

Abstract
Organophosphorus (OP) nerve agents and pesticides present significant threats to civilian and military populations. OP compounds include the nefarious G and V chemical nerve agents, but more commonly, civilians are exposed to less toxic OP pesticides, resulting in the same negative toxicological effects and thousands of deaths on an annual basis. After decades of research, no new therapeutics have been realized since the mid-1900s. Upon phosphylation of the catalytic serine residue, a process known as inhibition, there is an accumulation of acetylcholine (ACh) in the brain synapses and neuromuscular junctions, leading to a cholinergic crisis and eventually death. Oxime nucleophiles can reactivate select OP-inhibited acetylcholinesterase (AChE). Yet, the fields of reactivation of AChE and butyrylcholinesterase encounter additional challenges as broad-spectrum reactivation of either enzyme is difficult. Additional problems include the ability to cross the blood brain barrier (BBB) and to provide therapy in the central nervous system. Yet another complication arises in a competitive reaction, known as aging, whereby OP-inhibited AChE is converted to an inactive form, which until very recently, had been impossible to reverse to an active, functional form. Evaluations of uncharged oximes and other neutral nucleophiles have been made. Non-oxime reactivators, such as aromatic general bases and Mannich bases, have been developed. The issue of aging, which generates an anionic phosphylated serine residue, has been historically recalcitrant to recovery by any therapeutic approach—that is, until earlier this year. Mannich bases not only serve as reactivators of OP-inhibited AChE, but this class of compounds can also recover activity from the aged form of AChE, a process referred to as resurrection. This review covers the modern efforts to address all of these issues and notes the complexities of therapeutic development along these different lines of research.
Keywords: acetylcholinesterase, butyrylcholinesterase, organophosphorus, reactivation, resurrection
Graphical Abstract
In memory of Dr. Douglas Cerasoli, a beloved colleague and friend, who worked tirelessly to improve medical countermeasures against organophosphorus chemical nerve agents at the US Army Medical Research Institute of Chemical Defense and who left his family, friends and colleagues too early in life
1. Introduction and History
In this review, we will outline the development and uses of organophosphorus compounds and their biological targets, the cholinesterases.
1.1. Historical development of organophosphorus pesticides and nerve agents
Following the First World War, Germany found itself in a state of complete distress and devastation. The Third Reich strategists desired to become more self-sufficient during this time of desolation and strove to reduce Germany’s reliance on imported food. Gerhard Schrader, a chemist working at the I. G. Farben chemical company, had been assigned the task of designing and synthesizing new insecticides in efforts to protect food production.[1] The first pesticides were based on fluorine and sulfur, but proved to be ineffective, thus Schrader moved his focus to phosphorus and cyanide derivatives.
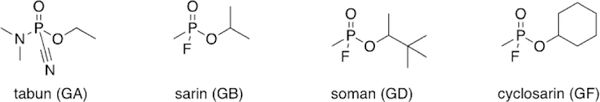
On December 23, 1936, Schrader synthesized “Preparation 9/ 91”, which proved to be extremely toxic. In fact, Schrader himself was hospitalized after working with small quantities of the sample, exhibiting a variety of symptoms including difficulty breathing, impaired vision, and dizziness.[2] Schrader’s co-workers were also inadvertently exposed to the compound and displayed similar symptoms—all who were exposed took weeks to recover. The sample to which they were exposed was ethyl(N,N-dimethylamido)-phosphoro-cyanidate, known today as tabun (Figure 1). The name is derived from the German word for taboo. Indeed, tabun affects the nervous system in a way that the victim’s bodily functions are no longer under the brain’s control. An early sample was administered as a vapor to apes and resulted in lethal effects. This lethality was primarily observed in mammals and not insects, making tabun a poor insecticide, despite Schrader’s initial intent. However, due to its obvious toxicity to humans, I. G. Farben alerted the German military concerning the potential of this compound to be weaponized. Schrader had inadvertently discovered a new class of toxic chemicals that are now more commonly referred to as nerve agents. Tabun (GA) became the first in the G-series, “G” standing for “German” (Figure 1).
Figure 1.
Select examples of G-series organophosphorus nerve agents.
Despite the peace accord laid out in the Treaty of Versailles, German scientists continued chemical weapon development. During WWI, some of the utilized chemical warfare agents, such as phosgene and mustard gas, took hours to days to cause lethality. The potential of this new organophosphorus nerve agent was recognized, as lethality occurred within 20 min. Scientists began studying the physiological effects of tabun and how to further increase its lethality. In June 1939, Schrader developed Substance 146, or isopropylmethylphosphonofluoridate. This compound is now more commonly referred to as sarin, an acronym for its four creators : Schrader, Ambos, RUdiger, and Van der Linde (Figure 1). Although more difficult to synthesize, sarin (GB) was found to be 500 times more lethal than cyanide.
In 1943, Richard Kuhn was hired to determine the mechanism of action of these compounds. Kuhn determined that these compounds inhibit acetylcholinesterase (AChE), resulting in the buildup of acetylcholine in synapses and the subsequent prevention of electrical termination of signals to muscles in the body, due to the muscle cells being overstimulated by excess neurotransmitter. Kuhn screened a wide variety of organophosphorus agents to test the various levels of inhibition of AChE. Upon replacing the isopropyl group with a pinacolyl group, he discovered an even more potent nerve agent than tabun, one that was twice as lethal as sarin. Compound 25 075 or 3,3-dimethylbutan-2-yl-methylphosphonofluoridate (soman, GD), deactivated AChE within two minutes and readily penetrated the skin to further increase lethality (Figure 1).[3]
Fortunately, Germany did not deploy these nerve agents during WWII, although they had significant stockpiles. Despite their chemical advantage, Adolf Hitler elected not to use the developed toxins; some suspect it was due to his fear of retaliation by the allies with similar weapons, while others suspect that due to his previous exposure to chemical weapons while a soldier in WWI, he did not want to subject other soldiers to the same fate.[4] Shortly after the war concluded, Russia found evidence of these chemical agents by uncovering old lab notebooks that detailed the synthesis of sarin. During the Cold War in the 1950’s, the United States and United Kingdom collaborated to develop analogous nerve agents. They screened nerve agents, like sarin and cyclohexylmethylphosphonofluoridate (cyclosarin, GF), and synthesized new sarin-like derivatives to determine which compounds would function as the best weapons (Figure 1). The most expensive agent, soman, was determined to be the most lethal agent, but as sarin was still sufficiently toxic and cheaper to synthesize, sarin was developed and stockpiled instead.
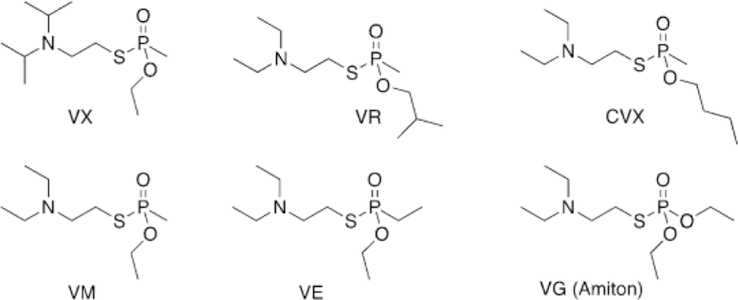
Following the conclusion of WWII, the field of insecticide and pesticide research became increasingly popular. In 1952, a researcher from the Imperial Chemical Industries, Ranajit Gosh, discovered a new class of nerve agents. Gosh and Newman were investigating the use of organophosphorus agents as potential pesticides by synthesizing nitrogen and sulfur analogues. One such compound was able to effectively kill lice and was placed on the market under the trade name Amiton.[5] Eventually, the company had to remove Amiton (O,O-diethyl-S[2-(diethylamino)ethyl] phosphorothioate) from the market due to its toxicity. This compound became part of what is known as the V-series, “V” meaning “venomous”, and is referred to as VG (Figure 2). Currently, structurally similar organophosphates, such as echothiophate, are used as anti-gluacoma treatments, despite having some toxicity.[6]
Figure 2.
Select examples of V-series organophosphorus nerve agents.
A similar compound, ethyl-N-2-diisopropylaminoethylmethylphosphonothiolate, was developed in the partner company in the US, later renamed VX (Figure 2). Some other well-known isomers include S-(diethylamino)ethyl-O-ethyl ethylphosphonothioate (VE) and S-2-(diethylamino)ethyl-O-ethyl methylphosphonothioate (VM, Figure 2). Studies then showed the V-series agents to be far deadlier than previously discovered agents. Under average temperature conditions, these compounds persist for days, have no odor, can be administered as a vapor or gas, and only require 20 mg kg-1 to be lethal, rendering these agents to be the mostly deadly known to mankind at the time of their discovery.[7] VX proved to be stable for months in cold temperatures, so it was selected as the US’s weapon of choice and thus stockpiled during the Cold War (Figure 2).
The Soviet Union began their own chemical weapon development to try and remain on an equal level to the US. At the Scientific Research Institute No. 42, Ivin, Soborovsky, and Shilakova developed Substance 33, an isomer of VX (Figure 2).[8] N,N-Diethyl-2-[methyl-(2-methylpropoxy)phosphoryl]sulfanylethanamine (VR or “Russian VX”, Figure 2) was found to have a similar lethal dose to VX, while reducing the treatment window due to its rapid inhibition of AChE.
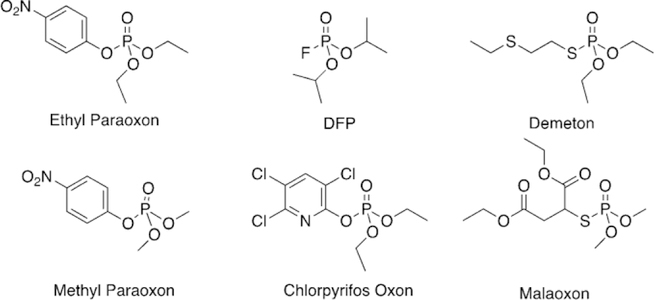
After WWII, the US began to develop organophosphorus pesticides in large quantities. Diethyl(dimethoxyphosphinothioyl) thiobutanedioate, also known as malathion, was discovered in 1950. In 1951, Schrader’s continued development of new insecticides resulted in the synthesis of the insecticide demeton (Figure 3). Demeton is a mixture of the thionoand thiolo-isomers of O,O-diethyl-2-ethylmercaptoethylphosphorothioate, thereby introducing a new class of insecticides possessing a thioether group. Today, a wide range of organophosphorus compounds is still available as pesticides. Subsequently, other compounds, such as chlorpyrifos, paraoxon, and tebupirimphos, were synthesized, showing lower environmental persistence as compared to previously synthesized pesticides. Their popularity increased after the ban of organochlorine insecticides in the 1970s. Organophosphorus pesticides are used in very low concentrations in order to pose less harm to users and food. Though dilute, there is still potential for these pesticides to cause harm to agricultural workers who handle these chemicals. The US Food and Drug Administration has lowered the limits of pesticides in foods sold to consumers in the US, but the world has varying levels of regulation, thus leading to 3 million global cases of pesticide poisoning per year, with an estimated 220 000 deaths annually.[9] The majority of these cases are in developing countries, and most cases are a result of poor working conditions, improper handling, inadequate regulation, or intentional self-harm.
Figure 3.
Select examples of organophosphorus pesticides in their oxon form.
1.2. Recent uses of organophosphorus pesticides and nerve agents for terrorism
During the Iran–Iraq War in the 1980s, Iraq used chemical warfare agents against Iran. Iraq claims to have used 600 tons of sarin and 140 tons of tabun against enemy forces.[10] The onslaught killed nearly 5000 Iranians and over 100 000 were hospitalized. In 1988, Iraq even attacked its own citizens in Halabja, killing over 5000 and injuring over 7000.
On March 20, 1995, cult members from the Aum Shinrikyo sect punctured bags of homemade sarin on a subway in Tokyo, Japan.[11] Five plastic bags of liquid sarin were deployed during rush hour in order to “speed up the pending apocalypse”. Though only a dozen people were killed, over 5500 were hospitalized. If the sarin had been deployed in a different manner, it is hypothesized that the release could have killed thousands.
Syria has made use of organophosphorus compounds as part of its civil war over the past few years. Damascus in Syria was the location of a sarin attack on the morning of August 21, 2013, when rockets filled with the agent struck the rebel suburbs of the capital, killing and injuring thousands.[12] Around 1429 were found dead, including 426 children. Syria was the site of another attack on April 4, 2017—more than 89 people were killed and another 541 injured in the rebel-held town of Khan Sheikhoun after the Syrian government’s air strike.[13] Traces of one of the decomposition products of sarin, isopropylmethylphosphonic acid, were detected in the urine and blood of some of the victims.
On February 13, 2017, Kim Jong-nam was murdered in an airport in Kuala Lumpur. Kim was the half-brother of Kim Jongun, the current leader of North Korea.[14] Kim Jong-nam was attacked by two women who smeared VX on his face, and he died shortly thereafter, while on the way to the hospital.
On March 4, 2018, a Russian spy, Sergei Skripal and his daughter were found unconscious on a bench in the UK.[15] Both were hospitalized and exhibited symptoms of organophosphorus nerve agent exposure. Reports detail that the Skripals were exposed to a nerve agent called Novichok, a compound that, at present, has an unconfirmed structure. The substance was reportedly placed on the front door of the Skirpal’s home using a modified perfume bottle where the Skripals later contacted the substance. An additional 21 doctors, first responders, and bystanders had to be treated for exposure as well. Later, on June 30, 2018, a second Novichok poisoning was determined that left Dawn Sturgess dead and hospitalized Charlie Rowley in Amesbury. A perfume bottle was again the mode of exposure. Rowley was exposed to some while putting the applicator and bottle together and Sturgess sprayed the perfume (agent) directly on her wrists.[15]
Although nerve agents receive the most publicity, organophosphorus pesticides still present significant risk for terrorist use, but they also continue to result in incidental ingestion or exposure. For instance, in 2013, 23 school children in India were killed after ingesting monocrotophos, which had contaminated their school lunch. The cooking oil used to prepare the meals was contaminated by the pesticide through storage of the cooking oil in a container that previously had been used for pesticide storage, resulting in the death of the children.[16] This problem seems to be of major concern as the diet of children in India has been found to contain almost 40 times higher concentrations of pesticides as compared to the US, based on the identification of metabolites in children’s urine.[17] The ease of access to such pesticides and the incidents caused by contamination present significant risk to civilian populations.
Additional incidental exposures have been noted in air travel professionals, in addition to passengers, and is now referred to as aerotoxic syndrome. The proposed cause of the toxicity is the exposure to tri-o-cresyl phosphate (TOCP), a component of jet hydraulic fluids and engine oils, which is converted physiologically to the toxic metabolite 2-(o-cresyl)-4H-1,3,2-benzodioxaphosphoran-2-one (CBDP). CBDP is a potent inhibitor of cholinesterases and is believed to lead to the neurological signs that are characteristic of aerotoxic syndrome.[18, 19]
1.3. Acetylcholinesterase structure and function in the native, inhibited, and aged states
Acetylcholinesterase (AChE) is an extremely efficient enzyme located throughout the body, namely at the synapses of the central nervous system, neuromuscular junctions in the peripheral nervous system, and bound to erythrocyte membranes in blood.[20] AChE is involved in the neurosynaptic communication process, specifically the process of maintaining proper levels of the neurotransmitter acetylcholine (ACh) at the synaptic cleft. The enzyme accomplishes this regulation by means of catalytic hydrolysis of ACh, forming acetate and choline, which are then used to regenerate ACh in the peripheral nerve. The catalytic efficiency (kcat/KM) for ACh hydrolysis in humans, a measure of the catalytic hydrolysis rate (kcat) corrected for binding affinity of the substrate (KM), is 1.50 × 109 m-1 s-1, and the turnover rate of the enzyme equates to hydrolyzing 25 000 molecules of ACh per second.[21]
The active site of AChE was confirmed by crystallographers in 1991, and it consists of a catalytic triad of serine (S203), histidine (H447), and glutamate (E334) (Figure 4). This catalytic triad is common to a variety of enzymes classified as serine hydrolases.[22, 23] In addition to the catalytic triad, two subsites aid in the hydrolysis of acetylcholine : the oxyanion hole and the cation-binding pocket (Figure 4). The oxyanion hole is comprised of the backbone peptide N-H groups of G121, G122, and A204. These residues are aligned to form strong hydrogen bonds with the carboxyl oxygen of ACh and aid in stabilizing the negative charge generated during the hydrolysis reaction. The cation-binding pocket is comprised of W86 and E202, and it stabilizes the choline moiety in the active site through cation–p interactions. Acetylcholine, upon reaching the catalytic site, undergoes nucleophilic attack at the carbonyl carbon by S203, forming a covalent bond with the enzyme. Through subsequent reactions with water activated in the active site, ACh is cleaved into acetate and choline, and the native AChE enzyme is regenerated (Figure 5). An additional peripheral binding site is located at the entrance of the AChE gorge and is capable of binding cationic and aromatic substrates. This site of AChE has inspired a great deal of research as a location by which a ligand can be tethered to a drug candidate to increase overall binding affinity.
Figure 4.
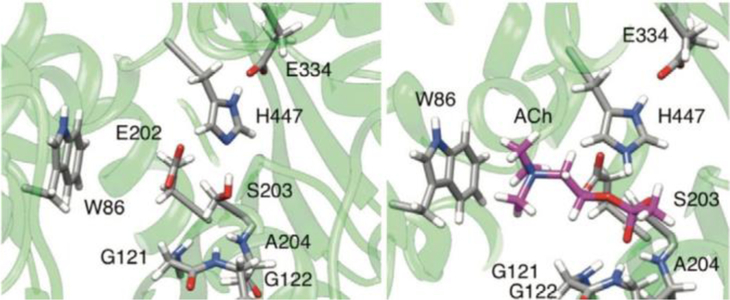
Catalytic active site of AChE (S203, H447, E334), the oxyanion hole (G121, G122, A204), and cation-binding pocket (W86, E202) without ACh being bound (left) and with ACh (right).
Figure 5.
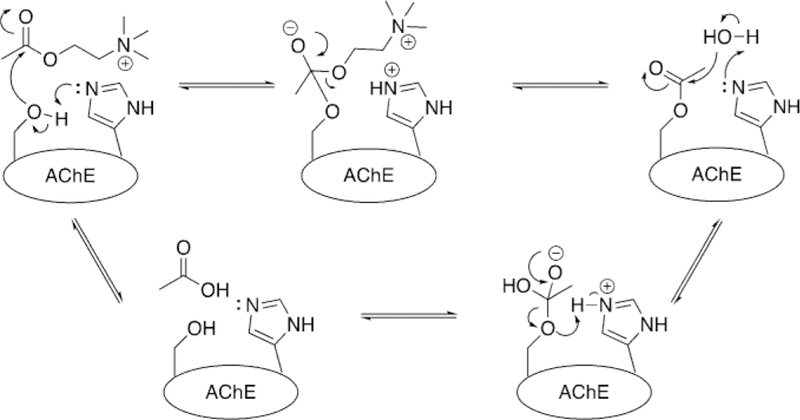
Hydrolysis of acetylcholine by native AChE.
Given the rapid hydrolysis rates of ACh, the cholinesterase community was surprised when researchers reported that the active site does not reside on the protein surface, but is in fact buried in the interior of the protein, connected to the exterior by a narrow gorge roughly 20 A deep.[23] Interestingly, the gorge is so narrow that in the crystallographic orientation, the narrowest point, referred to as the gorge bottleneck, is too compact to accommodate the movement of the substrate from the exterior of the protein to the buried catalytic site (Figure 6).[24, 25]
Figure 6.
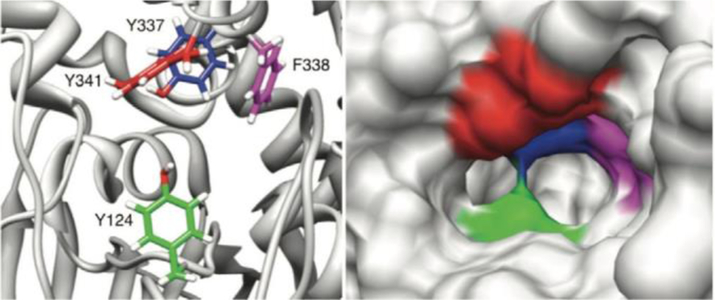
Aromatic residues (left) composing the gorge bottleneck and a space filling model (right) showing the width of the gorge.
In order for the enzyme to operate at near-diffusion limits with an active site buried at the bottom of a narrow 20 A deep gorge, substrate trafficking into the enzyme’s interior must be extremely efficient. Although in the crystallographic orientation, the gorge is too narrow for a substrate to pass from the protein surface into the catalytic site, molecular dynamics simulations have suggested a “breathing” motion that occurs on a 10 ns period, a frequency that is sufficient for the hydrolysis of 25 000 molecules of ACh per second.[26] This breathing motion is a combination of residue side chain motions as well as concerted motion of two loops in the AChE structure : the W-loop and acyl loop. These loops make up two of the walls that define the AChE gorge, spanning from the mouth of the gorge down to the catalytic site. As the two loops move, the gorge radius has been observed to fluctuate between 0.75 A and 2.5 A, although in the absence of substrate, the gorge is rarely wide enough to accommodate ACh transfer.[26]
As the gorge bottleneck can fluctuate to allow a substrate into the catalytic site of AChE, the mechanism to transfer the substrate from the bulk solution to the protein interior has been a subject of significant investigation. Association of ACh with the enzyme is an obvious requirement for rapid catalysis and occurs through a binding site at the mouth of the gorge, which has been named the peripheral anionic site (PAS).[27, 28] The PAS is a region of the protein with an abundance of aromatic residues that forms cation–p interactions with the choline portion of ACh, and includes W286, Y72, Y124 and the anionic D74. Previous studies have identified a large electrostatic dipole, calculated at 505 Debye, along the gorge axis of AChE that essentially pulls ACh from the PAS at the enzyme surface, past the gorge bottleneck, and into the catalytic site in the protein interior.[29] However, this electrostatic dipole would also seem to prohibit exit of the choline hydrolysis product, trapping it in the gorge.[30] Researchers have long debated the presence of a “back door” to the catalytic site, with most interest being focused on a thin area of the gorge wall along the W-loop region (C65–C92) and with W86 acting as a gate-keeper.[31] A computational study that modeled the behavior of the reaction product for acetylthiocholine hydrolysis in the AChE gorge indicated that there are three separate paths for thiocholine to exit the catalytic site. In the majority of simulations, totaling 27 out of 40 trajectories, thiocholine exited through the backdoor when the residues W86, V132, and G448 opened the pathway by means of concerted motions.[32] Recent work involving kinetic crystallography experiments have also confirmed the fluctuations leading to backdoor opening in the AChE gorge, thereby corroborating the computational hypothesis.[33, 34]
In understanding the catalytic cycle of acetylcholine hydrolysis by AChE, the reason for the toxicity of organophosphorus (OP) compounds becomes apparent. OPs, upon reacting with the catalytic serine and on dissociation of their leaving group they form a tetrahedral phosphonate (or phosphate) that resides in the catalytic site of AChE and prevents any further ACh hydrolysis (Figure 7). As nucleophilic attack at phosphorus is significantly slower than at carbon, water in the active site is not sufficiently nucleophilic to react with the phosphylated serine’s P center in order to cleave the covalently bound OP from the catalytic site. Thus, covalent modification of the catalytic S203 residue results in the inability of AChE to bind and hydrolyze ACh, thereby leading to a rapid buildup of ACh and subsequent overstimulation of ACh receptors.
Figure 7.
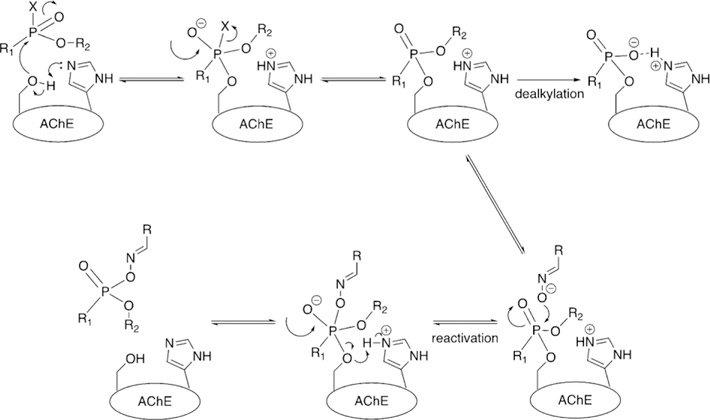
Inhibition and aging of AChE with a model phosphonate nerve agent.
