

SUMMARY
Aberrant apoptosis can lead to acute or chronic degenerative diseases. Mitochondrial outer membrane permeabilization (MOMP) triggered by the oligomerization of the Bcl-2 family proteins Bax/ Bak is an irreversible step leading to execution of apoptosis. Here, we describe the discovery of small-molecule inhibitors of Bax/Bak oligomerization that prevent MOMP. We demonstrate that these molecules disrupt multiple, but not all, interactions between Bax dimer interfaces thereby interfering with the formation of higher-order oligomers in the MOM, but not recruitment of Bax to the MOM. Small-molecule inhibition of Bax/Bak oligomerization allowed cells to evade apoptotic stimuli and rescued neurons from death after excitotoxicity, demonstrating that oligomerization of Bax is essential for MOMP. Our discovery of small-molecule Bax/Bak inhibitors provides novel tools for the investigation of the mechanisms leading to MOMP and will ultimately facilitate development of compounds inhibiting Bax/Bak in acute and chronic degenerative diseases.
In Brief
Niu et al. identified novel small-molecule inhibitors of pro-apoptotic Bax and Bak oligomerization which allow cells to resist apoptosis and that protect neurons from excitotoxicity, providing novel tools for preclinical target evaluation and drug development in degenerative diseases.
INTRODUCTION
Apoptosis is a highly regulated form of programmed cell death that serves to remove superfluous cells during development and its balanced regulation is of fundamental importance for homeostasis in all organisms (Danial and Korsmeyer, 2004). However, different forms of acute stress such as ischemia, ischemia-reperfusion, inflammation, degenerative diseases, as well as cancer chemotherapy in normal tissues can lead to the pathologic activation of apoptotic cascades (Hardwick and Soane, 2013; Leber et al., 2010; Mergenthaleretal., 2004; Szeto, 2008).
Apoptosis can be largely divided into two connected pathways ultimately leading to caspase activation and subsequent cellular disintegration. The extrinsic pathway is triggered by extracellular signals activating death receptors, whereas the intrinsic pathway is activated by intracellular stress and largely regulated at the mitochondrial outer membrane (MOM) by the pro- and anti-apoptotic members of the B cell lymphoma 2 (Bcl-2) family of proteins (Bogner et al., 2010; Danial and Korsmeyer, 2004; Moldoveanu et al., 2014). Cancer cells have evolved many strategies to evade apoptosis (Delbridge et al., 2012), therefore, pharmacological inhibition of anti-apoptotic proteins has been studied in great detail. However, the therapeutic potential of pharmacological inhibition of pro-apoptotic Bcl-2 proteins has been less explored.
Mitochondrial outer membrane permeabilization (MOMP) is the first irreversible step in apoptosis (Delbridge et al., 2012; Moldoveanu et al., 2014; Willis et al., 2007). MOMP results from an ordered series of steps beginning with activation of one or more Bcl-2 homology 3 proteins (BH3-proteins) or releasing previously activated pro-apoptotic proteins Bax or Bak from inhibition by an anti-apoptotic protein of the Bcl-2 family (Bogner et al., 2010; Willis et al., 2007; Wilson-Annan et al., 2003). Once activated, BH3-proteins translocate to the MOM and directly recruit and activate cytoplasmic Bax and the constitutively membrane-bound Bak (Lovell et al., 2008; Sarosiek et al., 2013) catalyzing insertion of the central helices 5–6 of the proteins into the lipid bilayer of the MOM as part of a yet to be fully defined structure (Andrews, 2014). Some data suggest that oligomerization of membrane-bound Bax or Bak ultimately culminates in MOMP (Dewson et al., 2012; Iyer et al., 2015; Ma et al., 2013; Zhang et al., 2016). Other results have been interpreted as suggesting that MOMP can be mediated by membrane-inserted monomers of Bax (Kushnareva et al., 2012; Xu et al., 2013). Thus, MOMP could be prevented by inhibiting any one of the individual steps that lead to the activation of Bax and Bak in the MOM, or possibly by preventing the oligomerization of the proteins.
Multiple structurally disparate BH3 proteins mediate activation of Bax and Bak, therefore directly inhibiting Bax and Bak would be a more efficient approach to inhibit MOMP. However, the lack of structural information about and the overall dynamic nature of Bax and Bak protein complexes in the MOM have prohibited rational design of small-molecule inhibitors.
Here, we identified small-molecule inhibitors active against both Bax and Bak oligomerization in the MOM that also inhibit apoptosis in live cells. Using a combination of biochemical in vitro assays and cellular studies, we demonstrate a specific mechanism of action for these inhibitors. In structural crosslinking studies we demonstrate that these small molecules partially disrupt normal Bax and Bak dimerization at similar interfaces, thereby preventing dimers from forming higher-order oligomers, and thus establish that proper Bax/Bak dimerization is necessary for MOMP. Importantly, we demonstrate that pharmacological inhibition of Bax and Bak with these small molecules allows cells to survive otherwise lethal stress and rescues neurons from prior excitotoxic damage. Finally, our studies provide novel tools to investigate the molecular mechanisms underlying MOMP and lay the ground for accelerated targeted development of refined Bax and Bak inhibitors that may be used for preclinical target validation.
RESULTS
Identification of Small-Molecule Bax Inhibitors
To identify novel Bax inhibitors, we screened a collection of 86 compounds based on structures previously shown to have a weak affinity for Mcl-1 (Prakesch et al., 2008) for inhibition of tBid/Bax-mediated membrane permeabilization (MP) in a MOMP-mimicking liposome dyerelease assay (Billen et al., 2008) (Figures 1A and 1B). A37-compound secondary collection based on the molecular structures of the compounds with the highest activities in the primary screen (BJ-1 and BJ-1-BP) was again screened for inhibition of Bax-mediated MP. From this collection, both MSN-50 and MSN-125 efficiently inhibited liposome permeabilization (Figures 1C and 2A).
Figure 1. Identification of Inhibitors of tBid-and Bax-Mediated Liposome Per-meabilization.
