

Abstract
LKB1/STK11 is a tumor suppressor gene responsible for Peutz-Jeghers syndrome, an inherited cancer disorder associated with genome instability. The lKB1 protein functions in the regulation of cell proliferation, polarization and differentiation. Here, we suggest a role of lKB1 in non-homologous end joining (NHEJ), a major DNA double-strand break (DSB) repair pathway. lKB1 localized to DNA ends upon the generation of micro-irradiation and l-Scel endonuclease-induced DSBs. lKB1 inactivation either by RNA interference or by kinase-dead mutation compromised NHEJ-mediated DNA repair by suppressing the accumulation of BRM, a catalytic subunit of the SWI/SNF complex, at DSB sites, which promotes the recruitment of an essential NHEJ factor, KU70. AMPK2, a major substrate of lKB1 and a histone H2B kinase, was recruited to DSBs in an lKB1 -dependent manner. AMPK2 depletion and a mutation of H2B that disrupted the AMPK2 phoshorylation site impaired KU70 and BRM recruitment to DSB sites. lKB1 depletion induced the formation of chromosome breaks and radials. These results suggest that lKB1-AMPK signaling controls NHEJ and contributes to genome stability.
Keywords: LKB1-AMPK signaling, non-homologous end joining, chromatin remodeling, tumor suppressor gene, genome instability
INTRODUCTION
LKB1/STK11 is a tumor suppressor gene that encodes a serine/threonine kinase whose inactivation is responsible for Peutz-Jeghers syndrome (PJS), an inherited cancer disorder associated with genome instability.1,2 LKB1 is also one of the most commonly mutated genes in sporadic cancers, including non-small cell lung carcinoma3–5 (http://www.sanger.ac.uk/genetics/CGP/cosmic/). lKB1 regulates multiple cellular functions, including cell proliferation, polarization and differentiation, by phosphorylating adenosine monophosphate-activated protein kinases (AMPKs), that is, lKB1-AMPK signaling, in the cytoplasm.6,7 However, LKB1 and AMPKs localize not only to the cytoplasm but also to the nucleus, suggesting a possible nuclear function for these proteins. In fact, lKB1-AMPK signaling was recently shown to function in the nucleus by regulating transcription through histone H2B phosphorylation in the cellular adaptation to metabolic stresses.8
Additional evidence for the intra-nuclear role of LKB1 was provided by results showing that it interacts with and activates BRG1, a catalytic subunit of the switch/sucrose non-fermentable (SWI/SNF) chromatin remodeling complex.9 The SWI/SNF complex was recently shown to act as a remodeler (that is, factor causing alteration of the nucleosomal position of chromatin) at DNA DSB sites.10 Several studies including ours have shown that chromatin remodeling at DSB sites is a critical step required for DSB repair, including non-homologous end joining (NHEJ).11–14 In the first step of NHEJ, the KU70/80 heterodimer binds to DSBs and forms a complex with DNA-dependent protein kinase, catalytic subunit (DNA-PKcs). The DNA ends are then ligated together by the LIGIV/XRCC4 complex.15,16 Here, we suggest a novel nuclear function of LKB1 in NHEJ.
RESULTS
Recruitment of LKB1 to DSB sites
Small interfering RNA (siRNA)-mediated depletion of LKB1 resulted in high basal levels of γ-H2AX (Figure 1 a). The LKB1 depletion also suppressed reduction of ionizing radiation (IR)-induced γ-H2AX foci 48 h after IR treatment (Figure 1a). In the neutral comet assay, LKB1-depleted cells retained longer comet tails (moments) after IR treatment than control siRNA-treated cells (Figure 1b). Concor-dantly, LKB1 depletion sensitized the cells to IR and other DSB-inducing agents (Supplementary Figure SI a). These results indicate that LKB1 has a positive effect on DSB repair activity.
Figure 1.
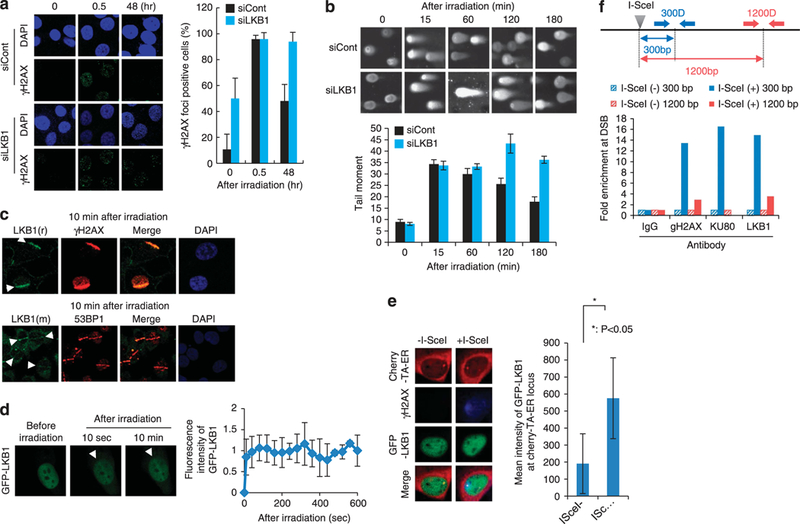
Recruitment of the LKB1 protein to DSB sites. (a) Effect of LKB1 depletion on the retention of IR-induced γH2AX foci. siRNA-treated H1299 cells were exposed to 2 Gy IR. Percentages of cells with γH2AX foci at 0, 0.5 and 48 h after irradiation are plotted on the right. Mean values ±s.d. from three independent experiments are shown. (b) Neutral comet assay of irradiated H1299 cells with or without LKB1 depletion. The tail moments of 100 cells treated with 20 Gy IR for each time point were measured and the mean values ( ± s.d.) are shown in the right graph. (c) Accumulation of endogenous LKB1 protein. At 10 min after laser micro-irradiation, H1299 cells were fixed and immunostained with anti-LKB1 (r: rabbit antibody), anti-γH2AX (upper panels); and anti-LKB1 (m: mouse antibody), and/or anti-53BP1 (lower panels). (d) Accumulation of GFP-LKB1 protein in H1299 cells subjected to laser micro-irradiation. Results obtained 10 s and 10 min after irradiation are shown. Increases in fluorescence intensities induced by micro-irradiation are shown (mean± s.d.) on the right. (e) Accumulation of GFP-LKB1 at DSB sites in U20S/TRE/1-Scel-19 cells. Left: Foci of GFP-LKB1 colocalized with those of Cherry-tTA-ER only in the presence of 1-Scel (that is, at DSB sites). Right The percentage of GFP-LKB1 foci colocalized with those of Cherry-TBP. Twenty cells showing a single focus of Cherry-TBP were examined for colocalization of GFP-LKB1. Detailed methods are described in Lan eta/. 19 (f) ChiP assay. Upper panel: locations of primers used for quantitative PCR. ChiP with a control lgG or antibodies against γH2AX, KU80 or LKB1 was performed 24 h after 1-Scel expression plasmid transfection. The graph shows the relative enrichment of examined proteins in l-Scel-treated cells versus untreated cells. The value for KuB0/1-Scel ( +) 1200 bp is almost zero.
