

SUMMARY
The integration of amidyl radicals with cross-coupling chemistry opens new venues for reaction design. However, the lack of efficient methods for the generation of such radical species has prevented many such transformations from being brought to fruition. Herein, the amidoarylation of unactivated olefins by a cascade process from non-functionalized amides is reported by merging, for the first time, photoredox proton-coupled electron transfer (PCET) with nickel catalysis. This new technology grants access to an array of complex molecules containing a privileged pyrrolidinone core from alkenyl amides and aryl- and heteroaryl bromides in the presence of a visible light photocatalyst and a nickel catalyst. Notably, the reaction is not restricted to amides - carbamates and ureas can also be used. Mechanistic studies, including hydrogen-bond affinity constants, cyclization rate measurements, quenching studies, and cyclic voltammetry were central to comprehend the subtleties contributing to the integration of the two catalytic cycles.
Keywords: Nickel catalysis, photoredox, PCET, dual-catalysis, amidoarylation, olefin difunctionalization, cross-coupling
eTOC
A rapid, highly diastereoselective amidoarylation of unactivated olefins was achieved to render medicinally privileged pyrrolidinone structures. Taking advantage of a photoredox proton-coupled electron transfer process, the amidyl radical was obtained from non-prefunctionalized N-H bond under mild conditions, and it was subsequently trapped by pendant olefins delivering alkyl radical for nickel-catalyzed cross-coupling. The mechanistic studies revealed the key balance between thermodynamic-driven radical transformation and kinetic-driven cyclization, which led to expanding the scope towards urea and carbamate substrates.
INTRODUCTION
The recent disclosure of nickel/photoredox dual-catalysis has made cross-coupling chemistry an even more important tool for synthetic chemists.1–7 Under mild conditions with excellent functional group tolerance, these powerful methods have facilitated the construction of numerous challenging Csp2-Csp31–7 and Csp3-Csp38 bonds - transformations that are crucial for success in modern drug discovery.9 Although powerful and enabling, most of the radical precursors contain a traceless redox handle previously introduced via multi-step synthesis1,2,6,7 that is homolytically cleaved, generating the desired radical species. Ideally, if radicals were generated from native functional groups benefitting from innate reactivity, excellent atom- and step-efficiencies could be achieved on top of the many other existing advantages associated with Ni/photoredox dual catalysis (i.e., mild reaction conditions, good chemoselectivity, reduced risks of side reactions, etc.).
In this vein, significant achievements have been reached by merging hydrogen atom transfer (HAT) of activated C-H bonds with Ni-catalyzed cross-coupling reactions.4,5 However, there has been a lack of reports in which heteroatom-centered radicals, derived from the homolytic cleavage of heteroatom-hydrogen bonds, are used as initial intermediates in a sequence of transformations, leading to subsequent Csp2-Csp3 bond construction.10 In fact, if the generation of such radicals could be coupled with a cyclization onto naturally-abundant olefins, one could not only maintain excellent atom-economy while accessing privileged heterocyclic scaffolds, but also transform relatively unreactive olefins to reactive, nucleophilic C-centered radicals, thereby providing new opportunities for rapidly increasing molecular complexity via novel radical cyclization/cross-coupling paradigms.
Owing to the prevalence of nitrogen-containing heterocycles in FDA-approved small- molecule drugs,11 an investigation was undertaken to explore the applicability of amidyl radicals, generated via homolytic N-H cleavage, to synthesize nitrogen-containing heterocycles via radical cyclization/cross-coupling paradigm. The generation of amidyl radicals from N-H bonds has previously posed significant challenges because of their high stability (BDFE ~100 kcal/mol).12–14,16–19 As a result, amidyl radical formation was only achieved using strong oxidants (e.g., DMP,16 IBX,17 di-tert-butyl peroxide18) at high temperatures (Scheme 1),16–19 seriously compromising the applicability of the developed methods. Alternatively, the use of prefunctionalized amides20–22 [e.g., LG = Cl,23 SPh,24 PTOC (pyridine-2-thione-N-oxycarbonyl),25 OAr,26 or SO2Ar27] made amidyl radicals more accessible under various conditions (i.e., UV light, radical initiators, reductants). However, this approach posed numerous disadvantages, such as the requirement for multistep syntheses of the amide derivatives, poor stability of the functionalized amides, and the use of hazardous reagents, among others.
Scheme 1.
Generation of Amidyl Radicals from Functionalized and Non-Functionalized Amides
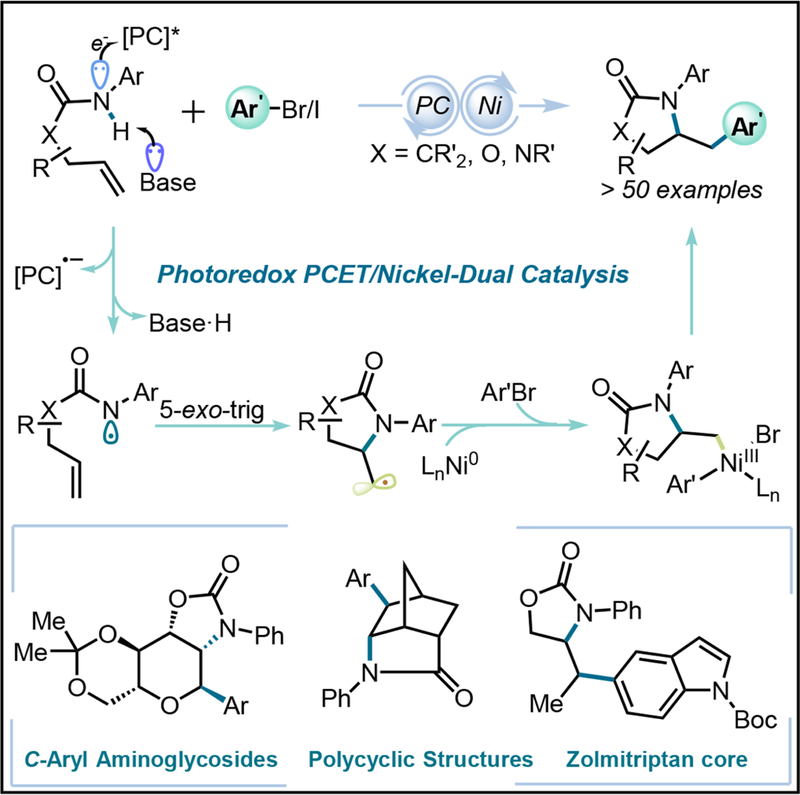
In a seminal publication, the Knowles group reported the generation of amidyl radicals by merging concerted Proton-Coupled Electron Transfer (PCET) with photoredox catalysis (Scheme 1).14,28–30 After adoption of the biologically ubiquitous PCET mechanism,15,31 this challenging bond disconnection was realized under very mild conditions by emphasizing a much lower-barrier concerted pathway, where charged intermediates are avoided.16–18 Mechanistic studies showed that upon hydrogen-bonding with a phosphate base, the amidyl radical was generated via SET oxidation by the excited photocatalyst. Subsequent addition of the amidyl radical across the pendant alkene resulted in a C-centered radical, which was quenched via hydrogen atom abstraction or Giese-type addition to a Michael acceptor.14,28,46
Being aware of the potential of alkene difunctionalization strategies for the rapid construction of molecular complexity,16,32–35 a means was sought to use PCET to expand the repertoire of current organic synthetic transformations in this realm. Indeed, this scenario to some extent mimics some stages of the dual-catalysis that we have been conducting with different radical precursors, where alkyl radicals generated via reductive quenching were merged with a nickel-catalyzed cross-coupling cycle with different aryl electrophiles.1,2 Based on our previous experience, a process was envisioned that would benefit from the rapid 5-exo-trig cyclization of amidyl radicals (k~105 s−1)36 generated via PCET to funnel the resulting alkyl radical into a nickel-catalyzed cross-coupling cycle, resulting in the introduction of biologically relevant pyrrolidinone cores11 onto aromatic backbones.