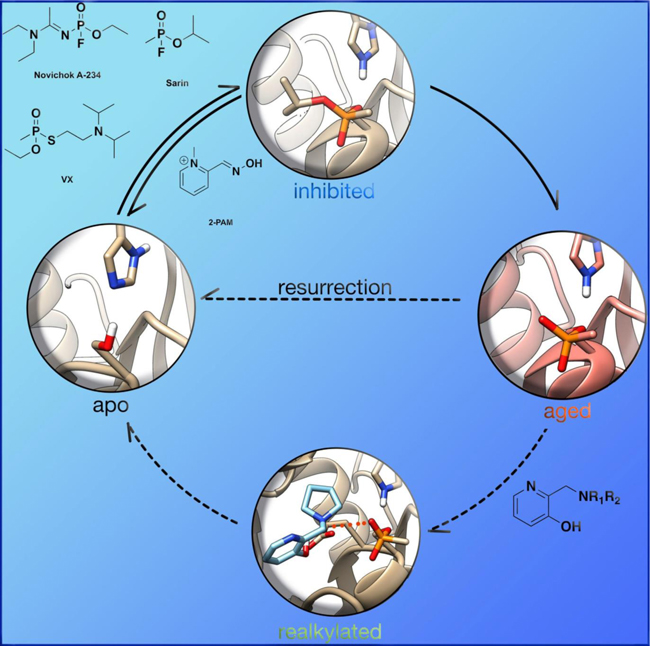
OP nerve agent deactivation of AChE occurs in two separate stages. The first stage, referred to simply as inhibition, is the formation of a covalent P-O(Ser) bond between the OP and catalytic serine of AChE (Figure 7). At this point, introduction of strong nucleophiles to the active site, mainly in the form of pyridinium oximes (Figure 8), can cleave the P-O(Ser) bond, regenerating the catalytic activity of the enzyme, a process called reactivation (Figure 7). The second inhibitory state was observed in early experimental studies, when the efficacy of oximes at cleaving the P-O(Ser) bond would decrease over time, and the enzyme was therefore referred to as “aged” (Figure 7).[36] Aging was later determined to correspond to a secondary reaction of the OP–AChE adduct, which for the common nerve agents is the O-dealkylation of the phosphylated center.[37] The O-dealkylation results in an anionic phosphylated serine residue, rendering the aged form to be resistant to nucleophilic attack by oximes. Strong hydrogen-bonding interactions with H447 have been postulated for stabilizing this anionic phosphylated serine residue.[38, 39] After the enzyme has undergone aging, it was considered to be un-reactivatable (until 2018, more on that later). Aging rates (Table 1) vary between OPs, but the range of aging half-times can span from roughly 37 h (VX) to a mere 4 min (GD).[40, 41]
Figure 8.
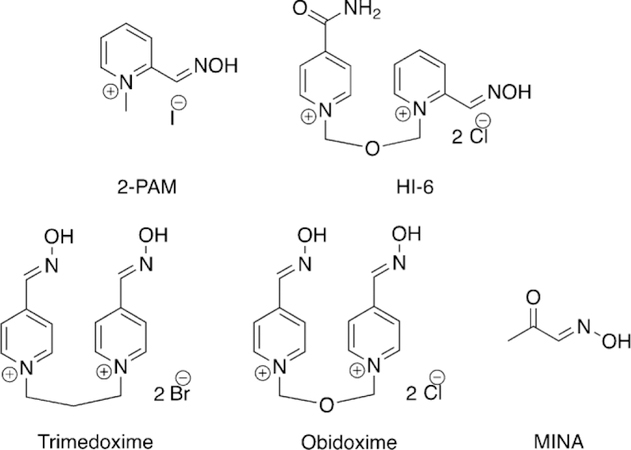
Examples of oximes administered for treatment of organophosphorus poisoning.
Table 1.
Aging half-times for various organophosphorus compounds.[35]
| OP | Aging half-time [h] |
|---|---|
| tabun (GA) | 19.2 |
| sarin (GB) | 3.0 |
| soman (GD) | 0.07 |
| cyclosarin (GF) | 7.0 |
| VX | 36.5 |
| VR | 138.6 |
| CVX | 32.2 |
| ethyl paraoxon | 31.5 |
| methyl paraoxon | 3.7 |
Wilson first reported the concept of reactivation in 1951 for OP-inhibited AChE, as he showed that incubation of two day old OP-inhibited AChE with concentrated solutions of choline or hydroxylamine for a few days regenerated about 75–90 % native AChE.[42] Wilson further showed that hydroxamic acids are better nucleophiles and can regenerate 96 % of the diisopropylfluorophosphate (DFP, Figure 3) bound AChE in 24 h, while hydroxylamine could only generate 19 % of the enzyme in the same period of time.[43] Following initial studies by Wilson, several other groups utilized different nucleophiles to further explore this concept.[44–50] However, for this approach to work effectively as a therapeutic, one needs to make a potent nucleophile that is selective for the OP-inhibited cholinesterase and shows fast reactivation, while minimizing any cross reactions and ensuring that extremely high concentrations are not required, so as to avoid severe immunological responses.
From the aforementioned reports on the reactivation of OPbound AChE, it could be concluded that other N-OH or oxime nucleophiles might also act as reactivators, and most oximes are mildly acidic. Based on this hypothesis, Rutland et al. evaluated several acidic oximes against GB-inhibited AChE, which led to the discovery of pyrimidine-2-hydroxamic acids and monoisonitrosoacetone (MINA) (Figure 8) that showed about 90 % reactivation of AChE after 15 min with modest concentrations.[49] Simultaneously, Holmes and Robins showed that treatment with MINA and pyridine-2-aldoxime methiodide (2-PAM, Figure 8) showed reactivation of AChE in rats after OP exposure.[50]
Although these early results opened an exciting avenue for finding better oxime-based reactivators, it should be noted that the OP compounds utilized in some of these experiments for AChE inhibition were pesticides, and not authentic nerve agents. It turned out to be very challenging to develop a selective nucleophile that can reactivate nerve-agent-inhibited AChE before the rapid aging step takes place. Another challenge in designing an oxime is that the considered candidates should be able to cross the blood–brain barrier (BBB) so as to reach OP-inhibited AChE in the central nervous system (CNS). Nonetheless, many attempts have been made in the last few decades to develop oximes that will work against a wide spectrum of nerve-agent-inhibited AChE, and in a timely manner.[51–55]
Nerve agents, and some pesticides, possess a stereogenic center at phosphorus, with GD possessing a second stereogenic center at the pinacolyl carbon atom. It has been observed experimentally that one stereoisomer, generally the SP (P-) isomer, is much more toxic than the RP (P + ) isomer.[56] This specificity is imparted through the arrangement of groups on the OP in relation to the protein structure in the active site of AChE, leading to more favorable binding of the Pstereoisomers.
In addition to the immediate symptoms of OP toxicity that are tied to the increase in ACh concentrations at the neuromuscular junction, exposure to AChE inhibitors has also demonstrated long-term side effects, especially in cases of chronic exposure such as agricultural settings.[57, 58] One such study has suggested the cause of these long-term symptoms may be due to removal of the inhibited enzyme from the synapse following prolonged inhibition.[59] The exact implication of chronic exposure and their effects are still being actively investigated.
1.4. Butyrylcholinesterase structure and function in the native, inhibited, and aged states
Butyrylcholinesterase (BChE), first discovered as pseudocholinesterase, is a native enzyme that is closely related to AChE.[60] Both enzymes belong to the a/b-hydrolase family, have the same catalytic triad located inside an approximate 20 A deep gorge; have similar structural features, such as the oxyanion hole and choline binding pocket; have almost identical number of residues and very similar tertiary structure; possess very high catalytic efficiency (almost at the diffusion-controlled limit); and are inhibited by OP nerve agents through a similar inhibition mechanism.[61] In addition to having almost the same number of amino acid residues, their sequence similarity is also quite high at 54 %. Critical components of the respective active sites of AChE and BChE have almost the same structure.
Despite the mentioned consistencies, there are significant differences among the two enzymes. AChE has fewer glycosylation sites than BChE has, thereby affecting the enzymes’ circulatory lifetime in the body as well as the folding, stability, and many other properties. BChE is so heavily glycosylated that four of the glycosylation sites were deleted in order to get a good X-ray crystal structure.[61] A major difference in their quaternary structure is the formation of a homo-dimer in the case of AChE at high concentrations, whereas BChE is homo-tetrameric.[61–63] Furthermore, the subunits of the homo-dimer in AChE are anti-parallel, while the openings of BChE subunits are parallel. Thus, the orientation of the helices forming the oligomerization motif are at an angle of about 458 in the case of BChE, rather than being anti-parallel for AChE. Recently, cryoEM techniques have been used to determine the three-dimensional structure of a hBChE tetramer demonstrating that the base of the tetramers are oriented like a propeller rather than being situated flat as has been previously proposed.[64]
The most important difference at the molecular level is that the BChE lacks six aromatic amino acids out of the fourteen that line the catalytic gorge of AChE. This disparity makes the gorge of BChE nearly double the width (ca. 13 A) of AChE’s gorge (ca. 6 A), thereby accounting for almost 300 A3 of extra available volume in BChE (Figure 9).[24, 25] For this reason, the gorge and active site domain for BChE is more accessible for a wide range of substrates and inhibitors. For example, the catalytic turnover for ACh is much higher for AChE than BChE.[65, 66] However, for a larger substrate, such as butyrylthiocholine (BCh), the catalytic efficiency for BChE is about 100 times greater than for AChE.[67]
Figure 9.

Visual comparison of the different widths and volumes of the (left) AChE and (right) BChE gorges.
There have been several suggestions regarding the physiological role of BChE such as neuronal function, hydrolysis of gamino-butyrylcholine; morphogenesis, cytogenesis, and tumorigenesis; hydrolysis of ACh at central nervous system synapses replacing AChE function; and converting the ß-amyloid form into more toxic forms in Alzheimer patients. However, none of these roles have been conclusively determined.[68–74] In fact, a very early study showed that dogs treated with selective inhibitors of BChE (about 95 % serum BChE inhibition) had no signs of toxicity. This report was followed by the discovery of a genetic variation named silent BChE, in which it was shown that 1 out of 100 000 people in Europe and America do not have BChE in their bodies.[75, 76] Further, discovery of the Vysya community in Coimbatore, India showed that about 1 in 24 people have this genetic variation.[77] Despite the absence of BChE in these individuals, there were no signs of any physical or brain disability in this population, supporting the suggestion that BChE may not have a direct physiological role.[78] Yet, recently it has been determined that BChE may serve to regulate the hydrolysis of the peptidic hormone ghrelin. A study evaluated the increased plasma levels of BChE in mice for cocaine hydrolysis and the mice showed less aggressive behavior than BChE knockout mice and control mice. The study concluded that the reason for this decrease in aggression, stress, and anxiety was lower levels of ghrelin in the plasma as a result of the hydrolysis facilitated by BChE.[79]
BChE is inhibited by OP compounds in a similar manner as AChE and also undergoes a similar aging process after OP inhibition (Figure 7).[80] The aging process is even faster for BChE than for AChE,[81, 82] and aged BChE is similarly recalcitrant to oxime reactivation.
To design oximes that can reactivate OP-bound BChE at a desirable rate, it is crucial to understand how an oxime binds to BChE, and which residues are the key players in this process. We carried out parallel studies in which oxime binding to AChE and BChE was studied by the same molecular docking protocol.[83, 84] We utilized several of the known oximes and BChE crystal structures with and without an OP molecule being bound in the active site for these simulations and identified important interactions between the different components of various oximes and BChE.
We found that monopyridinium oximes, such as 2-PAM (Figure 8), could have two binding sites with native BChE, that is, in the absence of an OP molecule at the catalytic serine. The first binding site is located at the choline binding pocket, aligned parallel to the W82 residue, while the second binding site utilizes hydrogen-bonding interactions at the oxyanion hole and simultaneously face-to-edge interactions with W231. Although in the presence of an OP in BChE, the second binding site is blocked, we found two binding sites for 2-PAM. Relatively more stable binding utilizes p–p interactions with the W82 residue sitting close to the bound OP molecule, while the second binding site is located at the mouth of the gorge. These data were in agreement with some experimental data obtained by Cerasoli and co-workers at the US Army Medical Research Institute of Chemical Defense that showed that two molecules of 2-PAM interact with BChE at the same time.[85] Furthermore, the first binding site of the 2-PAM-BChE complex is in excellent agreement with previous experimental findings from Lockridge and co-workers.[86]
These docking simulations also predicted that bis-pyridinium oximes (Figure 8) have relatively high affinities of binding to BChE when compared with monopyridinium oximes. Indeed, bis-pyridinium oximes utilize two p–p stacking interactions in the active site of BChE, with W82 and with F329 or Y332 residues, which suggests that these aromatic residues are essential for binding of the oximes with BChE. Out of all the oximes, HI-6 was predicted to have very good binding affinity to OP-inhibited BChE and with an appropriate orientation of the oxime unit to reactivate the OP-bound BChE. We also found that the D70 residue often interacts with the positively charged oximes at the mouth of the gorge and is essential for binding, which is also in accordance to extant experimental results.[86]
As part of the reactivation process of the OP-bound cholinesterases, phosphylated oximes (POXs) are generated that are known to re-inhibit the free cholinesterases.[87] Alternatively, POXs can undergo a decomposition process in which a corresponding cyano compound is generated. Thus, it is important to minimize the re-inhibition process of cholinesterases after reactivation and to accelerate the decomposition of POXs in order to provide effective oxime therapy. We studied the POX formation and decomposition pathways using ab initio quantum chemical calculations and atomistic charge analysis. It was found that the location of the oxime group should be ortho to the pyridinium center as it increases the charge separation at the oxime bond that may enhance the efficiency of the reactivation. Additionally, POXs formed from an ortho-substituted pyridinium oxime were found to be least stable, that is, more susceptible to decomposition. As a result, our calculations suggest that ortho oximes would be most efficient for the reactivation process, while minimizing the re-inhibition of the cholinesterases.
2. Reactivation of Acetylcholinesterase
Since the discovery of chemical nerve agents, significant efforts have been placed to develop novel and efficient therapeutics for their treatments. Most medical countermeasures for the “treatment” of OP exposure aim to minimize the cholinergic crisis upon deactivation of acetylcholinesterase (AChE) at the neurosynaptic and neuromuscular junctions or to remove the OP toxicant by some scavenging process.[88] Clinically, OP poisoning is currently “treated” by a combination of anti-cholinergic drugs (e.g., atropine) and oximes (e.g., 2-PAM). Atropine acts as an antagonist of muscarinic acetylcholine receptors, while oximes (more likely, their deprotonated forms, the oximates) substitute the phosphylated serine in a nucleophilic manner, thereby reactivating the OP-inhibited form of AChE.[89] Although significant progress has been made to date in the development of such treatments, researchers are still plagued by three main problems : 1) there is no broad scope reactivator of OP-inhibited cholinesterases, 2) current small-molecule therapeutics possess a permanent charge that limits their effectiveness in the central nervous system, and 3) to date, no therapeutics treat the aged form of AChE (or BChE).[35, 90, 91]
2.1. Recent results from efforts to develop broad scope reactivators of acetylcholinesterase
Currently, for effective treatment of a patient who has been exposed to an OP, one needs to know the OP toxicant. This problem is extremely difficult to address in a mass-exposure event because by the time patients develop symptoms, the therapeutic window has significantly decreased. Patients can be tested in efforts to determine the OP exposure, while racing the clock to provide adequate treatment. Therefore, significant effort has been placed into developing novel oximes and reactivators that are capable of treating multiple OPs, and in an effective manner, so that a single treatment can be provided that will provide coverage against a range of OPs. In theory this solution sounds trivial but the complexities of the enzyme’s active site after inhibition by various OPs has proven troublesome with the different classes of OPs, thereby requiring different therapeutic approaches due to the size and orientation of the attached Oor N-alkyl groups (Figure 7).[35, 90, 92] Despite these complexities, significant progress has been made in this field.
The focus of this review is primarily on addressing the issues of non-permanently charged reactivators and the development of treatments for aged AChE. Earlier in this review, we noted the seminal reports by Wilson and by Rutland on reactivation of OP-inhibited AChE.[43, 49] An extensive review of reactivators by Kuca et al. covering the significant developments since the discovery of 2-PAM in 1955 to 2016 has been published and covers this topic in great detail.[93] It has been estimated that a therapeutic with some effectiveness should have a kr > 1 min-1 and a KD < 100 mm based on models representative of OP exposure.[90]
In order to facilitate reactivation in the central nervous system (CNS), Chambers et al. took the unique approach of continuing to use pyridinium-based oximes but lengthened the phenoxylalkyl chains such that the lipophilicity increases, thereby providing for anticipated penetration into the CNS.[94, 95] Although previously outlined by Kuca et al., recent work using lethal doses of nerve agent analogues have been conducted in which two compounds show an increase in survival rates and earlier cessation of symptoms compared to those of 2-PAM.[93, 95] The Chambers study made use of two nerve agent surrogates—one for sarin and one for VX—and evaluated four novel lipophilic oximes, along with 2-PAM, to test survival rates after LD99 challenges. It was found that the use of 1 and 2 (Figure 10) increased the survival rate to 65 and 55 %, respectively, and when combined with 2-PAM, the survival rates increased to 73 and 80 %, respectively, relative to the 40 % 2-PAM control for a sarin analogue. When the same study was repeated using a VX analogue, recovery was determined to be 33 % with 2-PAM alone, 75 and 65 % for 1 and 2 alone, respectively, and interestingly, only 53 and 53 % when the novel oximes were used in combination with 2-PAM. Additionally, all animals treated with only 2-PAM displayed seizure-like symptoms after 8 h, whereas all the animals treated with 1 or 2, both alone and with 2-PAM, showed cessation of seizure-like symptoms after 6 h. Brain cholinesterase levels were also measured but no statistically significant differences were determined between the 2-PAM and novel oxime treated samples. These results provide only indirect evidence for the penetration of the novel oximes into the brain.
Figure 10.
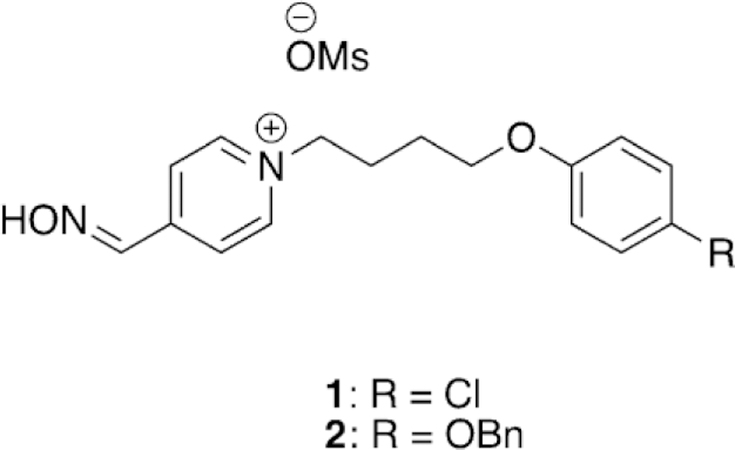
Novel lipophilic pyridinium oximes.[95]
The work of de Bruijn et al. made use of a multicomponent reaction, the Ugi reaction, to generate a small diverse library of both charged and uncharged oxime reactivators of OP-inhibited AChE (Figure 11).[96] The Ugi multicomponent reaction is a condensation reaction that combines an aldehyde or ketone, carboxylic acid, amine, and an isocyanide in a single reaction. Such a highly efficient reaction then allows the synthesis of a diverse library of compounds rapidly and easily, as was exploited by de Bruijn et al. In their work, the carboxylic acid moiety was attached to various oximes and possessed various linker lengths. The other components, that is, the amine, aldehyde/ ketone, and isocyanide, were then used to generate a moiety to bind in the peripheral site of AChE. The oxime moieties were chosen to cover a broad range of nucleophiles and included pyridinium oximes, imidazole oximes, ketoximes, and hydroxyiminoacetamides. Each oxime-containing compound was initially combined with methoxybenzylamine, isopropyl isocyanide, and formaldehyde to generate the peripheral site ligand; special attention was paid to the amine, and an aromatic amine was specifically selected to try and maximize peripheral site interactions. The initial library of compounds was tested against human AChE (hAChE), by using erythrocyte ghosts,[81] after inhibition by sarin, cyclosarin, and tabun at various concentrations. The library was ineffective at the reactivation of the cyclosarinand tabun-inhibited hAChE; however, the pyridinium and imidazole oximes, in addition to the hydroxyiminoacetamides, were able to reactivate sarin-inhibited AChE. In agreement with previous research efforts, the compounds that were the most efficient at reactivation of sarin-inhibited AChE were those that possessed a pyridinium oxime. The pyridinium oxime Ugi product with the longest linker length showed superior reactivation to that with a shorter linker length since it allows for more appropriate orientation of the peripheral site ligand in the enzyme. The influence of the peripheral site ligand was further tested by analyzing the reactivation ability of just the oxime functionality, which showed much more limited reactivation than the Ugi-linked product, further supporting the benefit of the peripheral site ligand. Interestingly, the uncharged imidazole oxime Ugi products also showed sarin reactivation potential, whereas just the imidazole oxime showed no reactivation.
Figure 11.
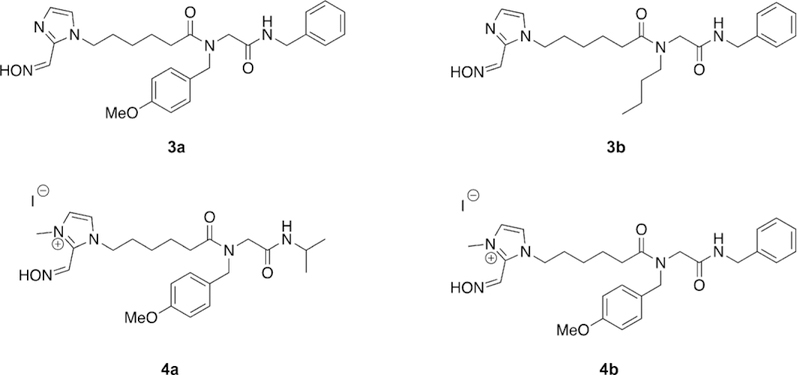
Novel oxime structures synthesized using the Ugi multicomponent reaction.[96]
In the interest of further exploring optimization of the peripheral site ligand, a representative imidazole Ugi product was carried through to the next phase of testing by expanding the Ugi multicomponent library to include additional imidazole oxime compounds as well as three different amines and two isocyanides. In addition to the normal characterization methods, pKa values were determined for representative Ugi products. The pKa for the imidazole oxime Ugi products were determined to be 10.2, for the pyridinium Ugi products to be 8.5, and for 4-PAM, the 4-substituted regioisomer of 2-PAM, to be 8.6. The two unit pKa difference between the products was addressed by alkylating the imidazole oxime Ugi products, thereby reducing the pKa from 10.2 in the non-alkylated product to 8.3–8.4 in the alkylated product. Despite the now permanent positive charge on the imidazole Ugi products, the chain length was anticipated to increase the BBB penetration, which was supported by in silico calculations. All newly synthesized compounds were screened for their reactivation potential versus sarin-, cyclosarin-, and tabun-inhibited hAChE. It was determined that the longer linker length, ranging from 1 to 3 to 5, the greater the reactivation efficacy of the tested oximes. The imidazolium compounds were found to reactivate faster than their imidazole counterparts, while demonstrating reactivation efficacy against cyclosarin-, sarin-, and tabun-inhibited hAChE. They also reported that aromatic amines and isocyanides have a positive effect on the reactivation efficacy of the oximes, and they asserted that binding affinity for the peripheral site was increased. Although none of the synthesized reactivators outperformed known oxime treatments, four of the synthesized Ugi products (3 a, 3 b, 4 a, and 4 b, Figure 11) were carried through to in vivo testing. These studies were conducted using rats challenged with sarin, and notably, 4b showed signs of toxicity at the original dosage and was then lowered to prevent lethality. Even so, compound 4b was the only oxime capable of preventing seizures in all test animals. Of the tested oximes, 4b also showed higher brain cholinesterase levels than that of 2-PAM and the other tested oximes. Although none of the developed Ugi oxime products showed superior reactivation to that of the reference oximes, the utility of the Ugi reaction to rapidly generate large libraries of structurally diverse oximes was effectively demonstrated. The Ugi products, even when permanently positively charged, did show increased brain cholinesterase activity and a reduction in seizures after sarin exposure. The synthetic utility of the Ugi multicomponent reaction can be further used to develop and modify reactivation efficacy of oxime derivatives.
Musilek and co-workers recently evaluated the ability of tetroxime, a bis-pyridinium compound with four nucleophilic oximes (Figure 12),[97] for its reactivation ability with AChE inhibited by VX, sarin, cyclosarin, and tabun. The in vitro study was conducted using rat brain homogenate and at two different concentrations, 1 mm and 10 mm. Unfortunately, tetroxime proved to not be a viable broad scope reactivator, and only showed superior reactivation relative to 2-PAM in the case of cyclosarinand VX-inhibited AChE. For sarin-inhibited AChE, 2PAM was shown to be a more effective reactivator especially at higher concentrations. Both oximes, even at high concentrations, were determined to be very poor reactivators of tabuninhibited AChE. Their in silico studies showed that at the tested pH of 8.0, the 2-positioned oxime is 88 % deprotonated and the 4-positioned oxime is only 6 % deprotonated, suggesting that the 2-position is the nucleophilic site in tetroxime. However, on the basis of molecular docking studies, the 4positioned oxime was shown to be in closer proximity to the reaction center suggesting that binding may be the reason for the poor performance of tetroxime. This study concluded that simply increasing the number of nucleophilic moieties on the reactivator compounds does not directly result in increased reactivation potential but may need to be further balanced by additional structural modifications.
Figure 12.