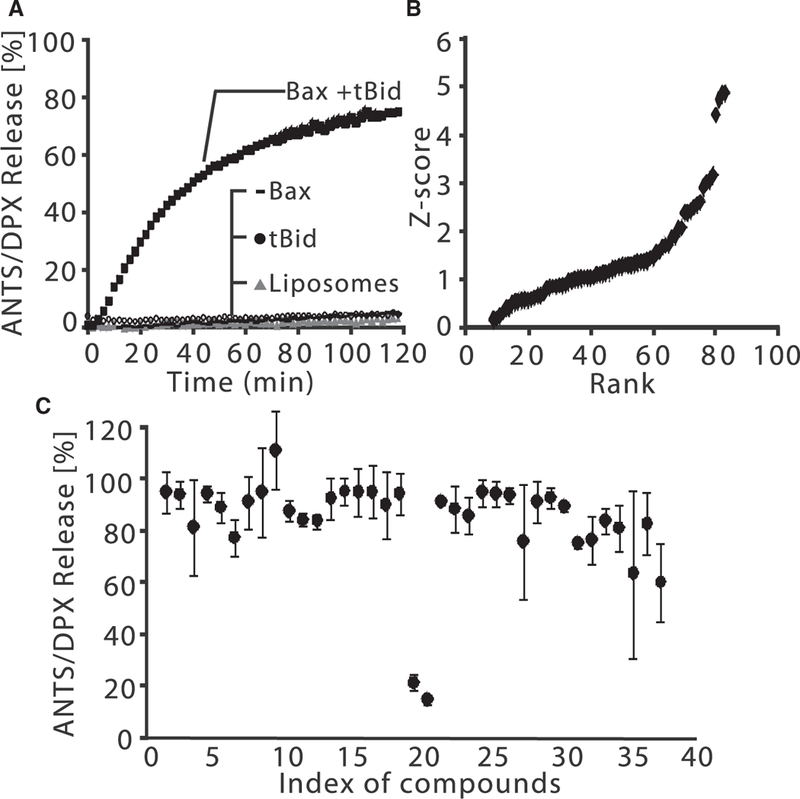
(A) Bax (100 nM) and tBid (20 nM) permeabilize liposomes releasing ANTS and DPX, resulting in a fluorescence increase. Fluorescence increase is converted to percentage release by comparing to liposomes solubilized with detergent.
(B) Z score versus rank for 86 compounds screened in triplicate at 10 μM final concentration. All compounds with a Z score > 0 for inhibiting tBid/Bax-mediated liposome permeabilization are shown.
(C) Primary ANTS/DPX release screen for 37 compounds designed based on BJ-1 and BJ-1-BP, the top hits from (B). MSN-125 and MSN-50 (index 19 and 20) inhibited liposome per-meabilization normalized to dye release by tBid and Bax alone. Endpoint release was measured after incubation for 120 min, n = 2, mean ± range. Unless otherwise specified in all figures, n indicates the number of independent experiments.
Figure 2. Small-Molecule Inhibitors that Prevent Bax-Mediated Permeabilization of Liposomes and Mitochondria.
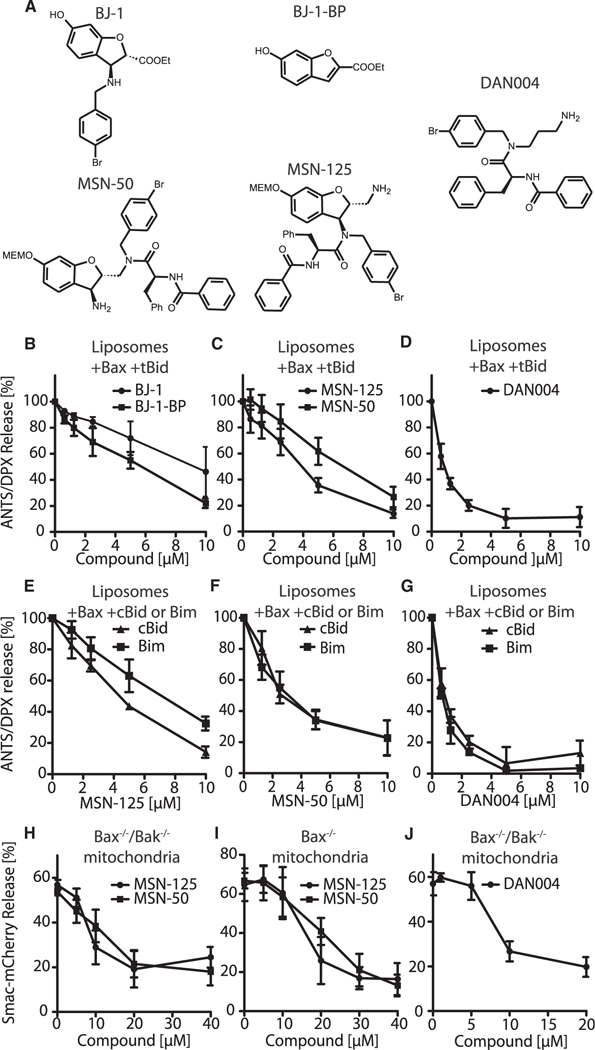
(A) Molecular structures of BJ-1, BJ-1-BP, MSN-125, MSN-50, and DAN004. MEMO. β-methox-yethoxymethyl ether.
(B-D) Concentration-dependent inhibition of liposome permeabilization by tBid and Bax by the indicated concentrations of (B) BJ-1, BJ-1-BP,(C) MSN-125, MSN-50, and (D) DAN004. Results are normalized to reactions without compound. Mean ± SD, n ≥ 3.
(E-G) Liposome permeabilization by Bax activated by either 5 nM cBid or Bim is inhibited equally by thecompound and concentrations indicated below the panels. Results are normalized to reactions without compound.
(H-J) Permeabilization of mitochondria isolated from (H and J) Bax–/–/Bak–/– or (I) Bax–/– BMK cells expressing Smac-mCherry incubated with (H and J) 2 nM tBid and 20 nM Bax or (I) 0.5 nM tBid and compounds as indicated. Release of SmacmCherry was determined by pelleting mitochondria and measuring mCherry fluorescence in the supernatant and pellet. Mean ± SD, n ≥ R 3. See also Figure S1 and Table S1.
