

Abstract
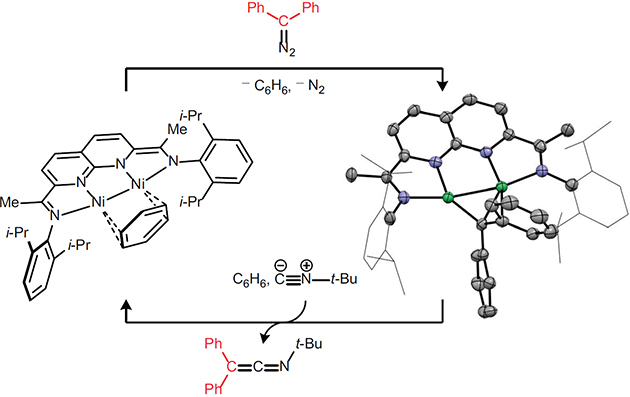
The synthesis and reactivity of a dinickel bridging carbene is described. The previously reported [i-PrNDI]Ni2(C6H6) complex (NDI = naphthyridine–diimine) reacts with Ph2CN2 to generate a metastable diazoalkane adduct, which eliminates N2 at 60 °C to yield a paramagnetic Ni2(μ-CPh2) complex. The Ni2(μ-CPh2) complex undergoes carbene transfer to t-BuNC via an initial isonitrile adduct, which, upon heating, releases free t-BuNCCPh2. Based on this sequence of stoichiometric reactions, a catalytic carbene transfer reaction is demonstrated.
Graphical Abstract
INTRODUCTION
Transition metal carbene complexes are implicated as key intermediates in catalytic cyclopropanation and bond insertion reactions.1 Synthetic studies targeting isolable carbene complexes have largely focused on accessing terminally coordinated M=CR2 species due to their potential to exhibit high reactivity.2 It is well-established, however, that carbene ligands can induce the spontaneous formation of dinuclear M2(μ-CR2) assemblies, particularly when supporting ligands lack sufficient steric protection.3 Because many catalytically-relevant systems may exhibit such monomer–dimer equilibria, it is of interest to examine whether dinuclear bridging carbenes are capable of participating in carbene transfer reactions and whether they might access unique pathways that involve the direct participation of two metals.
Dinuclear M2(μ-CR2) complexes are known to function as carbene transfer reagents, but they generally do so through an initial dissociation to form a more reactive mononuclear M(CR2) species (Figure 1a). For example, Warren reported that a β-diketiminate Cu(I) precursor reacts with Ph2CN2 to generate a Cu2(μ-CR2) dimer, which exists in solution equilibrium with a mononuclear Cu(CR2) species.3a,3b This mixture is capable of effecting the cyclopropanation of alkenes, and mechanistic studies indicated that the carbene transfer occurs through the minority mononuclear species. In a related study, the dimeric [(IPr)NiCl]2 complex (IPr = 1,3-(2,6-(i-Pr)2C6H3)2imidazolin-2-ylidene) was shown by Hillhouse to undergo a reaction with Ph2CN2 in the presence of NaB(ArF)4 to generate a cationic {[(IPr)Ni]2(Cl)(μ-CR2)}+ complex.4 This dinuclear carbene complex reacts with t-BuNC to yield the corresponding keteneimine product. It remains unclear, however, whether the dinuclear species is a kinetically competent carbene transfer reagent or whether a pre-equilibrium dissociation forms a more reactive terminal (IPr)Ni=CR2 species.
Figure 1.

(a) Examples of M2(μ-CR2) complexes. (b) A constrained dinuclear carbene complex using the [NDI]Ni2 platform.
Recently, our group described a redox-active naphthyridine–diimine (NDI) pincer ligand system that enables the synthesis of well-defined dinickel complexes featuring metal–metal single bonds.5 We hypothesized that the constrained environment of the [NDI]Ni2 system might suppress monomer–dimer equilibria in group transfer processes, allowing the reactivity of bridging carbene ligands to be studied in isolation.6 Here, we describe the synthesis of a Ni2(μ-CR2) complex from its R2CN2 precursor and a prototypical carbene transfer reaction to t-BuNC (Figure 1b).
RESULTS AND DISCUSSION
Synthesis of a Dinickel Carbene Complex.