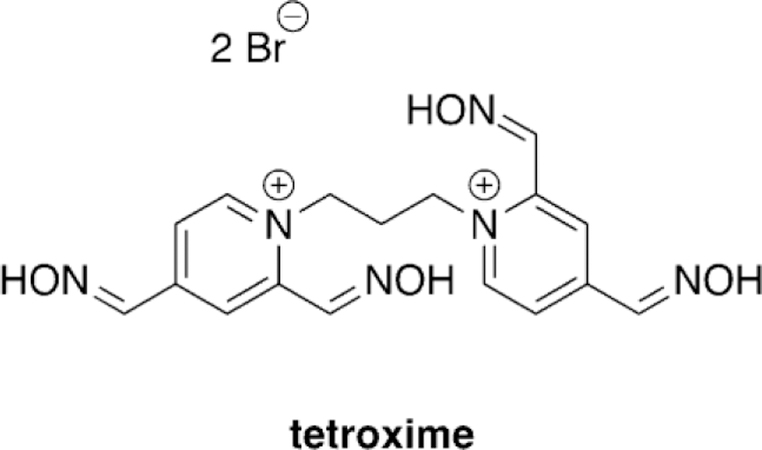
Tetroxime structure as studied by Musilek and co-workers.[97]
Franca and co-workers designed and synthesized novel oximes for the reactivation of (ethyl) paraoxon-inhibited AChE and evaluated them both in vitro as well as in silico.[98] In total, three different oximes (5, 6, and 7, Figure 13) were evaluated against the reference oximes (2-PAM, obidoxime, and HI-6, Figure 8) for their reactivation ability of OP-inhibited hAChE. Unfortunately, the best performing oxime 7, and with the shortest aromatic peripheral site linker, provided only 9.8 % of reactivation, and with lower efficacy than all the reference oximes. Indeed, obidoxime showed a near complete reactivation of 96.9 % in just 10 min when applied at a concentration of 100 mm. These results were analyzed in silico as well. The analysis method was unique in that not only was the energy of binding analyzed, but also what was referred to as the “near-attack conformations” (NAC). The NAC was determined to be those docked structures with a POP-Oox distance of less than 10 A and an angle of Oox-POP-OSer203 being 180 ± 608. When the docking data were analyzed strictly on the structure (pose) with the best docking energy, the only conclusion that could be drawn was that the oximes have affinity for the AChE active site. However, when the authors analyzed the NAC poses, there was a correlation between reactivator potency and the percentage of poses that satisfy the NAC criteria. Thus, although the reactivators themselves did not outperform the reference compounds, the NAC analysis showed some promise as an in silico diagnostic that may correlate to reactivator potency. Further investigation of in silico criteria, other than just qualitative interactions of binding, may further enhance the predictability of computational models.
Figure 13.
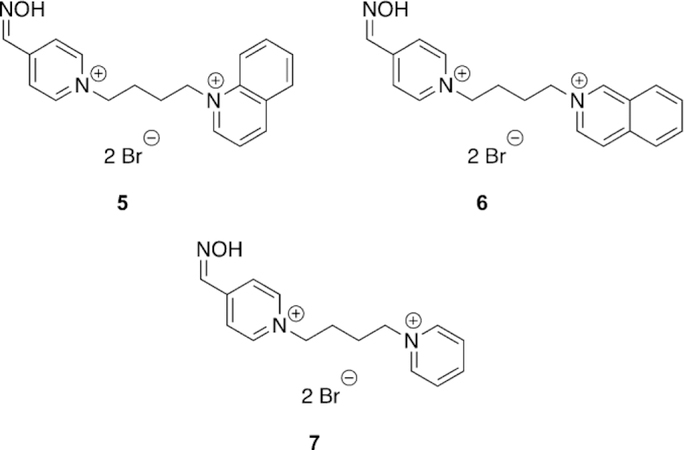
Bispyridinium compounds capable of reactivating AChE and BChE with (ethyl) paraoxon.[98]
Ghosh and co-workers synthesized bis-pyridinium oximes and evaluated their efficacy versus sarin-, VX-, tabun-, and paraoxon-inhibited human and electric eel AChE (hAChE and eeAChE).[99] The synthesized oximes were novel in that the spacer between the pyridinium moieties was an (E)-2-butene linker and the carbonyl groups at the 4-position of the pyridinium ring were varied from being a carboxylic acid, a methyl-between OP-inhibited hAChE and eeAChE did not appear to follow any clear trend. For instance, they determined that for paraoxon-inhibited hAChE, obidoxime was substantially more effective than 2-PAM; however, when the same OP was used for electric eel AChE (eeAChE), 2-PAM and obidoxime had nearly the same reactivation efficacy, albeit the efficacy for obidoxime was reduced. If one compares the previous results with tabun-inhibited hAChE and eeAChE, the reactivation efficacy increases in the electric eel isoform. Thus, organismal differences in the very well conserved AChE enzyme are nevertheless critical when evaluating oxime reactivation.
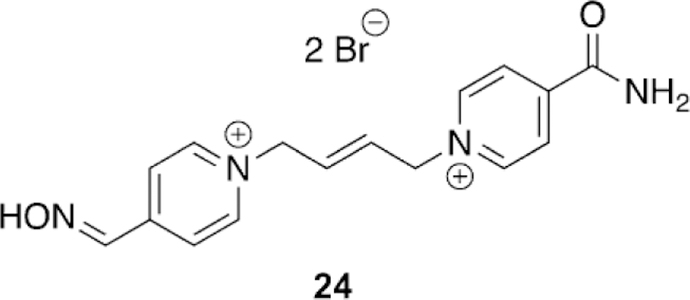
Continuing to adapt the K-oxime structures, Kuca and coworkers used the details from study of other K-oximes to design 15 new oximes for the reactivation of ethyl paraoxon-, methyl paraoxon-, tabun-, and DFP-inhibited hAChE. The authors noted that the but-1,4-diyl linkage worked the best in previous studies and focused instead on varying the aromatic group attached in the bis-pyridinium oximes.[100] The novel oximes were tested relative to a reference set of K-oximes in addition to known oximes : 2-PAM, HI-6, obidoxime, trimedoxime, and methoxime. Unfortunately none of the newly synthesized compounds were effective at tabun reactivation, but the previously observed affinity of 24 (Figure 21, see later) was again observed. Of the reference oximes, trimedoxime was the best performer at 31.5 % and 100 mm. Of the newly prepared oximes, only 11 and 12 showed any activity above that of the reference oximes and previously studied compounds for hAChE inhibited by ethyl paraoxon (Figure 15). In the case of ketone, and an ethyl ester (8, 9 and 10, respectively, Figure 14). Unfortunately, their oximes were only modestly better than the reference oximes (2-PAM and obidoxime) for tabun-inhibited hAChE. More intriguing, however, was that reactivation efficacy methyl paraoxon and DFP, the reference oximes were much better or all compounds were ineffective, respectively. The novel oximes in the study proved to be potent inhibitors of hAChE with IC50 values in the low micromolar range (6–90 mm). From the synthesized variants, they determined that the pyridinium ring with both hydrophobic and hydrophilic groups at the 4-position form more favorable interactions in the peripheral site owing to their increased reactivation potency. Unfortunately, none of these modifications resulted in any compound that had broad spectrum activity with multiple OP pesticides. Madura and co-workers made use of computational tools and the ZINC database to select a diverse library of commercial oximes to test for OP reactivation.[101] The authors started their study with a search of the ZINC database yielding thousands of compounds. Using a similarity and dissimilarity search, the authors narrowed the number of compounds down to just 60 compounds. The compounds were further reduced to 18 based on visual structural evaluation, yielding a final library of 18 untested oximes. To ensure that all the compounds could fit within the AChE active site, docking studies were conducted in which each compound was docked with mouse AChE inhibited by methamidophos, ensuring a docked structure was found with a P-oxime bond distance of less than 6 A. All 18 of the compounds satisfied this criterion and were continued forward. However, despite being extracted from the ZINC database, the authors could only purchase six of the 18 compounds for biological evaluation. All six of the oximes were compared relative to 2-PAM activity for their reactivation of ethyl paraoxon-, DFP-, fenamiphos-, and methamidophos-inhibited AChE. It is of interest that of the six oximes tested (Figure 16,13–18) for all the OP pesticides, the reactivation potency was similar or superior to that of 2-PAM in all cases. The most promising of the compounds is 14, which showed 41 % reactivation of fenamiphos and 58 % reactivation versus DFP in one hour; these OPs are two pesticides that are notoriously difficult to reactivate.
Figure 21.
Novel oxime structure that shows significant tabun reactivation.[107]
Figure 15.
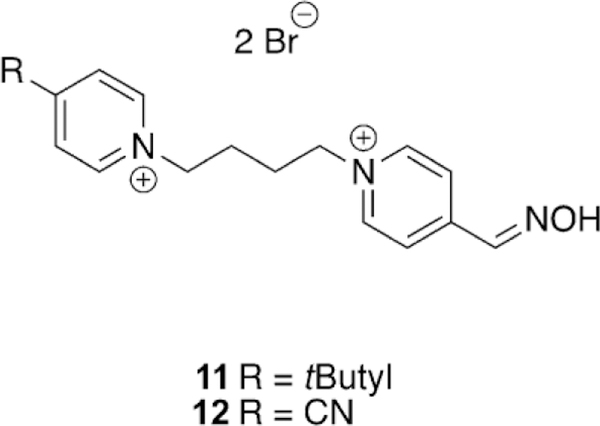
Two novel K-oximes showing some efficiency for the reactivation of ethyl paraoxon-inhibited hAChE. [100]
Figure 14.
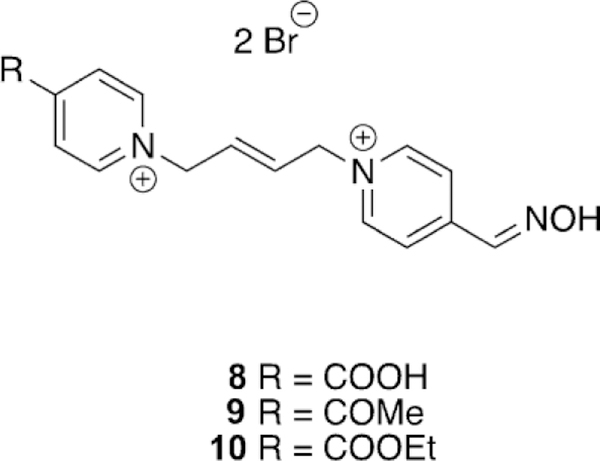
(E)-2-Butene-linked bispyridinium oximes for the reactivation of tabun-inhibited AChE.[99]
Figure 16.
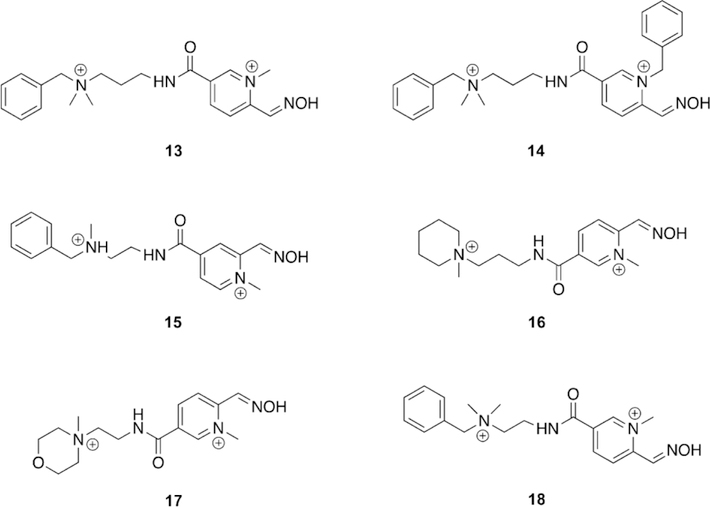
ZINC database oximes determined from virtual screening which are capable of reactivating multiple OP pesticides.[101]
Figure 18.
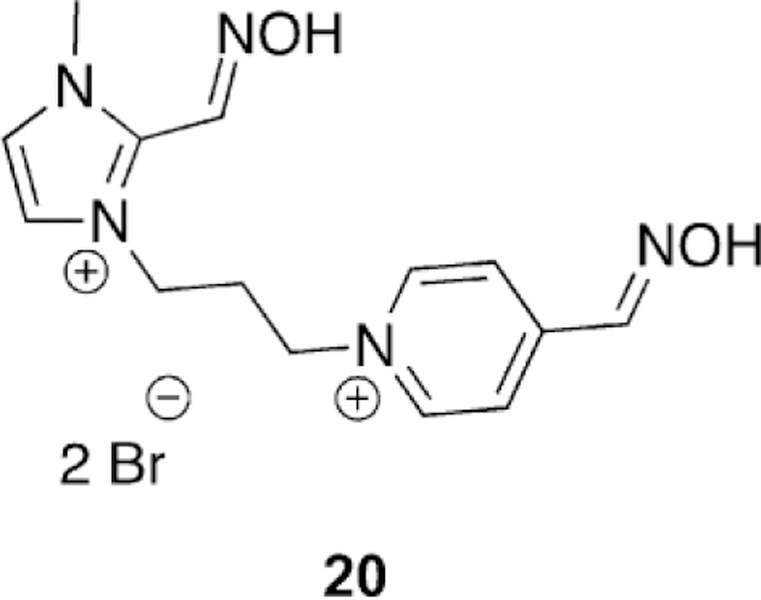
Imidazolium oxime for the reactivation of sarinand VX-inhibited AChE.[103]
Unfortunately, 13 and 14 (Figure 16) showed high inhibitory potency at the tested 100 mm concentration, and 25 and 15 % relative activity, respectively. Despite the high inhibition potency, this study shows the utility of database searches for provision of structural modifications that may not otherwise be made in drug design, offering a method for scaffold elucidation in future reactivation design.
Acharya and co-workers noted the efficiency of HI-6 and four different isonicotinamide derivatives of pyridinium substituted hydroxyiminoacetamides for the reactivation after sarin and VX exposure.[102] The four compounds were screened for their reactivation potency with sarinand VX-inhibited hAChE, by using erythrocyte ghosts, relative to the control oximes 2PAM and obidoxime. Oximes 19 a and 19 b (Figure 17) showed superior reactivation potency of sarin-inhibited AChE with effective rates of 29.77 and 23.94 mm-1 min-1, compared to 2PAM and obidoxime at 2.33 and 7.91 mm-1 min-1. However, for VX-inhibited AChE, 19 c (Figure 17) performed the best with an effective rate of 25.47 compared to 2-PAM and obidoxime at 3.50 and 8.23 mm-1 min-1, respectively. The oximes differed only in the length of the linker chain and the results suggest that steric effects of the inhibited serine play a role in the reactivation potency. This reiterates the difficulty in designing broad-spectrum reactivators. This study did prove that the oxime moiety itself does not need to be directly linked to the aromatic core of reactivators and can be pendent. Additionally, it confirms the utility of hydroxyiminoacetamides as potential nucleophiles for reactivation.
Figure 17.
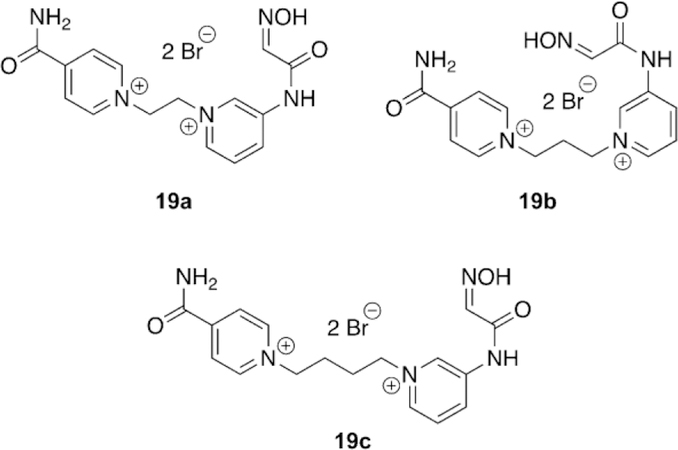
Hydroxyiminoacetamide nucleophiles using structural components of HI-6 for reactivation of sarinand VX-inhibited AChE.[102]
Ghosh and co-workers followed up the findings that imidazole-based reactivators are very effective for BChE reactivation by synthesizing and testing imidazole-based reactivators for AChE reactivation with sarin and VX.[103] In total eight imidazolium compounds were tested for their reactivation potency with erythrocyte ghost hAChE compared to the reference oximes 2-PAM, obidoxime, and HI-6. The pKa values of the compounds were determined to be within the expected 7–9 range as is usually observed for oxime nucleophiles. However, unfortunately the oximes were no more potent than the reference oximes HI-6 and obidoxime for both sarinand VX-inhibited hAChE. However, one compound, 20 (Figure 18), showed greater reactivation potency than 2-PAM for sarin and VX. This compound is an imidazolium linked to a 4-PAM moiety. It was determined from the reactivation studies that alkylimidazoliums are less effective reactivators when compared to pyridylimidazoliums, as may be expected based on aromatic interactions of the pyridinium ring with the peripheral site. For this set of compounds, it was observed that a three-carbon methylene linker between the imidazole and the pyridine was the best and extension of the chain length out to five carbons reduced reactivation potency. Thus, it appears as though the affinity and utility of imidazole-based nucleophiles for AChE is not the same as that for BChE and continued structural modifications may need to be made to achieve an imidazole-based reactivator of AChE.
Amitai et al. used a bifunctional drug concept to link together oximes with known ability to reactivate AChE with hydroxamic acids which showed a greater ability to scavenge and hydrolyze OPs in solution as a therapeutic capable of protection and reactivation.[104] The authors first investigated the ability of the conjugates to detoxify sarin, cyclosarin, soman, and VX in solution. It was determined that all the conjugates were faster at detoxifying the OP cyclosarin compared to 2-PAM; however, for sarin and soman, the rates of detoxification were slightly slower for two of the three conjugates. Of the tested compounds, 2-PAM-Me-BHA (Figure 19; BHA = benzylhydroxamic acid; 4-PHA = 4-pyridylhydroxamic acid; Me = methyl, Pr = propyl) was by far the best at detoxifying all the OPs compared to the reference compounds, having half-lives between 1.9–3.6 min. For all tested compounds, oximes and conjugates, VX detoxification was very slow. Next, the reactivation potency of the compounds was tested using the OPs sarin, VX, and cyclosarin and the reference oxime 2-PAM again. In the case of sarin, 2-PAM recovered the most activity of AChE and at the fastest rate when compared to the conjugates. For VX, 2-PAMMe-BHA showed the highest rate of reactivation but only reached a maximum of 56 % compared to 2-PAM which reached a maximum of 94 % AChE activity. This may be caused from re-inhibition of the phosphylated oxime. For cyclosarin-inhibited AChE, the fastest reactivator was 2-PAM-Pr-4-PHA (Figure 19), reaching a maximum of 70 % AChE activity relative to the maximum of 65 % for 2-PAM. Further analysis was conducted using HI-6 as a reference and comparing reactivation potency of 2-PAM-Pr-4-PHA with sarin, cyclosarin, and VX. In all cases HI-6 had superior reactivation ability. The synthesized conjugates proved to be relatively strong inhibitors with IC50 values of 34–90 mm, yet they were still weaker inhibitors than HI-6 with a measured IC50 of 11 mm. The ability of these compounds to serve as pre-treatments for OP exposure was explored on the premise of their high reversible inhibition. What was found was that 2-PAM-Me-BHA and 2,4-DiPAM-Me-BHA (Figure 19) were capable of significantly slowing down the inhibition of AChE by VX, sarin, cyclosarin, and soman—and significantly greater than by 2-PAM. This finding then suggested to these researchers that they can protect from OP inhibition much like has been observed for pyridostigmine. Prior to moving to in vivo testing, the LD50 values of the conjugates were determined. The conjugates had values between 179–200 mg kg-1 in rats, similar to 2-PAM, and 80–200 mg kg-1 in guinea pigs, with 2-PAM being about 170 mg kg-1. To determine the appropriate dosing of the conjugates for in vivo studies, cholinesterase activity in blood serum was measured following challenges with sublethal doses of sarin along with toxicology studies. At the highest dose of 142 mmol kg-1, 2PAM recovered 15 % of blood ChE activity, while 2-PAM-Pr-4PHA recovered 42 % at the same dose and 2-PAM-Me-BHA recovered 22 % when test animals were pre-treated. The results for post-treatment were slightly different with 2-PAM-Pr-4-PHA at 50 % and 2-PAM and 2-PAM-Me-BHA at 20 %, again at 142 mmol kg-1. The differences in results could be caused by reactivity with BChE in the blood, but it was demonstrated that the degradation and reversible binding did increase protection in the blood for these conjugates. The in vivo protective ratios were determined for each compound when mice were exposed to sarin, cyclosarin, VX, and soman. The best compound in all cases was 2-PAM-Pr-4-PHA. For sarin-treated mice, good protective ratios were observed for both 2-PAM-Pr-4-PHA and 2-PAM-Me-BHA in a pre-treatment regime, compared to 2-PAM, but post-treatment was similar to 2-PAM. The same results hold true for cyclosarin except that 2-PAM-Pr-4PHA was superior to other conjugates. In the case of VX, all the conjugates and 2-PAM had similar protective ratios for both preand post-treatment regimes. Finally, the protective ratios for 2PAM-Pr-4-PHA was better for soman exposure in both the preand post-treatment cases relative to 2-PAM. These same studies were repeated in a guinea pig model as well. Interestingly, for guinea pigs exposed to sarin, all the conjugates have lower protective ratios than 2-PAM for both preand post-treatment methods, in contrast to what was observed in mice; this may be attributed to having to lower the doses based on the findings that these conjugates were more toxic in the guinea pig model. For the cyclosarin-exposed guinea pigs, the best protective ratios were observed for 2PAM-Me-BHA and 2-PAM-Pr-4-PHA in pre-treatment and for post-treatment just 2-PAM-Pr-4-PHA. Unfortunately for VX exposure, all the conjugates performed worse than 2-PAM in terms of protection. Finally, for soman-exposed guinea pigs,2,4-DiPAM-Me-BHA showed the best pre-treatment protective ratios and 2-PAM-Pr-4-PHA and 2,4-DiPAM-Me-BHA showed the same protective ratios in post-treatment methods. Overall, the best conjugate for in vivo protection of both animals and multiple agents was 2-PAM-Pr-4-PHA, given its ability to reactivate, scavenge, and in vivo protection ratios, this conjugate method seems to present a unique way to treat and protect from OP exposure.
Figure 19.
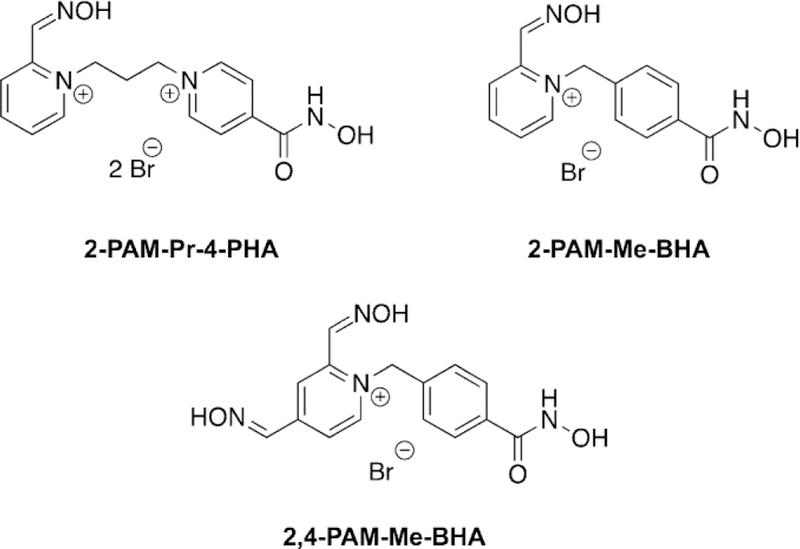
Hydroxyiminoacetamide nucleophiles using structural components of HI-6 for reactivation of sarinand VX-inhibited AChE.
2.2. Developing non-permanently charged oxime reactivators of OP-inhibited acetylcholinesterase
The pursuit to find an uncharged reactivator of OP-inhibited AChE has taken multiple different paths. One approach has been to use a pro-drug for the desired reactivator and to generate the active drug in the desired tissue, such as for pro-2PAM.[105] Some have tried to develop novel non-oxime reactivators, and some have attempted to take the existing oxime functionality from clinically approved therapeutics and to remove the need for the positive charge of the pyridinium moiety by tethering the oxime to a linker and peripheral site ligand that sufficiently enhances binding. The AChE active site has selectivity for positively charged species that closely resemble the native substrate, acetylcholine. Thus, the originally designed oximes, such as 2-PAM, trimedoxime, obidoxime, and HI-6 (Figure 8), share the positively charged pyridinium moiety that significantly increases the binding affinity. However, to develop a therapeutic that is capable of crossing the BBB, the permanent positive charge needs to be “removed”, and thus the affinity for the active site of AChE needs to be recovered in some manner, such as attachment to peripheral site ligands (PSL). A great deal of variability arises with selection of the peripheral site ligand, including linker type, linker length, and nucleophilic moiety. The difficulty in such an approach is that changing a single element, nucleophile, linker, and PSL, can have a profound impact on the reactivation potential requiring a complete structure–activity evaluation for each class of molecule to find the optimum combinations, not to mention the further complexities arising from stereochemistry, regiochemistry, and pKa. Thus, years of research have been conducted, and to date, no new therapeutics have been approved in decades. Nevertheless, the continued threat and worldwide crisis caused by OP compounds necessitates the continued efforts and developments of new therapeutics.