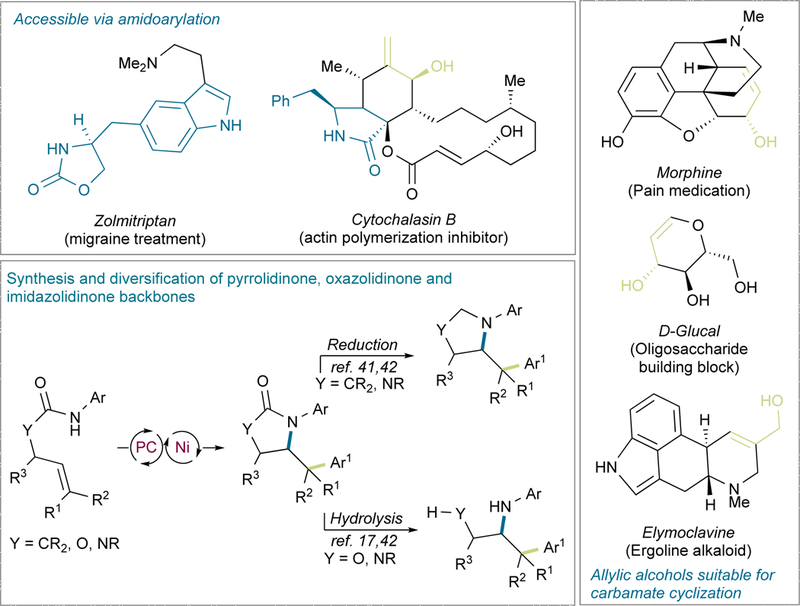
This novel photoredox PCET/Ni dual catalytic strategy would be expected to expedite the synthesis of medicinally privileged N-containing heterocyclic scaffolds. Importantly, there are several notable examples of bioactive molecules with this structure, such as Zolmitriptan and Cytochalasin B (Figure 1). It is also worth noting that, whereas other aminoarylation strategies rely on harsh reaction conditions37,38 or very reactive arylating reagents,39,40 this new strategy would benefit from the mild reaction conditions associated with photoredox catalysis and PCET mechanisms, providing ample opportunities for the high chemoselectivity profile required for the late-stage construction of densely functionalized molecules. Moreover, the proposed transformation would not necessarily be restricted to amides, so that carbamates and ureas could also be employed, expanding the array of heterocyclic cores within range. Carbamates are particularly interesting, considering the tremendous number of bioactive molecules containing allylic alcohols (Figure 1) that might be engaged in this transformation through conversion to the carbamate, as well as the ease of carbamate motif installation via quantitative alcoholysis of aryl isocyanates. Notably, thanks to the ease of oxazolidinone hydrolysis, ring-opened derivatives would be readily accessible,17,42 resulting in a formal three-component difunctionalization of unactivated olefins (Figure 1). Together with other derivatization possibilities such as reduction,41,42 a series of cyclic or acyclic aminoarylation backbones could be constructed.
Figure 1.
Biological Relevance, Diversification of “Lidinone” Derivatives, and Naturally Occurring Allylic Alcohols
RESULTS AND DISCUSSION
Reaction Discovery and Optimization
Prior to the search for suitable reaction conditions, an analysis was undertaken on the possible challenges in developing this demanding, yet appealing, transformation. Although 5-exo-trig cyclization was expected to be fast, a low thermodynamic driving force (ΔG° ≈ −3 to −5 kcal/mol in the transformation of amidyl- to 1º or 2º alkyl radicals) was anticipated to be non-optimal for the reaction progress.12,43 Indeed, in a previous hydroamination study, a thiophenol HAT catalyst was required to drive the transformation to completion.28 Moreover, the coexistence of amidyl- and alkyl radical species in the reaction medium could lead to selectivity issues, depending on the affinity of the nickel complexes for these species and the energy barrier of the subsequent step. This provided the potential for the generation of mixtures of N-arylated and olefin amidoarylated products.18 With these challenges in mind, an investigation was begun using pent-4-enylanilide S1 and 4- bromobenzonitrile as reaction partners. Initial screening using a mildly oxidizing Ir- photocatalyst [Ir-1] (E1/2* = 1.32 V vs. SCE) and the weak base Bu4N[OP(O)(OBu)2] (pKa = 1.72)44 led to a 29% yield of the expected product 1 (Figure 2, entry 1). Ir-based photocatalysts with higher oxidizing power (entries 2–3) gave lower yields or no product at all, likely owing to a poorer rate match between the two catalytic cycles or relative instability of the photocatalyst. Stronger bases favoring the formation of the corresponding amidate, such as KOt-Bu (entry 4), proved unsuccessful in this case. However, higher amounts of the phosphate base translated into higher yields (entry 6). The necessity for superstoichiometric amounts of Bu4N[OP(O)(OBu)2] was further validated after seeing failures at attempting to use it catalytically in combination with inorganic bases to regenerate the former over the course of reaction (entry 5). Supporting the initial mechanistic hypothesis, these conditions, along with the physical properties of the anilide [pKa of ~2113 and redox potential of 1.78 V (vs SCE)], suggest that the cleavage of the anilide N-H bond is most likely occurring via a concerted PCET pathway. Both Ni(II) and Ni(0) sources rendered similar yields (entries 6 and 9). Consequently, well-defined Ni(dMeObpy)(H2O)2Br2 was used as the catalyst precursor based on its stability, ease of preparation, and more convenient reaction set up. Among the several bipyridyl-type ligands tested, dMeObpy clearly stood out (entries 6–8), likely owing to easier oxidative addition with aryl halides because of its more electron-rich nature.
Figure 2. Optimization of Reaction Conditions.
