

Abstract
Infectious diseases due to multidrug-resistant pathogens, particularly carbapenem-resistant Enterobacteriaceae (CREs), present a major and growing threat to human health and society, providing an urgent need for the development of improved potent antibiotics for their treatment. We describe the design and development of a new class of aminoglycoside antibiotics culminating in the discovery of propylamycin. Propylamycin is a 4’-deoxy-4’-alkyl paromomycin whose alkyl substituent conveys excellent activity against a broad spectrum of ESKAPE pathogens and other Gram-negative infections, including CREs, in the presence of numerous common resistance determinants, be they aminoglycoside modifying enzymes or ribosomal RNA methyl transferases. Importantly, propylamycin is demonstrated not to be susceptible to the action of the ArmA resistance determinant whose presence severely compromises the action of plazomicin and all other 4,6-disubstituted 2-deoxystreptamine aminoglycosides. The lack of susceptibility to ArmA, which is frequently encoded on the same plasmid as carbapenemase genes, ensures that propylamycin will not suffer from problems of cross-resistance when used in combination with carbapenems. Cell-free translation assays, quantitative ribosome footprinting, and X-ray crystallography support a model in which propylamycin functions by interference with bacterial protein synthesis. Cell-free translation assays with humanized bacterial ribosomes were used to optimize the selectivity of propylamycin, resulting in reduced ototoxicity in guinea pigs. In mouse thigh and septicemia models of Escherichia coli, propylamycin shows excellent efficacy, which is better than paromomycin. Overall, a simple novel deoxy alkyl modification of a readily available aminoglycoside antibiotic increases the inherent antibacterial activity, effectively combats multiple mechanisms of aminoglycoside resistance, and minimizes one of the major side effects of aminoglycoside therapy.
Graphical Abstract
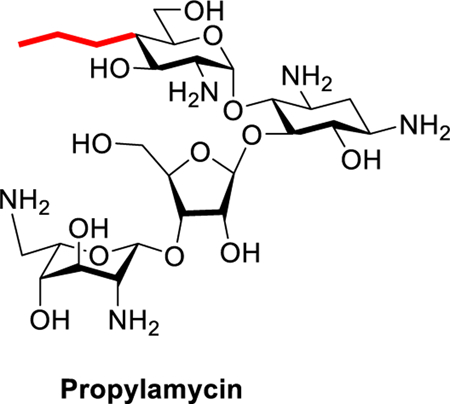
Introduction
Seventy years after their introduction, the aminoglycoside antibiotics (AGAs) remain some of the most efficacious and cost-effective treatments against life-threatening Gram-negative bacterial infections.1 Decades of clinical use, however, have led to the development of widespread resistance, which has resulted in diminished efficacy. The most common mechanism of resistance arises from AGA modification by aminoglycoside modifying enzymes (AMEs),2,3 which reduce AGA affinity for their target the decoding A site in helix 44 of the bacterial ribosome.4,5 A second mechanism of resistance, modification of the target by the ribosomal RNA methyltransferases (RMTs), especially the A1405 N7 methyltransferases, is an increasing threat, particularly as the RMT genes are frequently encoded on the same mobile genetic elements as the metallocarbapenemases.6–11 In addition, potentially serious side effects to the kidney and inner ear have contributed to the decline in use of AGAs. The potential for toxic side effects of AGAs is minimized in the clinic by limiting treatment to short duration regimens with once daily dosing.12 A subgroup of patients is rendered hypersusceptible to AGAs by mutations in the decoding A site of the mitochondrial ribosome.13–18
Astute chemical modification of AGAs enables circumvention of the action of the more problematic AMEs,3,19–24 as exemplified by the semisynthesis of the drugs amikacin, arbekacin, and most recently of plazomicin, a doubly modified version of the natural AGA sisomicin, that is active in the presence of most resistance determinants.25–27 However, recent analyses of the AGA resistome indicate that a much broader range of AMEs act on the 4,6-series of AGAs, typified by the clinical comparators gentamicin, tobramycin, and amikacin,19,28 suggesting that further development of the 4,6-AGAs will meet greater challenges than that of the 4,5-series that we favor. Guided by cell-free translation assays with bacterial and hybrid bacterial ribosomes carrying the decoding A sites of the wild-type and mutant human mitochondrial ribosomes,29 we have demonstrated that chemical modification of AGAs can also lead to reduced ototoxicity in a guinea pig model.30 An alternative approach to moderation of ototoxicity employs chemical modification of AGAs in such a way as to impair their uptake into inner ear hair cells by the mechanotransducer channels.31 AGA ototoxicity-reducing modifications, when installed at or proximal to the sites targeted by AMEs, can also suppress resistance due to the presence of AMEs. Unfortunately, while this duality of action has been amply demonstrated, it is typically accompanied by a reduction in antibacterial activity.30–33
We report here on a simple minimal modification of the 4,5-disubstituted 2-deoxystreptamine (DOS) AGA paromomycin 1 that blocks the action of multiple AMEs, and results in increased ribosomal selectivity and reduced ototoxicity in the guinea pig model, with no loss of and even increased antibacterial activity against Enterobacteriaceae and other Gram-negative pathogens for which the AGAs are often the preferred treatment. It is noteworthy that this simple modification does not cause any reduction in antibacterial activity in the presence of the G1405 RMTs, whose presence compromises the activity of all 4,6-DOS AGAs currently employed in the clinic including plazomicin.6–8,26,27 Based on structural studies of an AGA-ribosome complex, simple physical organic considerations, and exploration of structure-activity parameters, we further develop a structure-based model for increased AGA activity. This new modification and the model it stimulates open the way for the rational design of further AGAs displaying improved antibacterial activity in the presence and absence of AMEs and RMTs with minimization of hearing impairment, such as are increasingly recognized as necessary to combat the growing epidemic of multidrug resistant infectious diseases.
Design and Discovery of Propylamycin
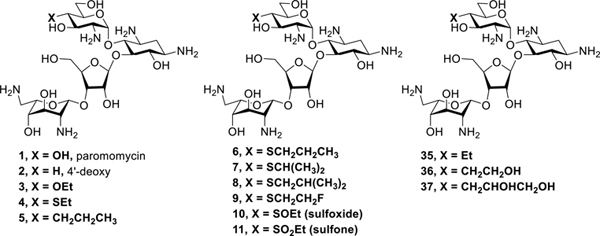
Earlier work from our laboratories revealed that 4’-O-ethylation 3 of the 4,5-DOS paromomycin 1 provokes a significant increase in ribosomal selectivity and a corresponding reduction in ototoxicity in guinea pigs, but is accompanied by a loss of antibacterial activity.30 4’-Deoxygenation 2 of paromomycin on the other hand causes only a minor loss in activity but no increase in selectivity.30 Combining these results we hypothesized that replacement of the 4’-ethoxy group of 3 by a similar and ideally isosteric substituent lacking the electron-withdrawing C-O bond would yield compounds with reduced ototoxicity and little or no loss of antibacterial activity. Accordingly, we targeted 4’-deoxy-4’-ethylthioparomomycin 4 and ultimately 4’-deoxy-4’-propylparomomycin 5 for synthesis (Figure 1).
Figure 1.
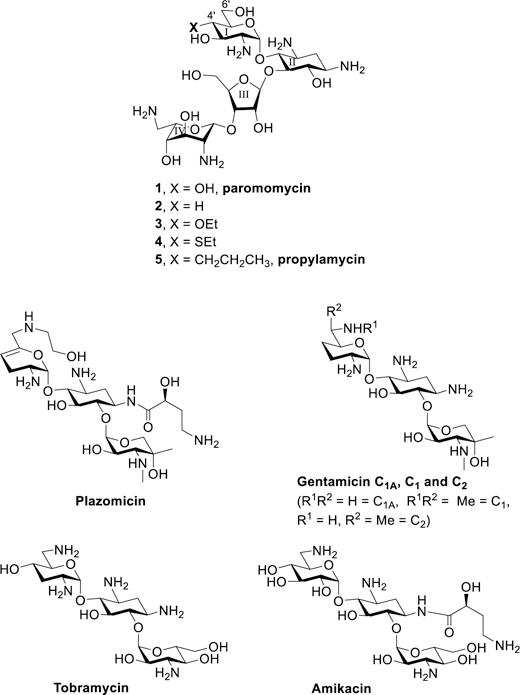
Paromomycin and selected derivatives at the 4’-position, and the current clinical drugs plazomicin, gentamicin, tobramycin and amikacin.
The synthesis of the 4’-deoxy-4’-ethylthio derivative 4 began with the previously described galacto-configured triflate 6,34 which was converted to the thioethers 7–10 and the thioester 11 by standard nucleophilic substitution reactions. Peroxide-mediated oxidation of 7 gave the sulfoxide 12 and the sulfone 13 in a temperature-dependent manner, while the thioester 11 served as precursor to the thioether 14. A two-step deprotection sequence of Staudinger reduction34 followed by hydrogenolysis then afforded 4′-deoxy-4’-ethylthio paromomycin 4 and the analogs 15-20 to which we return below (Scheme 1).
Scheme 1.