Li et al. took a unique approach of using an oxime, based on salicylaldoxime derivatives, a linker unit derived from the known indanone framework of anti-Alzheimer’s disease treatment donepezil, a piperidine linkage, and tetrahydroisoquinoline peripheral site ligands to construct new oxime reactivators, and then tested against OP-inhibited hAChE.[106] The peripheral site ligand alone showed no reactivation potential of OP-inhibited AChE as was expected. Additionally, the untethered oximes reactivated poorly, confirming the hypothesis that tethering an oxime to an appropriate PSL and linker can sufficiently enhance reactivation potential. Three of the studied oximes (21–23, Figure 20) were shown in vitro to outperform the reference oximes HI-6 and obidoxime in the reactivation of VXand tabun-inhibited AChE and had similar reactivation potential in the case of sarin-inhibited AChE. The authors concluded that piperidine-linked conjugates are more effective than methylene-linked analogues. Furthermore, the linkage at the ortho-position, relative to the phenol, outperformed those oriented in the para-position. Finally, increasing the hydrophobicity of the oxime rings by introduction of methyl, chloro, and bromo substituents also had a positive effect on the reactivation potential of the oximes. The study also conducted in silico evaluation of the BBB permeability using an ADMET program and showed that the newly synthesized conjugates have a much better probability of crossing the BBB compared to the reference oximes, though no other experimental data were presented.
Figure 20.
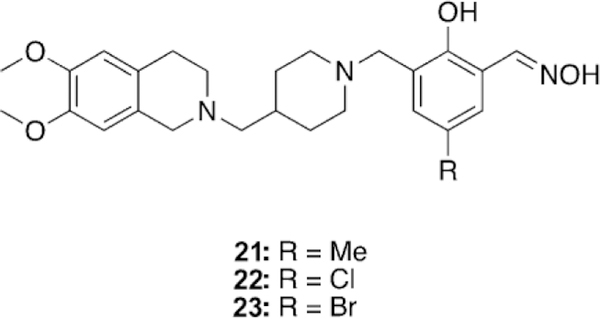
Novel oxime structures based on salicylaldoximes.[106]
Kuca et al. recently revisited the testing of 24 (Figure 21), which had shown promising tabun reactivation results in rodent models; however, no human data had been reported until recently. The study was unique in that they evaluated human AChE from human brain homogenate. Compound 24 reactivated over 60 % of tabun-inhibited hAChE, while for the three reference oximes (2-PAM, obidoxime, and HI-6), obidoxime was the best of three and only reached 33 % reactivation, and at a concentration higher than can be achieved in vivo. This same experiment was repeated in a rat model as verification of previous studies and the trends were consistent in both models. However, the effective reactivation rate was reduced by a factor of 7.5 for 24, but by a factor of 149 for obidoxime when comparing the ratio of reactivation rates between human brain homogenate and rat brain homogenate.[107]
Li and co-workers investigated the reactivation potential of novel ortho-hydroxylbenzaldoximes linked to phenols against soman-inhibited hAChE, which is extremely difficult to reactivate as the competing aging process is very rapid, on the order of just a few minutes.[108] Surprisingly, in their report, all of the synthesized oximes reactivated soman-inhibited hAChE to levels higher than that of 2-PAM, but not as high as HI-6. Despite being tested at multiple time points, the reactivation maxima by the oximes were fully achieved within the first 2 h, and then remained constant out to 24 h. The best performing compounds (25–27, Figure 22), showed reactivation percentages of 15.5, 19.7, and 13.4 %, respectively, compared to 36.9 % by HI-6 at 24 h. From the results in this study, the presence of inhibited and aged hAChE after soman exposure needs clarification.
Figure 22.
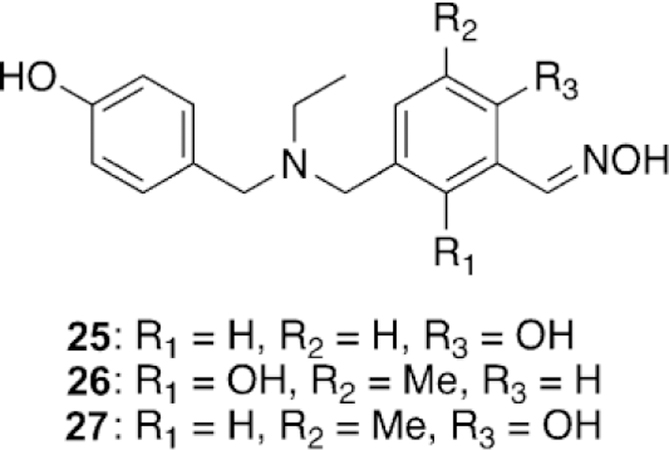
Novel oxime structures that show some soman reactivation.[108]
Renard and co-workers synthesized and tested uncharged oxime reactivators making use of the anti-Alzheimer’s disease drug donepezil as a peripheral site ligand.[109] The benzyl moiety of donepezil was replaced with a 3-hydroxypyridinaldoxime as the nucleophilic portion of the molecule (Figure 23). In total, four variants (28–31) were synthesized and evaluated for their reactivation ability versus VX-, sarin-, paraoxon-, and tabun-inhibited hAChE. Compounds 28–30 performed better than 2-PAM, but are not as efficient for reactivation of VX-inhibited AChE as obidoxime and HI-6. Comparing 28 to 29, they determined that the indanone carbonyl moiety increases binding affinity, but does not improve the overall reactivation efficacy. Comparing 30 and 31 showed that the positioning of the linkage has a significant effect on reactivation efficacy, with the 6-linkage having a better reactivation efficacy. Interestingly, replacement of the piperidine ring in the linkage seems to have no effect on the reactivation efficacy between 28 and 30. Sarin-inhibited AChE was tested with HI-6 and 28, but unfortunately the new oxime was not as efficient as HI-6. Oximes 29 and 30 were demonstrated to be better reactivators of paraoxon-inhibited AChE than HI-6. When tabun-inhibited AChE was analyzed, no reactivation was observed with the donepezilbased oximes. The strategy of linking the donepezil-based fragment to known oximes was shown to achieve binding affinity similar to that of monoand bis-pyridinium oximes for VX and sarin even though these synthesized oximes were uncharged, lending support to the peripheral site ligand strategy. Renard and co-workers subsequently published non-permanently charged oxime reactivators of various OP compounds using amine or tetrahydroisoquinoline moieties as peripheral site ligands.[110] All of the synthesized oximes (32–36, Figure 24) were shown to have moderate binding affinities in the mm range and similar to that of HI-6. The oximes were tested for their reactivation efficacy against VX, sarin, cyclosarin, tabun, and paraoxon. Compound 35 was significantly better at reactivating VX-inhibited AChE than HI-6, while the rest of the oximes were similar to HI-6. Unfortunately, for sarininhibited AChE, none of the oximes were shown to outperform HI-6, but their reactivation potencies were similar to HI-6. For cyclosarin-inhibited AChE, the oximes performed rather poorly compared to HI-6, and were 70–100 times less effective. Interestingly, tabun-inhibited AChE was reactivated more effectively for all the new oximes when compared to HI-6, and importantly, HI-6 showed very limited reactivation. Additionally, all the new oximes also outperformed HI-6 when tested against (ethyl) paraoxon-inhibited AChE. Although the reactivation potential for some of these compounds were below that of the reference, it is worth noting that at different concentrations, these newly synthesized oximes were capable of reactivating 50–100 % of the inhibited forms of each enzyme, and at various time points. The most notable is the recovery of 50–90 % of the activity of tabun-inhibited AChE, with tabun being known as an OP that is difficult to reactivate.
Figure 23.
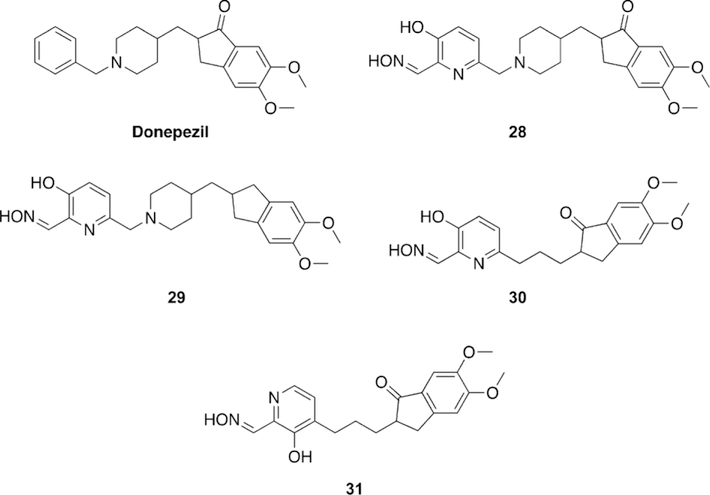
Novel oxime structures based on the Alzheimer’s disease drug donepezil by Renard and co-workers.[109]
Figure 24.
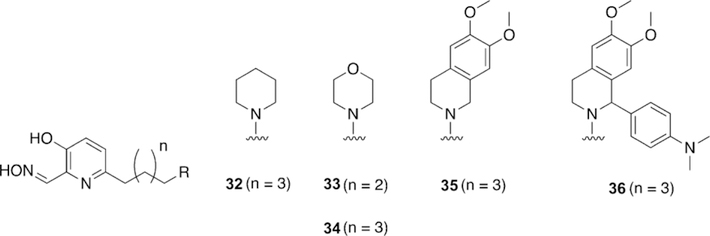
Uncharged reactivators of cholinesterases with the potential to cross the blood–brain barrier.[110]
These results are very promising and demonstrate a new class of compounds that are relatively broad scope in comparison to known oximes. This study further investigated the ability of the compounds to recover AChE activity in whole blood through both reactivation as well as scavenging of the OPs in the blood. Oxime 33 showed very promising results, recovering more than 80 % of the cholinesterase activity in whole blood in 5–15 min after VX, sarin, and paraoxon exposure and 60 min after cyclosarin exposure, thereby showing significant promise as a treatment, especially since whole blood cholinesterase activity is both a measure of the AChE and BChE activity. The pKa values of the oximes were tested using both in silico as well as in vitro methods, and for all oximes, the pKa of the oxime and amine moieties were determined to be about 8 and 10.5, respectively. These pKa results suggest that these compounds with protonation states near physiological pH should be able to penetrate the BBB. The ability to penetrate the BBB was further investigated using both in silico and in vitro approaches. The CNS multi-parameter optimization score for each of the oximes was determined and accounted for six physiochemical parameters and showed that all the new oximes are well within the high BBB permeation scores with the exception of 36, which is below the CNS cutoff. These results were confirmed using an in vitro parallel artificial membrane permeation assay PAMPA-BBB using lipid extract of porcine brain and a reference set of 14 commercial drugs. All the synthesized oximes were categorized based on the reference set to have high BBB permeation. Altogether, these newly synthesized and uncharged oximes reactivators of AChE are extremely promising, showing broad scope reactivation with Vagents, G-agents, and pesticides and with an ability to penetrate the BBB and be active in the CNS. Future in vivo testing is needed to confirm their utility.
Understanding the limitation of having a potent inhibitor of native AChE as a reactivator, Nachon and co-workers proposed structural modifications for a known tacrine-linked reactivator in efforts to reduce its interaction with the native enzyme using knowledge derived from crystal structures (Figure 25).[111] First, three X-ray crystal structures were solved after soaking 37 into one of native Torpedo californica AChE (TcAChE), a tabun-analogue-inhibited TcAChE, and an inhibited TcAChE with a rivastigmine analogue. For only the uninhibited TcAChE crystal structure was a molecule of 37 observed within the active site; for the other two, the ligand was observed to bind in the PAS. Interestingly, the uninhibited TcAChE structure has two molecules of 37 bound : one in the catalytic active site (CAS) and one in the PAS. The authors hypothesized that the interactions within the CAS are what leads to the high potency and they desired to interfere with these interactions present within the CAS by introducing a chlorine atom at the 7-position on the tacrine rings to generate a steric interaction with nearby residues, leading to the design of 38. To test the hypothesis of steric interference, molecular docking was performed with both molecules. The results supported their hypothesis as 38 was only observed in the PAS, and not the CAS. To determine how the reactivation efficiency was affected, in vitro studies were conducted with hAChE and compared to 2PAM, HI-6, and obidoxime. The IC50 values showed that the introduction of a Cl atom was successful in affecting the binding affinity of 37 (IC50 value of 0.25 mm) as compared to 38 with an IC50 of 2.3 mm. Interestingly, in addition to reducing the affinity for the native enzyme, the reactivation potency for tabun-inhibited hAChE was actually increased, while for VX, a sarin analogue (NIMP), and paraoxon, the potency remained approximately the same. Compounds 37 and 38 outperformed all three reference oximes for the reactivation of VX, tabun, and paraoxon and were very similar to HI-6 for the reactivation of the sarin analogue NIMP. To determine the new binding mode that 38 was adopting with AChE, the previous X-ray crystallography approach was repeated for native TcAChE, tabun-analogue-inhibited TcAChE, and rivastigmine-inhibited TcAChE. All the structures showed binding in only the PAS, confirming the disruption of interactions within the CAS. The decreased inhibition of native hAChE, combined with the similar reactivation profile and increased reactivation potency with tabun, suggests that 38 is a viable candidate for continued investigation. Additionally, this work supports the utility of using X-ray crystal structures of inhibited AChE to guide structure-based design of reactivators and inhibitors of AChE.
Figure 25.
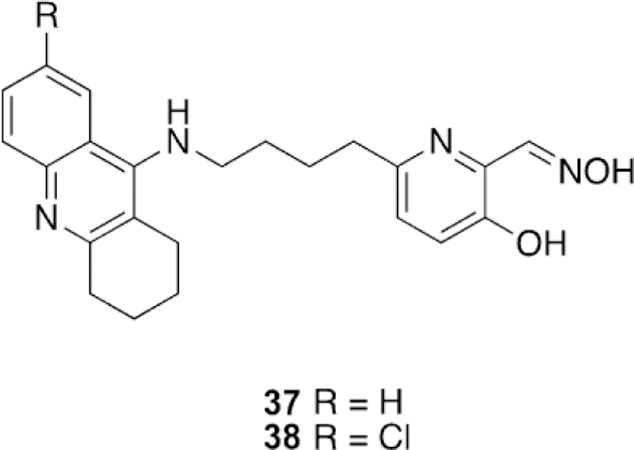
Tacrine-linked reactivators designed on the basis of X-ray crystallographic data.[111]
Using the structural scaffold of vitamin B6, Gaso-Sokac and workers designed a library of pyridoxal oxime derivatives and tested their reactivation potency with VX-, tabun-, and paraoxon-inhibited AChE and BChE.[112] Unfortunately, of the nine synthesized oximes (Figure 26), none performed better than the reference oximes (obidoxime, TMB-4, and HI-6). However, at slower rates of reaction, some compounds were able to recover 85 % of the hAChE activity in 23 h. The results for hBChE are slightly more promising, but still not successful overall in comparison to the observed rates by the reference oximes. Novel oximes 39 and 40 (Figure 26) showed the best results, recovering 90 and 95 % in 8 and 5 h, respectively. The binding affinities of all the compounds were tested in addition to the rates of reaction. They determined that for all compounds, the Ki is in the mm region, including the reference oximes, for recombinant hAChE and hBChE. It was postulated that the reason for the inactivity by hAChE is due to the increase in steric hindrance around the oxime moiety, which is reflected in the slower rate of oximolysis in the absence of enzyme.
Figure 26.
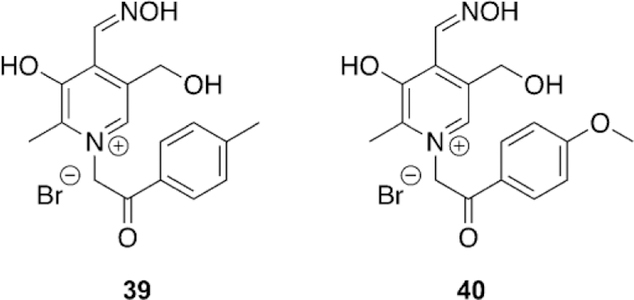
Vitamin-B6-based oximes for the reactivation of hAChE and hBChE.[112]
2.3. Developing non-oxime based reactivators of inhibited acetylcholinesterase
The continued need to develop non-permanently charged oximes for the treatment of OP exposure has started to see an increase in the investigation of non-oxime-based therapeutics. Pyridine-based therapeutics are effective, but their binding and efficacy are significantly increased when the therapeutics are alkylated to form the pyridinium. The positive charge increases affinity for the active site through cation–p interactions with Trp86.[113] Recently, more and more nucleophilic compounds, which are non-oximes, have been studied in order to find structural elements that provide similar binding affinities and rates of reactivation without the need to change the charge of the compound to facilitate binding and/or efficacy.
Stojanovic et al. took a unique approach of screening a library of 2000 bioactive and approved drugs for their ability to reactivate OP-inhibited AChE.[113] The screen resulted in two initial leads : the antimalarial drug amodiaquine and scopoletin (Figure 27). Amodiaquine was chosen as the compound of initial focus as scopoletin was effective against paraoxon-inhibited AChE, but not DFP-inhibited AChE. Investigation of the active structural features of amodiaquine led to the discovery that Mannich phenols were effective reactivators of organophosphate-inhibited AChE and in fact 4-amino-2-(diethylaminomethyl)phenol (ADOC, Figure 27), which is amodiaquine without the hydrophobic component, is the effective portion of the molecule. Further investigation of ADOC found that the para-relationship between the amine and hydroxyl group as well as the hydroxyl group itself are essential to reactivation ability. ADOC was shown to be an effective reactivator of multiple forms of both OP pesticide and nerve-agent-inhibited AChE. Mechanistic investigations postulated two separate mechanisms : 1) The phenol is participating in direct nucleophilic attack of the phosphylated serine, and 2) the Mannich base is providing an external base for reactivation within the active site. Further studies led to the discovery of chloroquine being a reactivator of inhibited AChE, but lacking the Mannich phenol. Instead, chloroquine possessed a hydrophobic anchoring moiety and a basic moiety. In fact, swapping of the linker length and the basic moiety led to the discovery of additional classes of AChE reactivators, such as 41 (Figure 27), all of which are non-oxime based. Further in vivo studies with both ADOC as well as 41 showed excellent protective effects against lethal doses of DFP. Most interestingly, when comparing cholinesterase levels in various tissues, the authors reported that tissues beyond the blood–brain barrier (BBB) in the CNS were more active when treated with ADOC and 41 than when mice were treated with 2-PAM or left untreated with any reactivator. This result suggests that these newly discovered classes of reactivators address directly the issue of permanent charge and the ability to penetrate the BBB. This discovery of two new classes of non-oxime reactivators of inhibited AChE opens new avenues of continued research and the potential development of more effective and BBB-penetrating therapeutics.
Figure 27.
Amodiaquine (Figure 27) was further investigated by Worek and co-workers for its reactivation potential, not only against DFP as was observed by Stojanovic and co-workers, but for its reactivation potential against tabun, sarin, cyclosarin, and VX.[115] Amodiaquine was determined to be a fully reversible inhibitor of erythrocyte ghost hAChE, but it is a highly potent one with a measured IC50 value of 669 nm. It was determined that amodiaquine is a mixed competitive/non-competitive inhibitor of hAChE in contrast to the results for eeAChE which showed amodiaquine to be a competitive inhibitor. As a reactivator, amodiaquine was able to recover small percentages of activity of sarin-, cyclosarinand VX-inhibited hAChE, but was ineffective versus tabun-inhibited AChE. Although reactivation was observed for these nerve agents, the rate of reactivation was significantly reduced in comparison to HI-6. Amodiaquine was shown not to scavenge OPs directly. These studies again confirm that amodiaquine has some functional moiety that is capable of reactivation of inhibited AChE.
Cerasoli et al. followed up on the study of ADOC with a focus on the importance of each structural element and the effectiveness versus OP nerve agents.[114] First, removal of the aniline and benzylamine moieties of ADOC significantly reduced the reactivation efficacy, but did not prevent reactivation. However, elimination of the hydroxyl functionality or blocking of the hydroxyl group (as a methyl ether) eliminated or significantly reduced reactivation, respectively. These results suggest that for ADOC, the portion of the molecule responsible for reactivation is the phenol acting as a nucleophile. It was found that altering the positioning of the benzylamine reduced reactivation ability. Interestingly, both decreasing and increasing the steric bulk of the benzylamine decreased inhibitory potential. If the size of the benzylamine was further increased reactivation potential was decreased. These results then suggest that the active mechanism for reactivation of inhibited AChE is unrelated to formation of the corresponding quinone methide as an intermediate, since all three variants would form the same intermediate—yet the results varied by structure.
Manipulation of the aniline to increase and decrease electron-donating effects resulted in decreased reactivation rates in both cases. The determined dissociation constants for the modification of the aniline moiety shows that the loss of reactivation is not related to binding, but instead to a reduced rate of reactivation. Changing the positioning of the aniline group from parato meta-position resulted in decreased reactivation potential, but not complete elimination, leading to the conclusion that the para-oriented aniline aids in proper positioning of ADOC within the AChE active site. The authors also conducted in vivo experiments with ADOC using an established guinea pig model; however, the results were contradictory to those previously reported in that there was no significant reactivation observed for ADOC. Notably, the authors report that the protocol used was based on pyridinium oximes and that administration time and method may have affected the results and also that there may be species differences between the in vitro human AChE, the previously studied mouse AChE, and guinea pig AChE. This species difference in reactivation continues to be a problem noted by multiple authors and studies.[99] Nonetheless, ADOC’s in vitro reactivation potential and its discovery as a new class of reactivator of OP-inhibited AChE can be explored by conventional medicinal chemistry techniques to develop optimized therapeutics.
Stojanovic and co-workers followed up their discovery of two classes of non-oxime reactivators in an additional expansion study. The first portion of the study focused on analyzing an analogue of amodiaquine called isoquine (Figure 28).[91] Isoquine was shown to have reduced toxicity as it does not form the iminoquinone; however, the structure still remains a Mannich base. Isoquine, like amodiaquine, showed reactivation potential versus paraoxon-inhibited AChE and actually performed at a level equal to that of amodiaquine. However, the results for DFP-inhibited AChE were reduced in comparison to amodiaquine. While Mannich bases are able to reactivate OP-inhibited AChE, the orientation of the Mannich base does have an effect on reactivation efficiency—this effect was also observed by Cerasoli and co-workers.[114]
Figure 28.
Additional reactivator classes.[91]
The Stojanovic study evaluated structure–activity relationships on the 41 (Figure 27) class of reactivators. The main modification was the removal of the p-bromophenyl ring resulting in deceased reactivation potency. Additional analogues were investigated in which the para-bromo moiety was replaced by a fluoro, chloro, phenyl, and hydroxyl group, all of which resulted in little change on reactivation potency leading to the conclusion that the para-position can be used to improve physical properties and formulation in future investigations. Unfortunately, none of the synthesized analogues outperformed 41. A similar set of compounds to that of 41 was tested drawing from the structure of 42 which was reported in their original study.[113] The new compounds replaced the imidazole moiety of 42 with a pyridine moiety, and three analogues were synthesized with methylene linkers ranging from 1–3 units. The newly synthesized compounds did show reactivation potential, but no increase in effectiveness, and the new compounds were all outperformed by 42. One of the reactivators (44, Figure 28) from the initial screen was noted for its features that were similar to donepezil and its reactivation potency was similar to 41 and 42. Additional donepezil-inspired analogues, such as 43, were synthesized and possessed either a piperidinyl or piperazinyl ring attached to the pyridyl ring. These compounds were evaluated for their reactivation potency but were ruled out as they, like donepezil itself, proved to be extremely potent inhibitors of native AChE.
The interest in reactivators with a Mannich phenol base continued as de Koning et al. expanded on the work conducted by Cerasoli and co-workers in synthesizing additional derivatives of ADOC and then did testing with human erythrocyte AChE.[114, 116] A total of 14 derivatives of ADOC were synthesized, with focus on variations in the amine leaving group. IC50 values were determined for all compounds and showed that simple modifications can have great effects. Compounds with 4–6 carbon atoms attached to the amine had some affinity for human erythrocyte AChE ranging from 6.3–482.5 mm. Lineweaver–Burk analysis showed that ADOC and compound 45 are mixed noncompetitive inhibitors (Figure 29). All the synthesized compounds were tested for their ability to reactivate human erythrocyte AChE at varying concentrations and after inhibition by VX, paraoxon, tabun, cyclosarin, and sarin. As with most reactivator studies, none of the tested compounds were able to in reactivity over time. It was hypothesized that this was due to re-inhibition by the phosphylated phenol after reactivation (perhaps, akin to POX re-inhibition,[117] as noted earlier in Section 1.4 of this review). The adduct was synthesized and found to be stable and a weak inhibitor of hAChE. Additional experiments using 1 mm reactivator and hAChE in the absence of OP or inhibition showed the same decrease in enzyme activity— the two offered explanations were that these high concentrations lead to denaturation of the enzyme or that the Mannich bases are forming quinone methides as intermediates and are then alkylating and altering the enzyme’s function. A second unusual finding was that some of the compounds, excluding 45, require up to 20 min to begin reactivation, in particular with VXand paraoxon-inhibited AChE. No explanation was offered for these results. Given the multiple studies based on Mannich phenols and their rapid reactivation for a number of OPs, it appears that significant focus should be shifted toward the investigation of these non-oxime, non-permanently charged reactivators, particularly trying to address some of the strange, or interesting, findings reported by van Grol and co-workers.
