public big data sources provides rich biological data for the compounds of interest (e.g., environmental compounds). This study shows that the hybrid read-across, which was based on the combination of chemical structure information and biological data, has certain advantages compared with the traditional read-across, especially for complex animal toxicities (i.e., acute oral toxicity). Although the integration of biological data into the read-across procedure brought new challenges (e.g. biased data and missing data), the development of new similarity approaches can make this practice applicable to predict new compounds. The bioprofiles generated from public biological data also provided new opportunity to reveal relevant toxicity mechanisms for potential toxicants. The hybrid read-across workflow developed in this study can be applied for other toxicity endpoints. The use of public big data sources in the predictive modeling can advance the computational toxicology into a big data era.
Supplementary Material
Table S1 The five representative compounds and their chemical nearest neighbors with high biosimilarity in Ames mutagenicity dataset.
Table S2 The five representative compounds and their chemical nearest neighbors with high biosimilarity in the acute oral toxicity dataset.
The highlights for this study are:
Two large toxicity datasets Ames and rat acute toxicity were used in this study for evaluating a hybrid read-across strategy based on both chemical descriptors and public biological data.
This hybrid read-across strategy showed improved accuracy of predictions compared to the traditional read-across.
This hybrid read-across strategy, which is based on public big data, can not only solve the “activity cliff’ issue of traditional read-across studies but also illustrate potential toxicity mechanisms.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (Grant Nos. 31100523), the Beijing Municipal Education Commission (Grant No. KM201410005030), and Beijing municipal colleges and universities young talents cultivation plan. This study was partially supported by the National Institute of Environmental Health Sciences [grant number R15ES023148], the Colgate-Palmolive Grant for Alternative Research, and the Johns Hopkins Center for Alternatives to Animal Testing (CAAT) grant.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Bakhtyari NG, Raitano G, Benfenati E, Martin T, Young D. 2013. Comparison of in silico models for prediction of mutagenicity. Journal of Environmental Science and Health, Part C 31:45–66. [DOI] [PubMed] [Google Scholar]
- Balls M 1994. Replacement of animal procedures: Alternatives in research, education and testing. Lab Anim 28:193–211. [DOI] [PubMed] [Google Scholar]
- Baumans V 2004. Use of animals in experimental research: An ethical dilemma? Gene Ther 11 Suppl 1:S64–66. [DOI] [PubMed] [Google Scholar]
- Belenky P, Bogan KL, Brenner C. 2007. Nad+ metabolism in health and disease. Trends in Biochemical Sciences 32:12–19. [DOI] [PubMed] [Google Scholar]
- Ciallella HL, Zhu H. 2019. Advancing computational toxicology in the big data era by artificial intelligence: Data-driven and mechanism-driven modeling for chemical toxicity. Chemical Research in Toxicology. In press [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz-Monteagudo M, Medina-Franco JL, Perez-Castillo Y, Nicolotti O, Cordeiro MN, Borges F. 2014. Activity cliffs in drug discovery: Dr jekyll or mr hyde? Drug Discov Today 19:1069–1080. [DOI] [PubMed] [Google Scholar]
- Devillers J, Devillers H. 2009. Prediction of acute mammalian toxicity from qsars and interspecies correlations. SAR QSAR Environ Res 20:467–500. [DOI] [PubMed] [Google Scholar]
- Dimitrov S, Mekenyan O. 2010. Chapter 15:An introduction to read-across for the prediction of the effects of chemicals. Issues in Toxicology:372–384. [Google Scholar]
- Dimitrov SD, Diderich R, Sobanski T, Pavlov TS, Chankov GV, Chapkanov AS, et al. 2016. Qsar toolbox – workflow and major functionalities. Sar & Qsar in Environmental Research 27:203–219. [DOI] [PubMed] [Google Scholar]