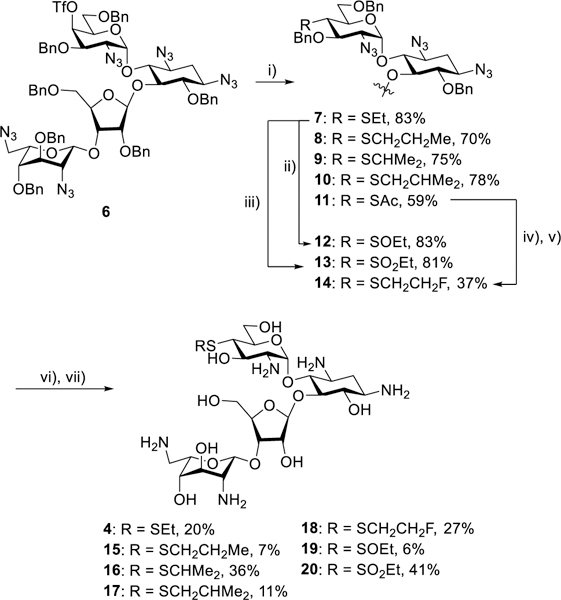
Synthesis of the sulfur-based paromomycin derivatives 4 and 15–20. i) RSNa or AcSK; ii) mCPBA, −78 °C; iii) mCPBA, room temp; iv) NH2NH2.HOAc; v) NaH, FCH2CH2OTf; vi) PMe3, NaOH; vii) H2, Pd/C.
In line with expectation, introduction of the ethylsulfanyl group 4 resulted in only a minor loss in activity against the bacterial ribosome compared to the parent, but reduced activity for the eukaryotic ribosomes and so excellent selectivity (Figure 2 and Table 1). Extrapolating further, we designed the 4’-deoxy-4’-propyl derivative of paromomycin 5, that retains the beneficial three atom chain at the 4’-position but replaces the 4’-oxygen by a methylene group, and which we name propylamycin.
Figure 2.
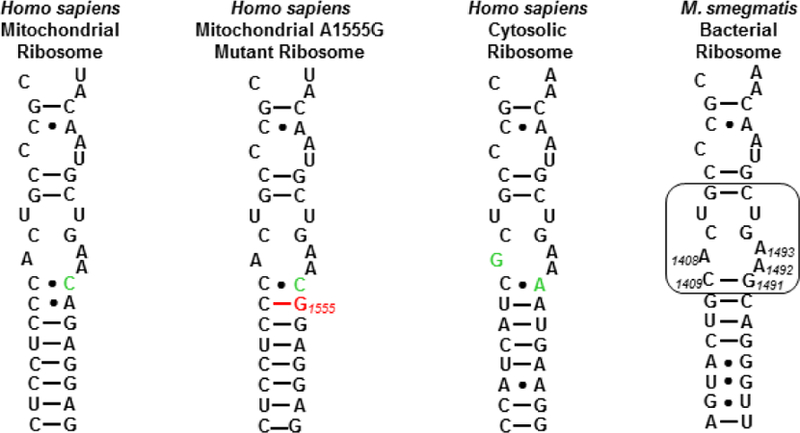
Decoding A sites of the human mitochondrial, A1555G mutant mitochondrial and cytoplasmic ribosomes, and of the bacterial ribosome. The bacterial AGA binding pocket is boxed. The bacterial numbering scheme is illustrated for the AGA binding pocket. Changes from the bacterial ribosome binding pocket are coloured green. The A1555G mutant conferring hypersusceptibility to AGA ototoxicity is coloured red.
Table 1.
In Vitro Translation Inhibitory Activity (μg.mL−1) and Selectivity.
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| IC50 (μg.Ml−1) | Selectivity | |||||||
| Cmpd | 4’-Subs | wt | Mit13 | A1555G | Cyt 14 | Mit 13 | A1555G | Cyt14 |
| 1 | OH | 0.02 | 50 | 5.4 | 9.4 | 2500 | 270 | 470 |
| 2 | H | 0.05 | 74 | 24 | 28 | 1480 | 480 | 560 |
| 3 | OEt | 0.08 | 86 | 95 | 88 | 1075 | 1188 | 1100 |
| 4 | SEt | 0.04 | 111 | 46 | 60 | 2775 | 1150 | 1500 |
| 5 | CH2CH2CH3 | 0.03 | 167 | 52 | 64 | 5567 | 1733 | 2133 |
| 15 | SCH2CH2CH3 | 0.07 | 125 | 110 | 130 | 1788 | 1571 | 1857 |
| 16 | SCH(CH3)2 | 0.10 | 121 | 125 | 111 | 1210 | 1250 | 1110 |
| 17 | SCH2CH(CH3)2 | 0.10 | 180 | 150 | 161 | 1800 | 1500 | 1610 |
| 18 | SCH2CH2F | 0.06 | 102 | 52 | 69 | 1700 | 867 | 1150 |
| 19 | S(O)Et | 0.50 | 118 | 139 | 123 | 236 | 278 | 246 |
| 20 | SO2Et | 8.5 | 207 | 175 | 136 | 24 | 21 | 16 |
| 35 | CH2CH3 | 0.06 | 182 | 129 | 112 | 3033 | 2150 | 1867 |
| 36 | CH2CH2OH | 0.03 | 31 | 25 | 51 | 1033 | 833 | 1700 |
| 37 | CH2CHOHCH2OH | 0.10 | 32 | 33 | 59 | 320 | 330 | 590 |
| Gentamicin | 0.02 | 9 | 0.5 | 33 | 600 | 33 | 2200 | |
| Tobramycin | 0.02 | 15 | 0.5 | 27 | 750 | 25 | 1350 | |
| Amikacin | 0.02 | 12 | 0.42 | 90 | 600 | 21 | 4500 | |
| Plazomicin | 0.06 | 50 | 2.8 | 419 | 833 | 47 | 6983 | |
Propylamycin 5 was obtained by sequential conversion of paromomycin 1 to the pentatrifluoroacetamide 21, its benzylidene acetal 22, and the subsequent hexabenzoate 23 in essentially quantitative yield on a 100 g scale. Cleavage of the acetal gave the diol 24, which was selectively benzoylated at the primary position with benzoyl cyanide35 to afford 25. Triflation followed by displacement with sodium iodide then gave the iodide 26 in 39% yield. In the key C-C bond forming step, reaction of 80 g of 26 with allyl phenyl sulfone36 in α,α,α-trifluorotoluene37 at 0 °C with initiation by triethylborane and air38–40 afforded 92% of crude 27 as a single diastereoisomer. The purification of 27 on a large scale was complicated by the formation of significant amounts of the byproduct 29. Accordingly, the mixture of 27 and 29 was taken forward to the next step when saturation of the double bonds in the two substances gave a still difficult-to-separate mixture of 28 and 30. Finally, removal of the benzoate esters from 28 with magnesium methoxide in methanol and subsequent cleavage of the trifluoroacetamides with barium hydroxide gave 5 in 64% yield for the three steps after purification by Sephadex chromatography (Scheme 2). Noteworthy features of this synthesis include the use of the trifluoroacetamide amine protecting group, which was selected because it: i) rendered all intermediates solid and easy to handle on a large scale; ii) afforded sharp and readily interpreted 1H-NMR spectra at all stages; and iii) could be cleaved under relatively mild basic conditions, yet was resistant to the conditions for the removal of the benzoate esters with magnesium methoxide,41 thereby avoiding potential problems of O→N benzoate migration during deprotection. The excellent equatorial selectivity of the radical C-C bond forming step is consistent with the precedent for such reactions at the 4-position of glucopyranosides.42,43
Scheme 2.
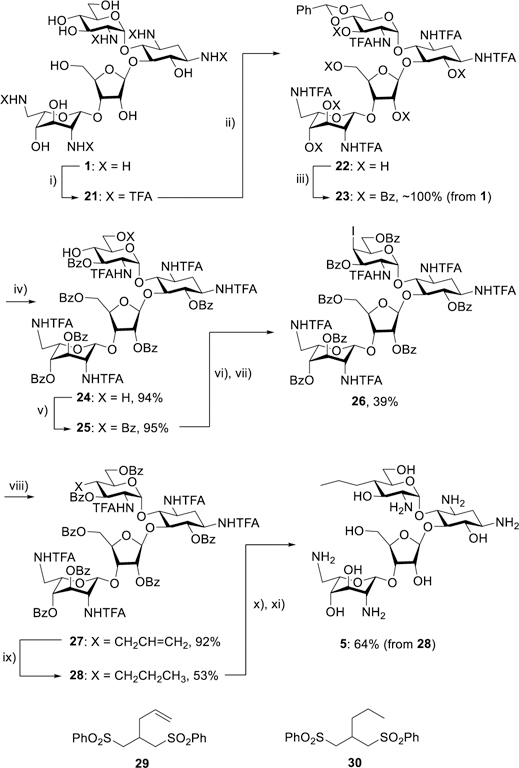
Synthesis of the 4′-C-alkyl paromomycin derivative 5 and structures of the byproducts 29 and 30. i) (CF3CO)2O; ii) PhCH(OMe)2, CSA; iii) Bz2O, py; iv) AcOH, H2O; v) BzCN, Et3N; vi) Tf2O, py; vii) NaI; viii) PhSO2CH2CH=CH2, Et3B, air, 0 °C; ix) Pd(OH)2, H2; x) Mg(OMe)2; xi) Ba(OH)2.