Figure 29.
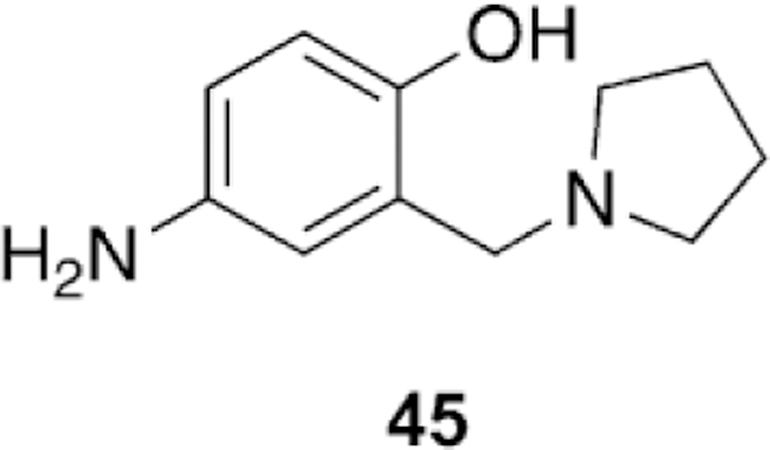
More reactive Mannich base for the reactivation of AChE.[116]
2.4. Difficulties with evaluation of developed reactivators
To date, thousands of oximes have been synthesized for the reactivation of various types of OP-inhibited AChE; however, the treatment methods still being used are those that were discovered in the 1950s. Outlined above is the current advancement in the field of oxime and reactivator development, but it is worth noting that all current and future studies will encounter the same problems in terms of advancing therapeutic development.
These difficulties begin with the structure-based design approach making use of X-ray crystal structures of known inhibitors and therapeutics with AChE. To date 178 structures have been reported with a majority being for T. californica and M. musculus and fewer for human forms. Of those structures reported, 146 are co-crystallized with various ligands.[118] X-ray crystallography is conducted at low temperatures and may not sample the appropriate conformations exhibited by AChE for binding under physiological conditions. This is particularly related to binding of some large ligands in which the crystallographic structure gives no inclination as to how the ligand can make its way into or out of the narrow AChE gorge, despite its high binding affinity. Additionally, crystallization additives have reactivate tabun-inhibited AChE. Of the tested compounds, the best were ADOC and 45. All other compounds showed negligible to some reactiva-been shown to bind within and on the periphery of AChE, which can alter positioning of the enzyme structure itself or the binding within the enzyme. It has also been noted that significant structural changes occur on ligand binding, most notation ability at high concentrations, but not in comparison to ADOC and 45. Most interestingly, compound 45 showed superior reactivation efficiency to ADOC for all tested OPs. Compound 45 displayed rapid reactivation as VX-inhibited AChE was reactivated in 20 min with only 10 mm 45. Some strange observations were made regarding some of the Mannich bases. For six of the compounds at 1 mm, they observed that there was initial rapid reactivation, followed by a slow decreasebly in the W loop. These structural changes cannot be predicted directly from the crystal structures of native AChE compared to the ligand-bound AChE due to the above-mentioned conditions.[92] In fact, a 20 ns simulation of hAChE shows upwards of 1400 A3 variation in the pocket volume resulting from significant structural movements.[118]
The most troubling issue is the comparison of data from various research groups with one another. Experimental details are critical, and different results can be obtained based on conditions including : AChE sources, in particular human compared to non-human, and the chosen experimental conditions including time of reactivation, buffer choice, pH, temperature, concentration, dilution, and calculation method.[35, 99, 107, 119] These experimental parameters are only those associated with in vitro experiments and the issues become even more widespread when moving to in vivo models. Issues of particular note for in vivo studies include animal species (additionally whether these are knockout or knock-in variants), OP exposure routes, dosage of OP, comparison to oximes or additional therapeutics, combination of therapeutic administration (whether atropine or benzodiazepines are included), time intervals in the study, and different methods for evaluating success (survival rate, AChE tissue levels, symptoms, etc.).[35] The only solution to these extensive problems seems to be a single testing protocol, although the implementation of such an idea is difficult as each individual laboratory has different access and resources to synthesize, express, or purchase sources of OPs, AChE, and animal models. At a minimum, these issues need to be acknowledged when comparing data from different sources when trying to determine how to further one’s own research.
3. Reactivation of Butyrylcholinesterase
Since BChE is a native human enzyme, has no assigned physiological role, and has high affinity for OP compounds, it has been suggested that BChE can act as a countermeasure that can scavenge OP nerve agents.[120–122] Additionally, the availability of BChE in blood serum with high concentrations (ca. 5 mg L-1, about 700 times more than AChE) makes it a suitable candidate to be used as a therapeutic against OP exposure.[78, 123] Indeed, Ashani and co-workers carried out in vivo experiments in mice and other models to show that stoichiometric quantities of BChE provide protection against nerve agent exposure.[120, 121, 124] Lenz et al. reported that a recombinant human form of BChE called ProtexialM, which was expressed in the milk of transgenic goats, can be a potential bioscavenger against nerve agent poisoning.[122, 125, 126] Lenz and co-workers showed that plasma-derived hBChE is a better candidate to be a therapeutic, and this formulation was granted investigational new drug status by the US Food and Drug Administration in 2006.[127–129]
Although several reports suggested BChE as a therapeutic approach against nerve agents, stoichiometric quantities of the enzyme are needed. Thus, an enormous quantity of BChE is needed to protect a large human population. Furthermore, recombinant hBChE does not have a sufficient serum lifetime as compared to plasma-derived hBChE, and subsequent dosages are needed for continuous protection, potentially causing several side effects. Regarding post-treatment, that is, BChE administration post-exposure to an OP agent, it was shown that the post-treatment of BChE, 120 min after exposure, provided protection against several dosages of the nerve agent VX.[130, 131] However, if one waits until the signs of OP poisoning are visible, only 1 out of 6 animals survived after BChE treatment.[132] As a result, despite many favorable properties of BChE in its native form, further improvements are needed for hBChE to be an effective treatment for OP poisoning. Ideally, the OP scavenger needs to have a long serum life that sufficiently overlaps with the therapeutic circulatory lifetime and should be able to hydrolyze the nerve agents catalytically so that large quantities of the scavenger are not needed for administration.
Once an OP inhibits BChE, there is no potent nucleophile available in the active site that can attack the (Ser)O-P bond to hydrolyze the OP molecule and reactivate the enzyme. However, if a potent nucleophile could reactivate the OP-inhibited BChE, then BChE and the nucleophile can act as a pseudo-catalytic bioscavenger to hydrolyze an OP.
However, none of the developed oximes for AChE demonstrated desirable efficiency of reactivation of nerve-agent-inhibited BChE. Since most of the oximes were optimized to interact with AChE, they did not have very effective binding affinities for BChE. Taylor et al. observed that reactivation of phosphylated BChE is not as successful with two common oximes, 2-PAM and HI-6, when compared to phosphylated AChE.[133] Furthermore, Jun and co-workers tested more than 20 oximes against AChE and BChE inhibited by the pesticide paraoxon.[134] Their work showed that none of the oximes were able to reactivate BChE more effectively than AChE. Similar results were reported recently for several other OPs bound to AChE and BChE.[135, 136] These results suggest that currently available oximes are not able to reactivate OP-bound BChE, even though AChE and BChE have extremely similar tertiary structure and active site surroundings.
The rate-limiting step for a pseudo-catalytic bioscavenger is dephosphylation, which must be more rapid then the competing aging reaction in BChE.[137] The effectiveness of treatment is also dependent on the method and agents to which an individual is exposed. For instance, the inhalation of a G-agent causes rapid distribution into the target tissues, so as a postexposure treatment, bioscavengers will likely not suffice. However, the slow distribution of V-agents from tissue after exposure offers a longer and more viable treatment window.[138] An additional issue with regard to catalytic bioscavengers is their specificity for classes, and even specific OPs. Thus, a mixture (cocktail) of multiple bioscavengers or mutants of the same enzyme might be needed to cover a broad spectrum of current and potential OPs.[138]
3.1. Recent results for reactivation of butyrylcholinesterase
Kovarik and co-workers developed a library of non-pyridinium based oximes for the generation of a BChE pseudo-catalytic bioscavenger.[139] A total of 39 imidazole and benzimidazole oximes were synthesized and screened for reactivation of BChE and AChE, inhibition potency of BChE, cytotoxicity as well as ex vivo and in vivo efficacy. The reactivators were designed to have a methylene linkage from the imidazole to which a hydrophobic group was attached as a means by which to increase the binding affinity of the reactivators for BChE. To determine the amount of the nucleophilic oximate form present in solution, pKa studies were conducted to determine the acidity of the oxime functional group. Of the compounds, neutral imidazoles were determined to have pKa values of 10.3–10.4, neutral benzimidazoles of 9.7–9.8, cationic imidazoles of 8.0–8.3, and cationic benzimidazoles of 7.4–7.6. These compounds were then screened with BChE after inhibition by tabun, VX, and (ethyl) paraoxon. Unfortunately, the only recovery of activity was demonstrated for VX-inhibited BChE. From the initial screen, a total of five compounds showed significant potency 46–50 (Figure 30). These compounds were further tested to ac- conducted using human whole blood and showed that 0.1 or 0.5 mm 49 and 0.3 mm BChE could degrade 1.5 mm of VX in 25 min. Following a few dosing trials, they determined that 2 mg kg-1 49 and 0.4 mg kg-1 BChE was capable of protecting animals up to 6.3 LD50 of VX. These results are very promising and are great proof of principle that BChE can be paired with an appropriate oxime to be pseudo-catalytic. However, as has been seen in AChE oxime research, there may be difficulty in finding a broad scope reactivator.
Figure 30.
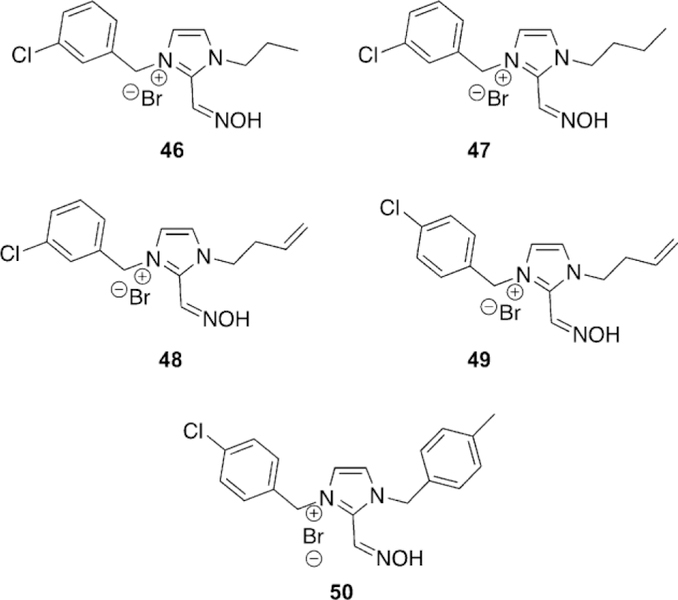
Imidazolium-based oximes for the reactivation of OP-inhibited BChE.[139]
Kovarik and co-workers took the known affinity of cinchonidine for inhibiting cholinesterases and synthesized and tested three novel oximes for their AChE and BChE reactivation potential (Figure 31).[140] Three different Chichona oximes were synthesized, and the resulting products were characterized fully, but left in their appropriate epimeric ratios, 51. The compounds were initially screened after human AChE inhibition by VX, sarin, cyclosarin, tabun, and paraoxon. Of the tested compounds, none of them reactivated more than 25 %, and there was no obvious trend based on structure of oxime. However, for hBChE reactivaquire detailed kinetic parameters to further refine their selection. These compounds were ineffective at recovering the activity of paraoxonand tabun-inhibited BChE, in comparison to obidoxime. However, for VX-inhibited BChE, all the compounds showed effective reactivation constants greater than that of HI-6 and obidoxime. All the tested compounds had reactivation maximum of 2: 85 % in 2 h or less. From the determined IC50 values, all the compounds had a high affinity for the enzyme with values :S 20 mm, significantly lower than traditional oximes. The imidazolium ring, especially with 1,3-substituted aromatic moieties, showed the highest affinity, and additional modifications may lead to improved oximes. One intriguing result is that the KOX and IC50 for the best oxime 49 were similar, suggesting that the size of the bound OP has no effect on the fit of the oxime, potentially allowing for structural design to be determined from un-inhibited BChE structures. AChE screening was also conducted, but none of the five lead compounds showed potency above that of the reference oximes. Toxicity studies were then conducted using HepG3 and THP-1 cell lines using the MTS assay. They determined that these imidazolium oximes possessed higher cytotoxicity than traditional pyridinium oximes. In particular the compounds with 1,3-aromatic substituents were the most toxic. However, given the promise and lower toxicity of compound 49, it was carried through to ex vivo and in vivo tests. The ex vivo study was tion, efficacy reached as high as 75 % and the best of the synthesized compounds for all OPs was 51. A derivative of 51, lacking the quaternary ammonium, performed the worst of all of the compounds, suggesting that like AChE, BChE has preference for positively charged groups. Most interestingly, 51 showed relatively broad scope reactivation with reactivation maxima of 60, 70, 45, 30, and 60 % for VX, sarin, cyclosarin, paraoxon, and tabun, respectively. When compared to the reference oximes for all the OPs, the reactivation maxima are lower than the reference; however, this broad scope reactivation with all OPs is unique to this set of compounds.
Figure 31.
Cinchona-based oxime showing broad scope reactivation potential. [140]
Additional studies to determine more detailed kinetic parameters were conducted as the compounds proved to be potent inhibitors of hBChE. Further investigation of the Ki’s for these compounds showed selectivity for hBChE over hAChE and that the values for hAChE were higher than literature values for pyridinium oximes and lower for hBChE. A Hunter– Downs analysis showed that these compounds are mixed inhibitors of cholinesterases and showed a preference for the 51 derivative with an N-benzylammonium group, suggesting preference for increased aromaticity. Toxicity studies using HepG2 and SH-SY5Y cell types showed that the compounds have an IC50 of about 800 mm suggesting they are not highly toxic. Given their broad scope reactivation potency and their moderate binding affinity and toxicity, this class of oximes requires further development in an attempt to generate hBChE as a pseudo-catalytic therapy.
Along with testing their non-oxime reactivators with AChE, Stojanovic and co-workers tested the same library of compounds with paraoxon-inhibited BChE.[91] Indeed, some of the AChE were capable of reactivating OP-inhibited BChE, most notably 52 (Figure 32), 42 and amodiaquine (ADQ) (Figure 27). Compound 52 is unique in that it only shows activity for BChE approaching 75 % recovery, yet it is inactive against AChE. More interestingly, compounds 42 and ADQ are active for both the reactivation of AChE and BChE, approaching 75 and 70 % reactivation, respectively. These initial findings are very promising since as has ods each giving a slightly different result. The hydroxyl pKa was determined to be 7.50, 7.65, or 7.21 dependent on the method used. For the oxime, the pKa was determined to be 10.6, 10.1, or 9.3 dependent on the method, showing that the oxime is significantly protonated at physiological pH. The authors also confirmed that 2-PAM was a better nucleophile than 53 as demonstrated by oximolysis studies. Oxime-assisted degradation of cyclosarin, VX, sarin, and soman were all found to be rapid, within the first few minutes of reaction, and in the case of cyclosarin and VX to be even more rapid. Similar experiments were conducted with human whole blood in ex vivo studies, showing near complete cholinesterase activity recovery, 80 %, after 100 min. Within the first 10 min following 53 been demonstrated with AChE, Mannich bases or other general bases can reactivate both cholinesterases, thereby opening up a number of new frameworks outside those of traditional oximes. These new routes present increased opportunity as the exhaustive search for oximes has thus far proven unfruitful for BChE reactivation.
Figure 32.
Mannich base for the selective reactivation of paraoxon-inhibited BChE.[91]
The very efficient and broad spectrum reactivators developed by Renard and co-workers proved to be efficient in the reactivation of BChE as well.[110] The synthesized compounds were tested with hBChE after inhibition by VX, sarin, cyclosarin, tabun, and paraoxon (the same OPs as the AChE study, Figure 24). The compounds showed good reactivation potential with all OPs, except sarin, and only modestly for tabun. Of the tested compounds, 35 (Figure 24) had the highest effective reactivation rate in the study. Also, all of the synthesized compounds outperformed HI-6 for reactivation of paraoxon-inhibited hBChE. Despite their reactivation potential, it is worth noting that all the tested compounds displayed higher dissociation constants than that for hAChE, suggesting further optimization with regard to binding may further increase their potency.
Using the molecule edrophonium as a scaffold for development, Taylor and co-workers synthesized a library of novel oximes in efforts to develop a pseudo-catalytic bioscavenger of OPs.[141] Structural adjustments on the molecules included changes to the position of the oxime, the presence or absence of a hydroxyl group, and the size of the alkyl substituents on the aniline group. All compounds were tested for their reactivation efficiency with both hAChE and hBChE. The authors determined that for hAChE, most of these compounds were inactive. In fact, only a single oxime showed any activity relative to 2-PAM for cyclosarin-inhibited hAChE, yet still performed significantly worse than the reference oxime HI-6. In contrast, one molecule, 53 (Figure 33), showed faster reactivation for OP inhibition by paraoxon, cyclosarin, and VX, when compared to the reference oximes, but not sarin. Any structural modification of 53 reduced the reactivation poten-addition up to 60 % activity for VX, 30 % activity for cyclosarin, and 15 % activity for sarin was recovered. To continue toward in vivo testing, the toxicity of the compound was determined to be 100 mg kg-1 in near direct agreement with that of 2PAM. The in vivo protective indices showed that they were actually higher for 2-PAM rather than for 53, in contradiction to the results of the in vitro tests. When mice were dosed with BChE, 53, and atropine, however, protective indices increased for paraoxon and sarin. Further tests show that small amounts of hBChE and 53 when administered together with atropine preand post-exposure could increase protection for paraoxon further demonstrating the utility of the combination in generating a pseudo-catalytic system. Unfortunately, the authors determined that 53 was 18-fold less concentrated in the brain than in the blood plasma, suggesting a poor ability to penetrate the BBB. In total, the results of the study are promising as the toxicity of 53 is similar to known therapeutics and shows potential as a reactivator with a catalytic bioscavenger.
Figure 33.
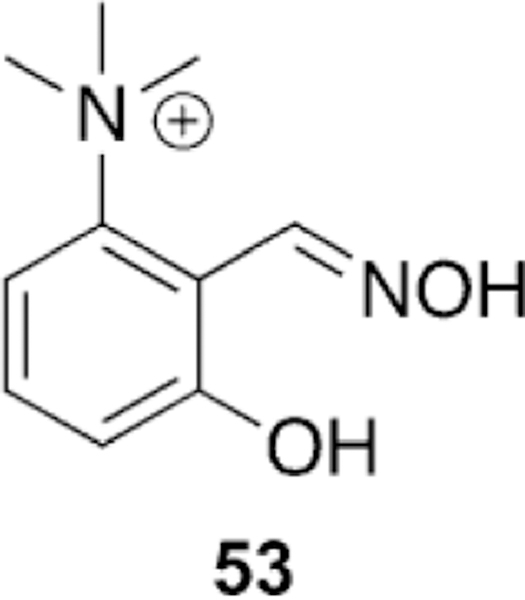
Edrophonium-derived derivative 53 for the reactivation of hBChE.[141]
Radic and co-workers followed up their discovery of the efficient hBChE reactivator 53 by investigating additional derivatives of N-alkylimidazole derivatives of similar compounds.[142] The investigation began with review of the screening of six imidazole aldoximes for their potency of reactivation of hBChE inhibited by low-toxicity analogues of sarin, cyclosarin, and VX in addition to (ethyl) paraoxon. The most efficient oxime was identified as 54 for all four OPs (Figure 34). Due to its universal nature of reactivation, additional derivatives of the 54 scaffold were prepared and evaluated with the same set of four OPs. Of the new oximes synthesized, compound 55 and 56 were identified as having a good, broad spectrum capacity as reactivators (Figure 34). The trends identified by the investigators were that the enzyme seems to have preference for hydrophobic groups positioned four methylene units away from the cy. Analysis of the reactivation efficiency showed that 53 was superior for paraoxon, cyclosarin, and VX, but slightly lower for sarin and tabun when compared to 2-PAM. imidazole ring, which may be used in future oxime development. The most interesting trend noted, and one that is contradictory to most previous findings, is that introduction of charge, by converting the imidazole to an imidazolium, reduced reactivation efficiency. The best compounds from the structural screen were then further subjected to additional modification of the imidazole group. Again, the finding was confirmed that a positive charge on these structures does not increase reactivation efficiency, and the only exception was for a single oxime in which its reactivation potency with a cyclosarin-surrogate-inhibited hBChE was increased 50-fold by the charge. At the time of this report, 55 and 56 are the fastest reactivators of inhibited hBChE. All of this work was repeated with hAChE, but unfortunately these structural modifications provided no benefit and all compounds were either comparable or slower than the reference oxime 2-PAM. Ultimately, this study shows that imidazole-based oximes can have very rapid rates of reactivation of hBChE inhibited by a variety of OPs. Given the lack of permanent positive charge on these molecules, it is hypothesized that these oximes will better cross biological barriers such as the BBB, making in vivo trials of great interest.
Figure 34.
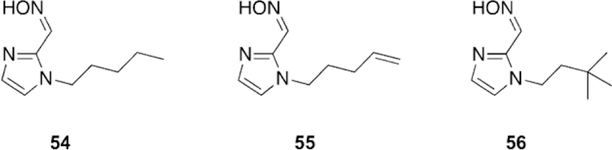
Imidazole-based oximes found by conducting structural modifications from the originally tested imidazole aldoximes by Radic and co-workers.[142]
Kovarik et al. continued the investigation of K-oximes for the application to generate pseudo-catalytic bioscavengers.[143] They determined that both oximes 57 and 58 (Figure 35) were capable of reactivating tabun-inhibited hBChE, with 57 having a maximum of 80 % reactivation in 50 min. They noted that reactivation kinetics deviated towards the end of the reactivation period, and this was attributed to three possibilities : 1) tabuninhibited BChE is aging, 2) reinhibition by the phosphylated oxime, and 3) different reactivation kinetics of the two enantiomers of tabun-inhibited BChE. Molecular modeling of the docked oximes within AChE and BChE noted that the major difference in affinity was attributed to the lack of aromatic groups in the peripheral site leading to differential binding. Overall, the authors concluded that introduction of an oxygen into the linker chain generates advantageous interactions in the BChE enzyme leading to reactivation.
Figure 35.
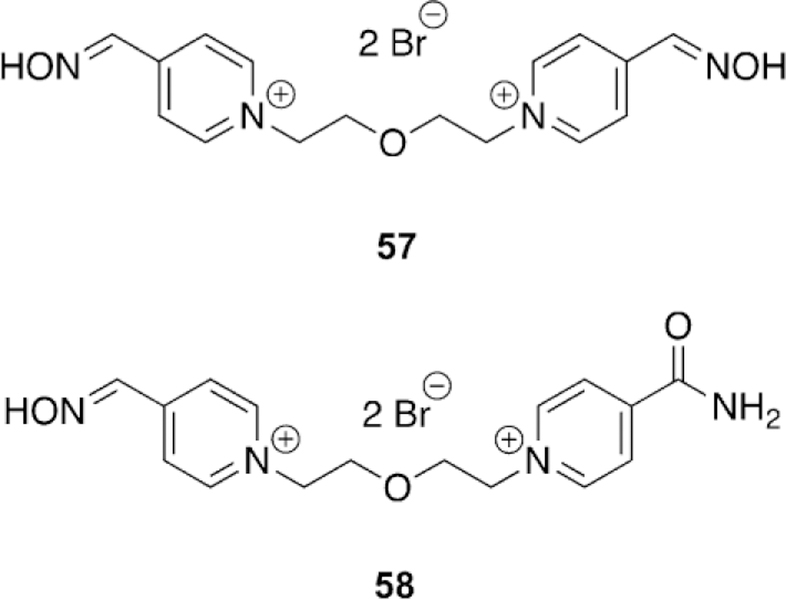
K Oximes capable of reactivating tabun-inhibited BChE.[143]
Worek and co-workers approached the pseudo-catalytic bioscavenger problem from a different angle, taking the currently available oximes, obidoxime and HI-6, and testing their ability to scavenge VX using different blood sources.[144] An interim solution was proposed that uses augmentation of AChE and BChE using blood products and currently approved oximes to scavenge OPs in the blood as more effective BChE reactivators have not yet been approved and are still in development. The samples of blood used were whole blood, human erythrocyte ghosts, plasma, packed red blood cells, and fresh frozen plasma. The whole blood system proved to be a better scavenger when paired with the oxime HI-6, and this trend was also observed for human erythrocyte ghosts. On the other hand, plasma showed a comparable effectivness for obidoxime and HI-6. When looking at storable blood sources, they determined that HI-6 and packed red blood cells worked better than with obidoxime. The observed rate of detoxification for freshly frozen plasma, however, proved to be slower and incomplete. All of these studies were conducted ex vivo and further in vivo trials are required.