We next examined the accumulation of the LKB1 protein at DSBs generated by laser micro-irradiation in HI299 human lung cancer cells expressing endogenous wild-type (WT) LKB1.4,17 LKB1 colocalized with γH2AX and 53BP1, a marker for DSB sites,18 at laser micro-irradiated regions (Figure 1c). Consistently, in a cell fractionation experiment, the LKB1 protein was detected in the chromatin fraction (Supplementary Figure S1b). Exogenous expression of GFP-LKB1 showed that the protein accumulates at irradiated sites within 10 s (Figure 1 d), as reported for other DSB repair proteins.19,20 LKB1 accumulation at DSB sites was confirmed by an experiment showing LKB1 accumulation at l-Scel-induced DSB sites in U20S/TRE/l-Scel-19 cells19 (Supplementary Figure Sic).
EGFP-LKB1 proteins colocalized with the Cherry-tTA-ER protein (a marker protein for l-Scel recognition sites in cell nuclei) upon l-Scel expression (Figure 1e). The recruitment of LKB1 to DSB sites was also examined by chromatin immunoprecipitation (ChIP) analysis using an anti-LKB1 antibody. H1299dA3–1#1 cells, which carry a plasmid containing recognition sites for the l-Scel endonuclease integrated into the chromosomal DNA, were used.13 Quantitative PCR analysis of the immunoprecipitated DNA showed that LKB1 was enriched in the 300-bp region from the l-Scel site, as observed for γH2AX and KU80 (Figure 1f). Taken together, these results suggest that LKB1 is recruited to DSB sites.
LKB1 regions required for recruitment to DSB sites were investigated. The LKB1 protein consists of an N-terminal kinase domain and a C-terminal non-catalytic domain6 (Figure 2a). LKB1 C-terminus fragments accumulated at irradiated sites to the same level as full-length LKB1, whereas the N-terminus segment did not. Mouse lkbl protein was previously shown to be phosphorylated atThr366 by ataxia telangiectasia mutated (ATM) and DNA-PKcs.21 Our results showed the phosphorylation of LKB1 at Thr363 (corresponding to Thr366 in mouse Lkbl) by the ATM, ATR and DNA-PKcs kinases (Figures 2b and c). Treatment with an ATM/ATR inhibitor suppressed LKB1 recruitment to DSB sites (Figure 2d). However, Thr363 phosphorylation was not required for LKB1 recruitment to DSB sites (Figure 2e). These results suggested that LKB1 recruitment to DSB sites may be mediated by its C-terminus under the regulation of ATM/ATR signaling pathways that do not operate via LKB1-Thr366 phosphorylation; LKB1 might be recruited to DSB sites through ATM/ATR-mediated phosphorylation of substrates other than LKB1. Concordantly, we did not detea any interaction between LKB1 and ATM (Supplementary Figure S2).
Figure 2.
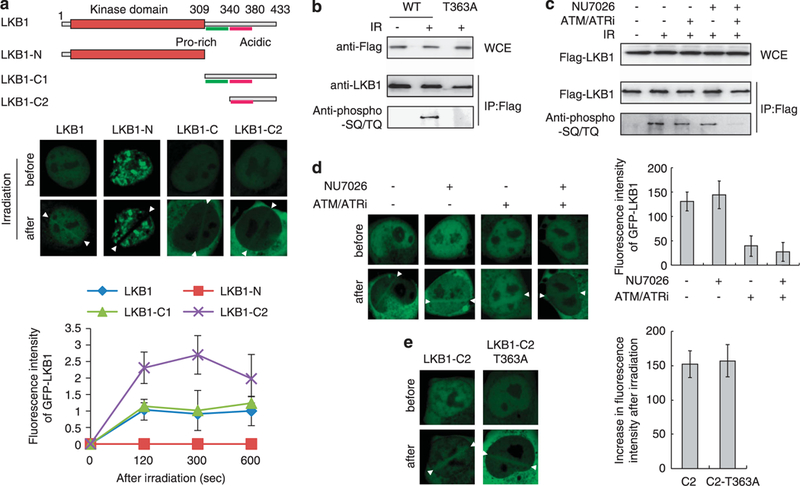
Analysis of LKB1 mutant proteins. (a) Accumulation of LKB1 mutant proteins at micro-irradiated sites. H1299 cells transfected with WT or mutant GFP-LKB1 expression plasm ids were examined for accumulation before and 2 min after laser micro-irradiation. Relative fluorescence intensities are shown as a graph. (b) IR-induced phosphorylation of LKB1 at T363. H1299 cells transfected with Flag-LKBl or Flag-LKB1-T363A mutant expression plasm ids for 24 h were irradiated or not with 10 Gy. Whole cell extracts (WCEs) were prepared 1 h after radiation treatment and immunoprecipitated with an anti-Flag antibody. The immunoprecipitates were subjected to western blot analysis with anti-Flag and anti-phospho-(S/T)Q antibodies. (c) LKBl phosphorylation by ATM, ATR and DNA-PKcs kinases. H1299 cells transfected with Flag-LKBl expression plasmids for 24h were irradiated or not with lOGy. WCEs from H1299 cells transfected with Flag-LKBl expression plasm ids were immunoprecipitated with an anti-Flag antibody and subjected to western blot analysis with anti-Flag and anti-phospho-(S/T)Q antibodies. Treatment with a DNA-PKcs inhibitor (NU7026; 100 μM) and/or an ATM/ ATR inhibitor (ATM/ ATRi; 10 μM) was performed for 3 h before IR treatment. (d) ATM/ ATR signaling-dependent LKBl accumulation. H 1299 cells expressing the GFP-LKB1-C2 protein were treated with a DNA-PKcs inhibitor (NU7026; 100 μM) and/or ATM/ ATR inhibitor (ATM/ ATRi; 10 μM) for 3 h before micro-irradiation. The graph shows fluorescence intensity at 10 min after irradiation. (e) Accumulation of GFP-LKB1-C2 proteins. H1299 cells transfected with WT and T363A mutant GFP-LKB1-C2 expression plasm ids for 24 h were micro-irradiated. The graph shows the mean ( ± s.d.) increase in fluorescence intensity in micro-irradiated regions 10 min after irradiation.