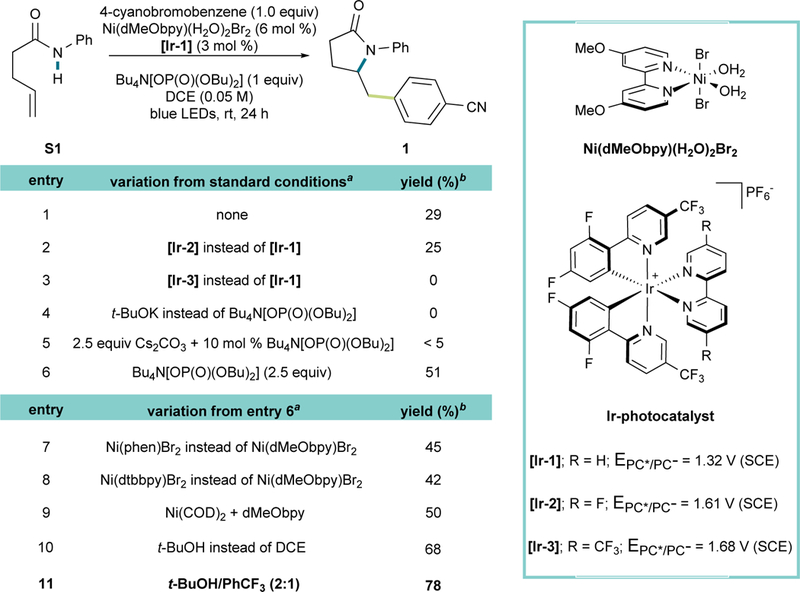
aReactions were performed on a 0.3 mmol scale. bYield of isolated product after column chromatography.
Attempting to increase the reaction yield, we investigated the byproducts generated over the course of the reaction. Among the few identified, chlorobenzonitrile was, by far, predominant. We reasoned that the 1,2-dichloroethane (DCE) solvent was responsible for the formation of this byproduct, being the only chloride source present in the reaction medium.45 After confirming that aryl chlorides are inert under reaction conditions, we examined other non-chlorinated solvents to avoid the consumption of the aryl bromide and its deleterious effect on yield. Upon testing numerous options, t-BuOH proved to be an excellent replacement, affording considerably enhanced yields (entry 9). We suggest that this improvement is likely due to an acidity enhancement from the H-bond acceptor property of t-BuOH. Finally, in an attempt to match DCE’s polarity, different binary solvent systems with t-BuOH were tested. In the event, a 78% yield was obtained using a 2:1 t- BuOH/PhCF3 mixture, which came as no surprise considering the beneficial effects of α,α,α -trifluorotoluene in radical reactions.46 As expected, control experiments showed the key role played by all of the reaction components.47
Substrate Scope Studies
Subsequently, the limits of this reaction were explored via substrate scope study. As shown in Figure 3, substitutions from the α- to the γ- positions on the amide are all accommodated, allowing the synthesis of a tetra-substituted carbon center (7). Notably, no significant Thorpe-Ingold effect was observed when comparing differently α-substituted substrates (1–3). A series of bicyclic (8, 10, 11), tricyclic (9), and spirocyclic structures (4) were accessible from simple precursors. Remarkably, when an endocyclic double bond was involved, the reaction in all cases showed diastereoselectivities greater than 20:1 in favor of the trans product (8–11). Protic functional groups such as hydroxy (10) and carbamate (5) did not pose a problem, nor did activated α-protons. Excellent regioselectivity was achieved, with no 6-endo-trig cyclization observed in any of the substrates examined. A notable yield drop was observed using an α,α-difluorosubstituted amide (12), most likely because of the higher oxidation potential of this more electron-deficient amide (2.03 V vs 1.78 V for non-substituted pentenamide S1). In line with this observation, when a more electron-deficient anilide was involved, a significantly lower yield was observed (16, 17). The more oxidizing photocatalyst [Ir-2] had to be applied to form 16, because [Ir-1] failed to give any product. We reasoned that the former favored the amidyl radical formation, whereas the latter was not oxidizing enough to accomplish the formation of that intermediate. Electron-neutral and electron-rich anilines or heteroaryl amines (13-15, 18) delivered the corresponding products in moderate to good yields. Considering that N-aryl amides are required to facilitate the PCET step, PMP was incorporated as the aryl motif, which allowed the synthesis of unsubstituted pyrrolidinones upon removal of the PMP group with CAN (19).48 Moreover, a gram-scale reaction was carried out without further optimization on the PMP-protected substrate (14) with no erosion in the yield, thus establishing the scalability of this reaction.
Figure 3. Scope of N-Arylated Alkenyl Amides.
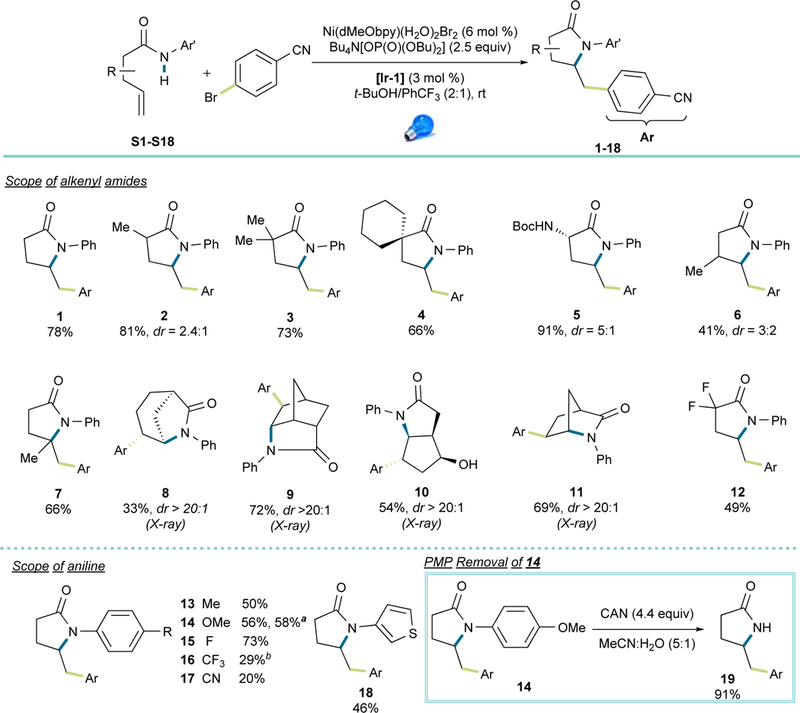
Reaction conditions: amide S1-S18 (0.36 mmol, 1.2 equiv), 4-bromobenzonitrile (0.3 mmol, 1.0 equiv), Bu4N[OP(O)(OBu)2] (0.75 mmol, 2.5 equiv), Ni(dMeObpy)(H2O)2Br2 (0.018 mmol, 6 mol %), [Ir-1] (0.009 mmol, 3 mol %), blue LED, 6 mL t-BuOH/PhCF3 (2:1). a Reaction run on 5 mmol scale. b [Ir-2] was used.
