

SUMMARY
To understand how the interaction between an intra-cellular bacterium and the host immune system contributes to outcome at the site of infection, we studied leprosy, a disease that forms a clinical spectrum, in which progressive infection by the intracellular bacterium Mycobacterium leprae is characterized by the production of type I IFNs and antibody production. Dual RNA-seq on patient lesions identifies two independent molecular measures of M. leprae, each of which correlates with distinct aspects of the host immune response. The fraction of bacterial transcripts, reflecting bacterial burden, correlates with a host type I IFN gene signature, known to inhibit antimicrobial responses. Second, the bacterial mRNA:rRNA ratio, reflecting bacterial viability, links bacterial heat shock proteins with the BAFFBCMA host antibody response pathway. Our findings provide a platform for the interrogation of host and pathogen transcriptomes at the site of infection, allowing insight into mechanisms of inflammation in human disease.
Graphical Abstract
In Brief
Montoya et al. utilizes dual RNA-seq to define the bacterial burden and transcriptional state within leprosy skin lesions. These molecular measures vary across patients and provide separate but informative metrics about the pathogenesis of mycobacterial disease, revealing that the immune system responds to bacterial states and not just their abundance.
INTRODUCTION
The interactions between the host immune response and an invading pathogen dictate the pathogenesis of an infectious disease. The spectrum of such interactions can be investigated in leprosy, caused by the intracellular bacterium Mycobacterium leprae, in which the clinical manifestations correlate with the host immune response to the pathogen (Ridley and Jopling, 1966). In the lesions of the self-limiting tuberculoid form (T-lep), bacilli are rare and the immune response is characterized by a CD4+ T cell infiltrate (Modlin et al., 1982) and an interferon γ (IFN-γ) transcriptional signature, resulting in an effective antimicrobial responses in macrophages (Fabri et al., 2011; Montoya et al., 2009; Yamamura et al., 1991). By contrast, the lesions of the disseminated lepromatous form (L-lep) are characterized by abundant bacilli, B cells, or plasma cells (Iyer et al., 2007; Ochoa et al., 2010), and the expression of a type I IFN gene program that suppresses macrophage antimicrobial activity (Teles et al., 2013). Some patients undergo a reversal reaction (RR) that manifests clinically as an upgrade from the L-lep to the T-lep form of the disease, associated with a change from a type I to type II IFN response.
Although M. leprae was the first human pathogen identified under the microscope, it has not been possible to grow this bacterium in vitro nor to develop a genetic system to study specific genes. Furthermore, research has been limited by the absence of animal models that mimic the clinical and immunologic spectrum of human disease (Kirchheimer and Storrs, 1971; Lahiri et al., 2005; Madigan et al., 2017a; Madigan et al., 2017b). Therefore, the study of human skin lesions from leprosy patients provides an opportunity to investigate the M. leprae determinants of the host immune response at the site of infection.
The advent of a dual RNA sequencing (RNA-seq) approach has recently been applied to simultaneously sequence the transcriptomes of both the host and pathogen organisms in vitro, with the subsequent isolation of each transcriptome in silico through alignment of the respective genomes of the organisms (Damron et al., 2016; Niemiec et al., 2017; Nuss et al., 2017; Thänert et al., 2017; Wesolowska-Andersen et al., 2017; Wester-mann et al., 2016; Zimmermann et al., 2017). The main challenge to this approach had been the size difference between bacterial and mammalian transcriptomes, which can lead to poor coverage of bacterial pathogens. This challenge has been addressed by molecular enrichment of bacterial RNA or by investigating cell cultures and animal models in which the multiplicity of infection is high (Damron et al., 2016; Niemiec et al., 2017; Nuss et al., 2017; Thänert et al., 2017; Wesolowska-Andersen et al., 2017; Westermann et al., 2016; Zimmermann et al., 2017). However, studies where dual RNA-seq has been employed to analyze lesions of a human disease are few, with one viral (Wesolowska-Andersen et al., 2017) and two microbiome dual RNA-seq studies (Franzosa et al., 2014; Pérez-Losada et al., 2015), but none of an infectious bacterial disease. We hypothesized that the high bacterial burden in L-lep lesions would provide an ideal opportunity to employ a dual-RNA-seq approach to investigate the host-pathogen interaction at the site of human infectious disease.
RESULTS
In Situ Dual RNA-Seq of Host-Pathogen Transcriptomes Identifies a Link between Pathogen Abundance and Type I Interferon Signaling
To investigate the host-pathogen interaction at the site of myco-bacterial infection, RNA-seq was performed on the total RNA from 24 leprosy skin biopsy specimens (9 L-lep, 6 T-lep, and 9 RR) (Figure 1A). Microbeads with consensus sequences to human and bacterial ribosomal RNA (rRNA) were utilized for depletion. The enriched mRNA was then converted into sequencing libraries by random hexamer priming, sequenced by standard Illumina protocol, and mapped to both the human and M. leprae genomes (see Methods Details). Hierarchical clustering of both human and M. leprae transcriptomes revealed a distinct gene expression profile in L-lep lesions compared to T-lep and RR lesions (Figure 1B). The co-clustering of T-lep and RR samples (which was also seen with human gene clustering alone) is consistent with their similar histologic features, as observed previously in microarray studies (Bleharski et al., 2003; Teles et al., 2013).
Figure 1. In Situ Dual RNA Sequencing of Host-Pathogen Transcriptomes.

(A) Experimental design for dual RNA-seq RNA extraction, library preparation, and sequence processing.
(B) Unsupervised clustering analysis of both M. leprae and human gene expression across leprosy skin lesions (coefficient of variation, >1.0). Heatmap represents expression of genes (rows) and patients (columns) in which red indicates a higher and green a lower expression Z score per gene across patients (n = 24).
(C) Ratio of the sum of all M. leprae gene abundances over the total sum of human or M. leprae gene abundance across L-lep patient lesions.
(D) Correlation plots of M. leprae abundance (log2 scale) against indicated Signature Visualization Tool (SaVanT) signature values (Z score) for IFN-b, IFN-g, CD4+ T cells, or plasma cells per L-lep patient. p value by Student’s t test (n = 9).
See also Table S1 and Figures S1 and S4.
With an average of ~50 million reads mapping to exons, M. leprae reads were abundantly detected in the L-lep lesions(0.53–3.2 million exonic reads) (Table S1), consistent with the histologic detection of bacilli in the different disease states (Ridley and Jopling, 1966). The M. leprae transcriptome had an exonic coverage (number of reads mapping to exons multiplied by the read length, then divided by the sum of length of all exons) of 22 ± 5.9 SEM times the total length of all exons in the bacterial genome. Furthermore, 95% ± 0.73% SEM of M. leprae genes had at least five average exonic reads across L-lep samples. By contrast, the RR or T-lep lesions had fewer than 1,100 reads mapping to M. leprae, except for three RR lesions that had 36,971, 34,213, and 9,109 reads detected. Because this represents less than 1× coverage of the transcriptome, they were omitted from analysis. Furthermore, the fraction of the transcript abundance (which accounts for gene length, see Methods Details) mapping to M. leprae relative to abundance of transcripts mapping to either human or M. leprae was variable across L-lep lesions and ranged from 0.06 to 0.38 (Figure 1C; Table S1). We next asked whether the variation in M. leprae abundance in the L-lep patients was linked to host immunologic features of the disease, through use of a gene-signature-based analysis measuring the IFN-activation or cell type profile, which was used to deconvolute the cellular composition of each lesion (Lopez et al., 2017). We found a significant correlation of the M. leprae abundance with an IFN-β-activation gene expression signature (r = 0.92, p = 4.4 × 10−4) but not an IFN-g signature (Figure 1D). By contrast, M. leprae abundance was not significantly correlated with a macrophage gene expression signature (Figure S1A), suggesting that the type I IFN response is related to the bacterial load independently of macrophage numbers. Finally, we also observed that the M. leprae abundance was inversely correlated with the CD4+ T cell signature (r = 0.87, p = 1.1 × 10−3) but did not correlate with a plasma cell signature.