LKB1 promotes NHEJ by facilitating KU70/80 accumulation at DSBs Because IR-induced DSBs are mainly repaired by NHEJ in human ceils15,16,22 we investigated the involvement of LKB1 in NHEJ using a chromosomal based-NHEJ assay.13 In this assay, the l-Scel endonuclease is exogenously expressed and NHEJ repair of DNA ends generated on chromosomal DNA is monitored by FACS (EGFP production) and quantitative genomic PCR (joined DNA formation) (Figure 3a). The PCR enabled us to detect the joining of two distal DNA ends generated at two l-Scel sites by amplifying the 150-bp region of DNA surrounding the two l-Scel sites.23 LKB1 depletion resulted in a reduction of EGFP-positive cell fractions similar to that caused by KU80 depletion (Figure 3b). LKB1 depletion also led to a reduction of DNA end joining without affecting DSB formation (Figure 3c). These results indicate that LKB1 is involved in NHEJ.
Figure 3.
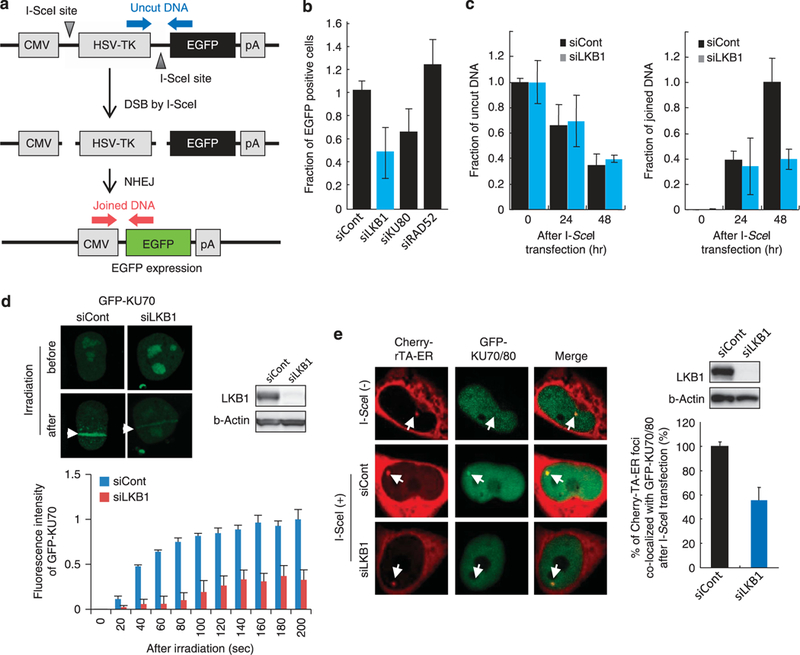
Impairment of NHEJ and KU70 recruitment to DSB sites by LKB1 depletion. (a) Assay design for NHEJ of chromosomal DSBs in vivo.13 Arrows indicate the locations of PCR primers used for quantitative PCR to monitor DSB introduction by 1-Scel (uncut DNA) and subsequent joining Ooined DNA). (b) Reduced NHEJ activity of LKB1-depleted cells. Fractions of EGFP-positive cells 24 h after transfection with the 1-Scel expression plasmid are shown. Values represent the ratio of EGFP-positive cells transfected with targeting siRNA to that of cells treated with non-targeting (siCont) siRNA. Mean values ( ± s.d.) for three independent experiments are shown. (c) Quantitative PCR analysis of uncut (left) and joined (right) DNAs. Left: Fractions of uncut DNA expressed as a ratio of the amount of uncut DNA present before 1-Scel expression plasmid transfection. Right: Fractions of joined DNA expressed as a ratio of the amount of joined DNA 48 h after transfection of the 1-Scel expression plasmid in non-targeting siRNA-treated cells. Mean values ( ± s.d.) for three independent experiments are shown. (d) LKB1- dependent GFP-KU70 recruitment. H1299 cells pre-transfected with control or LKB1 siRNA were transfected with the GFP-KU70 expression plasmid for 24 h. Fluorescence images of cells 3 min after micro-irradiation are shown on the left. Micro-irradiation induced increases in fluorescence intensity are shown (mean ± s.d.). (e) GFP-KU70/80 accumulation at DSB sites in U20S/TRE/1-Scel-19 cells. Left: Focus of GFP-KU70/ 80 colocalized with Cherry-tTA-ER (ie, DSB sites). Right: The percentage of GFP-KU70/80 foci colocalized with Cherry-TBP. Twenty cells showing a single focus of Cherry-TBP were examined for colocalization of GFP-KU70/80. The detailed method is described in Lan et al.19
The recruitment of KU70 to DSB sites is the first step in NHEJ, and LKB1 depletion suppressed this step as shown by the results of an exogenous GFP-LKB1 accumulation experiment (Figure 3d). On the other hand, recruitment of endogenous LKB1 was not suppressed by KU70 depletion (Supplementary Figure S2a and b). The LKB1 dependency of KU70 recruitment to DSB sites was validated by assessing the recruitment of KU70 to l-Scel-induced DSB sites in U20S/TRE/l-5cel-19 cells19 (Supplementary Figure Sic). EGFP-KU70/ 80 proteins colocalized with Cherry-tTA-ER (a marker protein for l-SceI recognition sites in cell nuclei) upon l-Scel expression in almost all control cells; however, LKB1 depletion reduced this colocalization (Figure 3e). These results suggest that LKB1 promotes NHEJ by facilitating KU70/80 accumulation at DSBs.
LKB1 promotes the accumulation of BRM at DSB sites
Recent studies by our group and others have shown that the SWI/SNF remodeling complex is required for the recruitment of KU proteins to DSB sites.13,14,24 The SWI/SNF complex contains either BRG1 or BRM as its ATPase catalytic subunit.25 LKB1 was previously shown to bind to BRG1.9 HI299 cells do not express intact BRG1 protein due to a homozygous truncation mutation in the BRG1 gene.26 Therefore, BRM was deduced to be the only catalytic subunit of the SWI/SNF complex in HI 299 cells, and the activity of the SWI/SNF complex should therefore be abolished only by BRM depletion in these cells. In H1299 cells, the involvement of BRM in NHEJ was confirmed by its recruitment to DSB sites, the enhancement of NHEJ repair of l-Scel-induced DSBs, and the recruitment of KU70 to DSB sites (Figure 4). Notably, LKB1 depletion reduced the accumulation of BRM, whereas BRM depletion did not reduce the accumulation of GFP-LKB1 and endogenous LKB1 at micro-irradiated sites (Figures 5a-c). These results suggested that the SWI/SNF complex may have a major role in NHEJ under the regulation of LKB1. This idea was supported by the fact that the extent of the decrease was similar in response to LKB1, BRM and BRM/LKB1 depletion (Figure 4c).
Figure 4.