4. “Resurrection” of Aged Acetylcholinesterase
While the reactivation of inhibited AChE has proven to be a challenging task for the past 70 years, the process is only more complicated by the competition of aging with the rate of reactivation. Since aging is the spontaneous dealkylation of the alkyl (R) group on the (inhibited) phosphylated serine residue, leading to an anionic phosphylated serine (Figure 7), aging of AChE removes the alkyl side chain of the original OP. Thus, the aging process leads to only three unique aged AChE structures—phosphonates (G and V agents, with the exception of GA), phosphates (paraoxon, DFP, and other pesticides), and phosphoramidates (GA).
Despite many attempts and across decades of effort, aging was considered irreversible, and until 2018, there were no in vitro or in vivo reports of any recovery of the activity of aged AChE. The enzyme was hypothesized to adopt a stabilized aged conformation, perhaps by forming a strong salt bridge between the phosphylated oxyanion at the serine residue and the histidinium ion of the catalytic triad, and perhaps also due to charge–charge repulsion between the phosphylated oxyanion and typical reactivating nucleophiles, such as pyridinium oximates or their neutral forms. Whether the nucleophile possesses a full negative charge or not, the increase in electron density leading to its nucleophilicity generates a repulsive interaction.[145, 146] Multiple attempts were made to convert the phosphylated oxyanion aged form to a re-alkylated (i.e., inhibited) form of AChE using a variety of alkylating reagents, including sulfonates, haloketones, methoxypyridniums, and sulfoniums.[88, 145–151] However, while some of these alkylating strategies were effective in model systems, these attempts proved to be unsuccessful when they were attempted in vitro with aged AChE.
In 2018, our research group published the first report that demonstrated the in vitro recovery of activity from aged AChE by the use of a small drug-like compound, a process that is termed “resurrection” as the aged (dead) form of AChE is brought back to life as the active, native form.[147] In the follow-ing, we will outline some of the major difficulties for the resurrection process with the aged form of cholinesterases.
4.1. Early attempts at the re-alkylation of aged AChE
One of the first reports to attempt re-alkylation of phosphonate anions, as models of aged AChE after exposure to authentic chemical nerve agents, was conducted by Stevens and coworkers.[150, 151] A library of alkylsulfonates was synthesized with the inclusion of a quaternary ammonium moiety to aid in the binding to AChE. Structural modifications were made with regards to the linker length between the alkylating moiety and the binding portion in addition to changing the alkylating groups and the binding moiety to triethylammoniums and pyridiniums. Although a report of their synthetic success was detailed, the only compound that appears to have been published, due to its poor reactivity with a model phosphonate anion, is 1-methyl-3-(methylsulfonate)pyridinium, 59 (Figure 36). The compound was tested for the alkylation of a number of anions including isopro-pylmethylphosphonate and p-nitrophenylmethylphosphonate. Unfortunately, the results of the model study were limited as the compounds reacted slower with phosphonates than with nearly any other anion, exceptnitrate. These initial results were troubling given the structural similarity to known binders, but more troublesome due to their clear propensity for off-target alkylations. This aspect is very important—many alkylating agents are possible, but the selective re-alkylation and eventual resurrection of aged AChE is critical, and off-target activity must be minimized or preferably eliminated.
Figure 36.
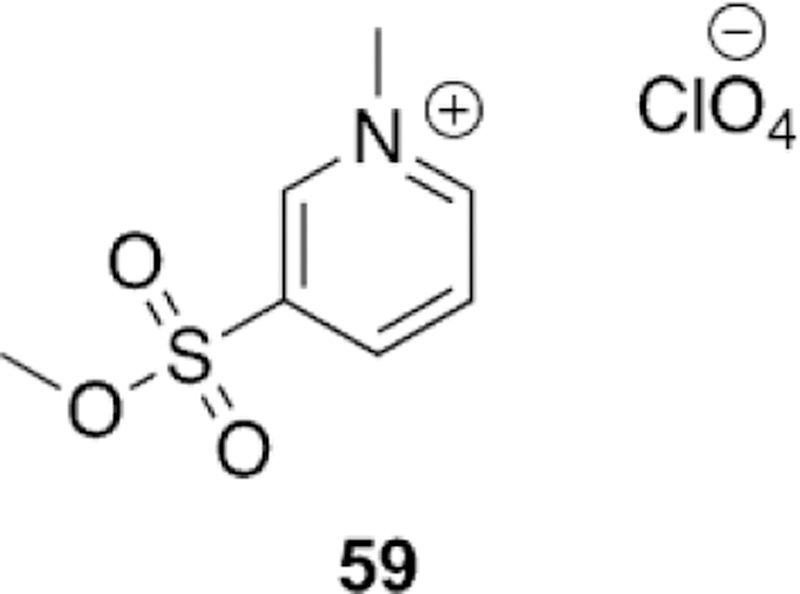
Sulfonate alkylator in first attempt to alkylate phosphonate anions.[150]
A second 20th century attempt to re-alkylate a model phosphonate and aged AChE showed only success with model nucleophiles, but the authors did draw some interesting conclusions based on rate changes for hydrolysis and alkylation due to distant carbonyl groups.[149] Boldt and co-workers used sodium p-nitrophenylmethylphosphonate as a model nucleo-phile for kinetic studies of alkylation and hydrolysis by strong alkylators composed of various functional groups including ahaloketones, a-halodiketones, alkyl and arylhalides, alkyl and arylmesylates, a-haloamides, a-haloximes, chloromethylphosphonic acid, a-halocarboxylic acids, a-haloalcohols, and epoxides. (Many of these “warheads” are often used for suicide inhibition of select biological targets.)[152] As was anticipated, compounds with electron-withdrawing groups resulted in both faster alkylation reactions due to an increase in electrophilicity, but also the rate of hydrolysis was increased for the generated mixed phosphoryl esters. The same trend holds true for hydrolysis of the alkylating agents themselves. One of the more interesting findings was the intramolecular participation of carbonyl groups in the hydrolysis of the generated alkylated phosphylated products. By placing either direct nucleophiles, such a phenols or oximes, on the ring or using activated carbonyl groups, with electron-deficient character favoring hydrate formation, or amides, the authors were able to promote hydrolysis, and likely through intramolecular catalysis. Following initial kinetic studies, in vitro reactivation of aged AChE was attempted with seven representative compounds : 60–65 (Figure 37). The compounds were incubated with soman-aged AChE, but none proved to be effective in vitro. Control studies did show that all the compounds were progressive inhibitors of the native enzyme, owing to their very high electrophilic character. Although no hits were identified by this study, their efforts did present some key themes regarding careful balance ing of the electrophilic character so as to not achieve offtarget activity, but also the inclusion of chemical groups to aid in the cleavage of the “re-alkylated” phosphylated product that is generated following an effective reaction.
Figure 37.
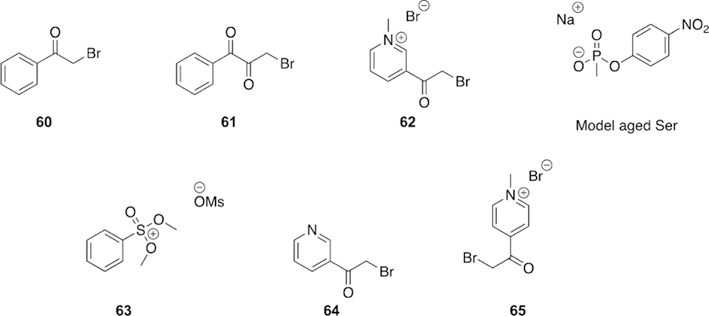
Strong alkylator groups used unsuccessfully in vitro to re-alkylate soman-aged AChE.[149]
One of the first modern approaches to alkylate aged AChE mimics was conducted by Quinn and co-workers through the use of N-methyl-2-methoxypyridiniums as methyl-transfer reagents.[147] Quinn was trying to recover what has long been treated as a dead enzyme and thus coined the phrase “resurrection” in which the aged adduct is reverted back to the native form of AChE. The selection of N-methyl-2-methoxypyridiniums was made based on the predicted balancing of selectivity, toxicity, and susceptibility to hydrolysis in addition to the similar structural features to that of the FDA approved oxime, 2-PAM. In total, nine different compounds were synthesized and their methyl-transfer rates were determined using the aged AChE model of sodium methyl methanephosphonate in [D6]DMSO using 1H NMR spectroscopy. The presence of small amounts of water also allowed for the competitive formation of methanol through the hydrolysis reaction. The rates of reaction were determined using numerical integration and displayed various methyl-transfer rates. They noted that the rate of methyl transfer, methoxy NMR shift, and Swain–Lupton field and resonance parameters allowed for estimation of the rate of reaction based on the substituent and the chemical shift of the methylmethoxypyridiniums. The effects of each parameter on the rate of reaction showed that the methyl transfer is sensitive to both resonance and field effects and can be tuned as such to increase or decrease the rate of reaction appropriately. The highest rate of methyl transfer was observed for the Nmethyl-2-methoxypyridinium, 66 (Figure 38), with a 3-F substituent, achieving 40 % methyl transfer in less than 10 min. Analysis of the rates of hydrolysis in D2O showed that some compounds were stable to hydrolysis for an entire week. In general, the rate of hydrolysis in D2O was two orders of magnitude slower than methyl transfer. However, when hydrolysis was measured in pH 7.2 phosphate buffer, the hydrolysis rates were faster, but still slower than methyl transfer in DMSO, and compound 66 showed methyl transfer to the phosphate buffer. Both the rates of methyl transfer and hydrolysis in all solvent systems showed that these compounds were stable under near physiological conditions and unselective alkylation should be minimal, making these compounds tunable alkylating agents of model phosphonate anions.
Figure 38.
N-Methyl-2-methoxypyridnium structure and model phosphonate anion used to study methyl transfer by Quinn and co-workers.[147]
In a subsequent study, Quinn and co-workers synthesized a larger library of methoxypyridinium compounds and drew inspiration from investigated and functional oximes. They determined the inhibitory potency of these methoxypyridiniums to ensure that inhibition was reversible with AChE.[148] In total, 32 different compounds, representing seven (67–73, Figure 39) different classes, were synthesized and their IC50 values were then determined with hAChE. For the 67 framework, potency varied from 7–70 mm, being slightly lower than structurally similar 2-PAM. The bis-methoxypyridinium moieties of the framework of 68 showed increased binding affinity with IC50 values below 1 mm, consistent with previous observations from numerous oxime studies. Interestingly, bis-substituted compounds for 69 and 70 did not show a reduction in IC50 when linked at the pyridinium, suggesting that the linking position can have a significant effect on potency. Other peripheral site linked compounds such as 71, the dimethoxyindanone core from donepezil, and 72, a tetrahydroisoquinoline, reduced the IC50 values to the nm range, further demonstrating the effectiveness of tethering the potential warhead to peripheral site ligands. The reduction was not as dramatic when using coumarin-linked compounds (73) with an observed reduction of only one order of magnitude. Although the binding affinities and re-alkylation efforts with model nucleophiles were successful, preliminary in vitro evaluations showed no recovery of activity for aged AChE. Yet, these compounds present a core framework and ideology to continue to investigate methods of re-alkylation of aged AChE.
Figure 39.
Various core frameworks and peripheral site ligands employed for hAChE inhibition by Quinn and co-workers.[148]
An approach making use of a sulfonium-based alkylating agent, selected for its observed stability, selectivity, and toxicity, for the re-alkylation and subsequent reactivation of methylphosphonate AChE was investigated by Ganguly and co-workers.[146] The authors made use of molecular docking studies followed by molecular dynamics simulations (MD), and subsequent steered molecular dynamics (SMD) to investigate the binding and egress pathways of three novel sulfonium alkylators (74–76, Figure 40). The final structure, 76, actually possessed a nucleophilic oxime moiety and was investigated to see if it adopted a pose that would allow for additional reactivation with the aged Ser residue. The first step was quantum mechanical investigation of the alkylation reaction between a model soman-aged phosphonyl adduct with the proposed alkylating agents using the MO5–2X/6–31G* level of density functional theory. For 74–76, the activation barriers were determined to be 31.4, 27.9, and 26.9 kcal mol-1 with all the reactions being exothermic with energies of −6.5, −9.9, and −11.4 kcal mol-1, respectively. The same method was used to determine the activation barrier for the aging of a model soman-inhibited Ser and found to be 37.8 kcal mol-1, suggesting that for these three compounds, the aging process is predicted to be reversible.[153]
Figure 40.
Computationally investigated sulfonium re-alkylators of aged AChE.[146]
Using the same methodology, an unsubstituted 2-methoxypyridinium was also evaluated and determined to have a larger barrier to re-alkylation than all the sulfonium compounds investigated at 32.3 kcal mol-1, only being exothermic by 0.6 kcal mol-1. These energetic results suggest why in vitro re-alkylation was not observed. There was an energetic gain by moving from 74 to 75 and incorporating a pyridine ring. This observation then inspired the design of 76, which still had the pyridine ring but was additionally tethered to a nucleophilic oxime as a means by which to accomplish the re-alkylation by the sulfonium and then subsequent reactivation by the oxime. The energetic study showed that 76 was capable of the re-alkylation so an additional investigation of the nucleophilicity was undertaken. The determined nucleophilicity index of 2PAM was 0.0027 eV and that of 76 was 0.0108 eV, thus implying that molecule 76 is capable of accomplishing both electrophilic and nucleophilic tasks. The affinity of 76 was then determined by analyzing the docking with both AChE and BChE followed by SMD simulations with TcAChE, a reference reaction was determined to be in good agreement with the crystal structure used for BChE and a 1-methyl-2-(pentafluorobenzyloxyimino)pyridinium. Compared to the reference compound, 76 was computed to have a shorter distance to the reactive phosphonate anion, being 3.840 A from the alkylator. To further investigate those interactions contributing to the binding and egress of 76, the ligand and enzyme were subjected to SMD. It was shown that during the course of the simulation, as 76 is pulled away from the enzyme, it breaks a number of hydrophobic interactions over the course of egress, pointing to significant stabilization within the enzyme active site. Most notably however, is the rotation of the drug candidate within the enzyme as 76 is pulled out as measured by multiple metrics. This is significant as it presents the possibility of reactivation the phosphylated Ser residue. However, the compound used in the SMD simulation is still a sulfonium and the enzyme itself is aged and not inhibited, yet according to the authors, the size of the enzyme active site does allow for rotational freedom. These results coupled together then offer computational evidence for the continued investigation of sulfonium-oxime compounds for the reactivation of aged AChE.
Wallqvist and co-workers used computational tools to develop novel reactivators of methylphosphonate-aged AChE in which the reactivation was direct and did not proceed through a re-alkylated intermediate.[145] Drawing inspiration from their research on CapD, which functions similar to serine proteases but having a single threonine residue rather than a catalytic triad, they noted that b-aminoalcohols may activate the catalytic threonine residue. Since b-aminoalcohols are neutral and become charged late into the nucleophilic attack, it was hypothesized that this functionality would serve as a sufficient nucleophile for aged AChE reactivation due to its proton-transfer ability to quench the negative charge of the phosphylated Ser, while being neutral should allow for BBB permeability. To achieve selectivity for the AChE active site, two molecules were then designed, 77 placed the b-aminoalcohol group onto a pyridinium ring, drawing inspiration from the oxime 2-PAM, and 78 was a constitutional isomer removing the nitrogen from the aromatic ring and placing it on the periphery (Figure 41).
Figure 41.
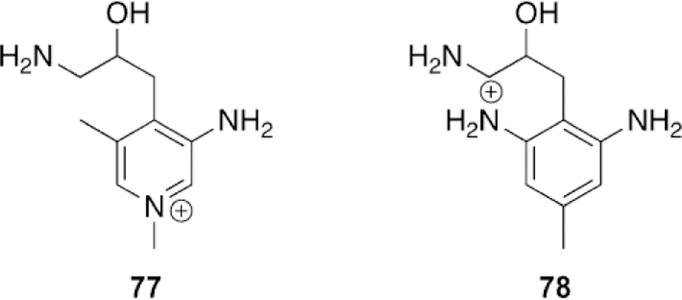
Computationally investigated aminoalcohol re-alkylators of aged AChE.[145]
To determine the ability to reactivate aged AChE, the molecules were first docked within the enzyme and then molecular dynamics simulations in solution were performed to allow for flexibility of the enzyme. Following these initial calculations, QM/MM studies were conducted in which the catalytic triad, oxyanion hole, E199, Y121, five water molecules, and the ligand were all modeled using quantum mechanics, while the remainder of the enzyme was then evaluated using a molecular mechanics force field. Despite the structural similarity to 2PAM, 77 was determined to have a 50 kcal mol-1 activation barrier with the more reactive S enantiomer. The authors concluded that for this particular molecule, the reaction proceeded in a single step; however, there was insufficient disruption of the salt bridge between the phosphylated oxyanion and the histidinium to allow for proton transfer from the ligand. It was thus concluded that 77 would not be an effective reactivator of aged AChE. In the case of 78, the overall reaction was exothermic by 1.1 kcal mol-1 and the reaction proceeded through an intermediate with an energy of 7.2 kcal mol-1. The overall activation barrier for this reaction was 25.3 kcal mol-1, proceeding in two steps : the first step being the activation of the alcohol by an intramolecular proton transfer and the second step being the nucleophilic displacement of the phosphylated moiety. The studied protonation state was not predicted to be the one observed in solution. The authors determined the pKa values of the groups to be 9.4 for the b-amine, then 5.1 and 2.8 sequentially for the anilines. The authors, however, justified these differences, noting that enzyme active sites can have significant effects on pKa values and also that structural modifications can further alter the pKa values of the studied molecule. Initial estimations of the rate of reaction were predicted to result in 50 % reactivation of aged AChE in approximately 4 min. This estimation makes multiple approximations and does not account for any binding energies but rather just activation barriers and lacking entropic contributions. Such b-aminoalcohol functional groups have not been tested experimentally, to our knowledge.
4.2. Quinone methides as alkylating agents of model nucleophiles
Initial attempts to find alkylating agents for aged AChE have proven difficult due to the primary issues regarding binding, selectivity, stability, and toxicity. During the 1990s to the 2000s, there was a number of publications noting the utility of quinone methides (QMs) as alkylating agents of various biologically relevant molecules including amino acids, DNA and nucleic bases, and structural modifications could be used to modulate the selectivity and reactivity of the alkylating agents to promote or repress specific parts of the reaction. In 1999 it was reported that these QMs could alkylate phosphodiesters, which are structural analogues of pesticide-aged AChE. The investigation of such molecules and reactions could then be used as a method to develop effective therapeutics for in vitro and in vivo re-alkylation of aged AChE.[154–156]
The first report of alkylation of a phosphodiester was published by Turnbull and co-worker in 1999.[154] This study made use of a p-quinone methide generated from mixing 2,4,6-trimethylphenol with silver oxide in organic solution (Figure 42). Upon the generation of the QM, an organic solution of tetrabutylammonium dibenzyl phosphate was added, but no product was observed by 1H NMR spectroscopy until the addition of MsOH. Studies showed that the reaction was completely dependent upon the addition of MsOH, and was in fact reversible upon manipulation of the pH through addition of potassium carbonate. Changes in acid strength, ensuring that the conjugate base was not nucleophilic enough to compete, proved to be unsuccessful. Through changes in the concentration of the phosphate nucleophile, it was determined that there was a pre-equilibrium with the added acid reducing nucleophilicity and lowering overall conversion. Additional experiments were conducted with dibenzyl and diethylphosphoric acid serving as both the acid and the nucleophile to observe that the equivalents of the phosphoric acid pushed the reaction to a high conversion of 83 % for 2.7 equivalents of dibenzyl and 3.9 equivalents of diethyl. These same reactions showed that the rate of reaction was dependent on both the acidity and nucleophilicity of the phosphoric acid, a finding that needs to be considered and evaluated when designing a therapeutic for re-alkylation of aged AChE. The authors concluded that the QM intermediate had to be activated to allow for attack by the weak phosphates and thus overall, the reaction was acid catalyzed. This initial study was an important and first demonstration of the potential for alkylation of aged AChE model compounds with QMs, but they did not address aqueous solubility, competitive hydrolysis or the generation of the QMs in solution.
Figure 42.
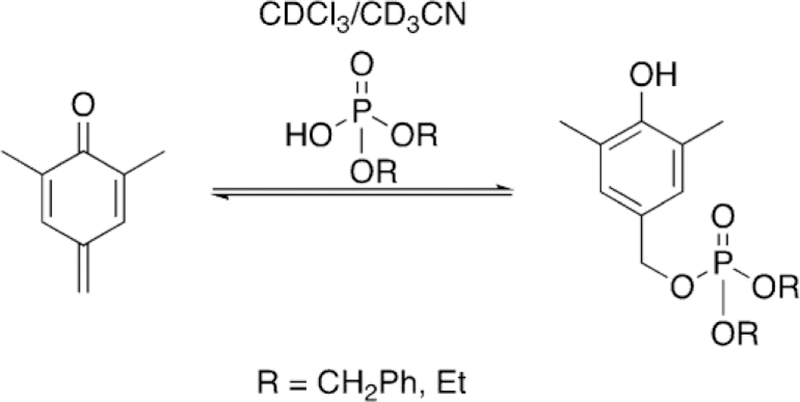
Initial reaction showing the potential of QM alkylation of phosphodiesters.[154]
The initial study was followed up two years later and addressed the competitive hydrolysis reactions by moving to mixed aqueous solutions (Figure 43).[155] The initial study was conducted in 10 % aqueous buffered solutions with the same p-QM used in the initial studies and diethylphosphate as the choice of nucleophile. The maximum conversion to the phosphodiester was 16 % within 9 min, and by 10 min, the conversion to the benzyl alcohol had reached 75 %. However, kinetic determination of the reaction showed that the hydrolysis rate was only 2.2 % that of the alkylation rate despite the concentration of water being 220 times that of the phosphate nucleophile. Additional studies were carried out in which 28.5 % aqueous buffered solutions were used and the nucleophile was varied between an inorganic phosphate and dibenzyl, dibutyl, and diethyl phosphodiesters at a pH of 4.0 and 7.0. It was determined for inorganic phosphate, that the hydrolysis of the QM is a specific acid catalyzed process. For the tested phosphodiesters, the authors reported that dibenzylphosphate alkylation is five times faster than dibutyl and 10 times faster than diethyl at pH 4.0, again showing that the nucleophile has a significant effect on the rate of reaction, which will be key to drug design. For the pH 4.0 studies, dibenzylphosphate reacted 3700 times faster than water and even the slowest phosphate (diethyl) still reacts 370 times faster than water. Unfortunately, when they used pH 7.0, it was determined that hydrolysis was the dominant reaction and almost no rate of change was seen when varying the concentrations of the phosphates in solution.
Figure 43.
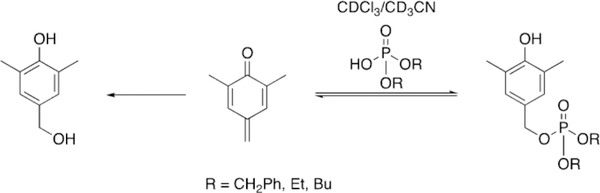
Subsequent study showing the potential of QM alkylation of phosphodiesters.[155]
Additional studies with phosphate nucleophiles were also reported by Turnbull and co-workers, using a cyclization reaction to increase the synthetic yield of phosphate reaction with the QM intermediate.[157, 158]
Thus, QMs are capable of making similar transformations to the one desired for the re-alkylation of aged AChE, yet the selectivity and reactivity still require substantial adjustment given high conversions to the benzyl alcohol previously reported. Rokita and co-workers reported on the ability to appropriately modify formation and stability of QMs using substituent effects.[156] Their study tested the effects that substituents have on product distribution as well as reversibility by changing the groups attached at the metaand para-positions relative to the QM methylene, while intentionally avoiding the steric complications caused by ortho substitution. The initial study used a silyl-protected phenol as a quinone methide precursor (QMP) which, upon treatment with a fluoride source, would cleave the Si-O bond, and then would be followed by the expulsion of the acetate leaving group, in order to generate the appropriate QM. Three different QMPs (79–81, Figure 44) were used with para substitution, relative to the methylene, and substituted by either a methyl, hydrogen, or methyl ester. Each of these QMPs was then reacted with deoxycytidine (dC) and the rates of formation and decomposition of the product were monitored. For 80, the more electron-rich precursor, initial formation of the product was rapid and was complete within 5 h, yet the decomposition was also rapid with near complete decomposition, half-life of 5 h. When this is compared to 79, product formation is slower, taking approximately 10 h with a slower rate of decomposition as well, half-life 24 h. Finally, 81 slowly formed product reaching a maximum at about 40 h and showed no decomposition. These results then suggested that compounds with increased electron density will more rapidly form the QM than those that are electron deficient. These results were further supported when the same set of compounds was incubated with four different deoxynucleosides in a competition experiment. It was again noted that electron-deficient 81 formed product at a slower rate; for example, for a dA complex with 81, it took 10 h to reach its maximum but for 80 only 0.5 h. The more interesting of the findings was that the overall product distribution changed based on the electronic substituents of the ring, reflecting that some adducts had such poor leaving groups that the QM was never regenerated in the case of 81, but was for 79 and 80. To further study the effects that electron-donating and electron-withdrawing groups have on QM generation, a new set of QMPs were used to ensure that the reaction was on a detectable time scale. To this end methoxy, carboxymethyl, and unsubstituted phenols were used with a morpholinoleaving group, 82–86. The stability of these QMPs was then monitored in aqueous solution for the generation of their benzyl alcohol derivatives and the results emphasized the previous observations even further.