Gratifyingly, 5 displayed excellent selectivity for bacterial over the humanized ribosomes (Figure 2) in a series of cell-free translation assays (Table 1) surpassing that of the earlier lead 3 and of the ethylsulfanyl derivative 4, without any significant loss of activity against the bacterial ribosome. The selectivity of 5 for the bacterial over the mitrochondrial and mutant mitochondrial ribosomes also exceeds that of the clinical comparators gentamicin, tobramycin, amikacin and plazomicin (Table 1). Screening against a panel of Gram-positive and Gram-negative reference strains (Table 2) revealed excellent levels of antibacterial activity for 5, comparable to and in some cases better than those of the clinical comparators gentamicin, tobramycin, amikacin, and plazomicin, and even across-the-board improved levels of activity when compared to the parent paromomycin.
Table 2.
Antibacterial Activity Against MRSA and ESKAPE Pathogens (μg.mL−1)
| MRSA | E. coli | K. pneumoniae | E. cloacae | A. baumannii | P. aeruginosa | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Cmpd | 4’-Subs | AG038 | AG042a | AG001 | AG003b | AG215 | AG290 | AG225 | AG031c | AG220c |
| 1 | OH | 4 | >256 | 2–4 | 8–16 | 1 | 2 | 2–4 | >64 | >64 |
| 2 | H | 16 | 8–16 | nd | 32 | 4 | 4–8 | 8 | nd | nd |
| 3 | OEt | 8–16 | 8–16 | 16 | 32 | 8 | 8 | 16–32 | >64 | >64 |
| 4 | SEt | 4 | 2–4 | 8 | 8 | 2 | 2–4 | 4 | 32 | nd |
| 5 | CH2CH2CH3 | 1–2 | 2 | 1–2 | 1–2 | 0.5 | 0.5–1 | 2 | 8 | 2–4 |
| 15 | SCH2CH2CH3 | 1–2 | 2 | 8 | 4 | 2–4 | 2 | 8 | 32 | 32 |
| 16 | SCH(CH3)2 | 4–8 | 2–4 | 16–32 | 16–32 | 8 | 8 | 16–32 | >128 | nd |
| 17 | SCH2CH(CH3)2 | 2 | 2 | 4–8 | 4–8 | 4 | 2 | 8 | 64 | 64 |
| 18 | SCH2CH2F | 8 | 4–8 | 16–32 | 16–32 | 4 | 4 | 8–16 | 128 | nd |
| 19 | S(O)Et | 64–128 | 64–128 | >64 | >128 | 64 | 64 | >128 | nd | nd |
| 20 | SO2Et | >64 | >64 | >64 | >64 | >128 | >128 | >128 | nd | nd |
| 35 | CH2CH3 | 8 | 4–8 | 8 | 8 | 4 | 4–8 | 16–32 | 64–128 | nd |
| 36 | CH2CH2OH | 1 | 2 | 2–4 | 4 | 2–4 | 1–2 | 2–4 | 16 | 32 |
| 37 | CH2CHOHCH2OH | 4 | 8 | 16 | 8 | 8 | 4–8 | 8 | 16–32 | 16 |
| Gentamicin | 0.25 | 64 | 1–2 | 32–64 | 0.25 | 0.25 | 0.5–1 | 1 | 1 | |
| Tobramycin | 0.5–1 | ≥256 | 1–2 | 4 | 0.5–1 | 0.5–1 | 1 | 1 | 1 | |
| Amikacin | 4 | 16–32 | 2 | 2 | 1 | 1 | 2 | 4 | 2 | |
| Plazomicin | 2 | 2 | 2 | 2–4 | 0.25–0.5 | 0.5 | 2 | 4 | 2–4 | |
strain AG042 carries the AAC(6)’, ANT(4’,4’’) and APH(3’) genes.
strain AG003 carries the AAC(3) gene.
strains AG031 and AG220 carry the APH(3’) gene.
Quantitative dimethyl sulfate footprinting44 of the E coli 70S ribosome in the presence of 5, paromomycin, and the early hit 3 afforded apparent Kd values of 0.34±0.05, 0.59±0.05, and 1.03±0.20 μM, respectively (Figures 3 and 4), consistent with the trend in IC50 values and confirming the tight association of the compounds with the decoding A site of helix 44.
Figure 3.
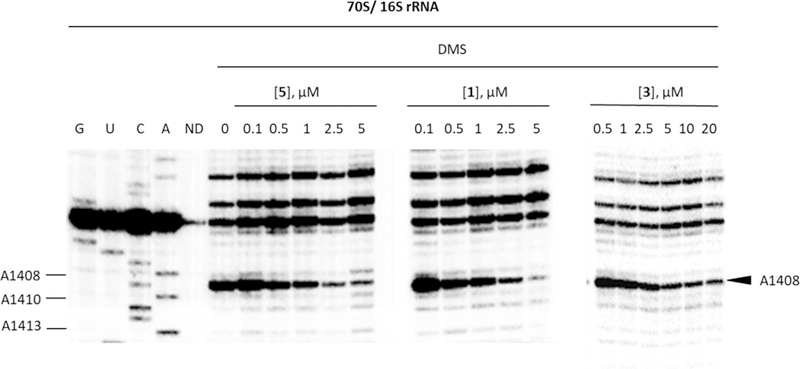
Autoradiogram for DMS footprinting of 5, 1, and 3 binding to the helix 44 region of E. coli 70S ribosomes. Autoradiogram of reacted rRNA followed by primer extension using a radiolabeled primer is shown (G, U, C, and A, dideoxy sequencing; DMS, dimethyl sulfate; ND, no DMS; 0, 0.1, 0.5, 1, 2.5, 5, 10, and 20 correspond to the compound concentrations (μM)).
Figure 4.
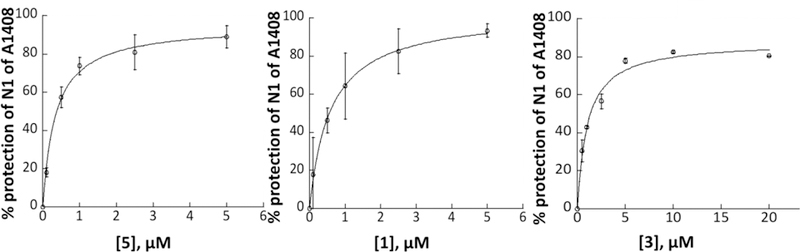
Quantification for DMS footprinting of 5, 1, and 3 binding to the helix 44 region of E. coli 70S ribosomes. Average calculated % protection of N1 of A1408 from three trials for 5 and two trials for 1 and 3 was plotted as a function of aminoglycoside concentration and fitted to a simple binding equation to obtain apparent Kd values of 0.34 ± 0.05 μM (R2=0.99), 0.59 ± 0.05 μM (R2=0.99), and 1.03 ± 0.20 μM (R2=0.99) for 5, 1, and 3, respectively.
Confirmation of the Bacterial Decoding A Site as Drug Target
To support the hypothesis that the antibacterial activity of propylamycin 5 arises from its interaction with the bacterial decoding A site, we tested for activity against the wild type Gram-positive eubacterium Mycobacterium smegmatis and a single allelic derivative with the A1408G mutation obtained by site-directed mutagenis and RecA-mediated gene conversion (Table 3).34,45–48 The A1408G mutation disrupts the canonical pseudobase interaction of ring I of the DOS class of AGAs with A1408 in the drug binding pocket and results in a significant loss of activity.47 Accordingly, the parent 1 and propylamycin 5 both displayed an approximately 100–200-fold loss of activity against M. smegmatis in the presence of the A1408 mutation. This loss of activity reveals the activity of 5 to be the result of binding to the decoding A site, not of an off-target effect, and analogous to that of the parent paromomycin. As expected mutation A1408G decreases the antibacterial activity of the comparators gentamicin, tobramycin, amikacin, and plazomicin, which were used as positive controls, to a greater extent than that of paromomycin or propylamycin. This is because gentamicin, tobramycin, amikacin, and plazomicin all carry an amino group at the 6’-position, whereas paromomycin and propylamycin are 6’-hydroxy AGAs. In the 6’-amino series the A1408G mutation results in a repulsive interaction between the base and the AGA with a correspondingly high loss of activity, whereas in the 6’-hydroxy series the loss of activity is smaller consistent with the simple loss of the canonical pseudobase interaction.47,49
Table 3.