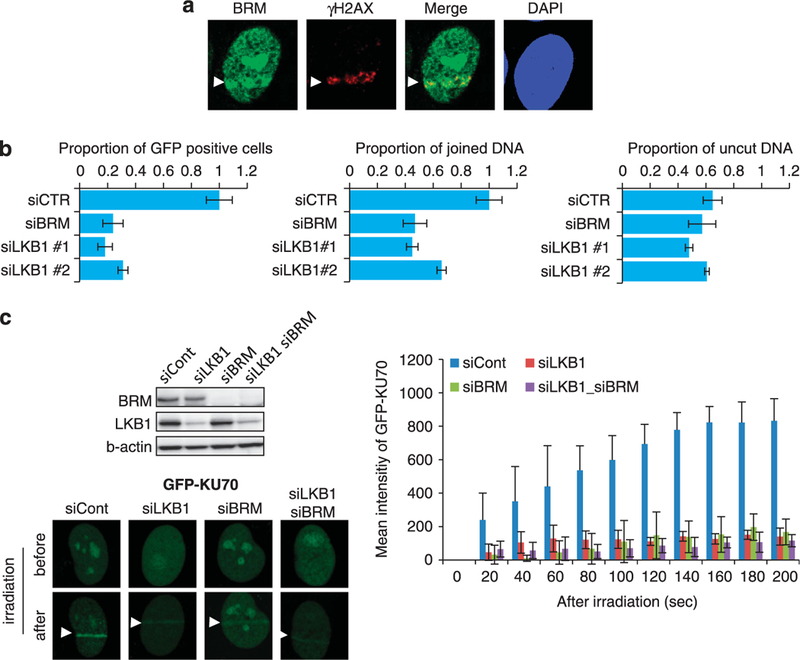
Involvement of BRM in NHEJ. (a) Accumulation of endogenous BRM at micro-irradiated sites. H1299 cells were fixed 15 min after laser micro-irradiation and immunostained with anti-BRM plus Alexa 488-conjugated anti-mouse lgG, and with anti-γH2AX antibodies plus Alexa 594-conjugated anti-rabbit lgG. Nuclei were stained with DAPI. The arrows indicate accumulated BRM. (b) Effect of depleting LKB1 and BRM on NHEJ activity. The mean fractions ( ± s.d., n = 3) of EGFP-positive cells (left) and joined DNA (middle) relative to the amounts in nontargeting siRNA-treated cells (siCont) 48 h after transfection of the l-Scel expression plasmid are shown. The mean fractions ( ± s.d., n = 3) of uncut DNA relative to the amounts present before transfection of the 1-Scel expression plasmid are shown (right). Data represent the mean ( ± s.d.) from three independent experiments. (c) Effect of LKB1 and/or BRM depletion on the recruitment of GFP-KU70 to DSB sites. H1299 cells were pre-transfected with the indicated siRNAs for 48 h and then transfected with the EGFP-KU70 expression plasmid for 24 h. A western blot (top left) and fluorescence images of cells 3 min after micro-irradiation (bottom left) are shown. The graph shows mean increases ( ± s.d.) in fluorescence intensity induced by micro-irradiation. Fluorescence intensity was expressed as a ratio relative to that of cells treated with nontargeting (siCont) siRNA at the last time point.
Figure 5.
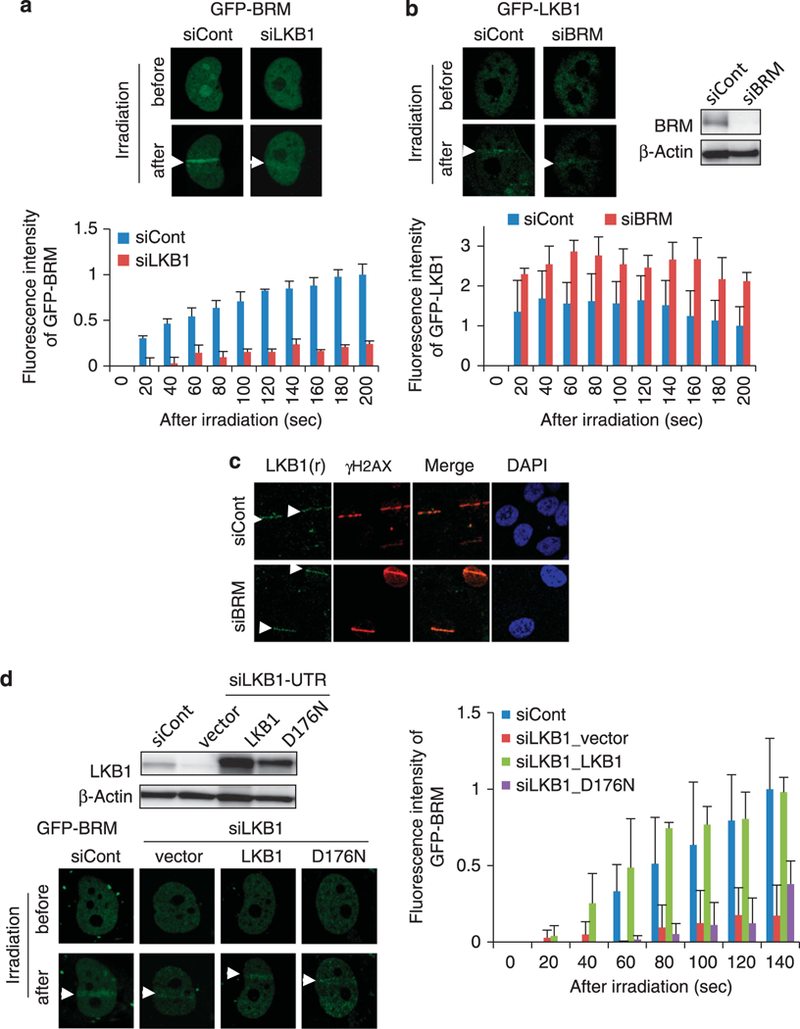
Involvement of LKBl in BRM accumulation at DSB sites. (a) LKBl-dependent GFP-BRM recruitment. H1299 cells pre-transfected withcontrol or LKB1 siRNA were transfected with the GFP-BRM expression plasmid for 24 h. Fluorescence images of cells 3 min after microirradiation are shown. Micro-irradiation induced increases in fluorescence intensity are shown (mean± s.d.). (b) BRM-independent GFP-LKBl recruitment. H1299 cells pre-transfected with control or BRM siRNA were transfected with GFP-LKB1 expression plasmid for 24 h. (c) Endogenous LKB1 recruitment. Control or BRM siRNA-treated H1299 cells were micro-irradiated, fixed 10 min after laser micro-irradiation, and immunostained with anti-LKB1 and anti-γH2AX antibodies. (d) LKB1 kinase-dependent GFP-BRM recruitment. LKB1–3’UTR-siRNA-treated H1299 cells were transfected with GFP-BRM and with empty, WT or D176N kinase-dead mutant LKBl expression plasm ids for 48 h. The result of western blot analysis is shown. Micro-irradiation induced increases in fluorescence intensity are shown on the right (mean ± s.d.).
We next addressed whether the kinase activity of LKB1 is required for the recruitment of KU70/80 and the SWI/SNF complex to DSB sites. We expressed cDNA encoding either WT LKB1 or a kinase-dead version of LKB1, D176N, derived from a PJS patient27 in cells in which endogenous LKB was knocked down by siRNA targeting the 3’-untranslated region of the LKB1 gene. Exogenously expressed WT LKB1, but not D176N LKB1, rescued the level of BRM protein recruited to DSB sites (Figure 5d). These results suggest that the kinase activity of LKB1 is required for the recruitment of the SWI/SNF chromatin remodeling complex to DSB sites.