Composition of the M. leprae Transcriptome
We next investigated whether more specific properties of the bacterial transcriptome might be associated with immune signatures. Examining only the lepromatous lesions in which significant M. leprae reads were detected, structural noncoding RNAs, including transfer messenger RNA, ribonuclease P, and 23S rRNA, accounts for a majority of the M. leprae transcriptome (5S and 16 s rRNA omitted) (Table S2). Abundant mRNAs included virulence proteins that compose the ESX1 secretion system (esxA and esxB), the ESX1-associated proteins (the apparently operonic transcripts, espA, espC, espD MLBr00411, and PE3), and stress protein transcripts (cspA, hsp18, groEL2, and groES), as well as the master transcriptional regulator whiB1. Interestingly, many of the most abundant M. leprae RNAs were encoded by pseudogenes (Table S2), even though pseudogenes are not translated into full proteins due to nonsense mutations.
Although we depleted bacterial rRNA using affinity beads, we observed that certain regions of 23S rRNA were retained at high coverage, presumably because the rRNA was fragmented and certain regions were not effectively captured by the beads (Figure S2A). Sequencing a separate set of two samples sequentially before and after rRNA depletion showed one 261-bp region in particular that was consistently retained, while the transfer messenger RNA (tmRNA) locus was unaffected (Figure S2B). We postulated that we could use this retained rRNA fragment as a surrogate for total 23S abundance. To confirm that the relative expression of this region matched 23S expression before rRNA depletion, cDNA, which was acquired from the nine L-lep lesions before depletion, was analyzed by qPCR. There was a high correlation (r = 0.71, p = 0.032) between the abundance of RNA-seq 23S and that of the 261-bp region (Figure S2C). As a control, M. leprae esxA, an mRNA presumed to be unaffected by rRNA depletion, also showed significant correlation (r =0.71, p = 0.014) before and after depletion.
M. leprae mRNA:rRNA Ratio as a Bacterial Metric Linked to a Plasma Cell Response
The viability of M. leprae cannot be assessed using the colony forming unit assay, as the bacterium cannot be grown in culture. Instead, M. leprae viability can be estimated from the ratio of rRNA transcripts to genomic DNA, an approach that leverages the difference in stability between rRNA and genomic DNA (gDNA) (Kralik et al., 2010; Liu et al., 2012; Martinez et al., 2009). Here we sought to extend this logic and estimate bacterial viability based on measurements of the RNA only, exploiting the difference in mRNA and rRNA stabilities. For example, in vitro mycobacterial mRNA has an average half-life of 9.5 minutes, whereas rRNA has an average half-life over 24 h (Cangelosi and Brabant, 1997; Rustad et al., 2013). To validate that the mRNA:23S ratio correlates with the M. leprae viability, we measured the ratio of mRNA:rRNA in M. leprae gene expression data from various conditions in which the physiologic state of M. leprae is known to vary. M. leprae bacteria grown in the mouse footpad of immunocompromised (athymic nu/nu) mice were purified and placed into axenic culture for 0–96 h. We calculated the M. leprae mRNA:rRNA ratio by dividing the sum of the transcript abundance of all bacterial mRNA genes by the transcription abundance of 23S (based on the 1343455–1343551 region). We find that in these conditions, the M. leprae mRNA:rRNA ratio significantly decreased with time (n = 2, p = 0.005 at 48 h, p = 0.006 at 96 h) (Figure 2A), indicating that the ratio tracks the viability of the bacteria. A second series of analyses were performed using data from conditions in which M. tuberculosis H37Rv was depleted of all nutrients and grown only in PBS or M. tuberculosis H37Rv during log or transcriptionally quiescent stationary phase (Arnvig et al., 2011; Cortes et al., 2013). The M. tuberculosis mRNA:rRNA ratio was significantly lower in bacteria after nutrient depletion (p = 0.027) or in bacteria harvested in stationary phase than that in exponential phase (p =0.002), further supporting our interpretation of this ratio (Figures S3A and S3B).
Figure 2. Measure of M. leprae mRNA:rRNA Ratio.

(A) M. leprae derived from the footpad of nu/nu mice were isolated and cultured in vitro under starvation conditions for 0 h, 48 h, or 96 h before RNA extraction and RNA sequencing. Data are shown as mean transcriptional activity ± SEM per time point (n = 2). *p < 0.05 by Student’s t test.
(B) Ratio of the log2(x + 1)-transformed sum of all M. leprae mRNA genes divided by 23S RNA abundance per L-lep lesion.
(C) Plot of RNA abundance versus mRNA:rRNAM. leprae measures per L-lep patient.
(D) Scatterplots of M. leprae transcriptional activity versus indicated SaVanT signature score per L-lep patient. p value by Student’s t test (n = 9).
See also Table S2 and Figures S2, S3, and S4.
To validate the two M. leprae (MLEP) RNA-seq bacterial measures, MLEP abundance, and the MLEP mRNA:rRNA ratio, we sequenced a separate set of seven lepromatous leprosy patient samples in which the current standard measures of bacterial burden and bacterial viability had been measured. Furthermore, to avoid 23S ribosomal depletion issues, these samples were not depleted for bacterial rRNA and the 23S transcriptional abundance was calculated both by the 1343455–1343551 region or the full length. The MLEP abundance, calculated by the 23S region or 23S full length, significantly correlated (r = 0.83, p = 0.02; r =0.79, p = 0.02, respectively) with the current clinical standard to assess leprosy bacterial burden, the skin bacillary index (Ridley, 1957) (Figure S4A). Similarly, the MLEP mRNA:rRNA ratio, measuring either the 23S region or 23S full length correlated significantly (r = 0.88, p = 0.004; r = 0.78, p = 0.02, respectively) with the M. leprae 16S rRNA to the MELP-specific repetitive element (RLEP) genomic DNA measured by quantitative real-time PCR (Kralik et al., 2010; Liu et al., 2012; Martinez et al., 2009) (Figure S4B).
Having established that the ratio of mRNA to rRNA provides a distinct metric of the M. leprae physiologic state, we next asked if its variation across L-lep lesions correlated with host gene expression signatures that are characteristic of L-lep patients. This value was found to vary across L-lep lesions (Figure 2B; Table S1), and was largely independent of M. leprae abundance (Figure 2C; r = −0.11, p = 0.79). We found that the M. leprae mRNA:rRNA ratio was significantly negatively correlated with the plasma cell signature (r = −0.84, p = 0.005) but not the IFN-β, IFN-γ and CD4+ T cell signatures in L-lep lesions (Figure 2D).
Identification of an M. leprae Gene Module Associated with Host Antibody Production in L-Lep Lesions
To identify the M. leprae gene modules associated with the variation in M. leprae mRNA:rRNA ratio, a weighted-gene correlation analysis (WGCNA) was performed on the bacterial transcriptome (Figure 3A) (Langfelder and Horvath, 2008; Montoya et al., 2014). WGCNA is an unsupervised clustering method that uses correlations across samples to group genes into modules, similar to traditional clustering analysis, but also accounts for the network neighborhood of a given set of genes, thus lending more weight to gene networks with more reliable correlations. Each resultant module is given an arbitrary color name and represents a set of highly interconnected genes that change expression coordinately across samples. To account for the varying abundance of bacilli across patient lesions, the relative percentage of total bacterial mRNA was input for each M. leprae gene. We found that among the 14 modules that we identified, the M. leprae RNA abundance measure correlated most highly with the average transcriptional abundance (module eigengene) of the MLEPpurple module, although this did not reach significance (r = 0.5, p = 0.2). The MLEPpurple module contained two major components of the genes of the ESX-1 secretion system esxA and esxB, which are required for type I IFN induction in macrophages by mycobacterial infection (Stanley et al., 2007). The relative expression of esxA was significantly correlated with the M. leprae RNA abundance (r = 0.71, p = 0.03; Figure 3B), and correlated with type I IFN gene signature but did not reach significance (r = 0.66, p = 0.053; Figure S1B). Meanwhile, the M. leprae mRNA:rRNA ratio positively correlates with the MLEPcyan module eigengene, (r = 0.84, p = 0.005), and the MLEPblue module eigengene composed of 257 genes inversely correlates with the M. leprae mRNA:rRNA ratio (r = −0.75, p = 0.02) (Figures 3A and 3C; Table S3).
Figure 3. Module Analysis of M. leprae Transcriptome.