Excellent functional group tolerance was observed when different aryl bromides were subjected to the reaction conditions, including electron-deficient and electron-neutral substrates (20, 22-35, 37), featuring functional units such as aldehydes, boronates, sulfones, amides, esters, trifluoromethoxy groups, and various halides. Electron-rich aryl bromides were challenging, probably because of the more demanding oxidative addition step, yet good yields could be achieved using the corresponding aryl iodides (21, 36 and 49). Similarly, because of the more challenging oxidative addition, relatively electron-neutral (hetero)aryl bromides (e.g., 23, 32 and 39) and ortho-substituted aryl bromides (33) exhibited diminished efficiency, with the remaining anilide starting material and (hetero)aryl bromides recovered after purification. Notably, aryl iodides only seemed to work on electron-rich systems, whereas electron-neutral or electron-poor substrates rendered compromised yields. Substructures with reduceable functional groups or activated hydrogens such as benzylic alcohols (40), saccharides (36) and aldehydes (29, 35) remained intact under the reaction conditions, demonstrating the highly chemoselective nature of this sequential transformation. In an effort to focus on more medicinally relevant scaffolds, several electron-deficient (37-39) or electron-rich (49) heteroaryl bromides were tested, resulting in modest to high yields of the desired products.
To broaden the utility of this transformation, β,γ-unsaturated aryl carbamates and ureas were attempted to provide an easy route toward oxazolidinone and imidazolidinone scaffolds. Unfortunately, initial attempts with an O-allyl aryl carbamate produced a mixture of N-arylation (41a) and amidoarylation (41) products (37% and 22% yield, respectively).47 To improve the reaction performance, an all-substituted carbon was placed at the β- position (42) to favor the cyclization rate by invoking the Thorpe-Ingold effect. In line with previous observations (1–3), the yield enhancement was poor. However, N-arylated product was suppressed in this case. Indeed, although a highly reversible amidyl radical addition to nickel has been described,18 such transformations for carbamate-N radicals appear to be unknown.
As a more electron-rich motif compared to amides, nitrogen-based radicals derived from carbamates would be expected to exhibit a higher nucleophilicity, therefore favoring bonding with nickel. To minimize this putative reactivity, a design motif was sought to enhance the rate of 5-exo-trig cyclization. Considering that the carbamate’s N-H BDE (~95 kcal/mol)13 is lower than that of amides (~100 kcal/mol),12,14 it becomes evident that, from a thermodynamic standpoint, generation of a primary alkyl radical via cyclization (BDE of primary C-H ~98 kcal/mol)43 is less favored for carbamates than for amides (ΔG° ≈ 3 kcal/mol vs −2 kcal/mol). Owing to the BDE difference between primary and secondary C-H bonds (~4 kcal/mol lower for secondary C-H),43 we anticipated that internal alkenes would react more smoothly. To test this hypothesis, an indirect kinetic study was carried out using N-(phenylthiol)amides as the amidyl radical precursors to determine the relative rate of radical cyclization (Scheme 2, Figure S1).24,36 As shown, the cyclization rate for O-crotyl phenylcarbamate (S55) was ~25 times faster than O-allyl phenylcarbamate (S56), thus supporting the notion that generation of a secondary alkyl radical leads to a more efficient cyclization step. Indeed, for both ureas and carbamates, satisfactory yields were obtained when internal alkenes were used (42-52, Figure 4). Along this line, amino aryl glycoside 50 derived from galactal was obtained in an 83% yield and excellent diastereoselectivity, suggesting a more efficient cyclization step that can most likely be attributed to stabilization of the generated α-oxy radical. The success of incorporating N-Boc-indole (49) provides an excellent starting point for the synthesis of Zolmitriptan analogs in a straightforward manner, thus highlighting a direct application of this method. Interestingly, for the urea 52, one of the double bonds remained unreacted without any observable intermolecular transformations.
Scheme 2. Relative Cyclization Rate Studies and Radical Clock Experiment.
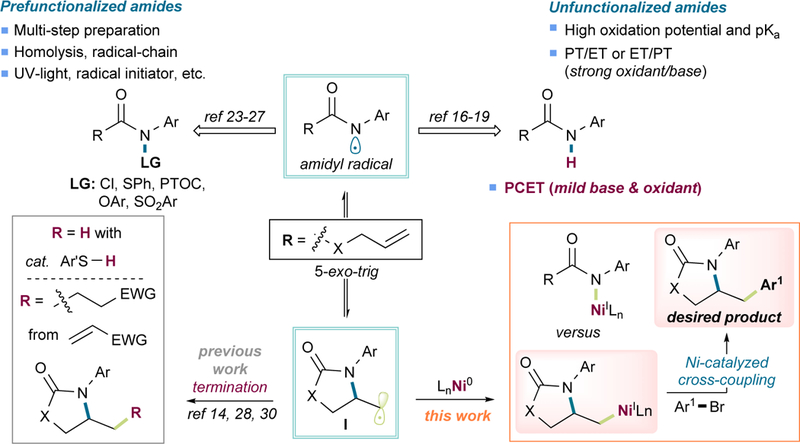
aThe values reported are relative to kT. See Figures S1 for further details.
Figure 4. Scope of (Hetero) Aryl Halides, Carbamates, and Ureas.