Recruitment of BRM and NHEJ proteins to DSB sites by LKB1-AMPK signaling via H2B phosphorylation
A recent study showed that the intra-nuclear function of LKB1 in the cellular adaptation to metabolic stress involves AMPK2 as a substrate for signaling.8 Therefore, we examined whether AMPK2 also functions as an LKB1 mediator in DSB repair. As shown in Figure 5a, AMPK2 was recruited to DSB sites upon DSB generation; however, this recruitment was reduced in LKB1-depleted cells, suggesting that LKB1 is required for the localization of AMPK2 to DSBs. Recruitment of KU70 and BRM protein to DSB sites as well as NHEJ activity was decreased in AMPK2-depleted cells (Figures 5b-d). This reduction in NHEJ activity was confirmed by depleting AMPK2 using another siRNA (Supplementary Figure S3). In addition, AMPK2 depletion sensitized cells to bleomycin (Figure 5e). These results suggest that the function of LKB1 in NHEJ involves AMPK2 as a substrate for signaling.
Because AMPK2-mediated phosphorylation of histone H2B at Ser36 in the nucleus leads to transcriptional regulation in the adaptation response to metabolic stresses,8 we examined the involvement of H2B phosphorylation in the recruitment of KU70 and BRM to DSB sites. Phosphorylated Ser36-H2B colocalized with γH2AX at DSB sites in micro-irradiated cells (Figure 5f). The recruitment of KU70 and BRM to DSB sites was impaired by the expression of the H2B S36A mutant (Figures 5g and h). These results suggested that the LKB1-AMPK signaling pathway promotes NHEJ via the phosphorylation of H2B.
Chromosome aberrations caused by LKB1 depletion
The results described above prompted us to examine whether LKB1 and AMPK2 ablation cause chromosome instability. The number of chromosome aberrations (breaks and radials) per cell detected by metaphase spread analyses was significantly increased by both LKB1 and AMPK2 depletion (Figure 7a; Supplementary Figure S4). The increase in the number of chromosome aberrations induced by LKB1 depletion was more evident than the increase induced by AMPK2 depletion, and was similar to that caused by LKB1 and AMPK2 double depletion. Therefore, LKB1 regulation of DNA repair is likely mediated by AMPK2 and other factors acting downstream of LKB1. Taken together, these results suggested that LKB1 has a role in the maintenance of genome integrity.
Figure 7.
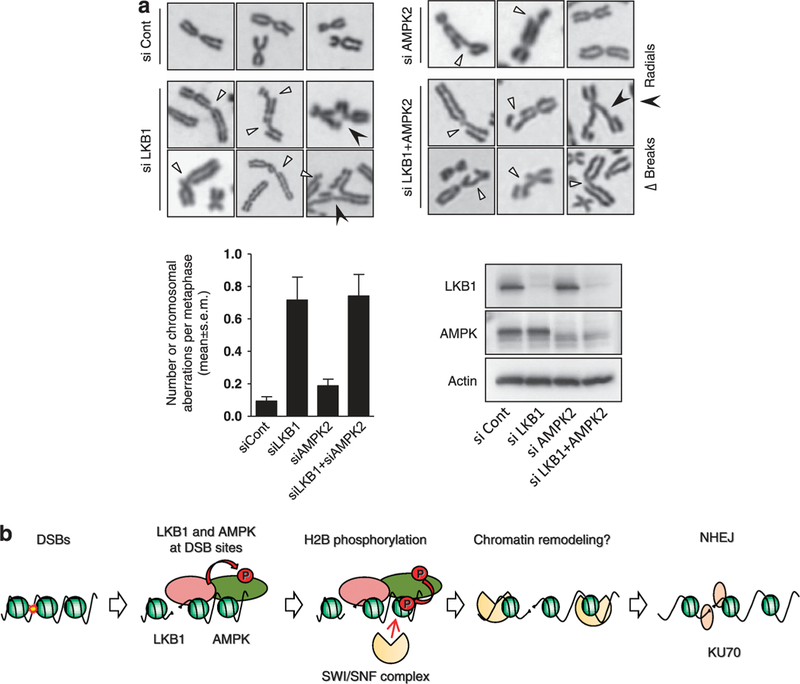
Chromosome aberrations caused by LKB1 and AMPK2 depletion. (a) Representative cells showing chromosome breaks and radials in H1299 cells transfected with control, LKB1 and/or AMPK2 siRNA. Slides were Giemsa stained and metaphase spreads were analyzed for the number of chromosomal aberrations. The numbers of chromosomal aberrations per metaphase and western blotting results are also shown (lower). (b) A model for how LKB1-AMPK signaling regulates NHEJ. Upon DSBs, LKB1 accumulates at DSB sites, and AMPK accumulates at DSB sites in an LKB1-dependent manner. AMPK is activated by LKB1-mediated phosphorylation and phosphorylates H2B. Then, the SWI/SNF complex is recruited in an H2B phosphorylation-dependent manner, probably resulting in the promotion of chromatin remodeling at DSB sites. This remodeling might promote the localization of KU proteins to DSB sites and trigger NHEJ.
DISCUSSION
The present study suggests a novel function for the LKB1 protein: that of a positive regulator of NHEJ. The present results allowed us to suggest a model of how LKB1 regulates NHEJ (illustrated in Figure 7b). lKB1 and AMPK2 (which is dependent upon lKB1) accumulate at DSB sites. AMPK2, which is phosphorylated and activated by LKB1, phosphorylates histone H2B. This histone modification promotes the recruitment of the SWI/SNF complex (BRM in this case), and chromatin remodeling (thought to occur at DSB sites) facilitates the accumulation of KU proteins.
The immediate ( < 10 s) accumulation of LKB1 upon the generation of DSBs, as well as the localization of LKB1 to DNA ends, resembled that observed for other proteins involved in NHEJ, such as KU70/80.19,20 Depleting LKB1 also inhibited the in vivo repair of l-Scel-induced DSBs in chromosomes, and suppressed the recruitment of KU70/80 to DSB sites. The depletion of LKB1 inhibited the repair of IR-induced DSBs, as determined by both γH2AX foci and neutral-comet assays, and sensitized cells to DSB inducing agents (as observed in cells depleted of key NHEJ genes).15,16,22 Based on these results, we conclude that LKB1 affects the efficiency of NHEJ.