Figure 44.
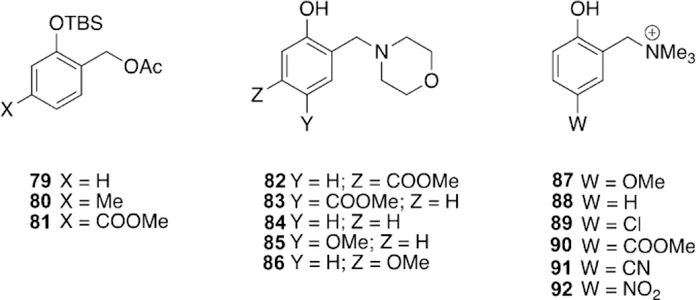
QMP structures used to study the aspects of QM reaction by Rokita and co-workers.[156]
The rate of formation of the QM is significantly affected by the ring’s electronic effects, and substitution is more sensitive to resonance effects as compared to field effects. To determine the effects that the same substituents had on nucleophile addition, a final set of 87–92 was used where para-substituted Mannich bases with trimethylammonium leaving groups were converted to their QMs by means of photolysis and the rate of product formation determined based on competition between three nucleophiles : water, morpholine, and 2-mercaptoethanol. It was determined by these studies that electron-withdrawing groups greatly increase the electrophilicity of the QM as is demonstrated by multiple orders of magnitude increase in the rate of reaction. The opposite is observed for electron-rich substituents in which the rate of reaction for nucleophiles is reduced by approximately a factor of four. Additionally, selectivity was reduced as stronger electron-withdrawing groups are included as is expected by the reactivity–selectivity principle.
Ultimately, this study showed that quinone methide precursors can generate their QMs in solution through multiple routes : thermally, by deprotection, and photochemically; moreover, the properties related to reactivity, selectivity, and reversibility can all be altered by changing the substituents on the aromatic ring. With these conclusions and the results with alkylation of phosphodiesters, QMPs may serve as viable options for the design of small-molecule re-alkylators for aged AChE.
4.3. Mannich bases for the alkylation of aged acetylcholinesterase
Given the proven potential of QMs and QMPs, Hadad and co-workers began an initial study to determine if these types of compounds would be sufficient to recover the activity of aged AChE.[159] A total of eight different compounds were synthesized on the basis of a vanillin framework with various amine leaving groups in both the protonated and deprotonated forms, 93–96 and 97–100 (Figure 45). First, the compounds were tested with model nucleophiles (4-methylbenzenethiol, piperidine, and benzyl al-cohol) for their re-alkylation abilities. The compounds were incubated with the nucleophiles at 100 8C for 3 h and the conversion was observed. For the strong sulfur-based nucleophile, conversion was about 85 %, the weaker piperidine nucleophile was 70–80 %, and the benzyl alcohol was very limited, only 0–16 %. Thus, the compounds were determined to be sufficient alkylators with modest activity and notably low hydrolysis. The alkylation with a model phosphonate was then attempted with 93 and 97 and monitored by HPLC/MS; however, only trace amounts of alkylation were observed.
Figure 45.
QMP structures used in reaction with nucleophiles for proof of principle alkylation.[159]
To determine if the QMPs have any affinity for the active site of hAChE, molecular docking studies were conducted in which the percentage of docking poses with the benzylic carbon within 5 A of the phosphylated oxyanion was used as a metric of a reactive conformation. The docking studies suggested a preference for the pyrrolidine and piperidine leaving groups. This result was further confirmed for pyrrolidine when the compounds were subjected to MD simulations, again using the benzylic distance to the oxyanion of the aged phosphonate as a metric of reactivity. The MD simulations showed that of the amines, the pyrrolidine compound spent the most time within the enzyme active site. The MD simulations also showed a strong preference for those compounds which are protonated at the amine, likely due to the similarity to ACh. To try and confirm the accuracy of the docking and MD results, IC50 values were determined for native AChE, as an assay to determine affinity for the aged enzyme is unknown. For the native enzyme, the preference for binding was determined to be piperidine > pyrrolidine > dimethylamine > morpholine, and IC50 values were on the order of 0.227–1.83 mm. Although the ordering is not the same as that concluded for the docking and MD, this is reasonable as the active sites of native and aged AChE are substantially different. All the compounds were shown to be competitive or near competitive mixed-type inhibitors and confirmed a preference for the aged active site. A re-alkylation assay of the HCl salts of all the compounds was conducted with eeAChE, but unfortunately no recovery of activity was observed in the initial study. Thus, it was proven that QMPs are capable of serving as moderate alkylating agents with model nucleophiles and the structures do have affinity for the AChE active site as was demonstrated by inhibition studies, molecular docking, and MD simulations. Given the tunability of the reactivity presented previously, the concept was further carried forward for additional optimization.
Inspired by the positive results of the nucleophilic alkylation and affinity for the AChE active site, Hadad and co-workers continued their investigation of computational libraries of QMPs.[160] These computational investigations showed that the AChE active site had a higher affinity for pyridyl QMPs over phenyl frameworks, and these results are certainly consistent with effective AChE reactivators being monoor bis-pyridinium compounds. A library of compounds was synthesized including 13 derivatives or 2-alkylamino-3-hydroxypyridines with variation of the amines. To test whether these compounds were capable of reactivation of aged AChE, both 101 (Figure 46), a 3cyano-4-methylcoumarin analogue of soman (GD),[161, 162] and a model phosphate (DFP) were selected to generate methyl-phosphonate-aged and isopropylphosphate-aged forms of AChE. The OPs were incubated with eeAChE for a sufficient time to ensure the enzyme was completely aged. In the case of 101, a double aging cycle was employed in which the enzyme was aged and then reactivated with 2-PAM and aged again with the same OP, and this procedure ensured a low baseline for detecting eventual resurrection of AChE activity. Using various concentrations, the 13 compounds were then incubated with aged eeAChE for 24 h at pH 8 in the presence of a nonselective reactivator NH4F, and to ensure nothing was trapped in the inhibited state following re-alkylation, a second 2-PAM treatment was conducted. Native AChE activity was then detected by Ellman’s assay. Surprisingly, a number of molecules showed activity above baseline and the results followed relatively similar trends. Interestingly, the methylphosphonateaged AChE showed higher levels of recovered activity than that of the isopropylphosphate-aged AChE. The results for the methylphosphonate-aged AChE show a trend related to steric effects of the amine. The N-methyl-N-ethyl substituted amine recovered about 9.5 % of native activity and decreased until it reached the 5-membered heterocycle pyrrolidine 103 (Figure 47) which was the most active at 12 %. The isopropylphosphate-aged AChE showed similar trends, but the results were not as pronounced and showed some tolerance for N-methyl-N-isopropylamine, N,N-di-ethylamine, and piperidine. The compound with the pyrrolidine ring was chosen as a lead compound and was pursued in subsequent studies.
Figure 46.
Nerve agent analogues used in biological assays.[160]
Figure 47.

Lead QMP structure for resurrection of aged AChe.[160]
Kinetics studies were conducted on an extended timeframe, although in this perturbation of the assay, an external reactivator was not included. Activity of the aged samples were recovered at linear rates out to four days and successfully resurrected 32.7 and 20.4 % of the activity for methylphosphonate-aged and isopropylphosphate-aged eeAChE, respectively. Importantly, these results also indicated that these compounds are not only resurrectors of aged to the native state but also reactivators of the inhibited (or re-alkylated) form. To determine the reactivation potential of these compounds, four of them were selected and incubated with a VX analogue, 102 (Figure 46), for 24 h at high concentrations. Two of the compounds com- pletely recovered the activity of the enzyme up to the level of the positive control and the other two reached 60–70 %. These results are in good agreement with a number of studies which found that Mannich bases are reactivators of OP-inhibited AChE.[91, 113, 114, 116]
While the kinetic assays confirmed recovery of the native enzyme, bottom-up proteomics were conducted to try and isolate the enzyme in the native, inhibited (re-alkylated), and aged states. A solution of the lead compound 103 was incubated with aged eeAChE for 11 days before being digested with trypsin. Positive and negative controls were prepared in parallel. The negative control showed only aged AChE, the positive control revealed only native AChE, and the sample treated with a QMP showed both aged and native AChE following digestion, but not re-alkylated—and in a ratio consistent with the kinetic assay for resurrected activity. These results confirmed the kinetic recovery of activity of the aged enzyme to the native state, clearly showing resurrection to the active serine residue.
Since these molecules showed clear reactivation, a limited study was performed with various pH values to determine the effect on resurrection. Surprisingly, a very large pH dependence was noted for methylphosphonate-aged resurrection. The difference between pH 7 and 9 for resurrection was approximately 20 %. 103 was analyzed by UV/Vis and 1H NMR spectroscopy at various pH values to determine the pKa of between 7–8 and implicated various protonation and zwitterionic states of 103. Computational techniques were used to investigate the highest affinity for the enzyme active site, and the MD results of the study suggest both zwitterionic forms of 103 have affinity for the active site over other protonation states. The kinetic profile of 103 was finally compared for its ability to reactive hAChE aged with DFP and similarly reached about 20 % resurrection, confirming that these results are not just exclusive to eeAChE.
After almost 70 years of effort, the goal to recover activity from the aged form of AChE has been achieved, and resurrection was demonstrated after aging by both organophosphate pesticides and authentic organophosphonate nerve agents. However, further work remains to create a therapeutic drug with all the desired activity, efficacy and selectivity.
4.4. Problems with re-alkylation of aged AChE
Despite the recent success in resurrection by Hadad and coworkers, the reactivation or re-alkylation of aged AChE is challenging; a substrate has to bind selectively and efficiently in the active site, to bind in an active conformation so as to generate the critical reactive intermediate and/or to facilitate the critical transition state for the desired re-alkylation, and then to allow the re-alkylated phosphylated serine to be reactivated by a good nucleophile. In addition, all of this has to occur for an active site that is relatively compact and constricted.
A computational investigation conducted by Liu and coworkers evaluated the attempts to re-alkylate aged AChE with 2-methoxypyridniniums by Quinn and co-workers.[163] Liu evaluated two different mechanisms of action as proposed by Quinn as the mechanism by which re-alkylation occurs, and in total, the reaction of nine different 2-methoxypyridinium derivatives were investigated for their mechanism of action both in solution using density functional theory (DFT) and bound within the enzyme using QM/MM techniques. They determined that the mechanism of action is an SN2 displacement of the methyl moiety from the methoxy group. The calculated activation barriers in solution were in good agreement with experiment. The QM/MM methods confirmed that as in solution, the preferred reaction mechanism in the enzyme is an SN2 process, but with substantial energetic barriers. The activation barriers for the reaction have a mean of 30.4 kcal mol-1 and a Boltzmann-weighted average of 26.6 kcal mol-1, suggesting that the reaction will not proceed in vitro. The authors reported that the significant energy barrier is due to the strong p–p interaction between W86 and the pyridinium substrates. The authors proposed a few solutions, such as introducing a linker between the pyridinium and the electrophile, increasing the size and reactivity of the atom to S or Se, or increasing the size of the aromatic ring to use steric effects to push the compound away from W86, or increasing the electrophilicity of the methyl group through substitution. These results may suggest an additional reason why oximes have found limited use in reactivation of aged AChE since most are pyridinium species.
The aged state of AChE is a thermodynamically stable form of AChE, and resurrection presents some difficulties in overcoming the conformational changes and hydrogen-bonding networks within the active site of aged AChE. Quinn notes that the aged state of the enzyme makes four hydrogen bonds with the aged Ser residue, one between the phosphylated oxyanion and the histidinium residue of the catalytic triad and three additional hydrogen bonds with the oxyanion hole. Since the enzyme has evolved to turn over acetylcholine at near diffusion-controlled rates, the ability of the active site to stabilize the tetrahedral or negatively charged intermediate is high. Quinn and co-workers estimated the stabilization of the tetrahedral intermediate in the deacylation step to be at least 11 kcal mol-1.[164] Quinn and co-workers estimated that the strong hydrogen-bonding network drops the pKa of the phosphonic acid to −2 due to stabilization of negative charge that has evolutionarily developed for catalytic turnover.[38] This vast reduction in pKa would then have a significant effect on the anion’s ability to serve as a nucleophile, making the re-alkylation reaction to be more difficult than those conducted in bulk solution. A calculated estimate places the rate of reaction at 100 times slower than what would be observed with a model phosphonate in solution.[164]
Thus, a successful resurrector of aged AChE has to bind in order to disrupt this hydrogen-bonding network or to cause significant conformational changes within the active site, thereby reducing the strength of the hydrogen bonding to allow for reactivity of the phosphylated oxyanion, while also facilitating the desired transition state for electrophilic re-alkylation.
5. Perspective and Future Work
Despite extensive quantities of work with regard to reactivation of OP-inhibited and aged AChE and BChE, a new therapeutic solution has yet to be reached and we continue to rely on old oximes and management of the cholinergic crisis as the standard of care. However, recently significant discoveries have been made that have the potential to change the landscape for the reactivation and resurrection of cholinesterases.
Newly synthesized oximes that are uncharged have shown activity for a number of OP compounds with AChE and BChE, potentially addressing the issue of being broad spectrum therapeutics for OP poisoning and the permanent positive charge of most treatments preventing BBB permeability.[110] The linking strategy of oximes to peripheral site ligands has proven to be advantageous for increasing overall reactivity in a number of circumstances, yet the broad spectrum issue often continues to arise, in particular with efficiency for reactivation of phosphoramidates, such as after tabun (or Novichok) exposure, as has been noted in a number of studies. The approach of using peripheral site ligands to increase binding affinity continues to serve its purpose but the issue may lie in the nucleophile of choice that is being attached. For nearly 70 years, the nucleophile has been chosen as an oxime, and this trend has continued with some limited variation.
Attention needs to be placed on oximes that are outside those traditionally studied. The inhibition and aging of OP compounds such as CBDP with hAChE and hBChE has two different pathways and in the case of hBChE, even two different aging steps.[19] Although not traditionally considered as a target, the toxicity and challenge of treating aerotoxic syndrome is unique and requires further development. Moreover, entirely new and highly toxic classes of OP compounds, Novichoks, have emerged as a threat, yet to date, very little is known with regards to treatment, modeling, and even structure as much of the information is still classified. This presents a unique problem in that only those in the military or defense sector may have access to the appropriate information required to try and develop effective therapeutics for this class of OPs.
The recent discovery of novel non-oxime nucleophiles presents entirely new chemical frameworks to create improvement in standard of care. Two promising families of compounds have been most notable, including those with a pendant base attached to an aryl anchor and then Mannich bases.[91, 113, 114, 116, 160] While all of these compounds were discovered independently, the structural features continue to be similar. What is conserved between both types of compounds is the basic nitrogen attached to an aromatic core; for the Mannich bases, ortho substitution relative to a phenol is the most effective. Continued efforts are needed to better understand the mechanism of action for these particular types of molecules to aid in future drug design, whether those efforts are computational and crystallographic studies or structure–activity relationships. Another major benefit of both classes of compounds is that they exist as non-permanently charged compounds and this should allow for BBB penetration; however, this needs to be confirmed as these are only predicted or hypothesized.
The other major advantage of Mannich bases is that they address the additional issue of aging, having been shown to recover small quantities of activity from aged hAChE.[160] This may be slightly expected as computational investigations of the aged enzyme have noted that the hydrogen-bonding network needs to be altered in order for these compounds to function efficiently.[145, 164] Of the computationally proposed molecules, Mannich bases are simply aromatic b-aminoalcohols which were reported to disrupt this H-bonding network.[145] The very specific orientation of H-bond donors and acceptors may then explain the high affinity for the active site of AChE and the rapid rate of reactivation in addition to the ability to resurrect the anionic aged Ser residue.
Given the computational, crystallographic, and synthetic techniques developed during the course of these reported investigations, many of these answers can be determined and continued efforts toward the treatments and developments of OP exposed AChE can continue. All of the work that goes into the development of these non-oxime reactivators though can then be mirrored in the reactivation and development of BChE as a bioscavenger. Continued efforts support the potential of BChE to be an effective endogenous or therapeutically applied scavenger, but we are lacking means to express larger quantities of the enzyme and methods to make BChE to be catalytic or pseudo-catalytic. The most ideal situation would be to use endogenous BChE and to make this efficient scavenger to be pseudo-catalytic by administration of a therapeutic, but this solution may take longer than using doses of BChE combined with a reactivator in the more immediate future. Nonetheless, the field seems to be at a turning point as the exhaustive search for oxime-based therapeutics has proven unfruitful, but newer scaffolds have demonstrated great potential for addressing the continued problems of BBB permeability, reactivation after inhibition and also resurrection of the aged forms.
Acknowledgements
We acknowledge financial support from the National Institutes of Health (1U01-NS087983) and the Dr. Ralph and Marian Falk Medical Research Trust Bank of America, N.A., Trustee. A.J.F. acknowledges a Robert H. Edgerley Environmental Toxicology fellowship from the Ohio State University. We acknowledge generous allocations of computational resources from the Ohio Supercomputer Center.
Biography
Andrew J. Franjesevic received his B.Sc. degree in chemistry from Wittenberg University (USA) in 2014. He began his graduate studies that same year at The Ohio State University (USA) under the supervision of Prof. Christopher M. Hadad. He is finishing his Ph.D. studies at The Ohio State University with a research focus in the development of small molecule therapeutics for the treatment of aged acetylcholinesterase.

Sydney Sillart received her B.Sc. in 2018 from The Ohio State University (USA). Her undergraduate research focused on the development of small molecule therapeutics for the treatment of aged acetylcholinesterase. She has begun medical school studies at Washington University in St. Louis (USA).

Shubham Vyas received his M.Sc. in chemistry from IIT-Bombay (India) and then his Ph.D. in chemistry from The Ohio State University (USA) in 2011, under the supervision of Prof. Christopher M. Hadad, where he researched nerve agent inhibition of butyrylcholinesterase, improvements in oximes, paraoxonases, and ultrafast spectroscopy. Following his graduate work, he was a Camille and Henry Dreyfus postdoctoral fellow in Environmental Chemistry at Colorado School of Mines (USA) and then was appointed as an assistant professor in the Department of Chemistry at Colorado School of Mines, in Golden, CO.

Christopher S. Callam obtained his Ph.D. in Organic Chemistry from The Ohio State University (USA) under the supervision of Prof. Todd L. Lowary in 2003. Following one year of postdoctoral training at The University of Pittsburgh (USA) working with Prof. Dennis Curran, he became an associated faculty member at The Ohio State University (USA) in 2004. His research interests focus on the synthesis of small molecule reactivators and resurrectors for the treatment of aged acetylcholinesterase.

Jeremy M. Beck received his Ph.D. in chemistry from The Ohio State University (USA) in 2011, under the supervision of Prof. Christopher M. Hadad, where he researched nerve agent inhibition of acetylcholinesterase and therapeutics for reactivating acetylcholinesterase. Following his graduate work, he served as a postdoctoral researcher at the Novartis Institutes for BioMedical Research in Cambridge, MA (USA) under the supervision of Clayton Springer, implementing data mining tools and machine learning models for optimizing ADME properties of drugs. Jeremy now works as a data scientist based in Denver, CO (USA).

Christopher M. Hadad received his B.Sc. degree from the University of Delaware (USA) in 1987 and then completed his Ph.D. as a Hertz Foundation fellow under the guidance of Professor Kenneth B. Wiberg at Yale University (USA). After a NSF post-doctoral fellowship at the University of Colorado (Boulder, USA) with Professor Charles H. DePuy, he joined the faculty of The Ohio State University (USA) as an assistant professor of Chemistry in 1994. In 2006, he was promoted to full professor, was the vice chair for undergraduate studies in the Department of Chemistry from2006–2011, served the department as the interim chair in 2007, and served the College as Associate Dean (2011–2014) and then Dean (2014–2018) of the Natural and Mathematical Sciences division of the College of Arts and Sciences.

Footnotes
Conflict of interest
The authors declare no conflict of interest.
References
- [1].Everts S, Chem. Eng. News 2016, 94, 26–28. [Google Scholar]
- [2].Christianson S Fatal Airs : The Deadly History and Apocalyptic Future of Lethal Gases That Threaten Our World, Praeger, Santa Barbara, 2010, pp. 73–76. [Google Scholar]
- [3].Gillispie CC, Holmes FL, Koertge N, Complete Dictionary of Scientific Biography, Thomson Gale, Detroit, 2008. [Google Scholar]
- [4].Pruitt S, https://www.history.com/news/the-nazis-developed-sarin-gasbut-hitler-was-afraid-to-use-it, 2017, 18–21.
- [5].Minelle B, https://news.sky.com/story/vx-nerve-agent-what-is-it-wheredid-it-come-from-and-what-does-it-do-to-you-10780882, Sky News 2017, 1–12.
- [6].Nachon F, Asojo OA, Borgstahl GEO, Masson P, Lockridge O, Biochemistry 2005, 44, 1154–1162. [DOI] [PubMed] [Google Scholar]
- [7].Cotton S, http://theconversation.com/handle-with-care-the-worldsfive-deadliest-poisons-56089, Conversat 2016, 1–5.
- [8].“VR ( nerve agent ),” can be found under http://www.revolvy.com/page/VR-%28nerve-agent%29, accessed on January 8, 2018.
- [9].Cornell, “Parathion,” can be found under http://pmep.cce.cornell.edu/profiles/extoxnet/metiram-propoxur/parathion-ext.html, accessed on January 8, 2018.
- [10].Stone R, Science 2018; DOI: 10.1126/science.aas9157. [DOI]
- [11].Alfred C, https://www.huffingtonpost.com/2015/03/20/tokyo-subway-sarin-attack_n_6896754.html, Huffington Post 2015, 1–9.
- [12].Amarasingam A, https://www.theatlantic.com/international/archive/2017/04/sarin-syria-assad-chemical-nazi/522039. >, Atl 2017.
- [13].BBC News https://www.bbc.com/news/world-middle-east-39500947, 2017, 24–26.
- [14].Sephton C, Sky News https://news.sky.com/story/nerve-agent-classedas-weapon-of-mass-destruction-killed-kim-10779395, 2017.
- [15].BBC News https://www.bbc.com/news/uk-43315636, 2018.
- [16].Reuters, New York Times https://www.nytimes.com/2013/07/22/world/asia/pesticide-found-in-meals-that-killed-india-children-officialsays.html, 2013.
- [17].Nilesh V, New Indian Express http://www.newindianexpress.com/thesundaystandard/2018/jul/22/pesticide-in-food-40-times-more-than-inus-uk-1846721.html, 2018, 24–26.