- Fraczkiewicz R, Zhuang D, Zhang J, Miller D, Woltosz W, Bolger M. 2009. Busting the black box myth: Designing out unwanted admet properties with machine learning approaches. CICSJ Bulletin 27:96–102. [Google Scholar]
- Gallegos-Saliner A, Poater A, Jeliazkova N, Patlewicz G, Worth AP. 2008. Toxmatch-a chemical classification and activity prediction tool based on similarity measures. Regulatory Toxicology and Pharmacology 52:77–84. [DOI] [PubMed] [Google Scholar]
- Gibb S 2008. Toxicity testing in the 21st century: A vision and a strategy. Reprod Toxicol 25:136–138. [DOI] [PubMed] [Google Scholar]
- Gowri MS, Azhar RK, Kraemer FB, Reaven GM, Azhar S. 2000. Masoprocol decreases rat lipolytic activity by decreasing the phosphorylation of hsl. Am J Physiol-Endoc M 279:E593–E600. [DOI] [PubMed] [Google Scholar]
- Gregorc A, Alburaki M, Rinderer N, Sampson B, Knight PR, Karim S, et al. 2018. Effects of coumaphos and imidacloprid on honey bee (hymenoptera: Apidae) lifespan and antioxidant gene regulations in laboratory experiments. Sci Rep 8:15003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansch C, Hoekman D, Leo A, Zhang L, Li P. 1995. The expanding role of quantitative structure-activity relationships (qsar) in toxicology. Toxicol Lett 79:45–53. [DOI] [PubMed] [Google Scholar]
- Hansen K, Mika S, Schroeter T, Sutter A, Laak AT, Stegerhartmann T, et al. 2009. Benchmark data set for in silico prediction of ames mutagenicity. Chemistry Central Journal 3:1–1. [DOI] [PubMed] [Google Scholar]
- Hartung T 2009. Toxicology for the twenty-first century. Nature 460:208–212. [DOI] [PubMed] [Google Scholar]
- Hillebrecht A, Muster W, Brigo A, Kansy M, Weiser T, Singer T. 2011. Comparative evaluation of in silico systems for ames test mutagenicity prediction: Scope and limitations. Chemical Research in Toxicology 24:843–854. [DOI] [PubMed] [Google Scholar]
- Johnson AC, Donnachie RL, Sumpter JP, Jurgens MD, Moeckel C, Pereira MG. 2017. An alternative approach to risk rank chemicals on the threat they pose to the aquatic environment. Sci Total Environ 599-600:1372–1381. [DOI] [PubMed] [Google Scholar]
- Kim MT, Sedykh A, Chakravarti SK, Saiakhov RD, Zhu H. 2014. Critical evaluation of human oral bioavailability for pharmaceutical drugs by using various cheminformatics approaches. Pharm Res 31:1002–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MT, Huang R, Sedykh A, Wang W, Xia M, Zhu H. 2016. Mechanism profiling of hepatotoxicity caused by oxidative stress using antioxidant response element reporter gene assay models and big data. Environmental Health Perspectives 124:634–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labute P 2000. A widely applicable set of descriptors. Journal of Molecular Graphics & Modelling 18:464–477. [DOI] [PubMed] [Google Scholar]
- Lagunin A, Zakharov A, Filimonov D, Poroikov V. 2011. Qsar modelling of rat acute toxicity on the basis of pass prediction. Mol Inform 30:241–250. [DOI] [PubMed] [Google Scholar]
- Low Y, Sedykh A, Fourches D, Golbraikh A, Whelan M, Rusyn I, et al. 2013. Integrative chemical-biological read-across approach for chemical hazard classification. Chem Res Toxicol 26:1199–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maggiora GM. 2006. On outliers and activity cliffs--why qsar often disappoints. Journal of Chemical Information & Modeling 46:1535. [DOI] [PubMed] [Google Scholar]
- Modi S, Hughes M, Garrow A, White A. 2012. The value of in silico chemistry in the safety assessment of chemicals in the consumer goods and pharmaceutical industries. Drug Discovery Today 17:135–142. [DOI] [PubMed] [Google Scholar]
- Polishchuk PG, Kuz’Min VE, Artemenko AG, Muratov EN. 2013. Universal approach for structural interpretation of qsar/qspr models. Molecular Informatics 32:843–853. [DOI] [PubMed] [Google Scholar]