The recruitment of KU proteins by LKB1 may occur via the recruitment of SWI/SNF chromatin remodeling factors to DSB sites since LKB1 depletion suppressed the recruitment of both KU and BRM; the depletion of BRM also suppressed the recruitment of KU. Chromatin remodeling factors involved in DNA repair have been extensively studied in yeast, and SWI/SNF family proteins are important for NHEJ;11,12 therefore, in this study, we verified if the SWI/SNF complex also has a role in regulating NHEJ in human cells. Furthermore, LKB1 may regulate the recruitment of an SWI/ SNF protein, BRM, to DSB sites. WT LKB1, but not the LKB1-D176N kinase-dead mutant, promoted the recruitment of BRM, suggesting that the mechanism of regulation is dependent on the kinase activity of LKB1. The yeast serine/threonine kinases, Elml, Pakl and Tos3, which share sequence homology with LKB1, phosphorylate and activate Snfl, the yeast homolog of AMPK;28,29 however, the role of these proteins in regulating SWI/SNF chromatin remodeling during NHEJ is unknown. Therefore, the present study suggests the possible involvement of an LKB-dependent mechanism of SWI/SNF regulation during NHEJ. Interestingly, in our NHEJ assay, the structure of the breakpoint junctions resulting from the repair of l-Scel-induced DSBs was unchanged by the depletion of LKB1 and BRM (Supplementary Figure S5). Thus, suppression of LKB1 signaling is more likely to cause a reduction in NHEJ activity than to change the mode of joining.
Our data suggest that recruitment of AMPK2 to DSBs depends on LKB1; therefore, AMPK2 is likely to be a factor acting downstream of LKB1 in NHEJ. AMPK2 promoted the recruitment of the BRM and KU proteins to DSB sites. These results suggest that activation of the LKB1-AMPK pathway is involved in the first step of NHEJ probably by regulating SWI/SNF chromatin remodeling at DSBs, thereby allowing the subsequent recruitment of NHEJ proteins to DSB sites. LKB1-activated AMPK2 phosphor-ylates histone H2B Ser36 under conditions of metabolic stress, leading to transcriptional activation. In addition, AMPK2 is activated by IR, H202, and UV treatment.8 Histone phosphorylation disrupts the chromatin structure and provides an occasion of non-histone chromosomal proteins to chromatin.30 Therefore, we predicted that the phosphorylation of H2B-Ser36 also contributes to DSB repair. The H2B Ser36 mutation reduced the recruitment of BRM and KU70 to DSB sites. This suggests that phosphorylation of H2B by AMPK promotes the recruitment of the SWI/SNF complex to DSB sites for chromatin remodeling. The mechanism(s) by which H2B phosphorylation increases SWI/SNF recruitment remains unclear. One possibility is that H2B phosphorylation increases the incidence of other histone modifications, which in turn increases the affinity of the SWI/SNF complex for histones, since each histone modification has an effect on the others.31,32 The resulting histone modifications might cause the chromatin relaxation required for the DSB repair proteins to access the DSB sites. In addition, LKB1 interacts with BRG1 irrespective of its kinase activity, and increases the ATPase activity of BRG1 ? We also confirmed that LKB1 interacts with BRM, irrespective of its kinase activity (Supplementary Figure S6). Therefore, the LKB1 protein might further facilitate chromatin remodeling by increasing the remodeling activity of the BRG1 and BRM proteins. Notably, the effects of LKB1 ablation on the recruitment of KU and BRM to DSB sites, and on the suppression of chromosomal breaks/radials, were greater than those of AMPK2. These data suggest that AMPK2 may only partially mediate the effects of LKB1. In fact, siRNA-mediated ablation of AMPK1 also reduced NHEJ activity (Supplementary Figure S7). Other signal transduction mechanisms involving BRM (but not AMPK2) might also have a role. Further studies are needed to elucidate the detailed mechanisms by which LKB1 mediates signaling to increase NHEJ.
We recently showed that the human ACF complex, which belongs to the ISWI family, increases NHEJ by directly interacting with KU70 and promoting its localization to DSBs.19 In addition, we and others also identified histone acetyltransferases, TIP60, CBP and p300, as well as histone deacetylases (HDACs), HDAC1 and HDAC2, all of which are involved in NHEJ.13,33–35 The present study suggests that LKB1-AMPK signaling is involved in SWI/SNF chromatin remodeling during NHEJ. Therefore, NHEJ regulation may involve different types of chromatin remodeling/modification at DSB sites. Further research should be directed at understanding how the multiple steps involved in NHEJ are integrated to function harmoniously during DSB repair.
The role of LKB1 in maintaining genome integrity is suggested by the fact that inactivating this gene causes PJS, which is characterized by genomic instability. The LKB1-D176N kinase-dead mutant used in the present study was derived from a PJS patient,27 and showed a reduced ability to recruit the SWI/SNF remodeling complex, or KU70/80, to DSBs. Consistently, suppression of LKB1-AMPK signaling causes chromosome aberrations, as observed by the suppression of key molecules involved in DSB repair.36 Therefore, our results imply that inactivation of the LKB1 tumor suppressor contributes to carcinogenesis by disturbing not only known cellular functions such as cell proliferation, polarization and differentiation, but also the maintenance of chromosomal integrity. The purported role of LKB1 in DSB repair could be further strengthened by examining cells from individuals with PJS and cells with key NHEJ gene inactivation for deficiencies in NHEJ, V(D)J recombination and class switch recombination, as well as for IR hypersensitivity.
MATERIALS AND METHODS
siRNA
siRNAs against LKB1 were purchased from Dharmacon (Thermo Scientific, Chicago, IL, USA) (ON-TARGETplus SMARTpool), Invitrogen (Carlsbad, CA, USA, siLKB1#1, 12938–048) and Santa Cruz (Santa Cruz, CA, USA, siLKB#2, sc-35816). siRNA against BRM (sc-29831) was purchased from Santa Cruz. siRNAs against KU70, KU80, RAD52 and AMPK2 were purchased from Dharmacon (ON-TARGETplus SMARTpool, Yokohama, Japan). Non-targeting siRNAs were purchased from the same suppliers, Invitrogen (VHS50414), Santa Cruz (sc-35816) and Dharmacon (ON-TARGETplus SMARTpool). We also used another set of siRNA (QIAGEN SI02758595 and SI02758602) for AMPK2 in Supplementary Figure S3. siRNA transfection of HI 299 cells was performed using RNAiMAX (Invitrogen) and cells were analyzed after a 48-h culture.
Laser micro-irradiation
Laser micro-irradiation was performed using the FV-500 confocal scanning laser microscopy system (Olympus, Tokyo, Japan) as reported previously.19,37,38 Briefly, cells in glass-bottomed dishes were microirradiated with a 405-nm laser (Olympus), fixed and permeabilized with MeOH. The cells were analyzed by immunofluorescence using specific primary and secondary antibodies. Methods are described in detail in Supplementary Methods.
DSB repair and clonogenic assays
In vivo NHEJ, Comet and ChIP assays were performed as previously described.13 The methods are described in detail in Supplementary Methods.
Western and immunoprecipitation assays
The methods are described in detail in Supplementary Methods.