(A) Unsupervised weighted gene-correlation analysis (WGCNA) of the M. leprae gene expression across the nine L-lep patients. Heatmap represents the correlation of the module eigengene (rows) versus bacterial measure (columns) in which red indicates a positive and blue a negative Pearson correlation across patient samples. Number of genes per module indicated beside module color. Black outlines indicate highest and lowest correlation values.
(B) Correlation plot of the MLEPpurple module gene, esxA, and the M. leprae abundance per L-lep patient (n = 9).
(C) Heatmap of MLEPblue or MLEPcyan module protein-coding genes (pseudogenes excluded) across L-lep patients in order of M. leprae mRNA:rRNA (top). High or low relative expression indicated by red or blue, respectively.
(D) Functional category enrichment of genes within the MLEPblue or MLEPcyan modules. Heatmap color indicates the negative logarithm of the hypergeometric enrichment p value per functional category. *p % 0.05.
(E) Plot of the WGCNA module membership and the correlation to the M. leprae mRNA:rRNA per gene from the MLEPblue virulence and heat shock genes. p value by Student’s t test (n = 9).
The functional categories associated with the significantly correlated M. leprae gene modules were investigated using annotations from the Mycobrowser (Kapopoulou et al., 2011). The MLEPcyan and MLEPblue modules were significantly enriched in the “virulence, detoxification, adaptation” functional category defined by Mycobrowser, but we noted there were numerous heat shock proteins (HSPs); hence, we designate them as “virulence and HSPs” (Figure 3D). The MLEPblue module in particular was enriched in genes encoding HSPs, whereas MLEPcyan contained none. Furthermore, an additional enrichment in “intermediary metabolism” was identified for the blue module. The correlation of individual MLEPblue virulence genes with M. leprae mRNA:rRNA ratio correlated with module membership, a measure of the genes most highly connected to the module (Figure 3E).
Interactions between the M. leprae and Host Transcriptomes
To further understand the interaction between the bacterial and host transcriptomes in leprosy, the WGCNA module eigengenes derived from the M. leprae transcriptome and the metrics for bacterial abundance and the mRNA:rRNA ratio were correlated with the WGCNA modules derived from the human transcriptome within the same lesions. Of the 36 human modules generated, nine were highly correlated (p < 0.001) with the M. leprae mRNA:rRNA ratio, abundance, or module eigengenes of the MLEPblue or MLEPcyan modules (Figure 4A). The most significant correlation was between the HUMANgreenyellow and the MLEPblue module eigengenes (r = 0.78, p = 7 × 10−6). Gene set enrichment analysis (GSEA) (Subramanian et al., 2007) of the HUMANgreenyellow module genes identified “humoral immune response” as the top term, accounting for 27 of the 575 genes in the module. Overall, immunoglobulin genes accounted for 215 (37%) of the genes in module, including all of the immunoglobulin heavy chain constant genes (IGH), which determine antibody isotype (Figure 4B; Table S3). IGH transcripts were seven-fold more abundant in L-lep versus T-lep lesions, in part due to the dominance of IGHM transcripts. Of the class-switched isotypes, the total of the IGHG subtypes predominated in L-lep lesions followed in expression by IGHA (Figure 4B). Further investigation by ingenuity pathways analysis (IPA) of HUMANgreenyellow indicated the canonical pathway most significantly enriched to be “Communication between Innate and Adaptive Immune Cells” (hypergeometric enrichment p value, 1.71 × 10−16), identifying a pathway for immunoglobulin production. This pathway contains the ligands BAFF (B-cell-activating factor, also known as [aka] TNFSF13B) and APRIL (a proliferation-inducing ligand, aka TNFSF13), which activates the B cell receptor BCMA (B cell maturation antigen, aka TNFRSF17), leading to production of immunoglobulin G (IgG) and IgA, all of which were differentially expressed in L-lep versus T-lep lesions (Figure 4C). In addition to BCMA, the HUMANgreenyellow module genes IGHA1, IGHA2, and IGHG2 had significant inverse correlations with the M. leprae mRNA:rRNA ratio (r = −0.84, p = 0.005; r = 0.79, p = 0.011; r = 0.76, p = 0.017, respectively; Figure 4D). BCMA inversely correlated with the M. leprae mRNA:rRNA ratio (r = −0.77, p =0.008) but positively correlated with the plasma cell signature (r = 0.96, p = 4 × 10−5) (Figures 4D, 4E, and S5A).
Figure 4. Interactions between Host and Pathogen Transcriptomes.

(A) WGCNA of the human gene expression across all leprosy patients (n = 24). Heatmap represents the correlation of the host module eigengene (rows) versus bacterial measure (columns) in which red indicates a positive and blue a negative Pearson correlation across only the nine LL patient samples. Only modules with a significant correlation (p < 0.001) in at least one bacterial measure or module eigengene are shown. Black outline indicates most significant positive correlation. Number of genes per module indicated beside module color and top gene ontology terms for module genes listed on right with the false discovery rate (FDR) q-value for enrichment per module.
(B) Mean abundance of IGH immunoglobulin constant regions per leprosy clinical subtype (LL, n = 9; tuberculoid leprosy and reversal reaction leprosy [TL and RR], n = 15).
(C) Ingenuity pathways canonical pathway communication between innate and adaptive immune cells. Only the section of pathway containing enriched genes is shown. HUMANgreenyellow module genes shaded green and genes significantly higher in LL versus TL and RR patients (fold change, ≥1.5; padj ≤ 0.05 outlined in red; Bonferoni-adjusted p value by Wald test).
(D) Correlation heatmap of BAFF and APRIL ligands and BCMA receptor expression versus M. leprae bacterial measures, human SaVanT plasma cell and IFN-β signature score, and individual immunoglobulin IGH constant region genes across L-lep patients. High or low Pearson correlation indicated by red or blue, respectively.
(E) Correlation plots of M. leprae bacterial measures across L-lep skin lesions versus BAFF or BCMA expression. p value by Student’s t test (n = 9).
See also Figures S4 and S5 and Table S3.
Although BAFF and APRIL were not present in the HUMANgreenyellow, they were significantly higher in L-lep versus T-lep or RR patients (1.7 fold-change, p adjusted padj = 0.004, 1.6 fold-change, padj = 0.007, respectively). Although APRIL was expressed in L-lep lesions, it did not correlate with either M. leprae mRNA:rRNA ratio or abundance. By contrast, BAFF expression significantly correlated with the M. leprae abundance (r = 0.81, p = 0.008) but not the mRNA:rRNA ratio, the plasma cell signature, nor IGH expression (Figure 4D). BAFF expression did correlate with the type I IFN activation signature score (r = 0.71, p = 0.032) across L-lep patients (Figures 4D and 4E; Figure S5B). M. leprae abundance is likely linked to BAFF expression through type I IFN production, because in vitro M. leprae infection of monocyte-derived macrophages induces type I IFN (Teles et al., 2013) and BAFF (Figure S5C). Lastly, type I IFN stimulation induces BAFF in uninfected macrophages (Figure S5C).
Because BCMA expression appears to be linked to antibody expression, we investigated how the MLEPblue “virulence and HSP” genes were individually connected with BCMA expression. Six M. leprae genes (aac, ML0493, clpB, groEL1, groEL2, and hspR) significantly correlated with the expression of BCMA, the plasma cell signature, IGHA1, IGHA2, IGHG2, and the total amount of IGH (Figures 5A). GroEL1 and groEL2 also particularly demonstrated a strong correlation with the class-switched IGHA1 and IGHG2 antibody production (Figures 5A).
Figure 5. Bacterial Stress Proteins Are Linked to BAFF-BCMA-Induced Antibody Expression.

(A) Correlation heatmap of MLEPblue virulence and heat shock genes (columns) versus MLEP mRNA:rRNA and components of the host antibody response. Positive or negative Pearson correlation indicated by red or blue, respectively (n = 9).
(B) Heatmap of expression of human immune response components across the spectrum of leprosy clinical subtypes (n = 24). Color represents the Z score across each row with red as high and blue as low relative expression.
(C) Correlation plot of the total IGH abundance versus BCMA or BAFF expression across the spectrum leprosy samples (L-lep, T-lep, and RR). p value by Student’s t test (n = 24).