Antibacterial Activity (μg.mL−1) for Wild-type and A1408G Mutant M. smegmatis.
| Wild-type | A1408G | Activity Lossa | |
|---|---|---|---|
| Paromomycin 1 | 0.12 | 16 | 133 |
| Propylamycin 5 | 0.16 | 16–32 | 100–200 |
| Gentamicin | 0.12 | >128 | >1067 |
| Tobramycin | 0.12 | >128 | >1067 |
| Amikacin | 0.06 | >128 | >2133 |
| Plazomicin | 0.03 | >128 | >4267 |
Activity Loss = MIC Mutant/MIC wild-type
X-Ray Crystal Structure of Propylamycin 5 in Complex with the Bacterial Ribosome
To unambiguously identify the mode of binding of the 4’-deoxy-4’-propyl derivative of paromomycin, 5, to its target, we solved the crystal structure of the Thermus thermophilus 70S ribosome associated with mRNA, A-, P- and E-site tRNAs and 5 at 2.75 Å resolution (PDB ID 6O97, Supporting Information Table S1). In this study we used deacylated valine-specific tRNA as the A-site substrate and initiator methionine-specific tRNA as the P-site substrate. The E site of the ribosome contained tRNAVal. The difference electron density maps (Fobs Fcalc) were used to localize the antibiotic on the ribosome. A strong peak of positive electron density (Figure 5) resembling distinct features of 5 was observed in the decoding site at the top of the helix 44 of the 16S rRNA in both copies of the ribosome in the asymmetric unit. Atomic models of the ribosome-bound 5, generated from its chemical structure and the restraints based on idealized 3D geometry were used to fit the drug into the observed electron density (Figure 5).
Figure 5.
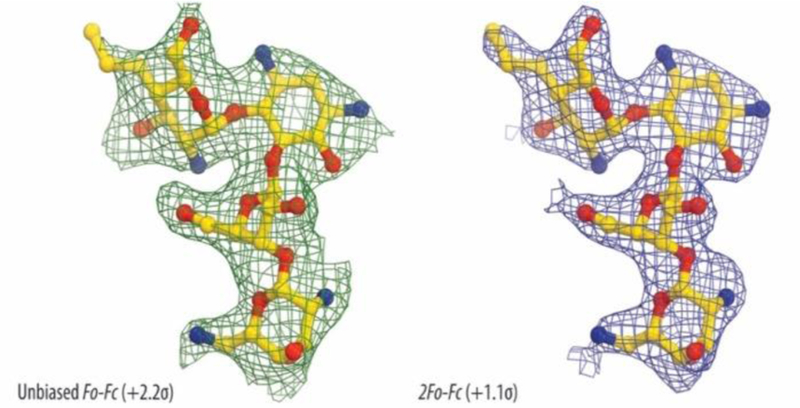
Unbiased Fo-Fc (A) and 2Fo-Fc (B) electron density maps of 5 in complex with the T. thermophilus 70S ribosome. The refined model of 5 is displayed in its electron density maps before and after refinement, respectively. Carbon atoms are colored yellow, nitrogen atoms are blue, and oxygen atoms are red.
The structure reveals that 5 binds to the 70S ribosome in the canonical AGA binding site located in the decoding region of the small ribosomal subunit 4,50 (Figure 6A and B) with bases A1492 and A1493 of the 16S rRNA in the flipped out conformation. The hydroxyl group at position 3’ of ring I forms a hydrogen bond with the phosphate groups of A1492 and A1493, thereby further stabilizing the location of ring I. Ring II of 5 forms hydrogen bonds with G1494 and U1495 as well as with the phosphate groups of A1493 and G1494. Rings III and IV of 5 are oriented towards base pairs 1409–1491 and 1410–1490, enabling the hydroxyl group at position 5’’ of ring III to contact N7 of G1491. The newly appended propyl group of 5 makes no direct contact with the ribosome but, like the alkyl group of the 4’-O-alkyl series of AGAs,49 extends into a highly hydrated area of the ribosome close to the backbone phosphates of G1491 and A1492. As with all other 4,5-DOS AGAs, ring I of 5 stacks upon G1491 and forms a pseudo base pair with A1408 (Figure 6 C,D).4,49,51 In addition to the primary site of action of 5, electron density peaks corresponding to this compound were observed in two additional locations on the large ribosomal subunit. Unlike the primary binding site in the decoding region, these two secondary sites are far from any known ribosome functional centers and are likely functionally irrelevant, because there are no known mutations around those secondary sites that can confer resistance to AGA antibiotics.
Figure 6.
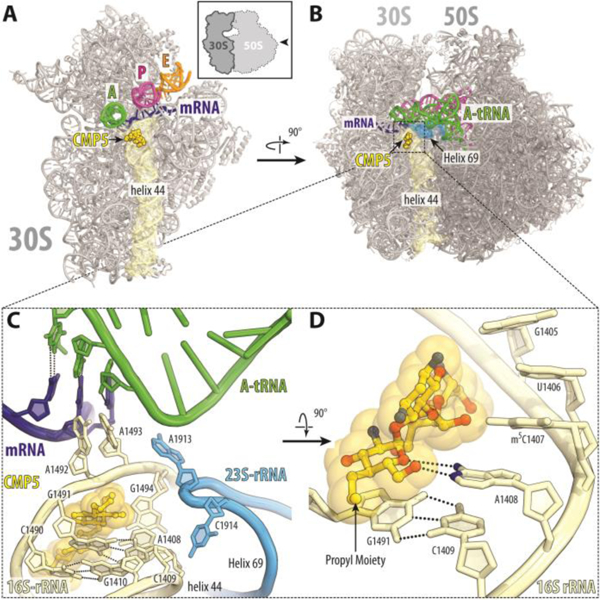
Overview of the propylamycin 5 (CMP5) binding site (yellow) on the T. thermophilus 30S subunit (A) and 70S ribosome (B) (PDB ID 6O97). The 30S and 50S subunits are, respectively, light and dark grey. mRNA is shown in blue and tRNAs are displayed in green for the A site, in magenta for the P site, and in orange for the E site. In (A), the 30S subunit is viewed from the subunit interface, as indicated by the inset; the 50S subunit and parts of tRNAs are removed for clarity. (C, D) Close-up views of the AGA canonical binding site showing the interactions of 5 with the 16S rRNA.
Rationale for Activity and Selectivity of Propylamycin and Structure-Activity Relation at the Target Level
We hypothesize that the increased activity of the 4’-deoxy-4’-C-propyl paromomycin derivative 5 as compared to the 4’-O-ethyl derivative 3 and to paromomycin arises from the influence of the modification on the strength of the pseudobase pair interaction with A1408. Thus, by analogy with the influence of β-bonds on the pKa values of amines,52 replacement of the 4’-C-O bond in 3 by a C-C bond results in enhanced basicity of the ring oxygen (O5’) in ring I rendering it a better acceptor for the hydrogen bond from N6 of A1408. This also accounts for the greater activity of 4’-deoxy paromomycin 2 over the 4’-O-ethyl derivative 3. The increased activity of 5 over 2 is necessarily due to the presence of the hydrophobic propyl group at the 4’-position, possibly retarding dissociation of the complex and/or interfering with the hydration shell.
In comparison to paromomycin, propylamycin 5 affords a significant increase in selectivity for the bacterial over the cytoplasmic ribosome. The same modification affords an increase in selectivity for the bacterial over the mutant mitochondrial ribosome for which the structural basis has yet to be determined.
We synthesized a small series of cognate derivatives to further explore the space available to the 4’-substituent and challenge our hypotheses. Specifically, compounds were designed to test the influence of length and branching on the alkyl group as well as the effects of electron-withdrawing groups and increased hydrophilicity. Working first in the alkylsulfanyl series − for ease of synthesis − we prepared three analogs of 4 with variation in the length and branching of the substituent (15–17, Scheme 1). Cell-free translation assays of these compounds with the wild-type bacterial ribosome revealed that each of these modifications is accompanied by a two- to three-fold reduction in inhibition of luciferase production (Table 1). Similar reductions in the inhibition of the humanized ribosomes were also observed, resulting in an overall comparable selectivity to that seen with the ethylthio derivative 4.
The availability of 27, a key intermediate in the synthesis of 5, in multigram amounts enabled the formation of further 4′-deoxy-4′-alkyl derivatives by standard manipulations of the alkene (Scheme 3). Thus, ozonolysis of the alkene 27 followed by reduction with sodium borohydride gave the alcohol 31, which, on treatment with iodine, triphenylphosphine and imidazole afforded the iodide 32. Hydrogenolysis of 32 over palladium on charcoal in the presence of triethylamine then gave the derivative 33. Treatment of 27 with N-methylmorpholine N-oxide and catalytic osmium tetroxide afforded the diol 34 as an inseparable mixture of diastereomers. Exposure of each of 33, 31, and 34 to magnesium methoxide and then barium hydroxide followed by Sephadex chromatography afforded the paromomycin derivatives 35-37, respectively.
Scheme 3.
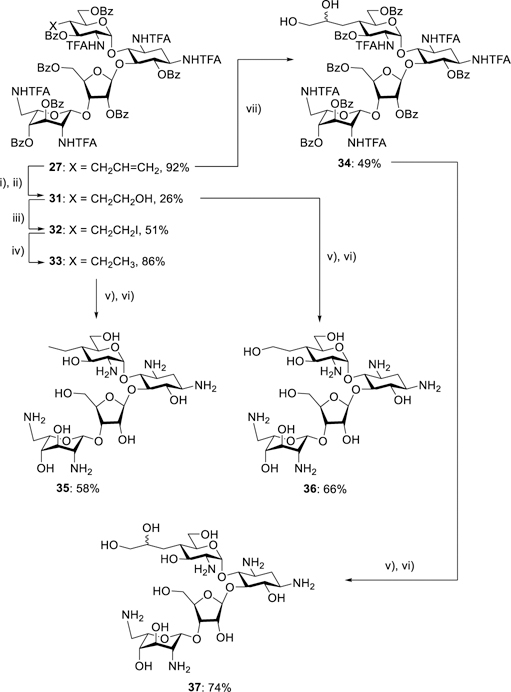
Synthesis of the 4′-C-alkyl paromomycin derivatives 35-37. i) O3; ii) NaBH4; iii) PPh3, imidazole, I2; iv) Pd/C, Et3N, H2; v) Mg(OMe)2; vi) Ba(OH)2; vii) OsO4, NMNO.