Chromosome aberration assay
HI299 cells were transfected with control, LKB1 and AMPK2 siRNAs and cultured for 3 days. Colcemid (GIBCO, Gaithersburg, MD, USA) was added at a final concentration of 0.15μg ml–1 for 45min before harvest. Collected cells were treated with 75 MM KCI and fixed onto slides with a 3:1 methanol/glacial acetic acid solution. Slides were then stained with Giemsa and metaphase spreads were analyzed for the number of chromosomal aberrations.
Supplementary Material
Figure 6.
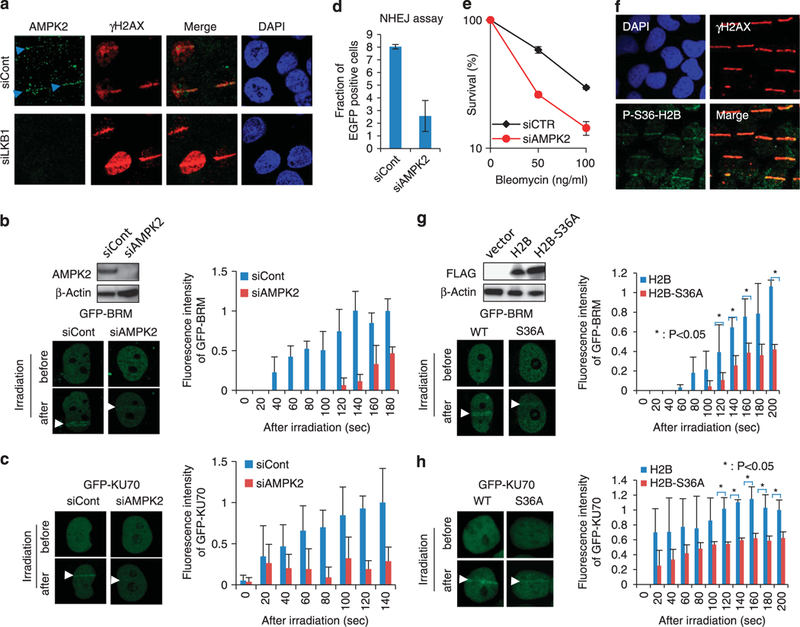
LKB1-AMPK signaling mediated the recruitment of BRM and NHEJ proteins to DSB sites via H2B phosphorylation. (a) Endogenous AMPK2 recruitment by LKB1 depletion. Control or LKB1 siRNA-treated H1299 cells were micro-irradiated, fixed 10 min after laser microirradiation, and immunostained with anti-LKB1 and anti-γH2AX antibodies. (b, c) AMPK2-dependent GFP-BRM and GFP-KU70 recruitment. Control or AMPK2 siRNA-treated H1299 cells were transfected with EGFP-BRM (b) or EGFP-KU70 (c) expression plasmid for 48 h. Fluorescence images of cells 3 min after micro-irradiation are shown on the left. Micro-irradiation induced increases in fluorescence intensity are shown on the right (mean ± s.d.). The top panel shows the results of western blot analysis. (d) Reduced NHEJ activity in AMPK2-depleted cells. (e) Clonogenic assay showing sensitization to bleomycin by AMPK2 depletion. (f) Colocalizations of phosphorylated histone H2B and γH2AX in micro-irradiated cells. At 10 min after laser micro-irradiation, H1299 cells were fixed and immunostained with anti-phospho-S36-H2B and anti-γH2AX. (g, h) GFP-BRM and GFP-KU70 accumulation by mutant (S36A) histone H2B expression. 293T cells stably expressing Flag-H2B or Flag-H2B-S36A proteins were transfected with GFP-BRM (g) or GFP-KU70 (h) expression plasmid for 48 h. Increases in fluorescence intensities induced by micro-irradiation are shown (mean ± s.d.). The differences in fluorescence intensity between the WT and the S36A mutant at certain time points (120 s and on) after irradiation were statistically significant (*P< 0.05; Student’s t-test). Western blot is shown in (g).
ACKNOWLEDGEMENTS
This work was supported by Grants-in-Aid from the Ministry of Education, Culture, Sports, Science and Technology of Japan for Scientific Research on Innovative Areas (22131006 to TK and HO; and 22131005 to AY), from the Japan Society for the Promotion of Science for Young Scientists (B) KAKENHI (23701110 to HO and 24710057 to AU) and Management Expenses Grants from the Government to the National Cancer Center. A part of this work was carried out under the Cooperative Research Project Program of the Institute of Development, Aging and Cancer, Tohoku University.
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supplementary Information accompanies this paper on the Oncogene website (http://www.nature.com/onc)
REFERENCES
- 1.Richard F, Muleris M, Dutrillaux B. Chromosome instability in lymphocytes from patients affected by or genetically predisposed to colorectal cancer. Cancer Genet Cytogenet 1994; 73: 23–32. [DOI] [PubMed] [Google Scholar]
- 2.Hemminki A, Markie D, Tomlinson I, Avizienyte E, Roth S, Loukola A et al. A serine/ threonine kinase gene defective in Peutz-Jeghers syndrome. Nature 1998; 391: 184–187. [DOI] [PubMed] [Google Scholar]
- 3.Sanchez-Cespedes M, Parrella P, Esteller M, Nomoto S, Trink B, Engles JM et al. Inactivation of LKB1/STK11 is a common event in adenocarcinomas of the lung. Cancer Res 2002; 62: 3659–3662. [PubMed] [Google Scholar]
- 4.Matsu moto S, Iwakawa R, Taka hash i K, Kohno T, Nakanishi Y, Matsu no Y et al. Prevalence and specificity of LKB1 genetic alterations in lung cancers. Oncogene 2007; 26: 5911–5918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wingo SN, Gallardo TD, Akbay EA, Liang MC, Contreras CM, Boren T et al. Somatic LKB1 mutations promote cervical cancer progression. PLoS One 2009; 4: e5137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer 2009; 9: 563–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roy BC, Kohno T, Iwakawa R, Moriguchi T, Kiyono T, Morishita K ef al. Involvement of LKB1 in epithelial-mesenchymal transition (EMT) of human lung cancer cells. Lung Cancer 2010; 70: 136–145. [DOI] [PubMed] [Google Scholar]
- 8.Bungard D, Fuerth BJ, Zeng PY, Faubert B, Maas NL, Viol let B ef al. Signaling kinase AMPK activates stress-promoted transcription via histone H2B phosphorylation. Science 2010; 329: 1201–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marignani PA, Kanai F, Carpenter CL. LKB1 associates with Brg1 and is necessary for Brgl-induced growth arrest. J Biol Chem 2001; 276: 32415–32418. [DOI] [PubMed] [Google Scholar]
- 10.Osley MA, Shen X. Altering nucleosomes during DNA double-strand break repair in yeast. Trends Genet 2006; 22: 671–677. [DOI] [PubMed] [Google Scholar]
- 11.van Attikum H, Fritsch O, Gasser SM. Distinct roles for SWR1 and IN080 chromatin remodeling complexes at chromosomal double-strand breaks. EMBO J. 2007; 26: 4113–4125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shim EY, Ma JL, Oum JH, Yanez Y, Lee SE. The yeast chromatin remodeler RSC complex facilitates end joining repair of DNA double-strand breaks. Mol Cell Biol 2005; 25: 3934–3944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ogiwara H, Ui A, Otsuka A, Satoh H, Yokomi I, Nakajima S et al. Histone acetylation by CBP and p300 at double-strand break sites facilitates SWI/SNF chromatin remodeling and the recruitment of non-homologous end joining factors Oncogene 2011; 30: 2135–2146. [DOI] [PubMed] [Google Scholar]