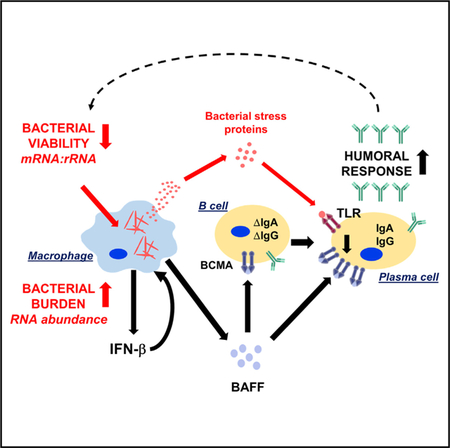
(D) Model for interaction between the pathogen and host humoral response. The model shows that bacterial abundance is correlated with induction of IFN-β leading to BAFF expression. At the same time, decreased M. leprae mRNA:rRNA is linked to the expression of M. leprae heat shock proteins, which can trigger TLR4 activation on plasma cells to upregulate the BAFF receptor BCMA. The BAFF-BCMA interaction results in maturation and survival of class-switched antibody plasma cells.
To further understand the relevance of BAFF and BCMA to antibody responses in L-lep patients, we investigated the expression of key immune response genes and signatures across the leprosy disease spectrum. As previously stated, BAFF, BCMA, IGH, as well as the individual immunoglobulin iso-forms were more strongly expressed in L-lep versus T-lep and RR lesions (Figure 5B). The plasma cell signature linked to BCMA and the type I IFN signature linked to BAFF were also greatest in L-lep lesions. Although BAFF and BCMA expression were highest in L-lep lesions, only the level of BCMA expression across all leprosy patients was tightly correlated with the total antibody expression (r = 0.94, p < 1 × 10−6; Figures 5B and 5C). Together, these data indicate that BCMA expression is central to immunoglobulin production at the site of disease in leprosy and is linked to the decreased M. leprae mRNA:rRNA ratio and a stress response gene module.
DISCUSSION
The locus of the battle between pathogens and host is primarily in tissue lesions, about which remarkably little is known for many pathogens. To define the molecular determinants of the host-pathogen interaction at the site of an easily accessible mycobacterial infection, we performed dual RNA-seq on leprosy lesions, simultaneously measuring both the human and M. leprae transcriptomes. The clinical spectrum of leprosy is defined by the number of skin lesions and microscopic quantification of bacilli, which grossly correlate with the host immunologic responses (Ridley and Jopling, 1966). Because M. leprae cannot be grown in vitro, it is not readily possible to determine the viability of the bacilli in lesions. Here, we define two independent molecular measures of M. leprae, each of which correlates with distinct aspects of the host immune response (Figure 5D). The first is the bacterial burden, represented by the fraction of bacterial transcripts, which correlates with a host type I IFN gene signature. However, the bacterial burden did not correlate with the host plasma cell gene signature. We overcome this limitation by considering a second property of bacilli, namely, the ratio of bacterial mRNA to rRNA, which was independent of the M. leprae RNA abundance. This second metric, the M. leprae mRNA:rRNA ratio, correlated with the bacterial viability in lesions and in in vitro culture of mycobacteria in different conditions. We find that the M. leprae mRNA:rRNA ratio in each individual is strongly correlated with the abundance of plasma cells, as measured by the plasma cell signature. Simultaneous measurement of both the human and M. leprae transcriptomes enabled us to identify associations between the host and pathogen, including a link between mycobacterial “stress response” and “human antibody response” gene networks, providing a precision medicine approach to identifying critical determinants of disease pathogenesis.
The clinical spectrum of leprosy defines a subgroup of patients, L-lep, presenting with disseminated lesions containing a high bacterial load. The current standard to assess bacterial burden in the clinical setting of leprosy is the bacillary index (BI), which was developed 61 years ago and determined through enumeration of acid-fast-stained bacteria per high-power microscopic field from blood slit smears from either the earlobe or the disease lesion (Ridley, 1957). Here, we developed a molecular definition of bacterial burden, measuring the M. leprae RNA abundance, finding that this metric correlated strongly with a host type I IFN signature. Previously, we found that IFN-b secretion by macrophages correlated with the multiplicity of infection by M. leprae in vitro (Teles et al., 2013). In addition, although the relative expression of mycobacterial esxA has been demonstrated in experimental models to be required for mycobacteria-induced type I IFN expression, we provide evidence linking M. leprae esxA to the human type I IFN signature at the site of infection. Although the type I IFN inducible program is a major immune factor for control of viral infection, it has been shown to suppress antibacterial responses, particularly to intracellular infections (Boxx and Cheng, 2016; Teles et al., 2013).
Although humoral immune responses are robust in multibacillary L-lep patients, little is known about antibody production at the site of infection (Iyer et al., 2007; Ochoa et al., 2010). We found that WGCNA was a powerful approach to associate bacteria and host gene networks, uncovering a molecular mechanism that links the M. leprae mRNA:rRNA ratio to the plasma cell response. A low bacterial mRNA:rRNA ratio correlated with an M. leprae network, MLEPblue, which was linked to a host gene module, HUMANgreenyellow, that was enriched for antibody genes and BCMA, a cell surface receptor on plasma cells known to induce IgA and IgG class-switched antibody production in B cells (Marsters et al., 2000). A ligand for BCMA, BAFF, correlated with M. leprae abundance and a type I IFN program. These data identify a mechanism connecting M. leprae abundance and the mRNA:rRNA ratio, likely reflecting bacterial viability, leading to the enhanced humoral response in L-lep patients. BCMA but not BAFF expression was tightly linked to the immunoglobulin expression across the spectrum of leprosy. Mining the correlated bacteria and human gene networks, we identified an association between the gene expression of M. leprae HSPs and BCMA. We suggest that M. leprae under immune attack are stressed and transcriptionally low, cutting back on protein synthetic functions and increasing the ratio of expression of proteostasis genes, such as HSPs (Lupoli et al., 2016). Furthermore, some stressed bacteria are dying, releasing their major cytoplasmic and nuclear contents. This would include DNA (Kim et al., 2011), known to trigger Toll-like receptor 9 (TLR9) on B cells, and plasma cells, leading to upregulation of BCMA, thereby allowing the BCMA ligand BAFF to augment antibody production. Alternatively, the increased ratio of HSPs can be released by dying bacteria (Vargas-Romero et al., 2016) and subsequently released from dying macrophages or secreted in exosomes (Giri et al., 2010). Of these proteins, GroEL1 and/or GroEL2 and DnaK, have been shown to activate TLR4 (Asea et al., 2002; Bulut et al., 2005), which is also present on plasma cells (Dorner et al., 2009). These HSPs are also known to be major antigens for the antibody response (Young et al., 1988). We noted that GroEL2 expression had the strongest correlation with an IgA1 antibody response, characteristic of a mucosal immune response, perhaps reflecting nasopharyngeal involvement in leprosy patients (Barton, 1975).
The link of GroEL1 and/or GroEL2 expression to an antibody response may reflect the immune system’s attempt to mount a host defense response. In tuberculosis, antibodies have been shown to contribute to an antimicrobial response against myco-bacteria (Lu et al., 2016). Targeting of HSPs has long been used for bacterial vaccine design and in particular, M. leprae GroEL2 (Lima et al., 2003; Lorenzi et al., 2010; Lowrie et al., 1999; Souza et al., 2008; Tascon et al., 1996); and intranasally administered M. tuberculosis DnaK (Chuang et al., 2018) has been shown to engender protective cell-mediated and humoral immune responses to tuberculosis in mouse models. Our data demonstrate a correlation between the decreased M. leprae mRNA:rRNA ratio with bacterial HSP expression and the host humoral response, suggesting the need for further investigation of the efficacy of mycobacterial HSPs in prophylactic vaccines. On the other hand, the link between M. leprae HSP gene expression and the host antibody response may reflect pathogenesis of the infection, indicating release of bacterial products that contribute to immunopathology. Although the expression of BAFF, BCMA, and antibody genes varied in L-lep patients, the levels were generally higher than in T-lep or RR patients. Excessive BAFF signaling of B cells has been shown to increase autoantibody production (Gross et al., 2000; Mackay et al., 1999; Zhang et al., 2001), which are prominent in L-lep patients complicating the differential diagnosis of systemic lupus erythematosus and syphilis (de Larrañaga et al., 2000; Loizou et al., 2003).