We initiated our studies by pursuing the characterization of a stable carbene complex using the [NDI]Ni2 platform. Addition of Ph2CN2 (1.0 equiv) to a THF solution of [i-PrNDI]Ni2(C6H6) complex 1 (1.0 equiv) yielded a dark brown product (2) featuring paramagnetically shifted 1H NMR signals spanning a range of –74 to 76 ppm. The identity of 2 was established by XRD analysis and features an intact Ph2CN2 ligand bridging the two Ni centers in a μ-η2:η1 fashion (Figure 2b). The N–N bond of the diazo ligand is significantly elongated (1.296(6) Å) relative to the characteristic distance for a free diazoalkane (1.146(2) Å for bis(4-dibromophenyl)diazomethane)7, indicating a high degree of back-bonding from the Ni2 fragment.7
Figure 2.

(a) Thermal conversion of [i-PrNDI]Ni2(N2CPh2) complex 2 to [i-PrNDI]Ni2(CPh2) complex 3. (b) Solid-state structure of 2. Ni1–Ni2: 3.135(1) Å. (c) Solid-state structure of 3. Ni1–Ni2: 2.372(1) Å.
Gaseous N2 is liberated from the Ph2CN2 adduct (2) upon heating in THF at 60 °C for 12 h (Figure 2a). The resulting dark purple carbene complex (3) possesses C2v symmetry on the 1H NMR chemical shift time scale (11 resolved signals ranging from –46 to 54 ppm) and exhibits a solution magnetic moment of 2.82 μB (Evans method, 298 K), consistent with an S = 1 ground state. In the solid state (Figure 2c), the symmetry observed in solution is broken by a weak interaction between one of the Ni centers and an ipso carbon of the carbene Ph substituent (Ni1–C10 = 2.487(6) Å). The two Ni–C distances are non-equivalent due to this interaction (Ni1–C9 = 1.874(7) Å vs. Ni2–C9 = 1.983(6) Å). A similar fluxional arene interaction was observed in Hillhouse’s {[(IPr)Ni]2(Cl)(μ-CR2)}+ complex.4
Given the redox-active nature of the NDI ligand, it was of interest to further examine the electronic structure of μ-CPh2 complex 3. In comparison to the metrical parameters for free i-PrNDI, complex 3 exhibits contracted C(imine)–C(ipso) distances and elongated C(imine)–N and C(ipso)–N distances, which are bond distortions characteristic of ligand-centered reduction.2j,5 By DFT (M06L/6–311G(d,p)), the spin density in 3 is highly delocalized between the NDI π-system and the Ni–Ni bond (Figure 3b). The two singly occupied molecular orbitals correspond to a predominantly ligand-centered SOMO and a SOMO–1 that is Ni2 in character (Figure 3a).
Figure 3.

(a) Calculated SOMO, SOMO–1, and (b) spin-density plot for the S = 1 state of [i-PrNDI]Ni2(CPh2) (3). Fractions of the total spin density for the two Ni atoms and the NDI ligand are shown. The i-Pr groups are truncated to Me groups in the model. M06L/6–311G(d,p) level of DFT.
Redox Chemistry of Dinickel Diphenyl Carbene.
We next carried out a series of experiments to probe the redox behavior of the carbene complex 3. In THF solution (0.4 M [nBu4N]PF6 electrolyte), 3 exhibits two reduction and two oxidation events spanning a potential range of −2.63 to −0.51 V vs. Cp2Fe/Cp2Fe+ (Figure 4a). The first reduction at −1.80 V is accessible by treating 3 with KC8 (1.0 equiv) in the presence of 18-crown-6 (Figure 4b). The resulting anionic complex 4 is isostructural to 3 in the solid state and features an outer-sphere K+ encapsulated by 18-crown-6 and two molecules of THF (Figure 4c). Relative to the metrical parameters for the neutral carbene complex (3), anion 4 displays an NDI ligand that is further reduced, suggesting that the electron being added to the system is located in a predominantly ligand-centered orbital (Table 1). By contrast, the Ni–Ni distance changes by only 0.01 Å upon reduction.
Figure 4.

(a) Cyclic voltammogram for [i-PrNDI]Ni2(CPh2) (3) (0.4 M [nBu4N][PF6] supporting electrolyte in THF, glassy carbon working electrode, 100 mV/s scan rate). The open circuit potential and direction of the scan is indicated by an arrow. (b) Synthesis and (c) solid-state structure of [K(18-Crown-6)(THF)2][(i-PrNDI)Ni2(CPh2) (4). [K(18-Crown-6)(THF)2] is omitted for clarity. Ni1–Ni2: 2.3816(9) Å.