Small-Molecule Inhibition of Liposome Permeabilization by tBid and Bax Is Concentration Dependent
In individual assays, BJ-1, BJ-1-BP, MSN-50, and MSN-125 all inhibited dye release from liposomes in a concentration-dependent fashion (Figures 2B and 2C). Structure-activity relationship (SAR) experiments suggested that most modifications of MSN compounds significantly reduced their activity (Table S1). The BJ-1-BP structure is included in all of the hit compounds suggesting that this might be the pharmacophore. Unexpectedly, we found that only MSN-50 and MSN-125 inhibited liposome permeabilization by a common mechanism (see below). Therefore, to identify the pharmacophore common to these compounds, we synthesized DAN004 (Figure 2A). Like the parent compounds, DAN004 showed concentration-dependent inhibition of liposome permeabilization but with improved activity in vitro (Figure 2D).
This assay does not permit determination of conventional kinetic parameters since new oligomers form in liposomes that have already been permeabilized, and existing Bax oligomers can continue to expand until they contain hundreds of Bax molecules (Satsoura et al., 2012). Neither of these events is recorded in our assay as liposome permeabilization is an “all-or-nothing” event. Nevertheless, this assay permits derivation of concentration response curves, thereby allowing quantitative comparison of different compounds under identical conditions. The concentration of compound required to inhibit 50% of the maximal dye release (i.e., IC50) obtained for MSN-125, MSN-50, BJ-1, and BJ-1-BP in this assay were approximately 4, 6, 9, and 6 μM, respectively (Figures 2B and 2C). DAN004 exhibited the most potent MP-inhibiting activity (IC50 ~0.7 μM, Figure 2D). Consistent with a previous report (Jiang et al., 2007), Nutlin-3a, a small-molecule inhibitor of MDM2, inhibited liposome permeabilization in a concentration-dependent fashion but was less active than DAN004 (Figure S1). MSN-125, MSN-50, and DAN004 inhibited activation of Bax mediated by either Bim or cBid more or less equivalently (Figures 2E-2G). Other than the BH3-sequence there is no structural similarity between these Bax activator-proteins consistent with the target of the inhibitors being Bax rather than the BH3 proteins. Moreover, DAN004 and Bcl-XL inhibited heat-activated Bax-mediated liposome permeabilization equivalently (Figure S11), further confirming that the compound does not function by inhibiting a BH3 protein. Finally, we took advantage of the fact that, when Bak is synthesized by in vitro transcription translation, the protein is constitutively active, releasing cytochrome c from mitochondria lacking endogenous Bax and Bak. Consistent with MSN-50 targeting Bak, adding the compound to constitutively active Bak prior to the addition of mitochondria reduced cytochrome c release, as measured by ELIZA in three independent experiments, from 43% to 19%. By comparison, in vitro synthesized Bcl-XL, used as a control in the same experiments, inhibited active Bak-mediated cytochrome c release from 45% to 3%.
MSN-50, MSN-125, and DAN004 Inhibit Bax- and Bak- Mediated MOMP
To determine whether the compounds inhibited tBid-activated Bax and/or Bak in mitochondria, the compounds were added to permeabilization assays containing mitochondria isolated from Bax–/– or Bax–/–lBak–/– baby mouse kidney (BMK) cell lines stably expressing Smac-mCherry in the mitochondrial intermembrane space (Shamas-Din et al., 2014). Release of the fluorescence protein measured by fluorimetry provided a quantitative assessment of MOMP (Figures 2H and 2I). In control experiments, none of the compounds alone resulted in significant mitochondrial permeabilization even at 40 μM (Figure S1C). Therefore, this concentration was chosen as the maximum for the assays used to measure inhibition of Bax and Bak. Both MSN-50 and MSN-125 inhibited tBid/Bax-mediated MOMP in a concentration-dependent manner (Figure 2H). However, BJ-1 and BJ-1-BP failed to inhibit Bax-induced MOMP (Figure S1D) and Nutlin-3a only poorly inhibited tBid/Bax-mediated MOMP (Figure S1B). The activity of Nutlin-3a in preventing mitochondrial permeabilization was so poor that, similar to BJ-1 and BJ-1-BP, it would not be possible to ascribe any activity observed in cells to inhibition of Bax and therefore these compounds were not pursued further. The reasons why these compounds are not active with mitochondria are not clear but may be related to non-specific binding to mitochondria or metabolism by enzymes in the crude mitochondrial fractions known to contain peroxisomes and vesicles derived from the secretory pathway.
As expected, mitochondria from Bax–/– cells were efficiently permeabilized by tBid alone due to activation of endogenous Bak in the MOM and/or release of activated Bak from endogenous anti-apoptotic proteins (Figure 2I). It was not possible to measure the amount of endogenous Bak on the MOM with sufficient accuracy to quantitatively compare compound-mediated inhibition of Bak with inhibition of the known quantities of Bax added to Bax–/–lBak–/– mitochondria. Nevertheless, MSN-50, MSN-125, and DAN004 showed concentration-dependent inhibition of Bak-mediated MOMP (Figures 2H-2J and S1F), whereas BJ-1 and BJ-1-BP did not show any activity in this assay (Figure S1E). Taken together the above results demonstrate that MSN-50, MSN-125, and DAN004 inhibit both Bax and Bak.