Previous studies seeking to gain insight into the battle between the host and a pathogen through dual RNA-seq have been limited to the study of infected cells in tissue culture or in mouse models of infection, often using genetically modified bacteria or different mouse strains. It is difficult to translate this approach to human infectious disease given that both the pathogen and the host cannot be genetically modified. Here, we were able to define host-pathogen transcriptional associations in lesions of a human disease. The complex cellular environment of tissue specimens provides a bioinformatics challenge to analysis. Previous dual RNA-seq studies of in-vitro-infected cells have been limited to only one cell type, whereas in vivo studies in mouse models lacked analysis of the tissue cellular environment (Damron et al., 2016; Nuss et al., 2017; Pittman et al., 2014; Thänert et al., 2017). Here, we developed a computational framework that reduces each disease lesion into a series of scores representing the host cellular immune pathways and pathogen abundance or physiologic state. This reduction enabled us to correlate the spectrum of interactions between the pathogen and host immune response, identifying human and bacterial gene modules that are co-regulated across patients. Together, the transcriptional state and bacterial burden that varied across patients provide separate but informative metrics about the pathogenesis of mycobacterial disease that are likely to be general for other intracellular pathogens, revealing that the immune system responds to bacterial states and not just their abundance. As such, this approach provides a paradigm for investigating additional host-pathogen interactions in human disease.
STAR★METHODS
CONTACT AND REAGENT RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, R.L. Modlin (rmodlin@mednet.ucla.edu).
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Human subjects
Adult patients over the age of 18 with leprosy were allocated into appropriate experimental groups through classification according to the criteria of Ridley and Jopling (1966); all T-lep patients (2 females, 4 male) classified as borderline tuberculoid, and all L-lep patients (1 female, 5 male, 3 gender not reported) had lepromatous leprosy. All L-lep and T-lep skin biopsies were taken at the time of diagnosis before treatment, and reversal reaction biopsies (5 female, 2 male, 3 gender not reported) were upgrading from patients originally diagnosed with borderline lepromatous leprosy, therefore starting from a different part of the disease spectrum than the L-lep group. In previous studies of the leprosy transcriptomes we have found five patient samples per patient group to provide sufficient power to discern clinical status through hierarchical clustering (Bleharski et al., 2003; Montoya et al., 2009; Teles et al., 2013), here more than six samples per leprosy subclassification were sequenced. For determining correlations across lepromatous leprosy patients, a sample size power analysis at 70% power (1-b) and a type I error rate of α = 0.05, revealed that at least eight subjects were needed at a pearson correlation coefficient r = 0.80 (Sample size = (standard normal deviate for a significance + standard normal deviate for β)/ln[(1+r)/(1-r)]). All leprosy patients were recruited with informed consent and approval from the Institutional Review Board of University of Southern California school of Medicine or the institutional ethics committee of Oswald Cruz Foundation, as well as the University of California, Los Angeles.
Monocyte derived macrophages experiments were derived from healthy human blood. Adult human donors were recruited with informed consent and approval from the Institutional Review Board of the University of California at Los Angeles, gender of cells was not reported under this protocol.
Mycobacterial strains
Mycobacterium leprae TN was grown in the footpad of nu/nu mice and harvested at log phase of growth (Lahiri et al., 2005) at the National Hansen’s Disease Program Laboratory. Mycobacterium tuberculosis H37Rv experimental data was derived from a re-analysis of previously published studies (Arnvig et al., 2011). Nutrient deprivation experiment was performed by growing M. tuberculosis H37Rv in Middlebrook 7H9 medium supplemented with 0.4% glycerol, 0.085% NaCl, 0.5% BSA, and 0.05% Tyloxapol. For nutrient depletion condition, exponentially growing bacteria were washed, resuspended in PBS supplemented with 0.025% Tyloxapol, and cultured for a further 24h. RNA was extracted and Illumina Sequencing libraries were constructed and sequenced as described (Cortes et al., 2013). In the exponential and stationary phase M. tuberculosis comparison, H37Rv was grown in Middlebrook 7H9 supplemented with 0.2% glycerol and 10% ADC. Exponential phase cultures were harvested at OD600 0.6 to 0.8; stationary phase cultures were harvested one week after OD600 had reached 1.0. RNA was from three independent exponential phase cultures of M. tuberculosis and two stationary phase cultures were used to generate cDNA preparations that were then analyzed by Illumina-based sequencing as described (Arnvig et al., 2011).
METHODS DETAILS
Dual RNA-seq of leprosy skin lesions
Frozen tissue sections of nine L-lep, nine RR, and six T-lep frozen skin lesions were cut into multiple ten micron sections and lysed in QIAGEN RLT buffer with 0.1% β-mercaptoethanol (Invitrogen). To ensure efficient lysis of mycobacterial cell wall, lysates were homogenized with 0.1mm silica beads (MP Biomedicals, Santa Ana, CA) in the FastPrep3000 twice at a setting of 6.5 for 45 s. RNA was extracted via QIAGEN AllPrep Kit (QIAGEN, Hilden, Germany). rRNA was depleted with the Illumina Ribo-Zero Gold rRNA Removal Kit (Epidemiology) before sequencing library preparation with the Illumina Truseq Stranded Total RNA Sample Preparation Kit. A second set of eight lepromatous leprosy lesions were isolated for validation by the same method above, however only the human but not bacteria rRNA was depleted by Ribo-Zero Gold rRNA Removal Kit (Human/Mouse/Rat). Libraries were sequenced on the HiSeq2000 Sequencer (Illumina) at single-end 50 bp reads. Reads with poor quality were trimmed by Trim Galore! (http://www.bioinformatics.babraham.ac.uk/projects/trim_galore) and high quality reads were mapped to the hg19 human genome via STAR (Dobin et al., 2013). Unmapped reads after human genome alignment were mapped to the Br4923 M. leprae genome (Assembly ASM2668v1). The sum of exonic reads per gene were counted using HTSeq (Anders, 2010) using the GENCODE GRCh37-mapped version Release 24 (Harrow et al., 2012). Although, M. leprae 16S rRNA, was depleted, a 96bp region (1343455–1343551) of the 23S rRNA remained abundant (Figure S2). Presumably, the approach to deplete rRNA did not include oligonucleotides that bound to these sequences and rRNA was fragmented. To confirm, the entire M. leprae 23S rRNA sequence and undepleted region were entered into the manufacturer website Epicenter Matchmaker (http://www.illumina.com/products/selection-tools/rna-matchmaker-tool.html) and indicated “No Match” for binding of Ribozero depletion probes, while the well-depleted 16S rRNA sequence indicated a 65%–85% match. Expression of M. leprae 23S rRNA and esxA were measured by quantitative real-time PCR KAPA SYBR FAST (MilliporeSigma, St Louis, MO) of cDNA synthesized from the same lesional RNA used in sequencing but before any ribosomal depletion. The relative quantities of the gene tested per sample were calculated against the GAPDH mRNA using the CT formula as previously described (Teles et al., 2013).
M. leprae axenic culture
For nutrient deprivation experiment of Mycobacterium leprae TN, bacteria were harvested from mouse footpad then cultured at 33°C, 5% O2 in National Hansen’s Disease Program (NHDP) medium for 48h or 96h post-harvest. NHDP medium components per liter are monopotassium phosphate 2.0 g, disodium phosphate 1.5 g, monosodium glutamate 0.5 g, sodium citrate 0.1 g, ammonium sulfate 0.5 g, pyridoxine 0.001 g, ferric ammonium citrate 0.04 g, magnesium sulfate 0.05 g, zinc sulfate 0.001 g, copper sulfate 0.001 mg, biotin 0.5 mg, calcium chloride 0.5 mg, bovine albumin (Fraction V) 0.5 g, dextrose 7.5 g, casitone 1.0 g. Total RNA of M. leprae was extracted and rRNA depleted with Illumina Ribo-Zero Gold rRNA Removal Kit (Bacteria). The Solid 5500 system was used for sequencing and quality control, alignment was performed by Maverix Biomics (San Mateo, CA). Gene counts (including 23S regional calculation) and mRNA:rRNA ratio were calculated as described for lesional RNaseq analysis.