The 4’-deoxy-4’-ethyl derivative 35 displayed a minor reduction in antibacterioribosomal activity compared to its higher homolog 5, which was accompanied by a small loss of selectivity (Table 1). Overall, it is clear that the optimal chain length is three linear non-hydrogen atoms from the 4’-position.
The replacement of the optimal ethylthio group in 4 by a 2-fluoroethylthio group with the intention of increasing hydrophilicity53,54 while maintaining the optimal chain length and shape provoked a minor decrease in inhibition of the bacterial ribosome and a small decrease in selectivity. This is consistent with a decrease in basicity of the ring I oxygen on incorporation of the strongly electron-withdrawing fluorine atom and so with reduced basicity of O5’. Oxidation of the thioether in 4 to either the corresponding sulfoxides 19 or the sulfone 20 resulted in a dramatic drop in antiribosomal activity suggesting that the combination of steric bulk with a strongly electron withdrawing group directly attached to ring I is not tolerated (Table 1). The 4’-deoxy-4’-(2-hydroxyethyl) derivative 36 had comparable activity to 5 against the bacterial ribosome, but significantly lower selectivity, whereas the 4’-deoxy-4’-(2,3-dihydroxypropyl) derivative 37 displayed reduced activity and lower selectivity (Table 1), indicating that increased hydrophilicity of the 4’-substituent is detrimental.
Overall, the optimum combination of high activity for the bacterial ribosome and high selectivity over the humanized ribosomes is found in the 4’-deoxy-4’-propyl derivative 5. The inhibitory effect on translation is affected by either lengthening or shortening of the three-atom backbone of the 4’-substituent, by the incorporation of branching, and by hydroxylation of the substituent as it results in a significant loss of selectivity.
Antibacterial Activity in the Presence and Absence of Specific Resistance Determinants
All compounds were screened for activity against the same panel of reference strains employed for 5 (Table 2), with results largely consistent with the levels of inhibition of the wild-type bacterial ribosomes.
Finally, 4’-deoxy-4’-C-propylparomomycin 5 was tested for antibacterial activity against a panel of wild-type E. coli, K. pneumoniae, and E. cloacae clinical isolates resulting in the distributions shown in Figure 7, and indicating MIC90s of 2, 1, and 1 μg.mL−1, respectively.
Figure 7.
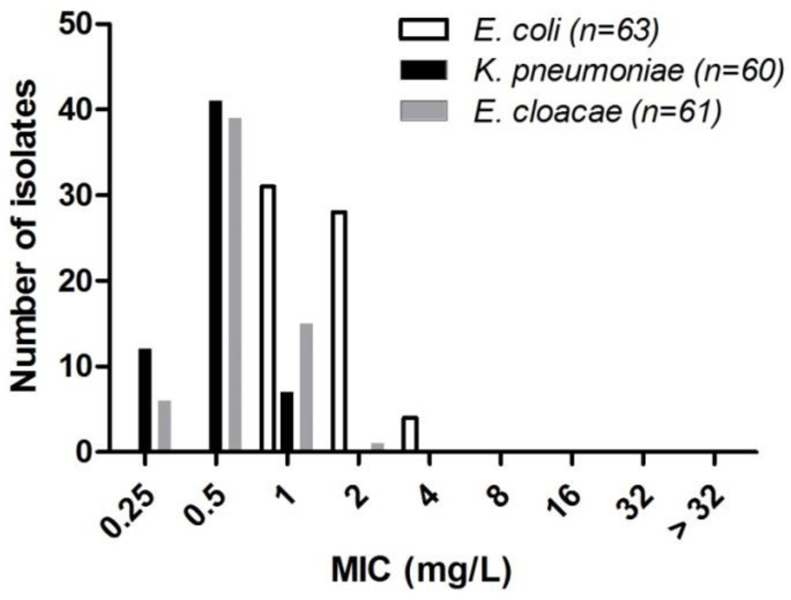
MIC distribution of 5 for clinical wild-type Enterobacteriaceae isolates
To determine the effectiveness of the novel 4’-modifications in thwarting the action of resistance determinants, selected compounds were screened for activity against a panel of wild-type and engineered E. coli carrying specific AMEs or RMTs (Table 4). While it is not surprising that the replacement of the 4’-hydroxy group in the parent paromomycin restores activity in the presence of ANT(4’,4’’) enzymes, it is of note that the 4’-C-propyl modification affords continued high levels of activity in the presence of the APH(3’). Indeed, the ability to circumvent the action of the APH(3’) obviates the need for modification at the 3’-position. The activity of 4’-deoxy-4’-C-propyl paromomycin 5 is also not affected by the presence of the AMEs AAC(3)-I, AAC(2’) and AAC(6’), consistent with the known resistome of the 4,5-AGAs.19 It is also of note that the structure of propylamycin 5 is such that it is not susceptible to the ANT(2’’) class of AMEs, which are the primary cause of resistance to the 4,6-AGAs in clinical use in North America.28 Importantly, the 4’-deoxy-4’-C-propyl modification does not interfere with the inherent resilience of the 4,5-AGAs to the ArmA ribosomal methyltransferase resistance mechanism that severely limits the action of all 4,6-DOS AGAs including the newly introduced plazomicin.6,7,26,27 Therefore, propylamycin will not suffer from problems of cross-resistance when used in combination therapy with carbapenems, unlike the 4,6-AGA plazomicin. This latter conclusion is borne out by a final set of screens in which a series of clinical isolates carrying two or more resistance determinants (AMEs and/or RMTs) were challenged with propylamycin 5 and the clinical comparators (Table 5).
Table 4.
Activity in the Presence of Specific Resistance Determinants (μg.mL−1)
| Strain | AG006 | AG007 | AG105 | AG009 | AG008 | AG036 | AG166 | AG103 | |
|---|---|---|---|---|---|---|---|---|---|
| Resistance Mechanism | WT | AAC(3) | AAC(2′) | AAC(6’) | ANT(2’’) | ANT(4′,4′′) | APH(3′) | armA | |
| 1 | OH | 1–2 | 4 | 2 | 4 | 2–4 | 32–64 | >128 | 4 |
| 3 | OEt | 8–16 | 16–32 | 8–16 | 16 | 8 | 8–16 | 64 | ≥64 |
| 4 | SEt | 1–2 | 4 | 2–4 | 4 | 1 | 1–2 | 4 | 8 |
| 5 | Pr | 0.5–1 | 2 | 1 | 1–2 | 0.5–1 | 1 | 1–2 | 2 |
| 15 | SPr | 1–2 | 4 | 2 | 2–4 | 1 | 2–4 | 8 | 4–8 |
| 17 | SCH2CH(CH3)2 | 2 | 4 | 2–4 | 4 | 1–2 | 1–2 | 8 | 8 |
| Gentamicin | 0.5 | 32 | 32 | 1–2 | 8–16 | 0.5 | 0.5 | >128 | |
| Tobramycin | 0.5 | 4 | 1 | 64–128 | 8 | 16–32 | 0.5–1 | >128 | |
| Amikacin | 1–2 | 2 | 2 | 32 | 1–2 | 4–8 | 2 | >128 | |
| Plazomicin | 0.5–1 | 2 | 1–2 | 2 | 0.5–1 | 1 | 1 | >128 | |
Table 5.
Activity of Propylamycin 5 and Clinical Comparators in the Presence of Multiple Resistance Determinants (μg.mL−1)
| Isolate | Resistance Mechanism | Propylamycin | Gentamicin | Tobramycin | Amikacin | |
|---|---|---|---|---|---|---|
| E. coli | 2014405344 | AAC(3), APH(3’), RmtB | 4 | >256 | >256 | >256 |
| 201430298 | AAC(3), AAC(6’) | 1 | 128 | 256 | 8 | |
| 2014251408 | AAC(3), AAC(6’) | 2 | 256 | 128 | 64 | |
| 201441114 | AAC(6’), ANT(2’’) | 1 | 16 | 32 | 64 | |
| K. pneumoniae | 2014310108 | AAC(3), AAC(6’), RmtB | 1 | >256 | >256 | >256 |
| 201440494 | AAC(3), AAC(6’) | 0.5 | 32 | 16 | 2 | |
| 2014310114 | AAC(3), AAC(6’) | 0.25 | 256 | 256 | 8 | |
| E. cloacae | 201385168 | AAC(3), AAC(6’), RmtC | 1 | >256 | >256 | >256 |
| 201333207 | AAC(3), AAC(6’), APH(3’) | 0.5 | 64 | 32 | 16 | |
| 201435487 | AAC(3), AAC(6’) | 0.5 | 64 | 16 | 4 | |
| 201430601 | AAC(6’), ANT(2’’) | 0.5 | 16 | 32 | 8 | |
The ability of the 4’-C-propyl modification to overcome the influence of the AAC(2’) resistance mechanism was further demonstrated in Mycobacterium abscessus. Thus, the MIC of 5 was either unchanged (4 μg.mL−1) or exhibited only a two-fold reduction (from 4 to 2 μg.mL−1) when the AAC(2’) gene was deleted from this increasingly problematic mycobacterium that presents a rapidly increasing threat in hospitalized patients with cystic fibrosis or chronic pulmonary disease.55 In contrast, the MIC of the parent paromomycin was reduced from 64 to 16 μg.mL-1. The antimycobacterial activity of 5 was further explored with clinical isolates of Mycobacterium tuberculosis, which showed MICs of 1 μg.mL-1. As the parent paromomycin had MICs of 2–4 μg.mL−1, the beneficial effects of the 4’-deoxy-4’-C-propyl modification potentially extend to the treatment of Mtb.