MSN-125 and MSN-50 Inhibit Apoptosis in Cells
To further characterize MSN-50, MSN-125, and DAN004 functionally, potency in inhibiting Bax/Bak-mediated apoptosis in human colon cancer (HCT-116) cells, BMK cells was investigated. To dissect the inhibition of Bax and Bak, wild-type (WT), Bax–/–, Bak–/–, and Bax–/–IBak–/– (DKO) HCT-116 cells and BMK cells were assayed for survival and proliferation after induction of apoptosis by treating these cells with actinomycin D (ActD) or staurosporine (STS). Since ActD and STS were added transiently and the cells allowed to recover for 4 days before replating, lack of growth indicates that the cells died. As expected, in the absence of the inhibitor compounds, only Bax–/–IBak–/– cells survived and proliferated after ActD or STS treatment and replating (Figures 3A–3C). Pretreatment of the cells with 5 or 10 μM MSN-50 or MSN-125 conferred survival to ActD-treated WT, Bax–/– and Bak–/– HCT-116, and BMK cells (Figures 3A and 3C, respectively). Concentrations of 5 and/or 10 μM of the MSN compounds are shown because lower concentrations were largely ineffective, while concentrations of 20 μM or higher had off-target toxicity that killed HCT-116 and BMK DKO cells. Similar results were also obtained for BMK cells treated with STS. Although the results with STS were more variable, the data are also consistent with the compounds inhibiting both Bax and Bak (Figures 3A–3C, respectively). Concentrations of 5 and/or 10 μM of the MSN compounds are shown because lower concentrations were largely ineffective, while concentrations of 20 μM or higher had off-target toxicity that killed HCT-116 and BMK DKO cells. Similar results were also obtained for BMK cells treated with STS. Although the results with STS were more variable, the data are also consistent with the compounds inhibiting both Bax and Bak (Figures 3A–3C). ILS- JRK-C95–182, a compound structurally similar to MSN-50 and MSN-125 that displayed only a minimal inhibitory effect in the liposome permeabilization assay (Table S1), also lacked protective activity in live cells (Figures 3A–3C). Control wells without ActD or STS treatment demonstrated that, when administered alone, 5 or 10 μM MSN-50 or MSN-125 had negligible effects on cell proliferation (Figure 3A).
Figure 3. MSN-50 and MSN-125 Protect HCT-116 Cells, BMK Cells, and Primary Cortical Neurons from Apoptosis.
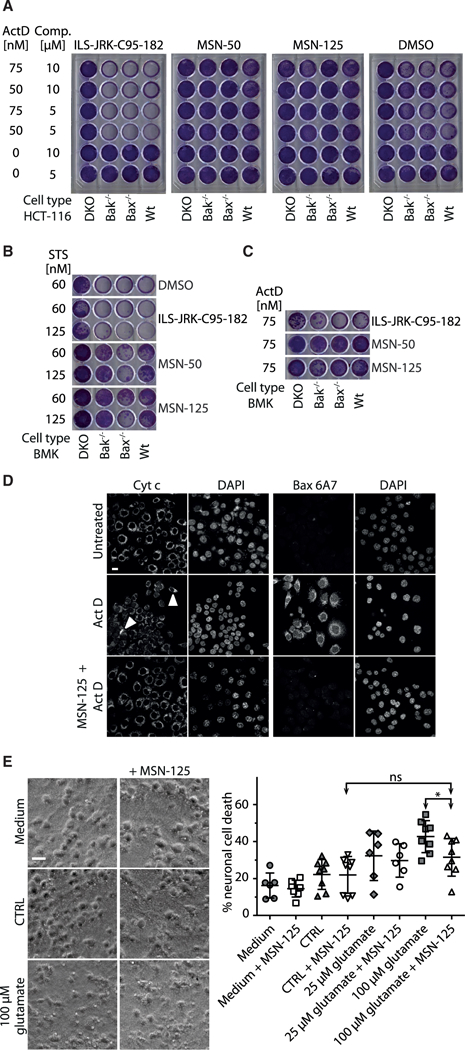
(A-C) HCT-116 or BMK cell lines were incubated with the inhibitor specified above the panels and ActD or STS. After replating, viable cells were stained with crystal violet. See STAR Methods for details.
(A) MSN-50 and MSN-125 (5 and 10 μM, indicated Comp.) but not the negative controls (ILS-JRK-C95–182, DMSO) conferred longterm survival and growth of HCT-116 cells exposed to actinomycin D (ActD).
(B) MSN-125 and MSN-50 (5 μM) conferred long-term survival and growth of BMK cells exposed to the indicated concentrations of STS.
(C) MSN-125 and MSN-50 (5 μM) conferred long-term survival and growth of BMK cells exposed to 75 nM ActD.
(D) In cells treated with ActD, MSN-125 prevented exposure of the N-terminal 6A7 Bax epitope and cytochrome c release. HCT-116 cells were treated with 10 μM MSN-125 for 3 hr before ActD treatment for 24 hr. Cells were fixed and immunofluorescence was performed with primary antibodies against activated Bax (6A7) or cytochrome c. Nuclei were visualized using DAPI. ActD treatment resulted in cytochrome c release from almost all cells. Two regions of mitochondria retaining cytochrome c in a field of ActD-treated cells are indicated with arrowheads. Scale bar, 10 μm. See also Figure S2C.
(E) MSN-125 (5 μM) protected neurons from prior excitotoxic treatment with 100 μM glutamate. Medium, continuous culture without wash; CTRL, neurons incubated in BSS0. Phase-contrast images (left panels) and lactate dehydrogenase-release assay (right panel) are shown. Individual data points and mean ± SD are displayed, n = 6–8 individual experiments. *p < 0.05, one-way ANOVA, Bonferroni post hoc; ns, not significant. Scale bar 25 μm. See also Figure S2.
Although DAN004 exhibited potent inhibitory activity in vitro, it exhibited marked toxicity at 10 μM in cell-replating assays (Figure S2A). Furthermore, at sub-toxic doses DAN004 did not confer protection against ActD treatment (Figure S2B). Due to the large number of potential reasons for the off-target toxicity of DAN004 the compound was not pursued further here.
To correlate the functional inhibition of apoptosis by MSN-125 to inhibition of Bax in cells, we performed immunofluorescence with the conformation-specific anti-Bax-6A7 antibody that binds to an N-terminal epitope of Bax that is exposed after activation (Hsu and Youle, 1998). As expected, ActD treatment triggered endogenous Bax to adopt the 6A7-positive conformation and cytochrome c release from almost all mitochondria in HCT-116 WT cells (Figures 3D and S2C). Addition of MSN-125 prevented both Bax-6A7 positivity and cytochrome c release (Figure 3D), but not morphological changes in cells and mitochondria (Figure S2C), consistent with the compound inhibiting both Bax and Bak but not the original damage caused by ActD.