M. leprae infection of macrophages in vitro
Peripheral blood mononuclear cells were isolated from healthy human blood through Ficoll gradient separation. Monocytes were then purified by CD14+ microbead enrichment (Miltenyi Biotec Bergisch Gladbach, Germany) according to manufacturer protocol. Monocytes were differentiated into macrophages for 5 days in M-CSF 50ug/ml in 10% fetal calf serum (FCS) in GIBCO RPMI 1640 medium 37°C (ThermoFisher Scientific, Waltham, MA) (Realegeno et al., 2016). Macrophages were infected at a multiplicity of infection of ten bacteria per cell from live M. leprae, in antibiotic-free RPMI medium with 10% FCS at 33°C. At indicated time points, RNA was extracted by QIAGEN AllPrep Kit (QIAGEN, Hilden, Germany). rRNA was depleted and Illumina stranded libraries prepared, sequenced, and raw data analyzed in same method as leprosy skin lesions.
QUANTIFICATION AND STATISTICAL ANALYSIS
RNA-seq data analysis
Filtered read counts (containing genes with ≥ 5 reads in at least one patient group) from bacterial and human transcriptomes were normalized via DESeq2 (Love et al., 2014) using default parameters, except for basing scaling factors on only human genes. DESeq2 normalized gene counts and rlog-transformed counts were output and used for subsequent data analysis. Unsupervised hierarchical clustering was performed on rlog gene counts filtered for adequate expression of ≥ 5 rlog of gene in at least 20% of samples and filtered for variance by having a CV ≥ 0.2 rlog. Transcript abundance per gene was calculated by dividing the number of normalized read counts per gene by the gene length (exons only). M. leprae RNA abundance was calculated by taking the sum of the transcript abundance of all bacterial genes was divided by the total abundance of bacterial or human genes per sample. The M. leprae mRNA:rRNA ratio was calculated by dividing of the sum of the transcript abundance of all bacterial mRNA genes by the transcription abundance of 23S (based on the 1343455–1343551 region). A log transformation (log2(x+1)) of the M. leprae mRNA:rRNA ratio was used for all analyses.
SaVanT cell signature calculation
Cell type signatures were calculated using the web-based Signature Visualization Tool (SaVanT; http://newpathways.mcdb.ucla.edu/savant) (Lopez et al., 2017). In brief, SaVanT calculates a cell type score based on averaging a set of pre-calculated signature genes for each cell type which is proportional to the cell abundance within the sample. Human data from skin lesions (rlog-transformed gene counts) were input into SaVanT with the setting to use the top 50 genes per signature for score calculation. Since the output scores are in arbitrary expression units, the z-score was calculated per sample relative to all samples to depict the same scale for all signatures.
Weighted gene correlation analysis (WGCNA) and gene enrichment analysis
Module analysis of bacterial or human correlation networks was performed using Weighted Gene Correlation Analysis (WGCNA) (Langfelder and Horvath, 2008). The fraction abundance of mRNA for M. leprae transcriptome was calculated by dividing the transcriptional abundance of each gene (DESeq2 normal counts/gene length) by the sum of all abundances of mRNA genes. Input for the M. leprae WGCNA was the mRNA fractional abundance with log-transformation (log2(x + 1)) and human WGCNA was rlog-transformed output from DESeq2 normalization. A signed gene correlation network was constructed using the “blockwiseModules()” function with a soft thresholding power of 12 for human and 16 for M. leprae and using a minimum module size of 50 genes, merged cut threshold of 0.3, and deepSplit of 0 for both analyses. The module eigengene, which represents a linear combination of genes that capture a large fraction of variance in each module, was calculated for each sample and correlated to M. leprae abundance or mRNA:rRNA ratio per sample.
Bacterial module gene enrichment analysis was performed through hypergeometric overlap analysis of the Mycobrowser functional categories (Kapopoulou et al., 2011) for each gene. Further enrichment analysis of the MLEPblue module virulence genes was performed by the Database for Annotation, Visualization and Integrated Discovery (DAVID) (Huang et al., 2009). Human module gene enrichment was performed using Gene Set Enrichment Analysis (GSEA) (Subramanian et al., 2005) of the Molecular Signatures Database (MSigDB) (Liberzon et al., 2011). Further enrichment analysis of the HUMANgreenyellow module was performed by Ingenuity Pathways Analysis (QIAGEN, Hilden, Germany). Canonical pathway ‘Communication between Innate and Adaptive Immune Cells’ was edited to include only the portion of the pathway which had enriched genes and for visual clarity by the Pathway Designer function.
Pearson correlation plots (Figures 4D and 5A) were constructed using the ‘corrplot’ R package (R Core Team, 2013; Wei, 2017). Correlation significance test for association between paired samples was performed by the R function “cor.test()” using Pearson’s product moment correlation coefficient. Three-way partial correlations performed using the “ppcorr” R package (Kim, 2015). The two-tailed Student’s t test was used when individual comparisons between two groups were performed. Individual details of statistical analyses are explained in the figure legends.
DATA AND SOFTWARE AVAILABILITY
The accession number for the dual RNaseq data for leprosy lesions reported in this paper is GEO: GSE125943. Signature Visualization Tool (SaVanT) can be accessed online at http://newpathways.mcdb.ucla.edu/savant.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| Mycobacterium leprae TN | R. Lahiri, National Hansen’s Disease Program | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| recombinant human M-CSF | R & D Systems (Minneapolis, MN) | 216-MC |
| Deposited Data | ||
| Leprosy dual RNaseq data | Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/) | GSE125943 |
| Oligonucleotides | ||
| M. leprae 23S forward primer | Integrated DNA Technologies, Inc. | GCA CCT GCC TTG TAT CAA TTC |
| M. leprae 23S reverse primer | Integrated DNA Technologies, Inc. | TCA TCG AGA AGT ATT TAG GCT TAC C |
| M. leprae esxA forward primer | Integrated DNA Technologies, Inc. | CAG CTT TGT GAG CGA TTC TTG |
| M. leprae esxA reverse primer | Integrated DNA Technologies, Inc. | TAC AGG CGT GGC ACT TTC |
| Software and Algorithms | ||
| Signature Visualization Tool (SaVanT) | Lopez et al., 2017 | http://newpathways.mcdb.ucla.edu/savant |
| Weight Gene Correlation Analysis | Langfelder and Horvath, 2008 | https://cran.r-project.org/web/packages/WGCNA/index.html |
| DESeq2 | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
Highlights.