Efficacy in vivo
In a mouse thigh infection model for E coli, 5 displayed a 1.5 log reduction in colony-forming units (CFUs) compared to vehicle with a dose of 3 mg.kg−1 similar to that observed with 6 mg of the parent paromomycin, and with 3 mg.kg−1 of plazomicin (Figure 8a). In a neutropenic mouse E. coli septicemia model a 4 mg.kg−1 dose of 5 reduced the bacterial burden in the blood by approximately 1.5 log units, 1 log more than the parent paromomycin at the same dose level (Figure 8b).
Figure 8.
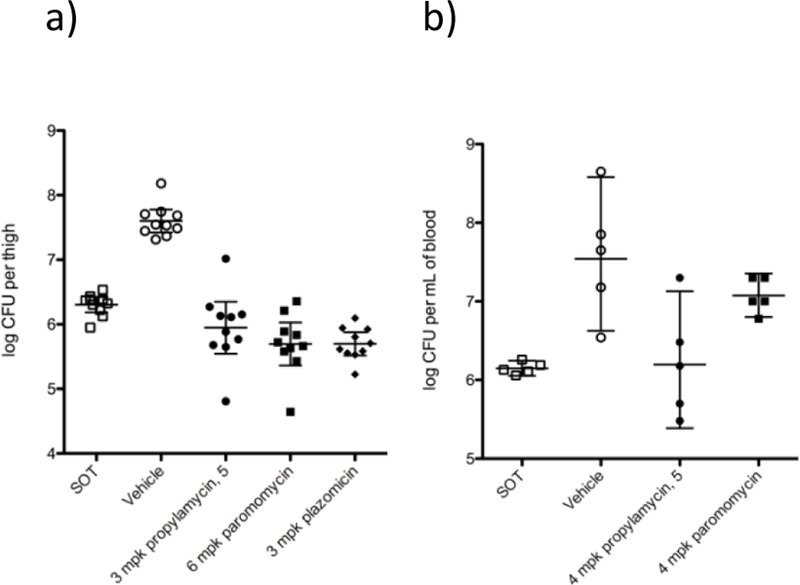
In-vivo efficacy of 5. a) CFU reduction in a neutropenic mouse thigh infection model. b) CFU reduction in blood in a mouse septicemia model (mpk = milligrams per kilogram)
Toxicity
Compounds 4 and 5 were screened for cytotoxicity against NIH 3T3 mouse embryonic fibroblast cells, with both showing only minimal loss of cell viability after 48 and 72 h at up to 2 M concentrations (Supporting Information, Figure S1).
To assess ototoxicity, 4’-deoxy-4’-C-propyl paromomycin 5 was administered once daily sub-cutaneously (SC) to guinea pigs for 14 days at dose levels of 80 and 100 mg.kg−1 with comparisons to controls (saline) and the clinical 4,6-AGA gentamicin (Figure 9, left column). Thresholds of auditory brain stem responses (ABR) were obtained before and 14 days after the end of treatment at 8, 16 and 32 kHz and the shift in thresholds was calculated as a measure of ototoxicity. At all three frequencies the threshold shifts for 5 are indistinguishable from the control with a dose of 80 mgkg−1 and only marginally increased over the control at 100 mg.kg-1. In contrast, gentamicin already shows significant threshold shifts at 80 mgkg−1, comparable to those with 5 at the more elevated dose, and displays even higher shifts at the 100 mgkg−1 dose level. This trend of the ABR thresholds is consistent with the pattern at the target level with 5 displaying very significantly greater selectivity for the bacterial ribosome over the mitochondrial, mutant mitochondrial and cytoplasmic ribosomes.
Figure 9.
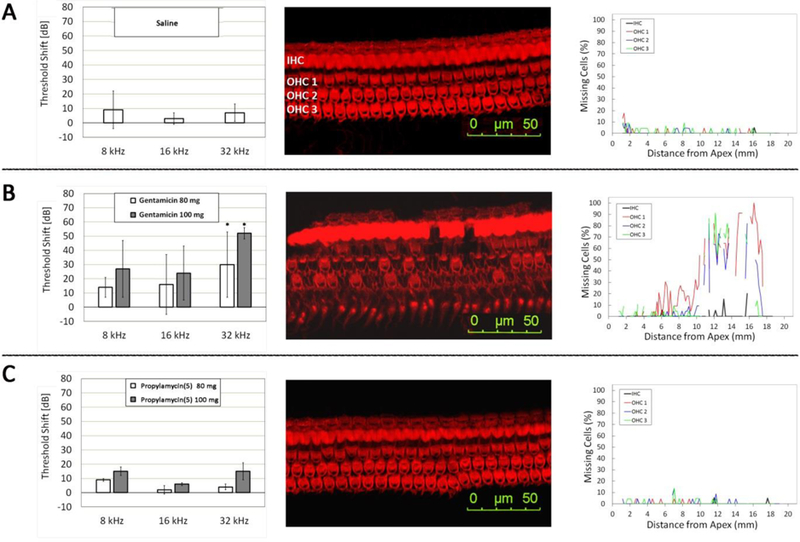
Comparative ototoxicity of gentamicin and propylamycin (5). Threshold shifts induced by treatment as determined by ABR (left column). Representative surface preparations outlining one row of inner hair cells (IHC) and three rows of outer hair cells (OHC) in sections from the base of the cochlea (Center column). Quantification of hair cell loss along the entire length of the cochlea (Right column). A: Control animals injected with saline for 14 days. B: Threshold shifts after treatment with gentamicin at 80 and 100 mg.kg−1 body weight, respectively, for 14 days; surface preparation and hair cell counts are from the same animal after treatment with 100 mg.kg−1 gentamicin. C: Threshold shifts after treatment with propylamycin (5) at 80 and 100 mg.kg−1 body weight, respectively, for 14 days; surface preparation and hair cell counts are from the same animal after treatment with 100 mg.kg−1 propylamycin. N = 3 for each drug treatment, n = 2 for controls. * significantly different from saline control and from propylamycin (p < 0.05).
The reduced ototoxicity of 5 compared to gentamicin is confirmed by microscopic examination of the cochlear hair cells (shown here at the higher 100 mgkg−1 dose level; Figure 9, center column), which are essentially unchanged from the control even in the basal turn, the most sensitive target of aminoglycosides, while gentamicin treatment results in extensive damage. Quantitative assessment of hair cell loss along the entire length of the cochlea confirms this pattern. Both saline and 100 mgkg−1 of 5 show no effective loss of inner or outer hair cells, whereas with gentamicin very high levels of outer hair cell loss are seen already beginning at 6 mm from the apex (Figure 9, right column). We have previously demonstrated by means of an ex vivo mouse cochlear explant study that both tobramycin and plazomicin display similar exvivo ototoxicity to gentamicin, consistent with their ribosomal selectivity patterns.27
Conclusion
A single modification to the 4,5-DOS-AGA paromomycin, replacing an hydroxyl group at the 4’-position by an ethylthio group, or optimally by a propyl group, has multiple beneficial effects including increased ribosomal selectivity, enhanced activity against bacterial pathogens, and retention of activity in the presence of multiple resistance determinants. These in vitro studies are borne out by preliminary efficacy studies in mice and the demonstration of lower ototoxicity in guinea pigs when compared to gentamicin.
The excellent antibacterial activity of these designed AGAs is rationalized by an enhanced pseudobase interaction of ring I with A1408 in the AGA binding decoding A site of helix 44, correlated with an increase in basicity of the ring oxygen on removal of the electronegative 4’-oxygen. Presumably, the enhanced basicity of the ring I oxygen in the absence of a C-O bond at the 4’-position also contributes to the high levels of antibacterial activity of other 4’-deoxy AGAs, including the gentamicins in the 4,6-DOS series and comparable synthetic compounds56 in the 4,5-DOS series. The same 4’-substituent that enhances antibacterial activity also protects against multiple AMEs that act on ring I, thereby circumventing several common mechanisms of AGA resistance, and is responsible for the increased ribosomal selectivity and consequent reduced ototoxicity. Overall, the 4’-deoxy-4’-alkyl sulfanyl and 4’-deoxy-4’-alkyl modifications establish a new paradigm for the rational design of improved semisynthetic AGAs with which to treat the growing threat of multidrug resistant infectious diseases.