- Raies AB, Bajic VB. 2016. In silico toxicology: Computational methods for the prediction of chemical toxicity. Wiley Interdiscip Rev Comput Mol Sci 6:147–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribay K, Kim MT, Wang W, Pinolini D, Zhu H. 2016. Predictive modeling of estrogen receptor binding agents using advanced cheminformatics tools and massive public data. Front Environ Sci 4:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rollin BE. 2003. Toxicology and new social ethics for animals. Toxicol Pathol 31 Suppl:128–131. [DOI] [PubMed] [Google Scholar]
- Russo DP, Kim MT, Wang W, Pinolini D, Shende S, Strickland J, et al. 2017. Ciipro: A new read-across portal to fill data gaps using public large-scale chemical and biological data. Bioinformatics 33:464–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo DP, Strickland J, Karmaus AL, Wang W, Shende S, Hartung T, et al. 2019. Nonanimal models for acute toxicity evaluations: Applying data-driven profiling and read-across. Environ Health Perspect 127:47001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz TW, Cronin MTD, Walker JD, Aptula AO. 2003. Quantitative structure–activity relationships (qsars) in toxicology: A historical perspective. Theochem 622:1–22. [Google Scholar]
- Schultz TW, Amcoff P, Berggren E, Gautier F, Klaric M, Knight DJ, et al. 2015. A strategy for structuring and reporting a read-across prediction of toxicity. Regul Toxicol Pharmacol 72:586–601. [DOI] [PubMed] [Google Scholar]
- Solimeo R, Zhang J, Kim M, Sedykh A, Zhu H. 2012. Predicting chemical ocular toxicity using a combinatorial qsar approach. Chemical Research in Toxicology 25:2763–2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi S, Hirayama K, Ueda K, Sakai H, Yonehara H. 1958. Blasticidin s, a new antibiotic. J Antibiot (Tokyo) 11:1–5. [PubMed] [Google Scholar]
- Tropsha A 2010. Best practices for qsar model development, validation, and exploitation. Molecular Informatics 29:476–488. [DOI] [PubMed] [Google Scholar]
- van Ravenzwaay B, Sperber S, Lemke O, Fabian E, Faulhammer F, Kamp H, et al. 2016. Metabolomics as read-across tool: A case study with phenoxy herbicides. Regul Toxicol Pharm 81:288–304. [DOI] [PubMed] [Google Scholar]
- Votano JR, Parham M, Hall LH, Kier LB, Oloff S, Tropsha A, et al. 2004. Three new consensus qsar models for the prediction of ames genotoxicity. Mutagenesis 19:365–377. [DOI] [PubMed] [Google Scholar]
- Wang W, Kim MT, Sedykh A, Zhu H. 2015. Developing enhanced blood-brain barrier permeability models: Integrating external bio-assay data in qsar modeling. Pharm Res 32:3055–3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Cheng F, Chen L, Du Z, Li W, Liu G, et al. 2012. In silico prediction of chemical ames mutagenicity. Journal of Chemical Information & Modeling 52:2840. [DOI] [PubMed] [Google Scholar]
- Zhao L, Wang WY, Sedykh A, Zhu H. 2017. Experimental errors in qsar modeling sets: What we can do and what we cannot do. Acs Omega 2:2805–2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Zhu H. 2018. Big data in computational toxicology: Challenges and opportunities In: Ekins S (Eds.) Computational Toxicology: Risk Assessment for Chemicals. 1111, John Wiley & Sons,Inc.page 291–312. [Google Scholar]
- Zhu H, Martin TM, Ye L, Sedykh A, Young DM, Tropsha A. 2009. Quantitative structure-activity relationship modeling of rat acute toxicity by oral exposure. Chemical Research in Toxicology 22:1913–1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Zhang J, Kim MT, Boison A, Sedykh A, Moran K. 2014. Big data in chemical toxicity research: The use of high-throughput screening assays to identify potential toxicants. Chem Res Toxicol 27:1643–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Xia M. 2016. High-throughput screening assays in toxicology. Springer Science+Business Media LLC, New York. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 The five representative compounds and their chemical nearest neighbors with high biosimilarity in Ames mutagenicity dataset.
Table S2 The five representative compounds and their chemical nearest neighbors with high biosimilarity in the acute oral toxicity dataset.