Dual RNA-seq of leprosy skin lesions defines two independent bacterial measures
The fraction of bacterial RNA relates to its burden and the host type I IFN pathway
Bacterial mRNA:rRNA correlates with its viability and the host antibody response
Mycobacterial stress response related to increased class-switched antibody production
ACKNOWLEDGMENTS
Financial support was provided by NIAID AAI15006–004 for provision of M. leprae. Funding was provided by UCLA CTSI (grant number UL1TR000124) and NIAMS (NIH3P50AR06302). This work used computational and storage services associated with the Hoffman2 Shared Cluster provided by UCLA Institute for Digital Research and Education’s Research Technology Group.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.02.109.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Anders S (2010). HTSeq: Analysing high-throughput sequencing data with Python. http://www-huber.embl.de/users/anders/HTSeq/doc/overview.html. [DOI] [PMC free article] [PubMed]
- Arnvig KB, Comas I, Thomson NR, Houghton J, Boshoff HI, Croucher NJ, Rose G, Perkins TT, Parkhill J, Dougan G, and Young DB (2011). Sequence-based analysis uncovers an abundance of non-coding RNA in the total transcriptome of Mycobacterium tuberculosis. PLoS Pathog. 7, e1002342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asea A, Rehli M, Kabingu E, Boch JA, Bare O, Auron PE, Stevenson MA, and Calderwood SK (2002). Novel signal transduction pathway utilized by extracellular HSP70: role of toll-like receptor (TLR) 2 and TLR4. J. Biol. Chem 277, 15028–15034. [DOI] [PubMed] [Google Scholar]
- Barton RP (1975). Importance of nasal lesions in early lepromatous leprosy. Ann. R. Coll. Surg. Engl 57, 309–312. [PMC free article] [PubMed] [Google Scholar]
- Bleharski JR, Li H, Meinken C, Graeber TG, Ochoa MT, Yamamura M, Burdick A, Sarno EN, Wagner M, Röllinghoff M, et al. (2003). Use of genetic profiling in leprosy to discriminate clinical forms of the disease. Science 301, 1527–1530. [DOI] [PubMed] [Google Scholar]
- Boxx GM, and Cheng G (2016). The Roles of Type I Interferon in Bacterial Infection. Cell Host Microbe 19, 760–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulut Y, Michelsen KS, Hayrapetian L, Naiki Y, Spallek R, Singh M, and Arditi M (2005). Mycobacterium tuberculosis heat shock proteins use diverse Toll-like receptor pathways to activate pro-inflammatory signals. J. Biol. Chem 280, 20961–20967. [DOI] [PubMed] [Google Scholar]
- Cangelosi GA, and Brabant WH (1997). Depletion of pre-16S rRNA in starved Escherichia coli cells. J. Bacteriol 179, 4457–4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang YM, Pinn ML, Karakousis PC, and Hung CF (2018). Intranasal Immunization with DnaK Protein Induces Protective Mucosal Immunity against Tuberculosis in CD4-Depleted Mice. Front. Cell. Infect. Microbiol 8, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortes T, Schubert OT, Rose G, Arnvig KB, Comas I, Aebersold R, and Young DB (2013). Genome-wide mapping of transcriptional start sites defines an extensive leaderless transcriptome in Mycobacterium tuberculosis. Cell Rep. 5, 1121–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damron FH, Oglesby-Sherrouse AG, Wilks A, and Barbier M (2016). Dual-seq transcriptomics reveals the battle for iron during Pseudomonas aeruginosa acute murine pneumonia. Sci. Rep 6, 39172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Larrañaga GF, Forastiero RR, Martinuzzo ME, Carreras LO, Tsarikt-sian G, Sturno MM, and Alonso BS (2000). High prevalence of antiphospholipid antibodies in leprosy: evaluation of antigen reactivity. Lupus 9, 594–600. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorner M, Brandt S, Tinguely M, Zucol F, Bourquin JP, Zauner L, Berger C, Bernasconi M, Speck RF, and Nadal D (2009). Plasma cell toll-like receptor (TLR) expression differs from that of B cells, and plasma cell TLR triggering enhances immunoglobulin production. Immunology 128, 573–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabri M, Stenger S, Shin DM, Yuk JM, Liu PT, Realegeno S, Lee HM, Krutzik SR, Schenk M, Sieling PA, et al. (2011). Vitamin D is required for IFN-gamma-mediated antimicrobial activity of human macrophages. Sci. Transl. Med 3, 104ra102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzosa EA, Morgan XC, Segata N, Waldron L, Reyes J, Earl AM, Giannoukos G, Boylan MR, Ciulla D, Gevers D, et al. (2014). Relating the metatranscriptome and metagenome of the human gut. Proc. Natl. Acad. Sci. USA 111, E2329–E2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giri PK, Kruh NA, Dobos KM, and Schorey JS (2010). Proteomic analysis identifies highly antigenic proteins in exosomes from M. tuberculosis-infected and culture filtrate protein-treated macrophages. Proteomics 10, 3190–3202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross JA, Johnston J, Mudri S, Enselman R, Dillon SR, Madden K, Xu W, Parrish-Novak J, Foster D, Lofton-Day C, et al. (2000). TACI and BCMA are receptors for a TNF homologue implicated in B-cell autoimmune disease. Nature 404, 995–999. [DOI] [PubMed] [Google Scholar]
- Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F, Aken BL, Barrell D, Zadissa A, Searle S, et al. (2012). GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 22, 1760–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Sherman BT, and Lempicki RA (2009). Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer AM, Mohanty KK, van Egmond D, Katoch K, Faber WR, Das PK, and Sengupta U (2007). Leprosy-specific B-cells within cellular infiltrates in active leprosy lesions. Hum. Pathol 38, 1065–1073. [DOI] [PubMed] [Google Scholar]
- Kapopoulou A, Lew JM, and Cole ST (2011). The MycoBrowser portal: a comprehensive and manually annotated resource for mycobacterial genomes. Tuberculosis (Edinb.) 91, 8–13. [DOI] [PubMed] [Google Scholar]
- Kim S (2015). ppcor: Partial and Semi-Partial (Part) Correlation. R package version 1.1. [Google Scholar]
- Kim J, Gross JA, Dillon SR, Min JK, and Elkon KB (2011). Increased BCMA expression in lupus marks activated B cells, and BCMA receptor engagement enhances the response to TLR9 stimulation. Autoimmunity 44, 69–81. [DOI] [PubMed] [Google Scholar]
- Kirchheimer WF, and Storrs EE (1971). Attempts to establish the armadillo (Dasypus novemcinctus Linn.) as a model for the study of leprosy. I. Report of lepromatoid leprosy in an experimentally infected armadillo. Int. J. Lepr. Other Mycobact. Dis 39, 693–702. [PubMed] [Google Scholar]
- Kralik P, Nocker A, and Pavlik I (2010). Mycobacterium avium subsp. para-tuberculosis viability determination using F57 quantitative PCR in combination with propidium monoazide treatment. Int. J. Food Microbiol 141, S80–S86. [DOI] [PubMed] [Google Scholar]
- Lahiri R, Randhawa B, and Krahenbuhl J (2005). Application of a viability-staining method for Mycobacterium leprae derived from the athymic (nu/nu) mouse foot pad. J. Med. Microbiol 54, 235–242. [DOI] [PubMed] [Google Scholar]
- Langfelder P, and Horvath S (2008). WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9, 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon A, Subramanian A, Pinchback R, Thorvaldsdóttir H, Tamayo P, and Mesirov JP (2011). Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima KM, dos Santos SA, Santos RR, Brandão IT, Rodrigues JM Jr., and Silva CL (2003). Efficacy of DNA-hsp65 vaccination for tuberculosis varies with method of DNA introduction in vivo. Vaccine 22, 49–56. [DOI] [PubMed] [Google Scholar]
- Liu PT, Wheelwright M, Teles R, Komisopoulou E, Edfeldt K, Ferguson B, Mehta MD, Vazirnia A, Rea TH, Sarno EN, et al. (2012). MicroRNA-21 targets the vitamin D-dependent antimicrobial pathway in leprosy. Nat. Med 18, 267–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loizou S, Singh S, Wypkema E, and Asherson RA (2003). Anticardiolipin, anti-beta(2)-glycoprotein I and antiprothrombin antibodies in black South African patients with infectious disease. Ann. Rheum. Dis 62, 1106–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez D, Montoya D, Ambrose M, Lam L, Briscoe L, Adams C, Modlin RL, and Pellegrini M (2017). SaVanT: a web-based tool for the sample-level visualization of molecular signatures in gene expression profiles. BMC Genomics 18, 824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzi JC, Trombone AP, Rocha CD, Almeida LP, Lousada RL, Malardo T, Fontoura IC, Rossetti RA, Gembre AF, Silva AM, et al. (2010). Intranasal vaccination with messenger RNA as a new approach in gene therapy: use against tuberculosis. BMC Biotechnol. 10, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowrie DB, Tascon RE, Bonato VL, Lima VM, Faccioli LH, Stavropoulos E, Colston MJ, Hewinson RG, Moelling K, and Silva CL (1999). Therapy of tuberculosis in mice by DNA vaccination. Nature 400, 269–271. [DOI] [PubMed] [Google Scholar]