MSN-125 Protects Primary Neurons against Glutamate Excitotoxicity
Glutamate excitotoxicity can be mediated by Bax-dependent pathways (D’Orsi et al., 2012) and excessive release of glutamate, as well as Bax-mediated apoptosis, contribute to neuronal cell death in stroke (D’Orsi et al., 2015; Mergenthaler et al., 2004). To determine whether MSN-125 would protect cultured primary embryonic mouse brain cortical neurons from glutamate excitotoxicity, neurons were treated with 25 or 100 μM glutamate for 30 min, which led to substantial neuronal cell death after 24 hr (Figure 3E). However, treatment with 5μM MSN-125 directly after the excitotoxic insult substantially reduced neuronal damage. At this concentration and time point, treatment with MSN-125 alone did not result in neuronal damage. Thus, unlike other neuroprotective agents MSN-125-medi- ated inhibition of Bax and Bak protects neurons when administered after the excitotoxic insult. Collectively, these data validate Bax and/or Bak as potential therapeutic and druggable targets.
Molecular Mechanism of Small-Molecule Inhibition of Bax
Bax-mediated MOMP proceeds as an ordered series of steps including tBid binding to the MOM, recruitment of Bax to tBid, insertion of Bax into the MOM, Bax oligomerization, and finally permeabilization of the MOM (Lovell et al., 2008). Given that MSN-125, MSN-50, and DAN004 inhibit both cBid- and Bim-mediated activation of Bax the most likely explanation for inhibition of MOMP by the small molecules is direct binding to Bax. However, it was not possible to measure direct binding of MSN-50 or MSN-125 to Bax by isothermal calorimetry or nuclear magnetic resonance due to the limited solubility of the compounds and protein when mixed in solutions containing more than 100 nM Bax as required for such studies.
Although a formal possibility, it is unlikely that inhibition of Bax was due to partitioning of the compounds into the liposome bilayer and thereby changing its physical properties, because adding the compounds to liposomes and then re-isolating the liposomes by gel filtration chromatography had no effect on Bax-mediated MP (Figure S3). Furthermore, many structurally similar compounds of equal or higher hydrophobicity had no detectable effect on inhibition of MP (Table S1). Finally, it is unlikely that inhibiting Bax and Bak by changing the lipid properties of subcellular or artificial mitochondria-like membranes as employed in our studies would function or be tolerated in the wide variety of systems reported here.
The molecular mechanism(s) by which the compounds inhibit Bax in membranes was investigated using fluorescence spectroscopic and chemical crosslinking approaches. Bax binding to membranes is the step immediately prior to oligomerization and is affected by all of the upstream steps. To measure Bax binding to membranes, the intensity increase that results when the environment-sensitive dye 7-nitrobenzene-2-oxa-1,3-diazol-4-yl-ethylenediamine (NBD) attached to Bax inserts into the bilayer was monitored (Lovell et al., 2008). Neither MSN-125 nor MSN-50 significantly decreased the rate or extent to which NBD-labeled Bax bound to membranes in response to cBid (Figure 4A). Furthermore, preincubation of Bax labeled with Alexa 568 with DAN004 did not inhibit Bax binding to membranes as measured by Forster resonance energy transfer (FRET) between the Alexa dye and liposomes labeled with the long-chain dialkylcarbocyanine (Figure S1G). Finally, preincubation of Bax or mitochondria with MSN-125 did not inhibit Bax binding to mitochondria measured by differential centrifugation (Figure S1H).
Figure 4. MSN-125 Restricts Bax Oligomerization beyond Dimer Formation.
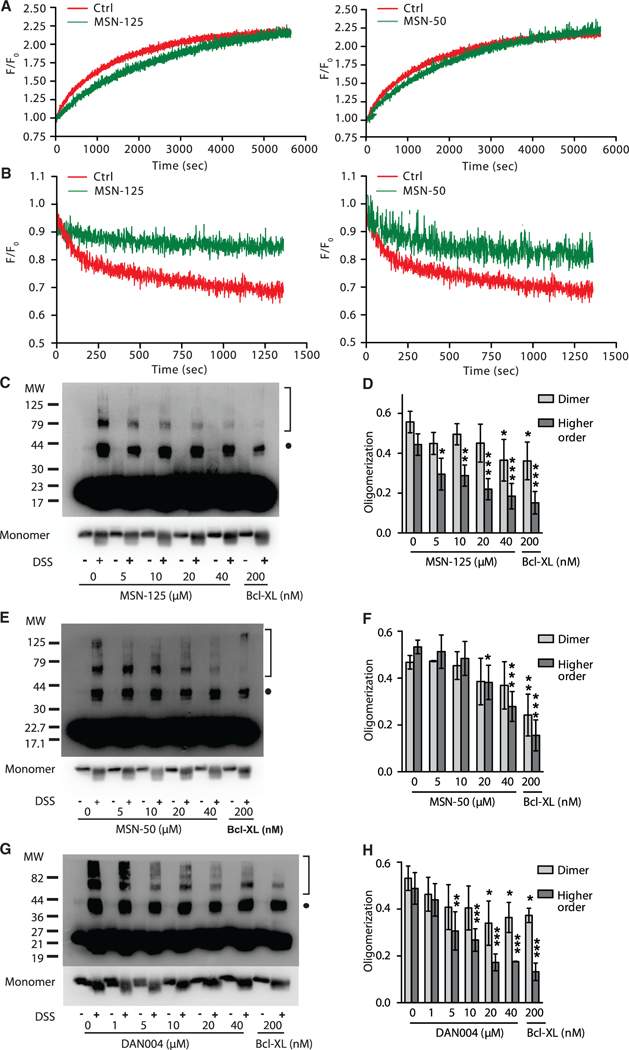
(A) MSN-125 and MSN-50 (10 μM) did not inhibit tBid-activated Bax binding to liposomes. MSN-125, MSN-50 (10 μM), or DMSO (as a control) was added to liposome incubations containing NBD-Bax-126C (100 nM) and tBid (20 nM). The time-dependent increase in fluorescence from NBD indicates Bax binding to liposomes. One representative measurement from three independent experiments is shown. See also Figures S1G, S1H, and S3.