Table 1.
Selected Bond Distances (Å) for 3 and 4.a
| 3b | 4 | |
|---|---|---|
| Ni1–Ni2 | 2.372(1) | 2.382(1) |
| Ni1–C9, Ni2–C9 | 1.877(4), 1.984(5) | 1.902(5), 1.975(5) |
| C1–N1, C8–N4 | 1.319(9), 1.305(9) | 1.342(7), 1.329(7) |
| C1–C2, C7–C8 | 1.434(9), 1.450(6) | 1.415(7), 1.432(7) |
| C2–N2, C7–N3 | 1.374(9), 1.369(8) | 1.411(7), 1.384(6) |
See Figures 2c and 4c for atom labels.
Average of two independent molecules in the asymmetric unit.
The reaction of carbene complex 3 with (Cp2Fe)PF6 (1.0 equiv) in MeCN yields an oxidized product that crystallizes as a homodimeric species (5) in 91% isolated yield (Figure 5a). The solid-state structure for 5 reveals the formation of a new C–C bond through the 4-position of the naphthyridine ring (Figure 5b). The C4–C4’ distance of 1.588(3) Å is modestly elongated relative to the typical distance of a C–C single bond. By comparison, a similar dimer generated from an Fe pyridine–diimine complex, [LMeFe(Py)]2(μ-C10H10N2) (LMe = 2,4-bis(2,6-diisopropylimino)pentyl)), features a C4–C4’ distance of 1.563(6) Å.8,9 The oxidized carbene complex 5 is NMR silent at room temperature and possesses a solution magnetic moment of 2.0 μB (Evans method, 298 K), suggesting that the solid-state dimer dissociates into monomeric S = 1/2 complexes when dissolved in THF. In accordance with this observation, 5 is EPR active and exhibits a sharp signal at g = 2.01 (2-Me-THF, 77 K) (Figure 5c). The minimal anisotropy and narrow line width of the frozen solution EPR spectrum are characteristic features of an organic radical.10
Figure 5.

(a) Synthesis and (b) solid-state structure of 5. Ni – Ni: 2.9843(5) Å. C4–C4’: 1.588(3) Å. (c) Solution EPR spectrum (2-Me-THF, 77 K) of 5.
Carbene Transfer with Isonitrile.
Having isolated a well-defined μ-CPh2 complex (3), we next sought to explore its carbene transfer reactivity. In this context, we recently found that [NDI]Ni2 complexes function as efficient cyclopropanation catalysts using methylene equivalents derived from CH2Cl2/Zn.11 The isolated Ni2(CPh2) complex 3, however, does undergo stoichiometric reactions with alkenes such as styrene, 1-octene, or ethylene. When t-BuNC (strictly 1.0 equiv) is added to a THF solution of 3, an isonitrile adduct (6) is generated (Figure 6a). The μ-CPh2 fragment is retained in this structure with one of the Ni atoms bearing the additional t-BuNC ligand. The dinuclear structure of the complex remains intact, but the Ni–Ni distance elongates to 3.0846(8) Å, which is longer than the sum of the van der Waals radii12 and suggests that there is no significant metal–metal bonding. The bound t-BuNC exhibits a C–N distance of 1.167(7) Å and a C–N stretching frequency (2156 cm–1) that is moderately red-shifted from that of free t-BuNC (2146 cm–1).13 The Ni center not bearing the isonitrile ligand forms a significant η2-arene interaction, and the Ni–Cipso distance shortens from 2.487(6) Å in carbene complex 3 to 2.072(8) Å in the isonitrile adduct 6. When isolated Ni2(μ-CPh2)(CNt-Bu) complex 5 is redissolved in C6H6 and gently heated at 80 °C for 1 h, Ni2(C6H6) complex 1 is regenerated, and free t-BuN=C=CPh2 is formed in >99% yield.
Figure 6.

(a) Stoichiometric Ph2C transfer to t-BuNC and solid-state structure of 6. Ni–Ni: 3.0846(8) Å. (b) Catalytic Ph2C transfer to t-BuNC using 10 mol% 1.