Supplementary Material
Acknowledgments
We thank Ariane Kanicki, Catherine Martin and Susan Deremer (Kresge Hearing Research Institute) for their expert help with the ototoxicity studies. We are grateful to Patrice Courvalin (Institut Pasteur) for the gift of engineered strains of E. coli, Evotec for the in vivo efficacy studies, and the NIH (AI123352) for support of this work. ECB thanks the Swiss National Science Foundation (407240_166998) for partial support of the work in Zurich. The crystallographic study was supported by Illinois State startup funds to YSP, and is based upon research conducted at the Northeastern Collaborative Access Team beamlines, which are funded by the National Institute of General Medical Sciences from the National Institutes of Health (P41 GM103403 to NE-CAT). The Pilatus 6M detector on 24ID-C beamline is funded by a NIH-ORIP HEI (S10-RR029205 to NE-CAT). The Eiger 16M detector on 24ID-E beamline is funded by a NIH-ORIP HEI grant (S10-OD021527 to NE-CAT]. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357.
Footnotes
Supporting Information Available.
Full experimental parts and copies of 1H and 13C NMR spectra for the synthesis of all new compounds.
Experimental protocols for the ribosome inhibition assays
Experimental protocols for the quantitative ribosome footprinting
Experimental protocol for crystallographic structure determination of propylamycin 5 in complex with the bacterial ribosome
Table of X-ray data collection and refinement statistics
Experimental protocol for the antimicrobial susceptibility testing
Experimental protocol for the animal efficacy studies
Experimental protocol for the cytotoxicity studies
Figure showing cytotoxicity of 4 and 5 in mouse fibroblasts in comparison to paromomycin and gentamicin.
Experimental protocols for the in‐vivo ototoxicity studies
References
- (1).Armstrong ES; Kostrub CF; Cass RT; Moser HE; Serio AW; Miller GH In Antibiotic Discovery and Development; Dougherty, Pucci TJ, M. J., Eds.; Springer Science+Business Media: New York, 2012, p 229–269. [Google Scholar]
- (2).Garneau-Tsodikova S; Labby KJ Mechanisms of resistance to aminoglycoside antibiotics: overview and perspectives. Med. Chem. Commun 2016, 7, 11–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Zárate SG; De la Cruz Claure ML; Benito-Arenas R; Revuelta R; Santana AG; Bastida A Overcoming Aminoglycoside Enzymatic Resistance: Design of Novel Antibiotics and Inhibitors. Molecules 2018, 23, 284, doi: 210.3390/molecules23020284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Carter AP; Clemons WM; Brodersen DE; Morgan-Warren RJ; Wimberly BT; Ramakrishnan V Functional Insights from the Structure of the 30S Ribosomal Subunit and its Interactions with Antibiotics. Nature 2000, 407, 340–348. [DOI] [PubMed] [Google Scholar]
- (5).Llano-Sotelo B; Azucena EF; Kotra LP; Mobashery S; Chow CS Aminoglycosides Modified by Resistance Enzymes Display Diminished Binding to the Bacterial Ribosomal Aminoacyl-tRNA Site. Chem. Biol 2002, 9, 455–463. [DOI] [PubMed] [Google Scholar]
- (6).Livermore DM; Mushtaq S; Warner M; Zhang J-C; Maharjan S; Doumith M; Woodford N Activity of aminoglycosides, including ACHN-490, against carbapenem-resistant Enterobacteriaceae isolates. J. Antimicrob. Chemother 2011, 66, 48–53. [DOI] [PubMed] [Google Scholar]
- (7).Doi Y; Wachino JI; Arakawa Y Aminoglycoside Resistance: The Emergence of Acquired 16S Ribosomal RNA Methyltransferases. Infect. Dis. Clin. North Am 2016, 30, 523–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Galani I; Souli M; Panagea T; Poulakou G; Kanellakopoulou K; Giamarellou H Prevalence of 16S rRNA methylase genes in Enterobacteriaceae isolates from a Greek University Hospital. Clin. Microbiol. Infect 2012, 18, E52–E54. [DOI] [PubMed] [Google Scholar]
- (9).Piekarska K; Zacharczuk K; Wołkowicz T; Rzeczkowska M; Bareja E; Olak M; Gierczyński R Distribution of 16S rRNA Methylases Among Different Species of Aminoglycoside-Resistant Enterobacteriaceae in a Tertiary Care Hospital in Poland. Adv. Clin. Exp. Med 2016, 25, 539–544. [DOI] [PubMed] [Google Scholar]
- (10).Taylor E; Sriskandan S; Woodford N; Hopkins KL High prevalence of 16S rRNA methyltransferases among carbapenenase-producing Enterobacteriaceae in the UK and Ireland. Int. J. Antimicrob. Agents 2018, 52, 278–282. [DOI] [PubMed] [Google Scholar]
- (11).Beauclerk AAD; Cundliffe E Sites of Action of Two Ribosomal RNA Methylases Responsible for Resistance to Aminoglycosides. J. Mol. Biol 1987, 193, 661–671. [DOI] [PubMed] [Google Scholar]
- (12).Drusano GL; Ambrose PG; Bhavnani SM; Bertino JS; Nafziger AN; Louie A Back to the Future: Using Aminoglycosides Again and How to Dose Them Optimally. Clin. Infect. Dis 2007, 45, 753–760. [DOI] [PubMed] [Google Scholar]
- (13).Prezant TR; Agapian JV; Bohlman MC; Bu X; Öztas S; Qiu W-Q; Arnos KS; Cortopassi GA; Jaber L; Rotter JI; Shohat M; Fischel-Ghodsian N Mitochondrial Ribosomal RNA Mutation Associated with Both Antibiotic-Induced and Non-Syndromic Deafness. Nat. Genetics 1993, 4, 289–294. [DOI] [PubMed] [Google Scholar]
- (14).Hobbie SN; Akshay S; Kalapala SK; Bruell C; Shcherbakov D; Böttger EC Genetic Analysis of Interactions with Eukaryotic rRNA Identify the Mitoribosome as Target in Aminoglycoside Ototoxicity. Proc. Natl. Acad. Sci., USA 2008, 105, 20888–20893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Hobbie SN; Bruell CM; Akshay S; Kalapala SK; Shcherbakov D; Böttger EC Mitochondrial deafness alleles confer misreading of the genetic code. Proc. Natl. Acad. Sci., USA 2008, 105, 3244–3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Talaska AE; Schacht J In Aminoglycoside Antibiotics: From Chemical Biology to Drug Discovery; Arya, DP, Ed.; Wiley: Hoboken, 2007, p 255–266. [Google Scholar]
- (17).Jiang M; Karasawa T; Steyger PS Aminoglycoside-Induced Cochleotoxicity: A Review. Front. Cell. Neurosci 2017, 11, 308-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Garinis AC; Cross CP; Srikanth P; Carroll K; Feeney MP; Keefe DH; Hunter LL; Putterman DB; Cohen DM; Gold JA; Steyger PS The cumulative effects of intravenous antibiotic treatments on hearing in patients with cystic fibrosis. J. Cyst. Fibros 2017, 16, 401–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Bacot-Davis VR; Bassenden AV; Berghuis AM Drug-target networks in aminoglycoside resistance: hierarchy of priority in structural drug design. Med. Chem. Commun 2016, 7, 103–113. [Google Scholar]
- (20).Chandrika NT; Garneau-Tsodikova S Comprehensive review of chemical strategies for the preparation of new aminoglycosides and their biological activities. Chem. Soc. Rev 2018, 47, 1189–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Takahashi Y; Igarashi M Destination of Aminoglycoside Antibiotics in the ‘Post-Antibiotic Era’. J. Antibiotics 2018, 71, 4–14. [DOI] [PubMed] [Google Scholar]
- (22).Wang J; Chang C-WT In Aminoglycoside Antibiotics; Arya DP, Ed.; Wiley: Hobeken, 2007, p 141–180. [Google Scholar]
- (23).Berkov-Zrihen Y; Fridman M In Modern Synthetic Methods in Carbohydrate Chemistry; From Monosaccharides to Complex Glycoconjugates; Werz DB, Vidal S, Eds.; Wiley: Weinheim, 2014, p 161–190. [Google Scholar]
- (24).Haddad J; Liu M-Z; Mobashery S In Glycochemistry: Principles, Synthesis and Applications; Wang PG, Bertozzi CR, Eds.; Dekker: New York, 2001, p 353–424. [Google Scholar]