(B) MSN-125 and MSN-50 (10 μM) reduced DAC-Bax-134C (20 nM) binding to NBD-Bax-126C(100 nM) on liposomes. F0 indicates DAC-Bax-134Cfluorescence before addition of NBD-Bax-126C. Binding of the proteins resulted in a time-dependent decrease in DAC-Bax-134C fluorescence (F) due to FRET between DAC and NBD. Addition of MSN-125 or MSN-50 decreased FRET compared with no compound control (Ctrl). One representative measurement from three independent experiments is shown.
(C) MSN-125 blocks tBid-induced Bax oligomerization in liposomes. Bax oligomers were detected after chemical crosslinking with DSS by immunoblotting for Bax. Bax monomers, ~20 kDa. The shorter exposure of the monomer fraction shown below the panel indicates that the changes in crosslinked species (dimers, solid dot; larger oligomers, brackets) are not due to differences in the amount of Bax monomer in each lane. MW, molecular weight marker (kDa). One representative immunoblot of n ≥ 3 is shown.
(D) MSN125 preferentially inhibits higher-order oligomerization of Bax. Quantification of crosslinking data from (C). Mean ± SD for n ≥ 3. *p < 0.05, **p < 0.01, ***p < 0.001; one-way ANOVA, post hoc, Dunnett.
(E–H) MSN-50 and DAN004 disrupt Bax oligomerization. Chemical crosslinking of Bax oligomers assayed as described in (C and D). (E and G) One representative immunoblot of n = 3 is shown. (F and H) Quantified immunoblot band intensities for three independent replicates.
To measure Bax oligomerization by FRET, single-cysteine mutants of Bax were labeled with diaminomethyl-coumarin (DAC-Bax-134C) as donor and with NBD (NBD-Bax-126C) as acceptor (Lovell et al., 2008). In reactions containing liposomes and tBid, addition of MSN-125 or MSN-50 reduced FRET between DAC-Bax-134C and NBD-Bax-126C (Figure 4B), demonstrating that both compounds inhibit Bax oligomerization.
However, FRET measurements do not distinguish between inhibition of dimer or oligomer formation. Therefore, the oligomerization state was further assessed by chemical crosslinking of purified Bax. Using the amine-specific crosslinker disuccinimidyl suberate (DSS), we clearly demonstrate that MSN-50, MSN-125, and DAN004 inhibit tBid-induced Bax oligomerization in liposomes with a more pronounced effect on inhibiting the formation of higher-order oligomers than dimers (Figures 4C-4H). At the highest concentration tested, MSN-125 also inhibited Bax dimerization to a limited extent, an effect not seen for MSN-50. High concentrations of MSN-50, MSN-125, or DAN004 inhibited large oligomers of Bax to a similar extent as the positive control Bcl-XL (Figures 4C-4H). Shorter exposures of the immunoblots indicated that changes in the oligomer bands were not due to inhibition of Bax binding to membranes leading to decreases in the amount of monomer at higher concentrations of the inhibitors. As expected (Dewson et al., 2012), crosslinking with DSS also generated intramolecular crosslinks resulting in faster migration and band smearing for the monomer species (Figures 4C, 4E, and 4G).
MSN-125 Inhibits Bax Oligomerization at Defined Dimer Interfaces
Previous disulfide crosslinking studies identified three dimer interfaces in Bax oligomers bound to mitochondria (Dewson et al., 2012; Zhang et al., 2010, 2016). Binding of the BH3 region of one Bax to the canonical BH1–3-groove of another Bax forms a BH3-groove interface, and is followed by binding of the two helices 6 and 9 in the neighboring Bax molecules that form the other two interfaces.
To determine whether MSN-125 inhibits the formation of a specific dimer interface in Bax oligomers, site-specific disulfide crosslinking was used. To this end, single-and double-cysteine Bax mutants predicted or previously shown to form disulfide-linked Bax homodimers when activated by an exogenous Bax BH3-peptide were targeted to mitochondria lacking Bax and Bak (Zhang et al., 2016) in the absence or presence of MSN-125. The resulting mitochondria-bound mutants were oxidized after incubation with mitochondria to induce disulfide crosslinking in the absence or presence of MSN-125. In the absence of MSN-125, single-cysteine Bax-E146C (mutation in helix 6) or Bax-A183C (mutation in helix 9) formed a disulfide-linked homodimer (Figure 5A, lanes 16/18, arrow). Addition of MSN-125 inhibited formation of both dimer interfaces (Figure 5A, lanes 14 and 20).
Figure 5. MSN-125 and MSN-50 Inhibit Some, but NotAll, Interactions in Bax and Bak Dimers.
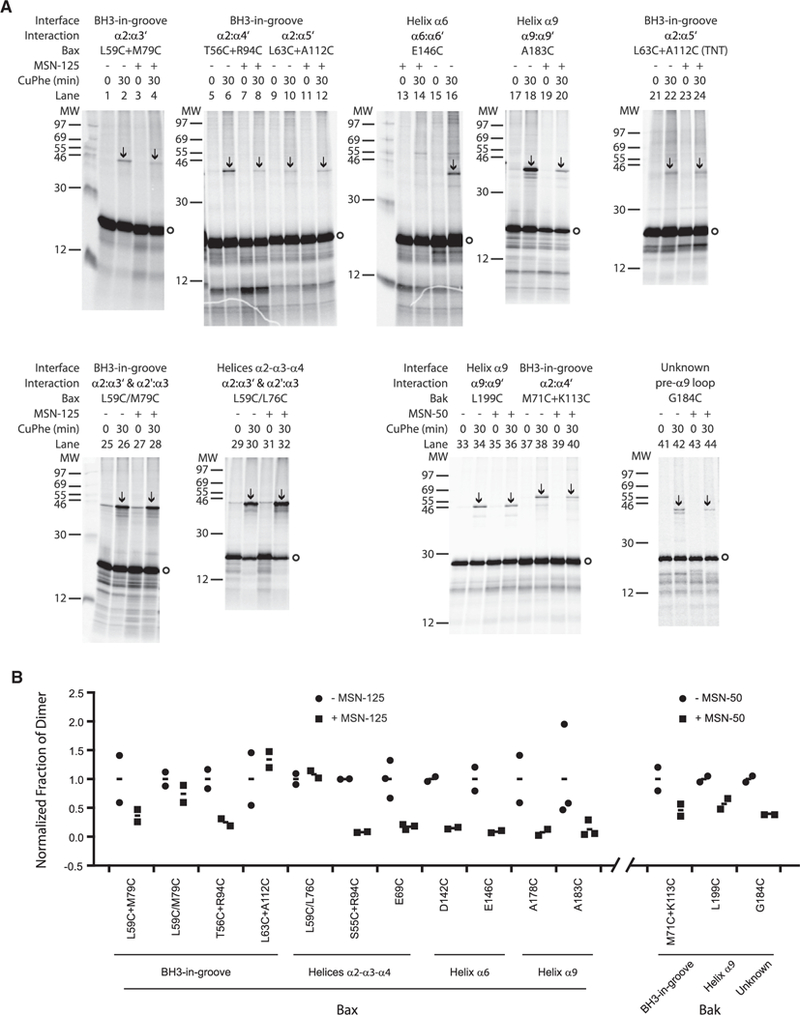
(A) Autoradiography of disulfide crosslinking of single- or double-cysteine Bax or Bak incubated with mitochondria lacking Bax and Bak in the absence or presence of MSN-125 or MSN-50 (40 μM) as indicated above the panels. MW, molecular weight marker (kDa); Bax monomers, circles; disulfide-linked Bax dimers, arrows. Addition of copper (II) (1,10-phenanthroline)3 (CuPhe) oxidant for 30 min to induce disulfide crosslinking is indicated (min). Representative data from n ≥ 2 experiments are shown.