The sequence of diazoalkane binding, N2 extrusion, isonitrile binding, and carbene transfer constitutes a complete set of elementary transformations required to carry out a catalytic group transfer reaction. Accordingly, slow addition of a t-BuNC (1.0 equiv) and Ph2CN2 (1.0 equiv) solution to Ni2(C6H6) complex 1 (10 mol%), heated at 80 °C in C6H6, provided t-BuN=C=CPh2 in 65% yield (Figure 6b). Slow addition of both reaction partners is critical for the catalytic process to be viable. When catalyst 1 is treated with excess t-BuNC, demetallation occurs to generate free i-PrNDI and Ni(CNt-Bu)4. Furthermore, exposure of 1 to excess Ph2CN2 leads to the catalytic consumption of the diazoalkane reagent to form azine dimers.
In conclusion, the naphthyridine–diimine (NDI) ligand framework enables the synthesis of a well-defined dinickel bridging carbene complex. The [i-PrNDI]Ni2(CPh2) complex (3) adopts an unusual paramagnetic ground state due to the presence of low-lying π-orbitals associated with the redox-active NDI ligand. Complex 3 engages in carbene transfer to t-BuNC by a mechanism in which the isonitrile initially coordinates to one Ni center, cleaving the Ni–Ni bond. The Ni–Ni bond is restored upon release of the keteneimine product. Together, these studies highlight a unique mechanism for carbene transfer from dinuclear M2(μ-CR2) species that involves the direct participation of both metals.
EXPERIMENTAL SECTION
General Information.
All manipulations were carried out using standard Schlenk or glovebox techniques under an atmosphere of N2. Solvents were dried and degassed by passing through a column of activated alumina and sparging with Ar gas. Deuterated solvents were purchased from Cambridge Isotope Laboratories, Inc., degassed, and stored over activated 3 Å molecular sieves prior to use. All other reagents and starting materials were purchased from commercial vendors and used without further purification unless otherwise noted. N2CPh2 was synthesized and purified by crystallization from cold pentane.14 18-Crown-6 was dried according to reported procedures prior to use.15 The [i-PrNDI]Ni2(C6H6) complex 1 was prepared according to previously reported procedures.5 Elemental analyses were performed by Midwest Microlab (Indianapolis, IN).
Complex 2.
To a 20-mL vial containing a stir bar was added [i-PrNDI]Ni2(C6H6) complex 1 (95 mg, 0.13 mmol, 1.0 equiv), THF (1 mL), and pentane (10 mL). The mixture was stirred for 30 min to generate a red-brown solution. To this stirring solution, N2CPh2 (25 mg, 0.13 mmol, 1.0 equiv) dissolved in pentane (2 mL) was added drop wise. An immediate color change to dark brown was observed. The stir bar was removed, and the reaction mixture was stored at –30 °C overnight to afford a microcrystalline solid. The mother liquor was decanted, and the solid was washed with cold pentane (3 × 1 mL) and dried under reduced pressure. Yield: 100 mg, 92%. Single crystals suitable for XRD were obtained from a saturated pentane solution cooled at –30 °C overnight. 1H NMR (400 MHz, 298 K, THF-d8) δ −73.6, −20.3, −14.0, −13.8, −0.5, 2.9, 7.9, 12.4, 20.3, 22.1, 29.6, 63.9, 66.4, 76.1. UV-Vis (THF, nm {M−1 cm−1}): 358 {30000}, 505 {9000}, 548{sh}, 750 {8000}. μeff = 2.86 μB (Evans method, 298 K, THF-d8). Anal. Calcd. for 2 (C49H54N6Ni2): C 69.70, H 6.45, N 9.95; found: C 69.28, H 6.63, N 9.94.
Complex 3.
To a 50-mL Schlenk flask containing a stir bar was added complex 2 (100 mg, 0.12 mmol) and THF (20 mL). The reaction mixture was stirred at 60 °C for 12 h, during which time the color of the solution turned from dark brown to dark purple. The volatiles were removed under reduced pressure to afford a dark purple sticky solid. Hexane (5 mL) was added to the residue, and the mixture was scraped with a spatula to obtain a uniform suspension. The volatiles were removed under reduced pressure to afford Complex 3. Yield = 95 mg, 98%. Single crystals suitable for XRD were obtained from a saturated pentane solution cooled at –30 °C overnight. 1H NMR (500 MHz, 298 K, THF-d8) δ −45.5 (2H), −40.1 (4H), −3.0(2H), 2.4 (12H), 2.5 (12H), 10.5 (4H), 20.5 (4H), 24.9 (4H), 37.4 (6H), 54.8 (2H). UV-Vis (THF, nm {M−1 cm−1}): 350 {30000}, 542 {9000}. μeff = 2.82 μB (Evans method, 298 K, THF-d8). Anal. Calcd. for 3 (C49H54N4Ni2): C 72.09, H 6.67, N 6.86; found: C 71.63, H 6.64, N 6.44.