- [18].Carletti E, Schopfer LM, Colletier JP, Froment MT, Nachon F, Weik M, Lockridge O, Masson P, Chem. Res. Toxicol 2011, 24, 797–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Carletti E, Colletier JP, Schopfer LM, Santoni G, Masson P, Lock-ridge O, Nachon F, Weik M, Chem. Res. Toxicol 2013, 26, 280–289. [DOI] [PubMed] [Google Scholar]
- [20].Quinn DM, Chem. Rev 1987, 87, 955–979. [Google Scholar]
- [21].Bajgar J, Jun D, Kuča K, Fusek J, Karasova JZ, Kassa J, Cabal J, Blaha V, J. Appl. Biomed 2009, 7, 201–206. [DOI] [PubMed] [Google Scholar]
- [22].Botos I, Wlodawer A, Curr. Opin. Struct. Biol 2007, 17, 683–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Sussman J, Harel M, Frolow F, Oefner C, Goldman A, Toker L, Silman I, Science 1991, 253, 872–879. [DOI] [PubMed] [Google Scholar]
- [24].Kovarik Z, Bosak A, Šinko G, Latas T, Croat. Chem. Acta 2003, 76, 63–67. [Google Scholar]
- [25].Saxena A, Redman AMG, Jiang X, Lockridge O, Doctor BP, Biochemistry 1997, 36, 14642–14651. [DOI] [PubMed] [Google Scholar]
- [26].Tai K, Shen T, Bo U, Philippopoulos M, Mccammon JA, Biophys. J 2001, 81, 715–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Taylor P, Lwebuga-Mukasa J, Lappi S, Rademacher J, Mol. Pharmacol 1974, 10, 703–708. [Google Scholar]
- [28].Changeux J-P, Mol. Pharmacol 1966, 2, 369–392. [PubMed] [Google Scholar]
- [29].Ripoll DR, Faerman CH, Axelsen PH, Silmanii I, Sussman JL, Proc. Natl. Acad. Sci. USA 1993, 90, 5128–5132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Botti SA, Felder CE, Lifson S, Sussman JL, Silman I, Biophys. J 1999, 77, 2430–2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Gilson MK, Straatsma TP, McCammon JA, Ripoll DR, Faerman CH, Axelsen PH, Silman I, Sussman JL, Science 1994, 263, 1276–1278. [DOI] [PubMed] [Google Scholar]
- [32].Xu Y, Colletier J, Weik M, Qin G, Jiang H, Silman I, Sussman JL, Biophys. J 2010, 99, 4003–4011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Colletier J, Royant A, Masson P, Zaccai G, Joel L, Goeldner M, Silman I, Weik M, Acta Crystallogr. Sect. D 2007, 63, 1115–1128. [DOI] [PubMed] [Google Scholar]
- [34].Colletier J, Bourgeois D, Sanson B, Fournier D, Sussman JL, Silman I, Weik M, Proc. Natl. Acad. Sci. USA 2008, 105, 11742–11747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Worek F, Thiermann H, Pharmacol. Ther 2013, 139, 249–259. [DOI] [PubMed] [Google Scholar]
- [36].Hobbiger F, Br. J. Pharmacol. Chemother 1955, 10, 356–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Wilson BW, Hooper MJ, Hansen ME, Neiberg PS, Organophosphates, Chemistry, Fate and Effects, Academic Press, New York, 1992. [Google Scholar]
- [38].Quinn DM, J. Med. Chem 2018, 61, 7032–7033. [DOI] [PubMed] [Google Scholar]
- [39].Wolthuis OL, Kepner LA, Eur. J. Pharmacol 1978, 49, 415–425. [DOI] [PubMed] [Google Scholar]
- [40].Sidell FR, Groff WA, Toxicol. Appl. Pharmacol 1974, 27, 241–252. [DOI] [PubMed] [Google Scholar]
- [41].Mage PP, Multidimensional Pharmacochemistry, Academic Press, San Diego, 1984. [Google Scholar]
- [42].Wilson IB, J. Biol. Chem 1951, 190, 111–118. [PubMed] [Google Scholar]
- [43].Wilson IB, Meislich EK, J. Am. Chem. Soc 1953, 75, 4628–4629. [Google Scholar]
- [44].Aldridge WN, Davison AN, Biochem. J 1953, 55, 763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Hackley BE, Plapinger R, Stolberg M, Wagner-Jauregg T, J. Am. Chem. Soc 1955, 77, 3651–3653. [Google Scholar]
- [46].Wilson IB, Ginsburg S, Arch. Biochem. Biophys 1955, 55, 569–571. [DOI] [PubMed] [Google Scholar]
- [47].Jandorf BJ, Crowell EA, Lebin AP, Fed. Proc 1955, 14, 231. [Google Scholar]
- [48].Michel HO, Fed. Proc 1955, 14, 255. [Google Scholar]
- [49].Childs AF, Davies DR, Green AL, Rutland JP, Br. J. Pharmacol. Chemother 1955, 10, 462–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Holmes R, Robins EL, Br. J. Pharmacol. Chemother 1955, 10, 490–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Dunn MA, Sidell FR, J. Am. Med. Assoc 1989, 262, 649–652. [PubMed] [Google Scholar]
- [52].Clement JG, Arch. Toxicol 1992, 66, 143–144. [DOI] [PubMed] [Google Scholar]
- [53].Dawson RM, J. Appl. Toxicol 1994, 14, 317–331. [DOI] [PubMed] [Google Scholar]
- [54].Eddleston M, Szinicz L, Eyer P, Buckley N, Q. J. Med 2002, 95, 275–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Jokanović M, Stojiljković MP, Eur. J. Pharmacol 2006, 553, 10–17. [DOI] [PubMed] [Google Scholar]
- [56].Ordentlich A, Barak D, Kronman C, Benschop HP, De Jong LPA, Ariel N, Barak R, Segall Y, Velan B, Shafferman A, Biochemistry 1999, 38, 3055–3066. [DOI] [PubMed] [Google Scholar]
- [57].Rosenstock L, Keifer M, Daniell WE, McConnell R, Claypoole K, Lancet 1991, 338, 223–227. [DOI] [PubMed] [Google Scholar]
- [58].London L, Nell V, Thompson ML, Myers JE, Scand. J. Work Environ. Health 1998, 24, 18–29. [DOI] [PubMed] [Google Scholar]
- [59].Krejci E, Valenzuela IM-PY, Ameziane R, Akaaboune M, J. Biol. Chem 2006, 281, 10347–10354. [DOI] [PubMed] [Google Scholar]
- [60].Mendel B, Rudney H, Biochem. J 1943, 37, 59–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Nachon F, Masson P, Nicolet Y, Lockridge O, J. C. Fonticilla-Camps in Butyrylcholinesterase Its Function and Inhibitors, Taylor & Francis, New York, 2003, pp. 39–54. [Google Scholar]
- [62].Bourne Y, Taylor P, Radić Z, Marchot P, EMBO J 2003, 22, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Dvir H, Silman I, Harel M, Rosenberry TL, Sussman JL, Chem. Biol. Interact 2010, 187, 10–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Boyko KM, Baymukhametov TN, Chesnokov YM, Hons M, Lushchekina SV, Konarev PV, Lipkin AV, Vasiliev AL, Masson P, Popov VO, Kovalchuk MV, Biochemie 2019, 156, 196–205. [DOI] [PubMed] [Google Scholar]
- [65].Rosenberry TL, Proc. Natl. Acad. Sci. USA 1975, 72, 3834–3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Lockridge O, Pharmacol. Ther 1990, 47, 35–60. [DOI] [PubMed] [Google Scholar]
- [67].Kaplan D, Ordentlich A, Barak D, Ariel N, Kronman C, Velan B, Shafferman A, Biochemistry 2001, 40, 7433–7445. [DOI] [PubMed] [Google Scholar]
- [68].Desmedt JE, La Grutta G, Proc. Physio. Soc 1955, 129, 46–47. [DOI] [PubMed] [Google Scholar]
- [69].Giacobini E, Drugs Aging 2001, 18, 891–898. [DOI] [PubMed] [Google Scholar]
- [70].Holmstedt B, Sjçqvist F, Acta Physiol. Scand 1960, 47, 284–296. [DOI] [PubMed] [Google Scholar]
- [71].Soreq H, Lapidot-Lifson Y, Zakut H, Cancer Cells 1991, 3, 511–516. [PubMed] [Google Scholar]
- [72].Giacobini E, Jpn. J. Pharmacol 1997, 74, 225–241. [DOI] [PubMed] [Google Scholar]
- [73].Li B, Stribley JA, Ticu A, Xie W, Schopfer LM, Hammond P, Brimijoin S, Hinrichs SH, Lockridge O, J. Neurochem 2000, 75, 1320–1331. [DOI] [PubMed] [Google Scholar]
- [74].Guillozet AL, Smiley JF, Mash DC, Mesulam MM, Ann. Neurol 1997, 42, 909–918. [DOI] [PubMed] [Google Scholar]
- [75].Hawkins RD, Gunter JM, Biochem. J 1946, 40, 192–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Primo-Parmo SL, Bartels CF, Wiersema B, van der Spek AF, Innis JW, La Du BN, Am. J. Hum. Genet 1996, 58, 52–64. [PMC free article] [PubMed] [Google Scholar]
- [77].Manoharan I, Wieseler S, Layer PG, Lockridge O, Boopathy R, Pharmacogenet. Genomics 2006, 16, 461–468. [DOI] [PubMed] [Google Scholar]
- [78].Manoharan I, Boopathy R, Darvesh S, Lockridge O, Clin. Chim. Acta 2007, 378, 128–135. [DOI] [PubMed] [Google Scholar]
- [79].Chen P, Gao Y, Geng L, Ping V, Gao Y, Geng L, Parks RJ, Pang Y, Brimijoin S, Proc. Natl. Acad. Sci. USA 2015, 112, 2251–2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Lenz DE, Yeung D, Smith JR, Sweeney RE, Lumley LA, Cerasoli DM, Toxicology 2007, 233, 31–39. [DOI] [PubMed] [Google Scholar]
- [81].Worek F, Thiermann H, Szinicz L, Eyer P, Biochem. Pharmacol 2004, 68, 2237–2248. [DOI] [PubMed] [Google Scholar]
- [82].Carletti E, Aurbek N, Gillon E, Loiodice M, Nicolet Y, FontecillaCamps J-C, Masson P, Thiermann H, Nachon F, Worek F, Biochem. J 2009, 421, 97–106. [DOI] [PubMed] [Google Scholar]
- [83].Beck JM, Organophosphorus Nerve Agent Chemistry; Interactions of Chemical Warfare Agents and Their Therapeutics with Acetylcholinesterase, The Ohio State University, 2011.
- [84].Vyas S, Computational and Experimental Studies Towards the Development of Novel Therapeutics Against Organophosphorus Nerve Agents: Butyrylcholinesterase and Paraoxonase, The Ohio State University, 2011.
- [85].Cerasoli D, Cadieux CL, USAMRICD, unpublished results.
- [86].Masson P, Froment MT, Bartels CF, Lockridge O, Biochem. J 1997, 325, 53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Ashani Y, Bhattacharjee AK, Leader H, Saxena A, Doctor BP, Biochem. Pharmacol 2003, 66, 191–202. [DOI] [PubMed] [Google Scholar]
- [88].Zhuang Q, Young A, Callam CS, McElroy CA, b. Doğ an Ekici, Yoder RJ, Hadad CM, Ann. N. Y. Acad. Sci 2016, 1374, 94–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Mercey G, Verdelet T, Renou J, Kliachyna M, Baati R, Nachon F, Jean L, Renard PY, Acc. Chem. Res 2012, 45, 756–766. [DOI] [PubMed] [Google Scholar]
- [90].Worek F, Thiermann H, Wille T, Chem. Biol. Interact 2016, 259, 93–98. [DOI] [PubMed] [Google Scholar]
- [91].Katz FS, Pecic S, Schneider L, Zhu Z, Hastings A, Luzac M, Macdonald J, Landry DW, Stojanovic MN, Toxicol. Lett 2018, 291, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Kovalevsky A, Blumenthal DK, Cheng X, Taylor P, Radic Z, Ann. N. Y. Acad. Sci 2016, 1378, 41–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Gorecki L, Korabecny J, Musilek K, Malinak D, Nepovimova E, Dolezal R, Jun D, Soukup O, Kuca K, Arch. Toxicol 2016, 90, 2831–2859. [DOI] [PubMed] [Google Scholar]
- [94].Chambers JE, Chambers HW, Meek EC, Pringle RB, Chem. Biol. Interact 2013, 203, 135–138. [DOI] [PubMed] [Google Scholar]
- [95].Chambers JE, Meek EC, Chambers HW, Ann. N. Y. Acad. Sci 2016, 1374, 52–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].De Koning MC, Joosen MJA, Worek F, Nachon F, Van Grol M, Klaassen SD, Alkema DPW, Wille T, De Bruijn HM, J. Med. Chem 2017, 60, 9376–9392. [DOI] [PubMed] [Google Scholar]
- [97].Kuca K, Korabecny J, Dolezal R, Nepovimova E, Soukup O, Gorecki L, RSC Adv 2017, 7, 7041–7045. [Google Scholar]
- [98].Kuca K, Jun D, Junova L, Musilek K, Hrabinova M, Alberto J, Ramalho TC, Valko M, Wu Q, Nepovimova E, Franca CTC, Molecules 2018, 23, 1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Sahu AK, Sharma R, Gupta B, Musilek K, Kuca K, Acharya J, Ghosh KK, Toxicol. Mech. Methods 2016, 26, 319–326. [DOI] [PubMed] [Google Scholar]
- [100].Malinak D, Nepovimova E, Jun D, Musilek K, Kuca K, Molecules 2018, 23, 2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Berberich JA, Stouch TR, Manepalli S, Esposito EX, Madura JD, Chem. Res. Toxicol 2016, 29, 1534–1540. [DOI] [PubMed] [Google Scholar]
- [102].Karade HN, Raviraju G, Acharya BN, Valiveti AK, Bhalerao U, Acharya J, Bioorg. Med. Chem 2016, 24, 4171–4176. [DOI] [PubMed] [Google Scholar]
- [103].Sharma R, Gupta B, Sahu AK, Acharya J, Satnami ML, Ghosh KK, Chem. Biol. Interact 2016, 259, 85–92. [DOI] [PubMed] [Google Scholar]
- [104].Amitai G, Gez R, Raveh L, Bar-Ner N, Grauer E, Chapman S, Chem. Biol. Interact 2016, 259, 187–204. [DOI] [PubMed] [Google Scholar]
- [105].Bodor N, Shek E, Higuchi T, J. Med. Chem 1976, 19, 102–107. [DOI] [PubMed] [Google Scholar]
- [106].Wei Z, Liu Y-Q, Wang S-Z, Yao L, Nie H-F, Wang YA, Liu XY, Zheng Z-B, Li S, Bioorg. Med. Chem 2017, 25, 4497–4505. [DOI] [PubMed] [Google Scholar]
- [107].Kuca K, Musilek K, Jun D, Zdarova-Karasova J, Nepovimova E, Soukup O, Hrabinova M, Mikler J, Franca TCC, Da Cunha EFF, De Castro AA, Valis M, Ramalho TC, BMC Pharmacol. Toxicol 2018, 19, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Wei Z, Liu Y, Wang Y, Li W, Zhou X, Zhao J, Huang C, Li X, Liu J, Zheng Z, Li S, Toxicol. Lett 2016, 246, 1–6. [DOI] [PubMed] [Google Scholar]
- [109].Renou J, Dias J, Mercey G, Verdelet T, Rousseau C, Gastellier A-J, Touvrey-Loiodice M, Baati R, Jean L, Nachon F, Renard P-Y, RSC Adv 2016, 6, 17929–17940. [Google Scholar]
- [110].Zorbaz T, Bräıki A, Marakovic N, Renou J, de la Mora E., Hrvat NM, Katalinic M, Silman I, Sussman JL, Mercey G, Gomez C, Mougeot R, Perez B, Baati R, Nachon F, Weik M, Jean L, Kovarik Z, Renard P-Y, Chem. Eur. J 2018, 24, 9675–9691. [DOI] [PubMed] [Google Scholar]
- [111].Santoni G, de Sousa J, De la Mora E., Dias J, Jean L, Sussman JL, Silman I, Renard P-Y, Brown RCD, Weik M, Baati R, Nachon F, J. Med. Chem 2018, 61, 7630–7639. [DOI] [PubMed] [Google Scholar]
- [112].Bušić V, Katalinić M, Šinko G, Kovarik Z, Gašo-Sokač D, Toxicol. Lett 2016, 262, 114–122. [DOI] [PubMed] [Google Scholar]
- [113].Katz FS, Pecic S, Tran TH, Trakht I, Schneider L, Zhu Z, Ton-That L, Luzac M, Zlatanic V, Damera S, MacDonald J, Landry DW, Tong L, Stojanovic MN, ChemBioChem 2015, 16, 2205–2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Cadieux CL, Wang H, Zhang Y, Koenig JA, Shih T, Mcdonough J, Koh J, Cerasoli D, Chem. Biol. Interact 2016, 259, 133–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Bierwisch A, Wille T, Thiermann H, Worek F, Toxicol. Lett 2016, 246, 49–56. [DOI] [PubMed] [Google Scholar]
- [116].de Koning MC, Horn G, Worek F, van Grol M, Eur. J. Med. Chem 2018, 157, 151–160. [DOI] [PubMed] [Google Scholar]
- [117].Luo C, Saxena A, Smith M, Garcia G, Radić Z, Taylor P, Doctor BP, Biochemistry 1999, 38, 9937–9947. [DOI] [PubMed] [Google Scholar]
- [118].Ochoa R, Rodriguez CA, Zuluaga AF, J. Mol. Graphics Modell 2016, 68, 176–183. [DOI] [PubMed] [Google Scholar]
- [119].Hrvat NM, Zorbaz T, Šinko G, Kovarik Z, Toxicol. Lett 2018, 293, 222–228. [DOI] [PubMed] [Google Scholar]
- [120].Ashani Y, Shapira S, Levy D, Wolfe AD, Doctor BP, Raveh L, Biochem. Pharmacol 1991, 41, 37–41. [DOI] [PubMed] [Google Scholar]
- [121].Raveh L, Grauer E, Grunwald J, Cohen E, Ashani Y, Toxicol. Appl. Pharmacol 1997, 145, 43–53. [DOI] [PubMed] [Google Scholar]
- [122].Cerasoli DM, Griffiths EM, Doctor BP, Saxena A, Fedorko JM, Greig NH, Yu QS, Huang Y, Wilgus H, Karatzas CN, Koplovitz I, Lenz DE, Chem. Biol. Interact 2005, 157 – 158, 362–365. [DOI] [PubMed] [Google Scholar]
- [123].Østergaard D, Viby-Mogensen J, Hanel HK, Skovgaard LT, Acta Anaesthesiol. Scand 1988, 32, 266–269. [DOI] [PubMed] [Google Scholar]
- [124].Raveh L, Grunwald J, Marcus D, Papier Y, Cohen E, Ashani Y, Biochem. Pharmacol 1993, 45, 2465–2474. [DOI] [PubMed] [Google Scholar]
- [125].Huang Y-J, Huang Y, Baldassarre H, Wang B, Lazaris A, Leduc M, Bilodeau AS, Bellemare A, Cote M, Herskovits P, Touati M, Turcotte C, Valeanu L, Lemee N, Wilgus H, Begin I, Rao K, Neveu N, Brochu E, Pierson J, Hockley DK, Cerasoli DM, Lenz DE, Karatzas CN, Langermann S, Proc. Natl. Acad. Sci. USA 2007, 104, 13603–13608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Huang YJ, Lundy PM, Lazaris A, Huang Y, Baldassarre H, Wang B, Turcotte C, Cote M, Bellemare A, Bilodeau AS, Brouillard S, Touati M, Herskovits P, Begin I, Neveu N, Brochu E, Hockley DK, Cerasoli DM, Lenz DE, Wilgus H, Karatzas CN, Langermann S, BMC Biotechnol 2008, 8, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Saxena A, Luo C, Chilukuri N, Maxwell DM, Doctor BP in Chemical Warfare Agents : Chemistry, Pharmacology, Toxicology, and Therapeutics, 2nd ed., CRC, New York, 2007, pp. 145–173. [Google Scholar]
- [128].Lenz DE, Broomfield CA, Yeung DT, Masson P, Maxwell DM, Cerasoli DM, in Chemical Warfare Agents : Chemistry, Pharmacology, Toxicology, and Therapeutics, 2nd ed., CRC, New York, 2007, pp. 175–202. [Google Scholar]
- [129].Lenz DE, Clarkson ED, Schulz SM, Cerasoli DM, Chem. Biol. Interact 2010, 187, 249–252. [DOI] [PubMed] [Google Scholar]
- [130].Saxena A, Viragh C, Frazier DS, Kovach IM, Maxwell DM, Lockridge O, Doctor BP, Biochemistry 1998, 37, 15086–15096. [DOI] [PubMed] [Google Scholar]
- [131].Duysen EG, Bartels CF, Lockridge O, J. Pharmacol. Exp. Ther 2002, 302, 751–758. [DOI] [PubMed] [Google Scholar]
- [132].Mumford H, Price ME, Cerasoli DM, Teschner W, Ehrlich H, Schwarz HP, Lenz DE, Chem. Biol. Interact 2010, 187, 304–308. [DOI] [PubMed] [Google Scholar]
- [133].Kovarik Z, Ciban N, Radić Z, Simeon-Rudolf V, Taylor P, Biochem. Biophys. Res. Commun 2006, 342, 973–978. [DOI] [PubMed] [Google Scholar]
- [134].Musilova L, Kuca K, Jung YS, Jun D, Clin. Toxicol 2009, 47, 545–550. [DOI] [PubMed] [Google Scholar]
- [135].Jun D, Musilova L, Pohanka M, Jung YS, Bostik P, Kuca K, Int. J. Mol. Sci 2010, 11, 2856–2863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Jun D, Musilova L, Musilek K, Kuca K, Int. J. Mol. Sci 2011, 12, 2077 –2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Masson P, Nachon F, J. Neurochem 2017, 142, 26–40. [DOI] [PubMed] [Google Scholar]
- [138].Worek F, Thiermann H, Wille T, Toxicol. Lett 2016, 244, 143–148. [DOI] [PubMed] [Google Scholar]
- [139].Katalinić M, Hrvat NM, Baumann K, Piperčić SM, Makarić S, Tomić S, Jović O, Hrenar T, Milič ević A., Jelić D, Zunec S, Primozic I, Kovarik Z, Toxicol. Appl. Pharmacol 2016, 310, 195–204. [DOI] [PubMed] [Google Scholar]
- [140].Katalinić M, Zandona A, Ramić A, Zorbaz T, Primožič I, Kovarik Z, Molecules 2017, 22, 1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Radić Z´, Dale T, Kovarik Z, Berend S, Garcia E, Zhang L, Amitai G, Green C, Radić B, Duggan BM, Ajami D, Rebek J Jr., Taylor P, Biochem. J 2013, 450, 231–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Sit RK, Fokin VV, Amitai G, Sharpless KB, Taylor P, Radic Z´, J. Med. Chem 2014, 57, 1378–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Kovarik Z, Katalinić M´, Šinko G, Binder J, Holas O, Jung YS, Musilova L, Jun D, Kuča K, Chem. Biol. Interact 2010, 187, 167–171. [DOI] [PubMed] [Google Scholar]
- [144].Wille T, von der Wellen J, Thiermann H, Worek F, Arch. Toxicol 2017, 91, 1309–1318. [DOI] [PubMed] [Google Scholar]
- [145].Khavrutskii IV, Wallqvist A, ChemistrySelect 2017, 2, 1885–1890. [Google Scholar]
- [146].Chandar NB, Lo R, Ganguly B, Chem. Biol. Interact 2014, 223, 58–68. [DOI] [PubMed] [Google Scholar]
- [147].Topczewski JJ, Quinn DM, Org. Lett 2013, 15, 1084–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Topczewski JJ, Lodge AM, Yasapala SN, Payne MK, Keshavarzi PM, Quinn DM, Bioorg. Med. Chem. Lett 2013, 23, 5786–5789. [DOI] [PubMed] [Google Scholar]
- [149].Steinberg GM, Lieske CN, Boldt R, Goan JC, Podall HE, J. Med. Chem 1970, 13, 435–446. [DOI] [PubMed] [Google Scholar]
- [150].Ash AB, Blumbergs P, Stevens CL, Michel HO, Hackley BE, Epstein JW, J. Org. Chem 1969, 34, 4070–4072. [Google Scholar]
- [151].Blumbergs P, Ash AB, Daniher FA, Stevens CL, Michel HO, Hackley BE, Epstein J, J. Org. Chem 1969, 34, 4065–4070. [Google Scholar]
- [152].Powers JC, Asgian JL, b. Ekici D, James KE, Chem. Rev 2002, 102, 4639–4750. [DOI] [PubMed] [Google Scholar]
- [153].Chandar NB, Ganguly B, Chem. Biol. Interact 2013, 204, 185–190. [DOI] [PubMed] [Google Scholar]
- [154].Zhou Q, Turnbull KD, J. Org. Chem 1999, 64, 2847–2851. [DOI] [PubMed] [Google Scholar]
- [155].Zhou Q, Turnbull KD, J. Org. Chem 2001, 66, 7072–7077. [DOI] [PubMed] [Google Scholar]
- [156].Weinert EE, Dondi R, Colloredo-Melz S, Frankenfield KN, Mitchell CH, Freccero M, Rokita SE, J. Am. Chem. Soc 2006, 128, 11940–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Zhou Q, Turnbull KD, J. Org. Chem 2000, 65, 2022–2029. [DOI] [PubMed] [Google Scholar]
- [158].Bakke BA, McIntosh MC, Turnbull KD, J. Org. Chem 2005, 70, 4338–4345. [DOI] [PubMed] [Google Scholar]
- [159].Yoder RJ, Zhuang Q, Beck JM, Franjesevic A, Blanton TG, Sillart S, Secor T, Guerra L, Brown JD, Reid C, McElroy CA, O. Doğ an Ekici, Callam CS, Hadad CM, ACS Med. Chem. Lett 2017, 8, 622–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Zhuang Q, Franjesevic AJ, Corrigan TS, Coldren WH, Dicken R, Sillart S, Deyong A, Yoshino N, Smith J, Fabry S, Fitzpatrick K, Blanton TG, Joseph J, Yoder RJ, McElroy CA, O. Doğ an Ekici, Callam CS, Hadad CM, J. Med. Chem 2018, 61, 7034–7042. [DOI] [PubMed] [Google Scholar]
- [161].Timperley CM, Casey KE, Notman S, Sellers DJ, Williams NE, Williams NH, Williams GR, J. Fluorine Chem 2006, 127, 1554–1563. [Google Scholar]
- [162].BriseÇo-Roa L, Hill J, Notman S, Sellers D, Smith AP, Timperley CM, Wetherell J, Williams NH, Williams GR, Fersht AR, Griffiths AD, J. Med. Chem 2006, 49, 246–255. [DOI] [PubMed] [Google Scholar]
- [163].An Y, Zhu Y, Yao Y, Liu J, Phys. Chem. Chem. Phys 2016, 18, 9838 –9846. [DOI] [PubMed] [Google Scholar]
- [164].Quinn DM, Topczewski J, Yasapala N, Lodge A, Molecules 2017, 22, 1464. [DOI] [PMC free article] [PubMed] [Google Scholar]