(B) MSN-125 does not completely block the Bax dimer interface. Quantification of data from two to three independent experiments demonstrates that disulfide crosslinking of the single-cysteine Bax mutants L63C + A112C and the double-cysteine mutants L59C/L76C and L59/M79C were not inhibited by MSN-125. Quantification ofall crosslinking pairs assayed asdescribed in (A) are shown as individual data points (circlesand squares) and means(dash). The single-cysteine Baxmutants L63C + A112C(TNT) were synthesized intheTNT system to increase proteinyield and reducethe impact of background. Inhibition ofcrosslinkingfor similar interfaces in Bax by MSN-50 is shown in Figure S4.
Quantification of the bands revealed that even though targeting of WT Bax (Figure S1H) and most of the Bax mutants to membranes is not affected by the inhibitors, targeting of some mutants was affected by the cysteine substitutions. Therefore, to quantify the effect of the compounds on crosslinking and correct for differences in the efficiencies of targeting of the various mutants to membranes, the mean fraction of dimer without inhibitor was set to one for each crosslinking pair (dash between circles) and compared with the fraction of dimer in the presence of inhibitor (dash between squares). Individual data points (circles and squares) were plotted to illustrate the spread in the data between independent replicates (Figure 5B).
To examine the BH3/helix 2 region binding to the canonical groove, two pairs of Bax mutants with single cysteines in the BH3/helix 2 region (Bax-L59C, Bax-T56C) and in the canonical groove (Bax-M79C in helix 3, Bax-R94C in helix 4) were analyzed. These mutants also generated disulfide-linked dimers (Figure 5A, lanes 2 and 6) that were inhibited by MSN-125 (Figure 5A, lanes 1–8). However, when two cysteines were included in this region of Bax (L59C/L76C) or in an adjacent area (L59C/ M79C) the crosslinks were not inhibited by MSN-125 (Figure 5A, lanes 25–32), suggesting that this part of the BH3-grove dimer interface (Figure 6) is only partially blocked by the compound.
Figure 6. MSN-125 and MSN-50 Interferes with Correct Dimer Formation for Bax and Bak thereby Inhibiting Further Oligomerization and Preventing MOMP.
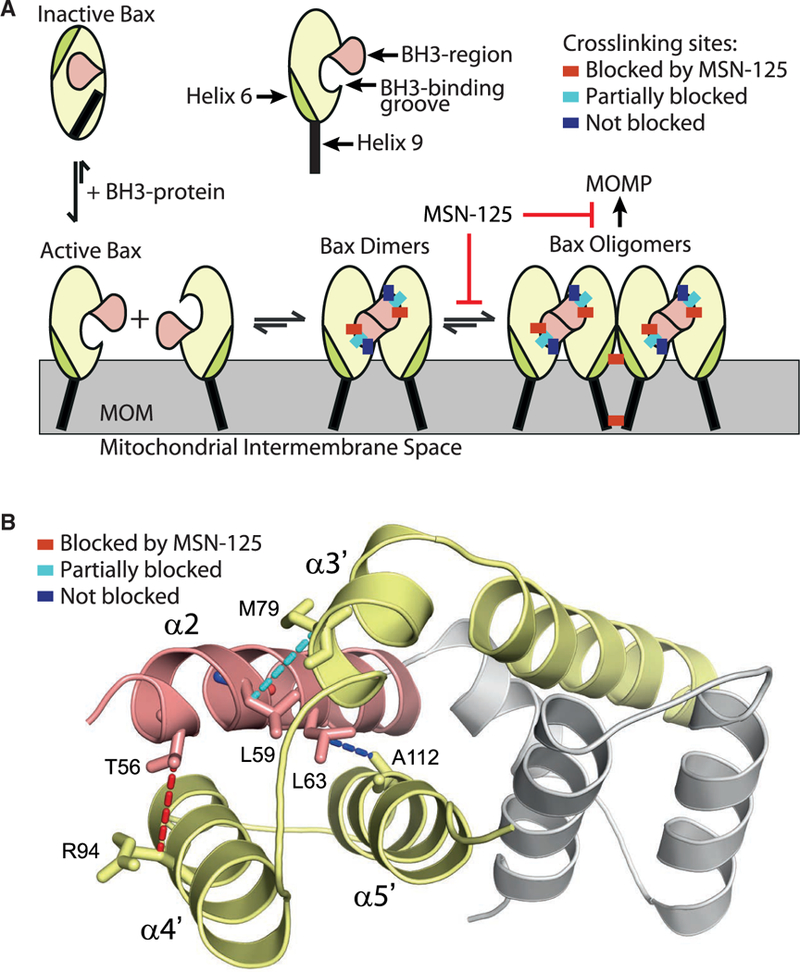
(A) Model of Bax oligomerization with schematic representation of the crosslinking positions (colored rectangles) that are not blocked (dark blue) or fully or partially blocked by MSN-125 (red and cyan, respectively); MOM, mitochondrial outer membrane. While the model depicts MSN-125 and Bax it is representative of the data for both MSN-125 and MSN-50 as well as both Bax and Bak.