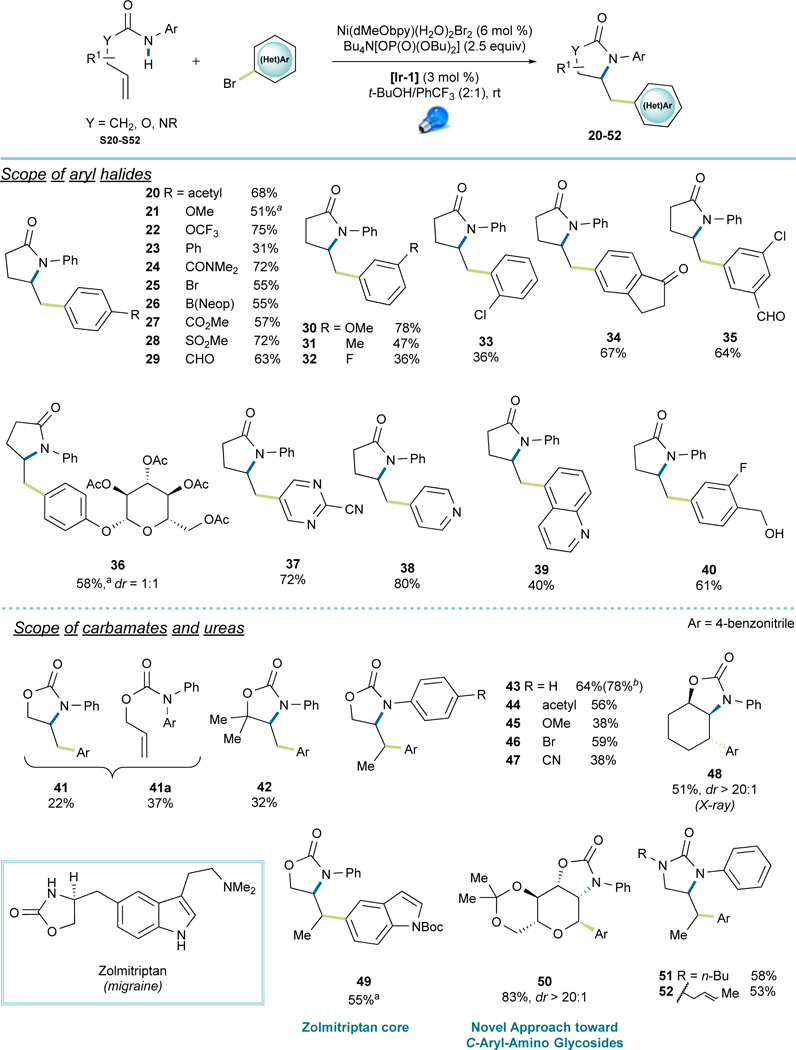
Reaction conditions: amide, urea or carbamate S20-S52 (0.36 mmol, 1.2 equiv), (hetero)aryl halide (0.3 mmol, 1.0 equiv), Bu4N[OP(O)(OBu)2] (0.75 mmol, 2.5 equiv), Ni(dMeObpy)(H2O)2Br2 (0.018 mmol, 6 mol %), [Ir-1] (0.009 mmol, 3 mol %), blue LED, 6 mL t-BuOH/PhCF3 (2:1). a(Hetero)aryl iodide was applied. b7.5 equiv of base were used.
Mechanistic Insights
Although preliminary results pointed toward a mechanistic pathway in line with our initial proposal (Figure 2), further experiments were conducted to substantiate the mechanism. As anticipated, control experiments excluding photocatalyst, nickel precatalyst, base, and light, gave no product (See Table S1).47 A radical clock experiment (Scheme 2, see also SI 4.2) led exclusively to rearranged product 53, indicating the unlikelihood of undergoing an energy transfer-mediated migratory insertion mechanism or alkene aminometalation.34 Still, the crucial role for 2.5 equiv of Bu4N[OP(O)(OBu)2] base remained unclear. Cyclic voltammetry measurements showed that after addition of 2.5 equiv of base, the redox potential of the amide dropped from 1.78 V to 1.27 V (vs SCE), which is within the [Ir-1] photocatalyst oxidative range, thus favoring a PCET mechanistic scenario. However, this potential was also achieved with only 1 equiv of base.47 Stern-Volmer studies were carried out (see SI 4.6) with a fixed ratio of 1:2.5 (substrate/base) to mimic the reaction medium. A linear relationship was observed. Although possessing a narrower linear range, the quenching efficiency of the carbamate-base mixture (KSV = 19000 ± 2000 M−1) was higher than that of amide-base (KSV = 8200 ± 700 M−1), consistent with the relative ease of oxidation of the carbamate compared to that of an amide (1.73 V vs SCE for allyl phenylcarbamate S41, 1.18V after addition of 2.5 equiv of base, see Figures S2-S4). A linear correlation was found between variable base loadings and quenching as well (KSV = 5700 ± 500 M−1 and 4800 ± 300 M−1 for amides and carbamates, respectively. See Figures S5-S11).47 The differences in quenching efficiency in these two experiment sets (with amide quenching being higher in the latter case) indicated that the base might have different affinity for amides versus carbamates. Hydrogen-bonding affinity constants were obtained via NMR titration (Figures 5, S12-S15), with an amide affinity almost 45-times higher than that of the carbamate (33 ± 4 M−1 for the amide and 0.74 ± 0.05 M−1 for the carbamate).49 Interestingly, NMR titration showed a behavior close to saturation for the amide at loadings higher than 2.5 equiv, whereas for the carbamate, even upon addition of 5 equiv of base, no signs of saturation were observed. Expecting that near-saturation conditions would be ideal for a PCET mechanistic scenario, a reaction of carbamate S43 with 7.5 equiv of Bu4N[OP(O)(OBu)2] was carried out,47 achieving a 14% yield increase within the same timeframe (Figure 4, 43). It is also noteworthy that, after adding t-BuOH to mimic the solvent system used in the reaction, a significant N-H chemical shift change from 7.19 to 9.14 ppm was observed, which suggested a beneficial N-H acidity enhancement in the presence of t-BuOH (see Figure S16).47 Under such reaction conditions, the substrate-base adduct would predominate and facilitate PCET, potentially via a proton wire-type mechanism.50 Finally, kinetic isotope effects were measured with an N-deuterated amide and carbamate. Although both the amide and the carbamate revealed a small secondary KIE, the value for carbamate (1.08 ± 0.04) was still more significant than that for amide (1.02 ± 0.02) (see Scheme S1, Tables S2 and S3). Although small, KIEs are consistent with PCET processes,14 and these data could indicate different rate-limiting steps between carbamates and amides. Further studies are required for a full understanding of such behavior.
Figure 5. Measuring the Hydrogen-Bonding Affinity of Amides and Carbamates.
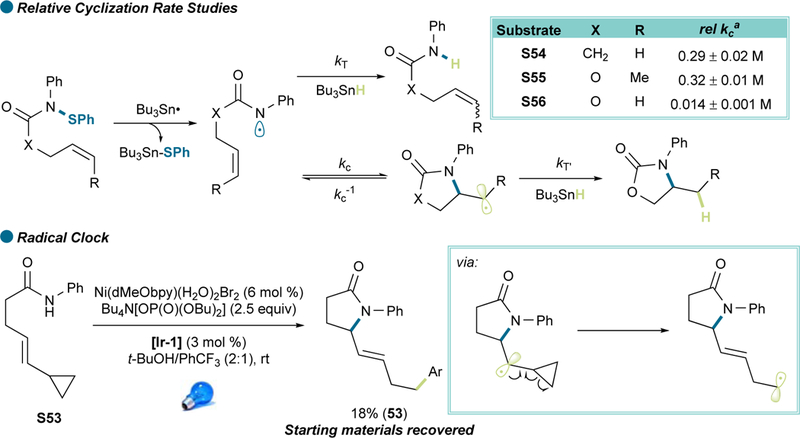
See also Figures S12-S15 for further details.