Complex 4.
To a 20-mL vial containing a stir bar was added complex 3 (49 mg, 0.060 mmol, 1.0 equiv), 18-crown-6 (16 mg, 0.060 mmol, 1.0 equiv) and THF (5 mL). KC8 (8 mg, 0.060 mmol, 1.0 equiv) was added with vigorous stirring. An immediate color change was observed from dark purple to dark green. The reaction mixture was stirred at ambient temperature for an additional 15 min then filtered through a glass fiber pad to remove black graphite. The dark green filtrate was quickly concentrated to a final volume of approximately 2 mL. The solution was stored at –30 °C overnight to yield dark green crystals of complex 4. The solvent was decanted, and the crystals were washed with Et2O (3 × 1 mL) and dried under reduced pressure. Yield: 75 mg, 88%. 1H NMR (400 MHz, 298 K, THF-d8) δ −64.1, −24.0, −19.7, −18.0, 0.7, 0.8, 2.5, 3.9, 5.6, 11.1, 12.6, 15.6. UV-Vis (THF, nm {M−1 cm−1}): 540 {6000}, 665 {5000}, 860 {5000}. Anal. Calcd. for 4.2 THF (C77H110KN4Ni2O10): C 65.67, H 7.87, N 3.98; found: C 65.66, H 7.68, N 4.03. Note: when crystalline 4 was redissolved in THF, rapid decomposition was observed to form red impurities.
Complex 5.
To a 20-mL vial containing a stir bar was added complex 3 (49 mg, 0.060 mmol, 1.0 equiv) and THF (5 mL). (Cp2Fe)PF6 (19.5 mg, 0.060 mmol, 1.0 equiv) dissolved in MeCN (0.5 mL) was added dropwise. An immediate color change from dark purple to brown was observed. The reaction mixture was stirred at ambient temperature for 1 h then concentrated to dryness under reduced pressure. Pentane (3 mL) was added to the residue, and the resulting suspension was stirred for 30 min. The pale yellow solution was decanted from the crude solid material. The brown solid was dissolved in THF (8 mL) and filtered through a glass fiber pad. Diffusion of Et2O vapor into the concentrated THF solution at ambient temperature yielded dark brown crystalline material. The solvent was decanted, and the crystals were washed with Et2O (3 × 1 mL) and dried under reduced pressure. Yield: 55 mg, 91%. Single crystals suitable for XRD were grown from a concentrated DME solution stored at –30 °C overnight. EPR (77 K, 2-Me-THF): giso = 2.01. UV-Vis (THF, nm {M−1 cm−1}): 335 {28000}, 470 {10000}. μeff = 2.0 μB (Evans method, 298 K, THF-d8). Anal. Calcd. for 5 (C102H112F12N10Ni4P2): C 61.17, H 5.64, N 6.99; found: C 60.79, H 5.74, N 6.81.
Complex 6.