- (25).Aggen JB; Armstrong ES; Goldblum AA; Dozzo P; Linsell MS; Gliedt MJ; Hildebrandt DJ; Feeney LA; Kubo A; Matias RD; Lopez S; Gomez M; Wlasichuk KB; Diokno R; Miller GH; Moser HE Synthesis and Spectrum of the Neoglycoside ACHN-490. Antimicrob. Agent. Chemother 2010, 54, 4636–4642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Cox G; Ejim L; Stogios PJ; Koteva K; Bordeleau E; Evdokimova E; Sieron AO; Savchenko A; Serio AW; Krause KM; Wright GD Plazomicin Retains Antibiotic Activity against Most Aminoglycoside Modifying Enzymes. ACS Infect. Dis 2018, 4, 980–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Sonousi A; Sarpe VA; Brilkova M; Schacht J; Vasella A; Böttger EC; Crich D Effects of the 1-N-(4-Amino-2S-hydroxybutyryl) and 6’-N-(2-Hydroxyethyl) Substituents on Ribosomal Selectivity, Cochleotoxicity and Antibacterial Activity in the Sisomicin Class of Aminoglycoside Antibiotics. ACS Infect. Dis 2018, 4, 1114–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Bassenden AV; Rodionov D; Shi K; Berghuis AM Structural Analysis of the Tobramycin and Gentamicin Clinical Resistome Reveals Limitations for Next-generation Aminoglycoside Design. ACS Chem. Biol 2016, 11, 1339–1346. [DOI] [PubMed] [Google Scholar]
- (29).Hobbie SN; Kalapala SK; Akshay S; Bruell C; Schmidt S; Dabow S; Vasella A; Sander P; Böttger EC Engineering the rRNA Decoding Site of Eukaryotic Cytosolic Ribosomes in Bacteria. Nucl. Acids Res 2007, 35, 6086–6093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Duscha S; Boukari H; Shcherbakov D; Salian S; Silva S; Kendall A; Kato T; Akbergenov R; Perez-Fernandez D; Bernet B; Vaddi S; Thommes P; Schacht J; Crich D; Vasella A; Böttger EC Identification and Evaluation of Improved 4’-O-(Alkyl) 4,5-Disubstituted 2-Deoxystreptamines as Next Generation Aminoglycoside Antibiotics. mBio 2014, 5, 10.1128/mBio.01827-01814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Huth ME; Han K-H; Sotoudeh K; Hsieh Y-J; Effertz T; Vu AA; Verhoeven S; Hsieh MH; Greenhouse R; Cheng AG; Ricci AJ Designer aminoglycosides prevent cochlear hair cell loss and hearing loss. J. Clin. Invest 2015, 125, 583–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Matsushita T; Chen W; Juskeviciene R; Teo Y; Shcherbakov D; Vasella A; Böttger EC; Crich D Influence of 4′-O-Glycoside Constitution and Configuration on Ribosomal Selectivity of Paromomycin. J. Am. Chem. Soc 2015, 137, 7706–7717. [DOI] [PubMed] [Google Scholar]
- (33).Mandhapati AR; Yang G; Kato T; Shcherbakov D; Hobbie SN; Vasella A; Böttger EC; Crich D Structure-Based Design and Synthesis of Apramycin-Paromomycin Analogues. Importance of the Configuration at the 6′-Position and Differences Between the 6′-Amino and Hydroxy Series. J. Am. Chem. Soc 2017, 139, 14611–14619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Pathak R; Perez-Fernandez D; Nandurdikar R; Kalapala SK; Böttger EC; Vasella A Synthesis and evaluation of paromomycin derivatives modified at C(4’). Helv. Chim. Acta 2008, 91, 1533–1552. [Google Scholar]
- (35).Peng P; Linseis M; Winter RF; Schmidt RR Regioselective Acylation of Diols and Triols: The Cyanide Effect. J. Am. Chem. Soc 2016, 138, 6002–6009. [DOI] [PubMed] [Google Scholar]
- (36).Barton DHR; Crich D Improved Methods for the Addition of Carbon Radicals to Substituted Allylic Groups. J. Chem. Soc., Perkin Trans 1 1986, 1613–1619. [Google Scholar]
- (37).Maul JJ; Ostrowski PJ; Ublacker GA; Linclau B; Curran DP In Top. Curr. Chem; Knochel P, Ed.; Springer: Berlin, 1999; Vol. 206, p 79–105. [Google Scholar]
- (38).Curran DP; McFadden TR Understanding Initiation with Triethylboron and Oxygen: The Differences between Low-Oxygen and High-Oxygen Regimes. J. Am. Chem. Soc 2016, 138, 7741–7752. [DOI] [PubMed] [Google Scholar]
- (39).Brown HC; Midland MM Organic Synthesis via Free-Radical Displacement Reactions of Organoboranes. Angew. Chem. Int. Ed 1972, 11, 692–700. [Google Scholar]
- (40).Nozaki K; Oshima K; Utimoto K Et3B-Induced Radical Addition of R3SnH to Acetylenes and Its Application to Cyclization Reaction. J. Am. Chem. Soc 1987, 109, 2547–2549. [Google Scholar]
- (41).Xu Y-C; LeBeau E; Walker C Selective deprotection of esters using magnesium methoxide. Tetrahedron Lett 1994, 35, 6207–6210. [Google Scholar]
- (42).Giese B; Witzel T Synthesis of “C-Disaccharides” by Radical C-C Bond Formation. Angew. Chem. Int. Ed 1986, 25, 450–451. [Google Scholar]
- (43).Gupta V; Kahne D Direct Introduction of CH2OH by Intermolecular Trapping of CO. Tetrahedron Lett 1993, 34, 591–594. [Google Scholar]
- (44).Moazed D; Noller HF Interaction of Antibiotics with Functional Sites in 16S Ribosomal RNA. Nature 1987, 327, 389–394. [DOI] [PubMed] [Google Scholar]
- (45).Sander P; Prammananan T; Böttger EC Introducing mutations into a chromosomal rRNA gene using a genetically modified eubacterial host with a single rRNA operon. Mol. Microbiol 1996, 22, 841–848. [DOI] [PubMed] [Google Scholar]
- (46).Prammananan T; Sander P; Springer BC; Böttger EC RecA-Mediated gene conversion and aminoglycoside resistance in strains heterozygous for rRNA. Antimicrob. Agent. Chemother 1999, 43, 447–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Pfister P; Hobbie S; Vicens Q; Böttger EC; Westhof E The molecular basis for A-Site mutations conferring aminoglycoside resistance: relationship between ribosomal susceptibility and X-ray crystal structures. ChemBioChem 2003, 4, 1078–1088. [DOI] [PubMed] [Google Scholar]
- (48).Hobbie SN; Bruell C; Kalapala S; Akshay S; Schmidt S; Pfister P; Böttger EC A genetic model to investigate drug–target interactions at the ribosomal decoding site. Biochimie 2006, 88, 1033–1043. [DOI] [PubMed] [Google Scholar]
- (49).Perez-Fernandez D; Shcherbakov D; Matt T; Leong NC; Kudyba I; Duscha S; Boukari H; Patak R; Dubbaka SR; Lang K; Meyer M; Akbergenov R; Freihofer P; Vaddi S; Thommes P; Ramakrishnan V; Vasella A; Böttger EC 4’-O-Substitutions Determine Aminoglycoside Selectivity at the Drug Target Level. Nature Commun 2014, 5, 3112/doi: 3110.1038/ncomms4112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).François B; Russell RJM; Murray JB; Aboul-ela F; Masquid B; Vicens Q; Westhof E Crystal structures of complexes between aminoglycosides and decoding A site oligonucleotides: role of the number of rings and positive charges in the specific binding leading to miscoding. Nucleic Acids Res 2005, 33, 5677–5690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Vicens Q; Westhof E Molecular Recognition of Aminoglycoside Antibiotics by Ribosomal RNA and Resistance Enzymes: An Analysis of X-Ray Crystal Structures. Biopolymers 2003, 70, 42–57. [DOI] [PubMed] [Google Scholar]
- (52).Morgenthaler M; Schweizer E; Hoffmann-Roder A; Benini F; Martin RE; Jaeschke G; Wagner B; Fischer H; Bendels S; Zimmerli D; Schneider J; Diederich F; Kansy M; Muller K Predicting and tuning physicochemical properties in lead optimization: amine basicities. ChemMedChem 2007, 2, 1100–1115. [DOI] [PubMed] [Google Scholar]
- (53).Böhm H-J; Banner D; Bendels S; Kansy M; Kuhn B; Müller K; Obst-Sander U; Stahl M Fluorine in Medicinal Chemistry. ChemBioChem 2004, 5, 637–643. [DOI] [PubMed] [Google Scholar]
- (54).Müller K Simple Vector Considerations to Assess the Polarity of Partially Fluorinated Alkyl and Alkoxy Groups. Chimia 2014, 68, 356–363. [DOI] [PubMed] [Google Scholar]
- (55).Nessar R; Cambau E; Reyrat JM; Murray A; Gicquel B Mycobacterium abscessus: A New Antibiotic Nightmare. J. Antimicrob. Chemothr 2012, 67, 810–818. [DOI] [PubMed] [Google Scholar]
- (56).Hanessian S; Giguere A; Grzyb J; Maianti JP; Saavedra; Aggen JB; Linsell MS; Goldblum AA; Hildebrandt DJ; Kane TR; Dozzo P; Gliedt MJ; Matias RD; Feeney LA; Armstrong ES Toward Overcoming Staphylococcus aureus Aminoglycoside Resistance Mechanisms with a Functionally Designed Neomycin Analogue. ACS Med. Chem. Lett 2011, 2, 924–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.