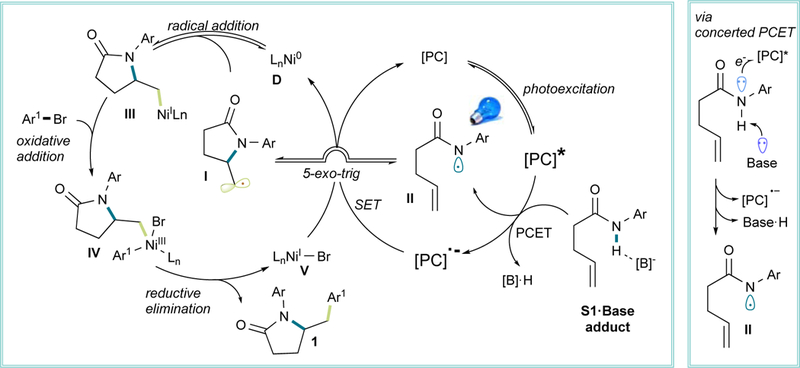
Based on the previous empirical evidence gathered and precedented literature,14,17,19,46,51 we propose a mechanistic scenario (Scheme 3) initiated by formation of an amidyl radical (II) via PCET as suggested by cyclic voltammetry, NMR experiments, and Stern-Volmer studies. Next, a fast 5-exo-trig cyclization follows, the rate of which is related to the nature of the newly-formed alkyl radical and N-H BDE, as evidenced by indirect kinetic studies. Once the alkyl radical I is formed, it enters the nickel-catalytic cycle, forming Ni(I)-complex III, which undergoes oxidative addition with the aryl halide.52 Next, the resultant high- valent Ni(III) intermediate IV undergoes reductive elimination, delivering the final product 1 along with a Ni(I)-halide complex (V). Both catalytic cycles are simultaneously closed by reduction of the Ni(I)-halide with the reduced form of the photocatalyst. Even though the key organonickel intermediates in the proposed mechanism proved challenging to isolate because of their highly reactive nature, their existence is in line with preliminary mechanistic investigations as well as previous computational studies on nickel/photoredox dual-catalyzed cross-coupling.52
Scheme 3. Proposed Catalytic Cycle and PCET Mechanism.
Data S1. X-Ray Crystal Structure of Nickel Precatalyst Ni(dMeObpy)(CH3CN)2Br2
Data S2. X-Ray Crystal Structure of Compound 8
Data S3. X-Ray Crystal Structure of Compound 9
Data S4. X-Ray Crystal Structure of Compound 10
Data S5. X-Ray Crystal Structure of Compound 11
Data S6. X-Ray Crystal Structure of Compound 48
Conclusion
A photoredox PCET/Ni dual-catalyzed amidoarylation of unactivated olefins has been disclosed, forging highly functionalized 5-membered heterocyclic systems under mild reaction conditions from readily available precursors. This method merges the concept of concerted PCET with a nickel-catalyzed cross-coupling process for the first time, taking advantage of photocatalytic activation of strong N-H bonds assisted by a phosphate H- bond acceptor, followed by rapid 5-exo-trig cyclization and alkyl-aryl single-electron cross- coupling. Relative cyclization rates for amides and carbamates were studied, the latter being disclosed for the first time, which allowed the introduction of carbamates and ureas into the protocol. Stern-Volmer studies, NMR titration experiments, cyclic voltammetry, and control experiments point toward a concerted PCET mechanism. This transformation adds to the repertoire of alkene difunctionalization processes in a manner that is exceedingly functional group tolerant, using readily available alkenyl amides, carbamates and ureas, and takes advantage of the tens of thousands of commercially available (hetero)aryl bromides and iodides to expand chemical space.
EXPERIMENTAL PROCEDURES
A general procedure for photoredox PCET/Ni-catalyzed amidoarylation of unactivated olefins: An 8.0 mL screw-cap vial containing a stirring bar was charged with Ni(dMeObpy)(H2O)2(Br)2 (8.5 mg, 0.018 mmol, 6 mol %), [Ir{dF(CF3)2ppy}2(bpy)]PF6 (9.0 mg, 0.009 mmol, 3 mol %), amide (0.36 mmol, 1.2 equiv), aryl bromide (if solid, 0.3 mmol, 1.0 equiv) and Bu4N[(n-BuO)2P(O)O] (338.8 mg, 0.75 mmol, 2.5 equiv). Next, the vial was closed, and three vacuum/argon cycles were carried out. Under an inert atmosphere, a 2:1 mixture of pre-degassed, anhydrous t-BuOH/PhCF3 was added (6.0 mL, 0.05 M) followed by addition of the aryl bromide (if liquid). After covering the cap with Parafilm®, the reaction was placed 3 cm from the light source in a blue LED bay (5 W, blue LED strip, ~5 W in total) and stirred at rt until complete (a fan was added ~30 cm from above to disperse any heat coming from the blue LEDs). When completed, the reactions were taken to dryness and purified by column chromatography using an automated system (hexane/EtOAc gradient), delivering the corresponding pure product.
The Bigger Picture
Rapid generation of molecular complexity and access to novel 3D chemical space is pivotal for successful and efficient drug discovery. Nickel/photoredox dual-catalysis has arisen as an appealing strategy towards such a goal by rapidly introducing Csp3 centers under mild reaction conditions. By taking advantage of a native amide group, we achieved an amidoarylation reaction of unactivated olefins, rendering a series of medicinally-privileged structures in a highly atom-economical way. The reaction takes advantage of a photoredox proton-coupled electron transfer event to cleave the strong amidyl N-H bond homolytically. Subsequent regiospecific 5- exo-trig cyclization generates an alkyl radical. High functional group tolerance has been achieved with excellent diastereoselectivities owing to reaction’s mild nature. Mechanistic studies showed the intricate relationship between the base stoichiometry and the N-H donor, as well as the key balance between kinetic and thermodynamic factors.
Supplementary Material
Highlights.
Generation of C-centered radicals via cleavage of strong X-H bonds for cross-coupling
Rapid formation of new N-Csp3 and Csp2-Csp3 bonds from olefins under mild condition
Highly diastereoselective synthesis of medicinally privileged pyrrolidinone structures
Mechanism-driven scope expansion towards carbamate and urea motifs
ACKNOWLEDGMENTS
The authors are grateful for the financial support provided by NIGMS (No. R01 GM 113878). We thank the NIH (No. S10 OD011980) for supporting the University of Pennsylvania (UPenn) Merck Center for High Throughput Experimentation, which funded the equipment used in screening efforts. Kai Chen (Caltech) is acknowledged for helpful discussion of the manuscript. The authors thank Dr. Jun Gu (UPenn) for assistance with the NMR studies, and Drs. P.J. Carrol and M.R. Gau (UPenn) for crystallographic analysis of compound 8–11, 48 and the Ni-precatalyst.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DATA AND SOFTWARE AVAILABILITY
Crystallographic data for the structures reported in this paper have been deposited at the Cambridge Crystallographic Data Center (CCDC), with accession numbers of CCDC 1854737 (Ni(dMeObpy)(H2O)(CH3CN)2Br2), 1854738 (8), 1854742 (10), 1854739 (48), 1854740 (11), and 1854741 (9).