To a 20-mL vial containing a stir bar was added complex 3 (82 mg, 0.10 mmol, 1.0 equiv) and THF (2 mL). t-BuNC (11.4 μL, 0.10 mmol, 1.0 equiv) dissolved in hexane (1 mL) was added dropwise with stirring. An immediate color change was observed from dark purple to dark brown. The reaction mixture was concentrated to dryness under reduced pressure. The dark brown solid was dissolved in hexane (5 mL) and stored at –30 °C overnight to yield a dark brown microcrystalline solid. The solvent was decanted, and the crystals were washed with pentane (3 × 1 mL) and dried under reduced pressure. Yield = 80 mg, 89%. Single crystals suitable for XRD were grown from a concentrated Et2O solution stored at –30 °C for 24 h. 1H NMR (800 MHz, 298 K, THF-d8) δ 0.51 (s, 9H, −C(CH3)3), 0.70 (d, J = 8 Hz, 3H, −CH(CH3)2), 0.84 (d, J = 8 Hz, 3H, −CH(CH3)2), 1.09 (d, J = 8 Hz, 3H, −CH(CH3)2), 1.12 (d, J = 8 Hz, 3H, −CH(CH3)2), 1.14 (d, J = 8 Hz, 3H, −CH(CH3)2), 1.32 (br s, 3H, −CH3), 1.34 (d, J = 8 Hz, 3H, −CH(CH3)2), 1.40 (br s, 3H, −CH3), 1.45 (d, J = 8 Hz, 3H, −CH(CH3)2), 1.57 (d, J = 8 Hz, 3H, −CH(CH3)2), 2.35 (sept, J = 8 Hz, 1H, −CH(CH3)2), 3.28 (sept, J = 8 Hz, 1H, −CH(CH3)2), 3.45 (sept, J = 8 Hz, 1H, −CH(CH3)2), 3.61 (sept, J = 8 Hz, 1H, −CH(CH3)2), 4.82 (br s, 2H, Ar H), 5.77 (br s, 1H, Ar H), 6.00 (br s, 2H, Ar H), 6.08 (d, J = 8 Hz, 1H, Ar H), 6.31 (t, J = 8 Hz, 1H, Ar H), 6.54 (t, J = 8 Hz, 1H, Ar H), 6.57 (t, J = 8 Hz, 1H, Ar H), 6.79 (d, J = 8 Hz, 1H, Ar H), 6.83 (d, J = 8 Hz, 1H, Ar H), 6.94 (m, 2H, Ar H), 7.00 (m, 2H, Ar H), 7.09 (d, J = 8 Hz, 1H, Ar H), 7.23 (m, 1H, Ar H), 7.28 (t, J = 8 Hz, 2H, Ar H), 8.11 (d, J = 8 Hz, 2H, Ar H). 13C NMR (200 MHz, 298 K, THF-d8) δ 15.3, 23.4, 24.0, 24.1, 24.2, 24.3, 24.4, 28.2, 28.3, 28.5, 28.7, 56.6, 91.5, 122.3, 122.6, 123.1, 123.3, 123.5, 124.8, 125.0, 125.4, 126.2, 127.9, 128.1, 129.1, 129.4, 130.2, 135.4, 140.6, 148.4. UV-Vis (THF, nm {M−1 cm−1}): 333 {21000}, 1125 {14000}. Anal. Calcd. for 5 (C54H63N5Ni2): C 72.10, H 7.06, N 7.79; found: C 72.07, H 7.19, N 7.66.
Catalytic Carbene Transfer Reaction with t-BuNC.
To a 50-mL Schlenk flask containing a stir bar were added complex 1 (9 mg, 0.0125 mmol, 10 mol%) and C6H6 (3 mL). The Schlenk flask was placed in an oil bath heated at 80 °C. The catalyst solution was stirred, and a mixture of N2CPh2 (24 mg, 0.125 mmol) and t-BuNC (14 μL, 0.125 mmol) dissolved in C6H6 (4 mL) was added dropwise over a period of 12 h using a syringe pump. Following the addition, the reaction was stirred for an additional 3 h at 80 °C. The solvent was removed under reduced pressure. Pentane (5 mL) was added to the residue, and the mixture was filtered through a glass fiber pad. The filtrate was concentrated to dryness under reduced pressure. The crude product was redissolved in C6D6, and mesitylene was added as a standard to determine the yield of t-BuN=C=CPh2.4 NMR Yield = 65%.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by the National Institutes of Health (R35 GM124791). XRD data were collected using an instrument funded by the NSF (CHE-1625543). CU is an Alfred P. Sloan Foundation Research Fellow.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Experimental details and characterization data. (PDF)
Cartesian coordinates for calculated structures. (XYZ)
The authors declare no competing financial interest.