- 14.Peng G, Yim EK, Dai H, Jackson AP, Burgt I, Pan MR et al. BRIT1/MCPH1 links chromatin remodelling to DNA damage response. Nat Cell Biol 2009; 11: 865–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Burma S, Chen BP, Chen DJ. Role of non-homologous end joining (NHEJ) in maintaining genomic integrity. DNA Repair (Amst) 2006; 5: 1042–1048. [DOI] [PubMed] [Google Scholar]
- 16.Lieber MR. The mechanism of human nonhomologous DNA end joining. J Biol Chem 2008; 283: 1–5. [DOI] [PubMed] [Google Scholar]
- 17.Blanco R, Iwakawa R, Tang M, Kohno T, Angulo B, Pio R et al. A gene-alteration profile of human lung cancer cell lines. Hum Mutat 2009; 30: 1199–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bonner WM, Redon CE, Dickey JS, Nakamura AJ, Sedelnikova OA, Solier S et al. GammaH2AX and cancer. Nat Rev Cancer. 2008; 8: 957–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lan L, Ui A, Nakajima S, Hatakeyama K, Hoshi M, Watanabe R et al. The ACF1 complex is required for DNA double-strand break repair in human cells. Mol Cell 2010; 40: 976–987. [DOI] [PubMed] [Google Scholar]
- 20.Rodrigue A, Lafrance M, Gauthier MC, McDonald D, Hendzel M, West SC et al. Interplay between human DNA repair proteins at a unique double-strand break in vivo. EMBO J 2006; 25: 222–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sapkota GP, Deak M, Kieloch A, Morrice N, Goodarzi AA, Smythe C et al. Ionizing radiation induces ataxia telangiectasia mutated kinase (ATM)-mediated phos-phorylation of LKB1/STK11 at Thr-366. Biochem J. 2002; 368(Pt 2): 507–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mladenov E, lliakis G. Induction and repair of DNA double strand breaks: the increasing spectrum of non-homologous end joining pathways. Mutat Res 2011; 711: 61–72. [DOI] [PubMed] [Google Scholar]
- 23.Ogiwara H, Kohno T. Essential factors for incompatible DNA end joining at chromosomal DNA double strand breaks in vivo. PLoS One 2011; 6: e28756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Park JH, Park EJ, Lee HS, Kim SJ, Hur SK, Imbalzano AN et al. Mammalian SWI/SNF complexes facilitate DNA double-strand break repair by promoting gamma-H2AX induction. EMBO J 2006; 25: 3986–3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reisman D, Glaros S, Thompson EA. The SWI/SNF complex and cancer. Oncogene 2009; 28: 1653–1668. [DOI] [PubMed] [Google Scholar]
- 26.Medina PP, Romero OA, Kohno T, Montuenga LM, Pio R, Yokota J et al. Frequent BRGl/SMARCA4-inactivating mutations in human lung cancer cell lines. Hum Mutat 2008; 29: 617–622. [DOI] [PubMed] [Google Scholar]
- 27.Mehenni H, Gehrig C, Nezu J, Oku A, Shimane M, Rossier C et al. Loss of LKB1 kinase activity in Peutz-Jeghers syndrome, and evidence for allelic and locus heterogeneity. Am J Hum Genet 1998; 63: 1641–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hong SP, Lei per FC, Woods A, Carling D, Carlson M. Activation of yeast Snfl and mammalian AMP-activated protein kinase by upstream kinases. Proc Natl Acad Sci USA 2003; 100: 8839–8843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Woods A, Johnstone SR, Dickerson K, Leiper FC, Fryer LG, Neumann D et al. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr Biol 2003; 13: 2004–2008. [DOI] [PubMed] [Google Scholar]
- 30.Perez-Cadahia B, Drobic B, Khan P, Shivashankar CC, Davie JR. Current understanding and importance of histone phosphorylation in regulating chromatin biology. Curr Opin Drug Discov Dev 2010; 13: 613–622. [PubMed] [Google Scholar]
- 31.Dover J, Schneider J, Tawiah-Boateng MA, Wood A, Dean K, Johnston M et al. Methylation of histone H3 by COMPASS requires ubiquitination of histone H2B by Rad6. J Biol Chem 2002; 277: 28368–28371. [DOI] [PubMed] [Google Scholar]
- 32.Kim J, Guermah M, McGinty RK, Lee JS, Tang ZY, Milne TA et al. RAD6-mediated transcription-coupled H2B ubiquitylation directly stimulates H3K4 methylation in human cells. Cell 2009; 137: 459–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Downs JA, Allard S, Jobin-Robitaille O, Javaheri A, Auger A, Bouchard N et al. Binding of chromatin-modifying activities to phosphorylated histone H2A at DNA damage sites. Mol Cell 2004; 16: 979–990. [DOI] [PubMed] [Google Scholar]
- 34.Murr R, Loizou Jl, Yang YG, Cuenin C, Li H, Wang ZQ et al. Histone acetylation by Trrap-Tip60 modulates loading of repair proteins and repair of DNA double-strand breaks. Nat Cell Biol 2006; 8: 91–99. [DOI] [PubMed] [Google Scholar]
- 35.Miller KM, Tjeertes JV, Coates J, Legube G, Polo SE, Britton S et al. Human HDAC1 and HDAC2 function in the DNA-damage response to promote DNA non- homologous end-joining. Nat Struct Mol Biol 2010; 17: 1144–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van Gent DC, Hoeijmakers JH, Kanaar R. Chromosomal stability and the DNA double-stranded break connection. Nat Rev Genet 2001; 2: 196–206. [DOI] [PubMed] [Google Scholar]
- 37.Lan L, Nakajima S, Komatsu K, Nussenzweig A, Shimamoto A, Oshima J et al. Accumulation of Werner protein at DNA double-strand breaks in human cells. J Cell Sci 2005; 118(Pt 18): 4153–4162. [DOI] [PubMed] [Google Scholar]
- 38.Nakajima S, Lan L, Kanno S, Usami N, Kobayashi K, Mori M et al. Replication- dependent and -independent responses of RADI 8 to DNA damage in human cells. J Biol Chem 2006; 281: 34687–34695. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.