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES AND NOTES
- 1.Tellis JC, Primer DN, Molander GA (2014) Single- electron transmetalation in organoboron cross-coupling by photoredox/nickel dual catalysis. Science 345, 433–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zuo Z, Ahneman DT, Chu L, Terrett JA, Doyle AG, MacMillan DWC (2014). Merging photoredox with nickel catalysis: Coupling of α- carboxyl sp3- carbons with aryl halides. Science 345, 437–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Primer DN, Karakaya I, Tellis JC, Molander GA (2015) Single- Electron Transmetalation: An Enabling Technology for Secondary Alkylboron Cross- Coupling. J. Am. Chem. Soc, 137, 2195–2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shields BJ, Doyle AG (2016). Direct C(sp3)–H Cross Coupling Enabled by Catalytic Generation of Chlorine Radicals. J. Am. Chem. Soc 138, 12719–12722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Heitz DR, Tellis JC, Molander GA (2016). Photochemical Nickel-Catalyzed C–H Arylation: Synthetic Scope and Mechanistic Investigations J. Am. Chem. Soc 138, 12715–12718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tellis JC, Kelly CB, Primer DN, Jouffroy M, Patel NR, Molander GA (2016). Single- Electron Transmetalation via Photoredox/Nickel Dual Catalysis: Unlocking a New Paradigm for sp3– sp2 Cross-Coupling. Acc. Chem. Res 49, 1429–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Matsui JK, Lang SB, Heitz DR, Molander GA (2017). Photoredox- Mediated Routes to Radicals: The Value of Catalytic Radical Generation in Synthetic Methods Development. ACS. Catal 7, 2563–2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Johnston CP, Smith RT, Allmendinger S, MacMillan DWC (2016) Metallaphotoredo x-catalysed sp3– sp3 cross-coupling of carboxylic acids with alkyl halides. Nature 536, 322–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lovering F, Bikker J, Humblet C (2009). Escape from Flatland: Increasing Saturation as an Approach to Improving Clinical Success. J. Med. Chem 52, 6752–6756. [DOI] [PubMed] [Google Scholar]
- 10.However, there has been one exception recently, which required prefunctionalized imines and was merely ristricted to dicyanophenyl derivatives: Nakafuku KM, Fosu SC, Nagib DA (2018). Catalytic Alkene Difunctionalization via Imidate Radicals. J. Am. Chem. Soc 10.1021/jacs.8b07578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vitaku E, Smith DT, Njardarson JT (2014). Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem 57, 10257–10274. [DOI] [PubMed] [Google Scholar]
- 12.The BDE of anlilide N-H is ~99kcal/mol. See: Bordwell FG Harrelson JA, Lynch TY (1990). Homolytic bond dissociation energies for the cleavage of .alpha.- nitrogen-hydrogen bonds in carboxamides, sulfonamides, and their derivatives. The question of synergism in nitrogen-centered radicals. J. Org. Chem 55, 3337–3341. [Google Scholar]
- 13.Zhang X-M, Bordwell FG. (1994). Acidities and Homolytic Bond Dissociation Enthalpies (BDEs) of the Acidic H-A Bonds in Acyclic and Cyclic Alkoxycarbonyl Compounds (Esters and Carbamates). J. Org. Chem 59, 6456–6458. [Google Scholar]
- 14.Choi GJ, Knowles RR (2015) Catalytic Alkene Carboaminations Enabled by Oxidative Proton- Coupled Electron Transfer. J. Am. Chem. Soc 137, 9226–9229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Warren JJ, Tronic TA, Mayer JM (2010). Thermochemistry of Proton-Coupled Electron Transfer Reagents and its Implications. Chem. Rev 110, 6961–7001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nicolaou KC, Baran PS, Zhong YL, Sugita K (2002). Iodine(V) Reagents in Organic Synthesis. Part 1. Synthesis of Polycyclic Heterocycles via Dess−Martin Periodinane- Mediated Cascade Cyclization: Generality, Scope, and Mechanism of the Reaction. J. Am. Chem. Soc 124, 2212–2220. [DOI] [PubMed] [Google Scholar]
- 17.Nicolaou KC, Baran PS, Zhong YL, Barluenga S, Hunt KW, Kranich R, et al. (2002). Iodine(V) Reagents in Organic Synthesis. Part 3. New Routes to Heterocyclic Compounds via o- Iodoxybenzoic Acid-Mediated Cyclizations: Generality, Scope, and Mechanism. J. Am. Chem. Soc 124, 2233–2244. [DOI] [PubMed] [Google Scholar]
- 18.Zhou L, Tang S, Qi X, Lin C, Liu K, Liu C, et al. (2014). Transition-Metal- Assisted Radical/Radical Cross-Coupling: A New Strategy to the Oxidative C(sp3)–H/N–H Cross-Coupling. Org. Lett 16, 3404–3407. [DOI] [PubMed] [Google Scholar]
- 19.Nicolaou KC, Baran PS, Kranich R, Zhong Y-L, Sugita K, Zou N (2001). Mechanistic Studies of Periodinane- Mediated Reactions of Anilides and Related Systems. Angew. Chem. Int. Ed 40, 202–206. [DOI] [PubMed] [Google Scholar]
- 20.Chen J-R, Hu XQ, Lu L-Q, Xiao W-J (2016). Visible light photoredox- controlled reactions of N- radicals and radical ions. Chem. Soc. Rev 45, 2044–2056. [DOI] [PubMed] [Google Scholar]
- 21.Zard SZ (2008). Recent progress in the generation and use of nitrogen-centred radicals. Chem. Soc. Rev 37, 1603–1618. [DOI] [PubMed] [Google Scholar]
- 22.Kärkäs MD (2017). Photochemical Generation of Nitrogen-Centered Amidyl, Hydrazonyl, and Imidyl Radicals: Methodology Developments and Catalytic Applications. ACS. Catal 7, 4999–5022. [Google Scholar]
- 23.Wolff ME (1963). Cyclization of NHalogenated Amines (The Hofmann-Löffler Reaction). Chem. Rev 63, 55–64. [Google Scholar]
- 24.Esker JL, Newcomb M (1993). Amidyl radicals from N- (phenylthio)amide s. Tetrahedron Lett 34, 6877–6880. [Google Scholar]
- 25.Esker JL, Newcomb M (1993). Chemistry of amidyl radicals produced from Nhydroxypyridine- 2- thione imidate esters. J. Org. Chem 58, 4933–4940. [Google Scholar]