REFERENCES
- (1).(a) Doyle MP Catalytic methods for metal carbene transformations Chemical Reviews 1986, 86, 919–939; [DOI] [PubMed] [Google Scholar]; (b) Brookhart M; Studabaker WB Cyclopropanes from reactions of transition metal carbene complexes with olefins Chem. Rev 1987, 87, 411–432; [Google Scholar]; (c) de Frémont P; Marion N; Nolan SP Carbenes: Synthesis, properties, and organometallic chemistry Coord. Chem. Rev 2009, 253, 862–892; [Google Scholar]; (d) Doyle MP; Duffy R; Ratnikov M; Zhou L Catalytic Carbene Insertion into C−H Bonds Chemical Reviews 2010, 110, 704–724; [DOI] [PubMed] [Google Scholar]; (e) Wang B; Qiu D; Zhang Y; Wang J Recent advances in C(sp3)–H bond functionalization via metal–carbene insertions Beilstein J. Org. Chem 2016, 12, 796–804; [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Keipour H; Carreras V; Ollevier T Recent progress in the catalytic carbene insertion reactions into the silicon-hydrogen bond Org. Biomol. Chem 2017, 15, 5441–5456. [DOI] [PubMed] [Google Scholar]
- (2).(a) Klose A; Solari E; Floriani C; Re N; Chiesi-Villa A; Rizzoli C Iron-carbene functionalities supported by a macrocyclic ligand: iron-carbon double bond stabilized by tetramethyldibenzotetraazaannulene Chem. Commun 1997, 2297–2298; [Google Scholar]; (b) Mindiola DJ; Hillhouse GL Synthesis, Structure, and Reactions of a Three-Coordinate Nickel-Carbene Complex, {1,2-Bis(di-tert-butylphosphino)ethane}NiCPh2 J. Am. Chem. Soc 2002, 124, 9976–9977; [DOI] [PubMed] [Google Scholar]; (c) Li Y; Huang J-S; Zhou Z-Y; Che C-M; You X-Z Remarkably Stable Iron Porphyrins Bearing Nonheteroatom-Stabilized Carbene or (Alkoxycarbonyl)carbenes: Isolation, X-ray Crystal Structures, and Carbon Atom Transfer Reactions with Hydrocarbons J. Am. Chem. Soc 2002, 124, 13185–13193; [DOI] [PubMed] [Google Scholar]; (d) Waterman R; Hillhouse GL Group Transfer from Nickel Imido, Phosphinidene, and Carbene Complexes to Ethylene with Formation of Aziridine, Phosphirane, and Cyclopropane Products J. Am. Chem. Soc 2003, 125, 13350–13351; [DOI] [PubMed] [Google Scholar]; (e) Mankad NP; Peters JC Diazoalkanes react with a bis(phosphino)borate copper(i) source to generate [Ph2BPtBu2]Cu([small eta]1-N2CR2), [Ph2BPtBu2]Cu(CPh2), and [Ph2BPtBu2]Cu-N(CPh2)(NCPh2) Chem. Commun 2008, 1061–1063; [DOI] [PubMed] [Google Scholar]; (f) Gutsulyak DV; Piers WE; Borau-Garcia J; Parvez M Activation of Water, Ammonia, and Other Small Molecules by PCcarbeneP Nickel Pincer Complexes J. Am. Chem. Soc 2013, 135, 11776–11779; [DOI] [PubMed] [Google Scholar]; (g) Marquard SL; Bezpalko MW; Foxman BM; Thomas CM Stoichiometric C=O Bond Oxidative Addition of Benzophenone by a Discrete Radical Intermediate To Form a Cobalt(I) Carbene Journal of the American Chemical Society 2013, 135, 6018–6021; [DOI] [PubMed] [Google Scholar]; (h) Bauer J; Braunschweig H; Damme A; Carlos JO; Kramer J-HT; Radacki K; Shang R; Siedler E; Ye Q Metathesis Reactions of a Manganese Borylene Complex with Polar Heteroatom–Carbon Double Bonds: A Pathway to Previously Inaccessible Carbene Complexes J. Am. Chem. Soc 2013, 135, 8726–8734; [DOI] [PubMed] [Google Scholar]; (i) Iluc VM; Hillhouse GL Three-Coordinate Nickel Carbene Complexes and Their One-Electron Oxidation Products J. Am. Chem. Soc 2014, 136, 6479–6488; [DOI] [PubMed] [Google Scholar]; (j) Russell SK; Hoyt JM; Bart SC; Milsmann C; Stieber SCE; Semproni SP; DeBeer S; Chirik PJ Synthesis, electronic structure and reactivity of bis(imino)pyridine iron carbene complexes: evidence for a carbene radical Chem. Sci 2014, 5, 1168–1174. [Google Scholar]