- Lu LL, Chung AW, Rosebrock TR, Ghebremichael M, Yu WH, Grace PS, Schoen MK, Tafesse F, Martin C, Leung V, et al. (2016). A Functional Role for Antibodies in Tuberculosis. Cell 167, 433–443.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupoli TJ, Fay A, Adura C, Glickman MS, and Nathan CF (2016). Reconstitution of a Mycobacterium tuberculosis proteostasis network highlights essential cofactor interactions with chaperone DnaK. Proc. Natl. Acad. Sci. USA 113, E7947–E7956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay F, Woodcock SA, Lawton P, Ambrose C, Baetscher M, Schneider P, Tschopp J, and Browning JL (1999). Mice transgenic for BAFF develop lymphocytic disorders along with autoimmune manifestations.J. Exp. Med 190, 1697–1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madigan CA, Cambier CJ, Kelly-Scumpia KM, Scumpia PO, Cheng TY, Zailaa J, Bloom BR, Moody DB, Smale ST, Sagasti A, et al. (2017a). A Macrophage Response to Mycobacterium leprae Phenolic Glycolipid Initiates Nerve Damage in Leprosy. Cell 170, 973–985.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madigan CA, Cameron J, and Ramakrishnan L (2017b). A Zebrafish Model of Mycobacterium leprae Granulomatous Infection. J. Infect. Dis 216, 776–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsters SA, Yan M, Pitti RM, Haas PE, Dixit VM, and Ashkenazi A (2000). Interaction of the TNF homologues BLyS and APRIL with the TNF receptor homologues BCMA and TACI. Curr. Biol 10, 785–788. [DOI] [PubMed] [Google Scholar]
- Martinez AN, Lahiri R, Pittman TL, Scollard D, Truman R, Moraes MO, and Williams DL (2009). Molecular determination of Mycobacterium leprae viability by use of real-time PCR. J. Clin. Microbiol 47, 2124–2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modlin RL, Hofman FM, Taylor CR, and Rea TH (1982). In situ characterization of T lymphocyte subsets in leprosy granulomas. Int. J. Lepr. Other Mycobact. Dis 50, 361–362. [PubMed] [Google Scholar]
- Montoya D, Cruz D, Teles RM, Lee DJ, Ochoa MT, Krutzik SR, Chun R, Schenk M, Zhang X, Ferguson BG, et al. (2009). Divergence of macrophage phagocytic and antimicrobial programs in leprosy. Cell Host Microbe 6, 343–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montoya D, Inkeles MS, Liu PT, Realegeno S, Teles RMB, Vaidya P, Munoz MA, Schenk M, Swindell WR, Chun R, et al. (2014). IL-32 is a molecular marker of a host defense network in human tuberculosis. Sci. Transl. Med 6, 250ra114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemiec MJ, Grumaz C, Ermert D, Desel C, Shankar M, Lopes JP, Mills IG, Stevens P, Sohn K, and Urban CF (2017). Dual transcriptome of the immediate neutrophil and Candida albicans interplay. BMC Genomics 18, 696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuss AM, Beckstette M, Pimenova M, Schmuhl C, Opitz W, Pisano F, Heroven AK, and Dersch P (2017). Tissue dual RNA-seq allows fast discovery of infection-specific functions and riboregulators shaping host-pathogen transcriptomes. Proc. Natl. Acad. Sci. USA 114, E791–E800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochoa MT, Teles R, Haas BE, Zaghi D, Li H, Sarno EN, Rea TH, Modlin RL, and Lee DJ (2010). A role for interleukin-5 in promoting increased immunoglobulin M at the site of disease in leprosy. Immunology 131, 405–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Losada M, Castro-Nallar E, Bendall ML, Freishtat RJ, and Crandall KA (2015). Dual Transcriptomic Profiling of Host and Microbiota during Health and Disease in Pediatric Asthma. PLoS One 10, e0131819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittman KJ, Aliota MT, and Knoll LJ (2014). Dual transcriptional profiling of mice and Toxoplasma gondii during acute and chronic infection. BMC Genomics 15, 806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2013). R: A language and environment for statistical computing (R Foundation for Statistical Computing).
- Realegeno S, Kelly-Scumpia KM, Dang AT, Lu J, Teles R, Liu PT, Schenk M, Lee EY, Schmidt NW, Wong GC, et al. (2016). S100A12 Is Part of the Antimicrobial Network against Mycobacterium leprae in Human Macrophages. PLoS Pathog. 12, e1005705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridley DS (1957). The use biopsies in therapeutic trials in leprosy. Trans. R. Soc. Trop. Med. Hyg 51, 152–156. [DOI] [PubMed] [Google Scholar]
- Ridley DS, and Jopling WH (1966). Classification of leprosy according to immunity. A five-group system. Int. J. Lepr. Other Mycobact. Dis 34, 255–273. [PubMed] [Google Scholar]
- Rustad TR, Minch KJ, Brabant W, Winkler JK, Reiss DJ, Baliga NS, and Sherman DR (2013). Global analysis of mRNA stability in Mycobacterium tuberculosis. Nucleic Acids Res. 41, 509–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souza PR, Zárate-Bladés CR, Hori JI, Ramos SG, Lima DS, Schneider T, Rosada RS, Torre LG, Santana MH, Brandão IT, et al. (2008). Protective efficacy of different strategies employing Mycobacterium leprae heat-shock protein 65 against tuberculosis. Expert Opin. Biol. Ther 8, 1255–1264. [DOI] [PubMed] [Google Scholar]
- Stanley SA, Johndrow JE, Manzanillo P, and Cox JS (2007). The Type I IFN response to infection with Mycobacterium tuberculosis requires ESX-1-mediated secretion and contributes to pathogenesis. J. Immunol 178, 3143–3152. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Kuehn H, Gould J, Tamayo P, and Mesirov JP (2007). GSEA-P: a desktop application for Gene Set Enrichment Analysis. Bioinformatics 23, 3251–3253. [DOI] [PubMed] [Google Scholar]
- Tascon RE, Colston MJ, Ragno S, Stavropoulos E, Gregory D, and Lowrie DB (1996). Vaccination against tuberculosis by DNA injection. Nat. Med 2, 888–892. [DOI] [PubMed] [Google Scholar]
- Teles RM, Graeber TG, Krutzik SR, Montoya D, Schenk M, Lee DJ, Komisopoulou E, Kelly-Scumpia K, Chun R, Iyer SS, et al. (2013). Type I interferon suppresses type II interferon-triggered human anti-mycobacterial responses. Science 339, 1448–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thänert R, Goldmann O, Beineke A, and Medina E (2017). Host-inherent variability influences the transcriptional response of Staphylococcus aureus during in vivo infection. Nat. Commun 8, 14268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas-Romero F, Guitierrez-Najera N, Mendoza-Hernández G, Ortega-Bernal D, Hernández-Pando R, and Castañón-Arreola M (2016). Secretome profile analysis of hypervirulent Mycobacterium tuberculosis CPT31 reveals increased production of EsxB and proteins involved in adaptation to intracellular lifestyle. Pathog. Dis 74, ftv127. [DOI] [PubMed] [Google Scholar]
- Wei TS, Villam (2017). R package “corrplot”: Visualization of a Correlation Matrix, Version 0.84.
- Wesolowska-Andersen A, Everman JL, Davidson R, Rios C, Herrin R, Eng C, Janssen WJ, Liu AH, Oh SS, Kumar R, et al. (2017). Dual RNA-seq reveals viral infections in asthmatic children without respiratory illness which are associated with changes in the airway transcriptome. Genome Biol. 18, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermann AJ, Förstner KU, Amman F, Barquist L, Chao Y, Schulte LN, Müller L, Reinhardt R, Stadler PF, and Vogel J (2016). Dual RNA-seq unveils noncoding RNA functions in host-pathogen interactions. Nature 529, 496–501. [DOI] [PubMed] [Google Scholar]
- Yamamura M, Uyemura K, Deans RJ, Weinberg K, Rea TH, Bloom BR, and Modlin RL (1991). Defining protective responses to pathogens: cytokine profiles in leprosy lesions. Science 254, 277–279. [DOI] [PubMed] [Google Scholar]
- Young D, Lathigra R, Hendrix R, Sweetser D, and Young RA (1988). Stress proteins are immune targets in leprosy and tuberculosis. Proc. Natl. Acad. Sci. USA 85, 4267–4270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Roschke V, Baker KP, Wang Z, Alarcón GS, Fessler BJ, Bastian H, Kimberly RP, and Zhou T (2001). Cutting edge: a role for B lymphocyte stimulator in systemic lupus erythematosus. J. Immunol 166, 6–10. [DOI] [PubMed] [Google Scholar]
- Zimmermann M, Kogadeeva M, Gengenbacher M, McEwen G, Mollenkopf HJ, Zamboni N, Kaufmann SHE, and Sauer U (2017). Integration of Metabolomics and Transcriptomics Reveals a Complex Diet of Mycobacterium tuberculosis during Early Macrophage Infection. mSystems 2, e0057–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the dual RNaseq data for leprosy lesions reported in this paper is GEO: GSE125943. Signature Visualization Tool (SaVanT) can be accessed online at http://newpathways.mcdb.ucla.edu/savant.