- 26.Davies J, Svejstrup TD, Fernandez Reina D, Sheikh NS, Leonori D (2016). Visible- Light-Mediated Synthesis of Amidyl Radicals: Transition-Metal- Free Hydroamination and N-Arylation Reactions. J. Am. Chem. Soc 138, 8092–8095. [DOI] [PubMed] [Google Scholar]
- 27.Fuentes N, Kong W, Fernandez-Sanchez L, Merino E, Nevado C (2015). Cyclization Cascades via NAmidyl Radicals toward Highly Functionalized Heterocyclic Scaffolds. J. Am. Chem. Soc 137, 964–973. [DOI] [PubMed] [Google Scholar]
- 28.Miller DC, Choi GJ, Orbe HS, Knowles RR (2015). Catalytic Olefin Hydroamidation Enabled by Proton-Coupled Electron Transfer. J. Am. Chem. Soc 137, 13492–13495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Choi GJ, Zhu Q, Miller DC, Gu CJ, Knowles RR (2016) Catalytic alkylation of remote C–H bonds enabled by proton-coupled electron transfer. Nature 539, 268–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhu Q, Graff DE, Knowles RR (2018). Intermolecular Anti-Markovnikov Hydroamination of Unactivated Alkenes with Sulfonamides Enabled by Proton-Coupled Electron Transfer. J. Am. Chem. Soc 140, 741–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weinberg DR, Gagliardi CJ, Hull JF, Murphy CF, Kent CA, Westlake BC, et al. (2012). Proton- Coupled Electron Transfer. Chem. Rev 112, 4016–4093. [DOI] [PubMed] [Google Scholar]
- 32.Lan X, Wang N, Xing Y (2017). Recent Advances in Radical Difunctionalization of Simple Alkenes. Eur. J. Org. Chem 39, 5821–5851. [Google Scholar]
- 33.Liu Z, Wang Y, Wang Z, Zeng T, Liu P, Engle KM (2017). Catalytic Intermolecular Carboamination of Unactivated Alkenes via Directed Aminopalladation. J. Am. Chem. Soc 32, 11261–11270. [DOI] [PubMed] [Google Scholar]
- 34.Hanley PS, Hartwig JF (2013). Migratory Insertion of Alkenes into Metal–Oxygen and Metal– Nitrogen Bonds. Angew. Chem. Int. Ed 52, 8510–8525. [DOI] [PubMed] [Google Scholar]
- 35.Schultz DM, Wolfe JP (2012) Recent Developments in Pd-Catalyzed Alkene Aminoarylation Reactions for the Synthesis of Nitrogen Heterocycles. Synthesis 44, 351–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martinez E, Newcomb M (2006). Rate Constants for Anilidyl Radical Cyclization Reactions. J. Org. Chem 71, 557–561. [DOI] [PubMed] [Google Scholar]
- 37.Ney JE, Wolfe JP (2004). Palladium‐ Catalyzed Synthesis of N‐Aryl Pyrrolidines from γ‐ (N‐Arylamino) Alkenes: Evidence for Chemoselective Alkene Insertion into Pd-N Bonds. Angew. Chem. Int. Ed 43, 3605–3608. [DOI] [PubMed] [Google Scholar]
- 38.Yip K-T, Yang D (2011). Pd(II)- Catalyzed Intramolecular Amidoarylation of Alkenes with Molecular Oxygen as Sole Oxidant. Org. Lett 13, 2134–2137. [DOI] [PubMed] [Google Scholar]
- 39.Sahoo B, Hopkinson MN, Glorius F (2013). Combining Gold and Photoredox Catalysis: Visible Light-Mediated Oxy- and Aminoarylation of Alkenes. J. Am. Chem. Soc 135, 5505–5508. [DOI] [PubMed] [Google Scholar]
- 40.Fumagalli G, Boyd S, Greaney MF (2013). Oxyarylation and Aminoarylation of Styrenes Using Photoredox Catalysis. Org. Lett 15, 4398–4401. [DOI] [PubMed] [Google Scholar]
- 41.Wu C, Luo X, Zhang H, Liu X, Ji G, Liu Z, et al. (2017). Reductive amination/cyclizat ion of levulinic acid to pyrrolidones versus pyrrolidines by switching the catalyst from AlCl3 to RuCl3 under mild conditions. Green Chem 19, 3525–3529. [Google Scholar]
- 42.Trost BM, Fandrick DR (2003). Dynamic Kinetic Asymmetric Cycloadditions of Isocyanates to Vinylaziridines. J. Am. Chem. Soc 125, 11836–11837. [DOI] [PubMed] [Google Scholar]
- 43.McMillen DF, Golden DM (1982). Hydrocarbon Bond Dissociation Energies. Annu. Rev. Phys. Chem 33, 493–532. [Google Scholar]
- 44.Kumler WD, Eiler JJ (1943). The Acid Strength of Mono and Diesters of Phosphoric Acid. The n-Alkyl Esters from Methyl to Butyl, the Esters of Biological Importance, and the Natural Guanidine Phosphoric Acids. J. Am. Chem. Soc 65, 2355–2361. [Google Scholar]
- 45.McNeece AJ, Mokhtarzadeh CC, Moore CE, Rheingold AL, Figueroa JS (2016). Nickel bismterphenylisocyanid e dihalide complexes formed from 1,2-alkyl dihalides: probing for isolable β- haloalkyl complexes of square planar nickel. J. Coord. Chem 69, 2059–2068. [Google Scholar]
- 46.Chu JCK, Rovis T (2016). Amidedirected photoredoxcatalysed C–C bond formation at unactivated sp3 C–H bonds. Nature 539, 272–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. See Supporting Information for further details.
- 48.Wakchaure VN, Zhou J, Hoffmann S, List B (2010). Catalytic Asymmetric Reductive Amination of α‐ Branched Ketones. Angew. Chem. Int. Ed 49, 4612–4614. [DOI] [PubMed] [Google Scholar]
- 49.Thordarson P (2011). Determining association constants from titration experiments in supramolecular chemistry. Chem. Soc. Rev 40, 1305–1323. [DOI] [PubMed] [Google Scholar]
- 50.An effective way to translocate protons in biological system where a longchain proton donor-acceptor complex is formed. For a reference, see: Cardenas DJ, Cuerva JM, Alias M, Bunuel E, Campana AG (2011). Water- Based Hydrogen- Atom Wires as Mediators in Long- Range Proton- Coupled Electron Transfer in Enzymes: A New Twist on Water Reactivity. Chem. – Eur. J 17, 8318–8323. [DOI] [PubMed] [Google Scholar]
- 51.Musacchio AJ, Nguyen LQ, Beard GH, Knowles RR (2014) Catalytic Olefin Hydroamination with Aminium Radical Cations: A Photoredox Method for Direct C–N Bond Formation. J. Am. Chem. Soc 136, 12217–12220. [DOI] [PubMed] [Google Scholar]
- 52.Gutierrez O, Tellis JC, Primer DN, Molander GA, Kozlowski MC (2015). Nickel- Catalyzed Cross- Coupling of Photoredox- Generated Radicals: Uncovering a General Manifold for Stereoconvergenc e in Nickel- Catalyzed Cross- Couplings. J. Am. Chem. Soc 137, 4896–4899. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.