- (3).(a) Dai X; Warren TH Discrete Bridging and Terminal Copper Carbenes in Copper-Catalyzed Cyclopropanation J. Am. Chem. Soc 2004, 126, 10085–10094; [DOI] [PubMed] [Google Scholar]; (b) Badiei YM; Warren TH Electronic structure and electrophilic reactivity of discrete copper diphenylcarbenes J. Organomet. Chem 2005, 690, 5989–6000; [Google Scholar]; (c) Hofmann P; Shishkov IV; Rominger F Synthesis, Molecular Structures, and Reactivity of Mono- and Binuclear Neutral Copper(I) Carbenes Inorg. Chem 2008, 47, 11755–11762. [DOI] [PubMed] [Google Scholar]
- (4).Laskowski CA; Hillhouse GL Synthesis and carbene-transfer reactivity of dimeric nickel carbene cations supported by N-heterocyclic carbene ligands Chemical Science 2011, 2, 321–325. [Google Scholar]
- (5).Zhou Y-Y; Hartline DR; Steiman TJ; Fanwick PE; Uyeda C Dinuclear Nickel Complexes in Five States of Oxidation Using a Redox-Active Ligand Inorganic Chemistry 2014, 53, 11770–11777. [DOI] [PubMed] [Google Scholar]
- (6).(a) Powers IG; Kiattisewee C; Mullane KC; Schelter EJ; Uyeda CA 1,2-Addition Pathway for C(sp2)−H Activation at a Dinickel Imide Chem.–Eur. J 2017, 23, 7694–7697; [DOI] [PubMed] [Google Scholar]; (b) Powers IG; Andjaba JM; Luo X; Mei J; Uyeda C Catalytic Azoarene Synthesis from Aryl Azides Enabled by a Dinuclear Ni Complex J. Am. Chem. Soc 2018, 140, 4110–4118. [DOI] [PubMed] [Google Scholar]
- (7).(a) Iikubo T; Itoh T; Hirai K; Takahashi Y; Kawano M; Ohashi Y; Tomioka H X-ray Crystal Structure Study of Sterically Congested Diphenyldiazomethanes Eur. J. Org. Chem 2004, 2004, 3004–3010; [Google Scholar]; (b) Kawano M; Hirai K; Tomioka H; Ohashi Y Structure Determination of Triplet Diphenylcarbenes by in Situ X-ray Crystallographic Analysis J. Am. Chem. Soc 2007, 129, 2383–2391. [DOI] [PubMed] [Google Scholar]
- (8).Dugan TR; Bill E; MacLeod KC; Christian GJ; Cowley RE; Brennessel WW; Ye S; Neese F; Holland PL Reversible C–C Bond Formation between Redox-Active Pyridine Ligands in Iron Complexes J. Am. Chem. Soc 2012, 134, 20352–20364. [DOI] [PubMed] [Google Scholar]
- (9).Nocton G; Lukens WW; Booth CH; Rozenel SS; Medling SA; Maron L; Andersen RA Reversible Sigma C–C Bond Formation Between Phenanthroline Ligands Activated by (C5Me5)2Yb J. Am. Chem. Soc 2014, 136, 8626–8641. [DOI] [PubMed] [Google Scholar]
- (10).(a) McMillan JA Electron paramagnetic resonance of free radicals Journal of Chemical Education 1961, 38, 438; [Google Scholar]; (b) Symons MCR In Advances in Physical Organic Chemistry; Gold V, Ed.; Academic Press: 1963; Vol. 1, p 283–363. [Google Scholar]
- (11).Zhou Y-Y; Uyeda C Reductive Cyclopropanations Catalyzed by Dinuclear Nickel Complexes Angew. Chem., Int. Ed 2016, 55, 3171–3175. [DOI] [PubMed] [Google Scholar]
- (12).Pauling L Atomic Radii and Interatomic Distances in Metals Journal of the American Chemical Society 1947, 69, 542–553. [Google Scholar]
- (13).Lukens WW; Speldrich M; Yang P; Duignan TJ; Autschbach J; Kogerler P The roles of 4f- and 5f-orbitals in bonding: a magnetochemical, crystal field, density functional theory, and multi-reference wavefunction study Dalton Trans 2016, 45, 11508–11521. [DOI] [PubMed] [Google Scholar]
- (14).Zhan M; Zhang S; Zhang W-X; Xi Z Diazo Compounds as Electrophiles To React with 1,4-Dilithio-1,3-dienes: Efficient Synthesis of 1-Imino-pyrrole Derivatives Org. Lett 2013, 15, 4182–4185. [DOI] [PubMed] [Google Scholar]
- (15).Gokel GW; Cram DJ; Liotta CL; Harris HP; Cook FL Preparation and purification of 18-crown-6[1,4,7,10,13,16-hexaoxacyclooctadecane] J. Org. Chem 1974, 39, 2445–2446. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.