(B) Schematic representation of the mechanism of MSN-125. Cartoon representation ofthe Bax BH3-groove dimer structure (PDB: 4BDU) shown with the BH3/α2 helix (pink) of one monomer (the rest of this monomer is colored gray) binding to the groove formed by α3’, α4’, and α5’ helices (yellow) of the other monomer. Disulfide crosslinking pairs are joined by dotted lines. Crosslinks blocked by MSN-125 (red), partially blocked (cyan), not blocked (blue) by MSN-125.
Consistent with this result, another pair of Bax mutants with single cysteines in the BH3 region/helix 2 (Bax-L63C) and in the groove/helix 5 (Bax-A112C) resulted in disulfide crosslinking that was not inhibited by MSN-125 (Figure 5A, compare lanes 10 to 12, and Figure 5B). Because the residues are positioned where the crosslinking was relatively weak the experiment was repeated with a fully coupled in vitro transcription/translation (TNT) system to produce more Bax-L63C and Bax-A112C protein (L63C + A112C) and confirmed that this crosslink is not inhibited by MSN-125 (Figure 5A, lanes 21–24, and 5B). Similar crosslinking results for the different Bax mutants were also obtained using MSN-50 (Figure S4), confirming that both MSN compounds inhibit some but not all Bax dimer interfaces.
To determine whether the inhibitors had a similar activity on Bak, crosslinking was performed for three previously known sites of interaction in Bak including the BH3-groove, helix 9, and the region upstream of helix 9 (Dewson et al., 2008; Iyer et al., 2015). As expected all of these crosslinks were inhibited by MSN-50 (Figure 5A, lanes 33–44, and 5B). Thus, correct formation of dimers is inhibited for both Bax and Bak by MSN-125 and MSN-50.
A schematic of the MSN-125-affected and -unaffected interaction sites in the BH3-groove interface is shown in Figure 6. These results suggest that when helix α2 of one Bax binds to the canonical groove of another Bax, MSN-125 blocks the binding of the N-terminal helical turn of α2 binding (T56), partially inhibits the binding of the next helical turn (L59), but does not affect binding of the third helical turn (L63) to the grove. Taken together, these results suggest that, although Bax and Bak homodimers form (Figures 4 and 5), the known dimer interfaces are at least partially disrupted by the MSN compounds (Figure 6).
As expected from our previous data suggesting that Bax oligomerization occurs after binding to membranes (Annis et al., 2005), cBid-mediated targeting of the WT and mutant Bax proteins to mitochondria in the crosslinking reactions was largely unaffected by MSN-125 (Figures S1H and 5A, open circles). Therefore, MSN-125 does not significantly interfere with targeting of Bax monomers to mitochondria, but rather inhibits assembly of Bax oligomers at the MOM by interfering with multiple, yet not all, Bax-Bax interactions at the three dimer interfaces of Bax. MSN-125, MSN-50, and DAN004 prevent Bax oligomerization predominantly beyond the formation of dimers, suggesting that correct formation of the dimer is required for oligomerization. Our data also support the notion that, even after activation by a BH3-protein, correct formation of Bax dimers and likely larger oligomers (Uren et al., 2017) is essential to execute MOMP.
DISCUSSION
Here, we have discovered novel small-molecule inhibitors of both pro-apoptotic Bax and Bak. Using these compounds, we provide new insight into unresolved questions in the regulation of the molecular mechanism of Bax/Bak oligomerization in the MOM. We establish that neither the Bax/Bak monomer nor an imperfect dimer is sufficient to permeabilize membranes. Our results clearly show that Bax binding to membranes does not result in MP unless the protein oligomerizes. Furthermore, we demonstrate that pharmacological inhibition of Bax and Bak protects cells from undergoing apoptosis after a variety of stimuli.
In most cell types, Bax and Bak can functionally substitute for each other (Degenhardt et al., 2002; Mikhailov et al., 2003). Therefore, to inhibit apoptosis in cells by blocking MOMP it is essential to inhibit both proteins. Unlike previously characterized molecules that inhibited Bax (Bombrun et al., 2003; Hetz et al., 2005; Peixoto et al., 2009; Polster et al., 2003), MSN-50, MSN-125, and DAN004 inhibit both Bax and Bak after activation by either cBid or Bim (Figure 2) protecting cells expressing either Bax or Bak (Figure 3) from ActD and STS by preventing oligomerization of Bax and Bak (Figures 4, 5, and S4).
These small molecules inhibit permeabilization ofthe MOM by altering the BH3-groove dimer interface of Bax and Bak (Figures 5,6, and S4). We speculate that partial disruption ofthis interface prevents the formation of the other interfaces required to form higher-order oligomers. Furthermore, we show that inhibition of oligomerization of both Bax and Bak is necessary and sufficient to inhibit genotoxic stress and broad kinase inhibition (STS) induced apoptosis such that at least a subset of drug-treated cells retained proliferative potential (Figures 3A–3D).
Our cellular studies using MSN-50 and MSN-125 indicate that it might be possible to exploit inhibition of Bax and Bak therapeutically. Neuronal cell death after excitotoxicity has been demonstrated to be at least in part mediated through Bax-dependent signaling mechanisms (D’Orsi et al., 2012). Excitotoxicity is one of the damaging mechanisms contributing to acute neuronal degeneration after stroke (Mergenthaler et al., 2004), traumatic brain injury, and epileptic seizures (Engel et al., 2011). Genetic models where deletion of Bax protected neurons from cell death after stroke in mice (D’Orsi et al., 2015) further support the notion that inhibition of both Bax and Bak may be of therapeutic potential in acute neurodegeneration. Our data demonstrating that MSN-125 protects neurons from cell death after glutamate excitotoxicity (Figure 3E) establishes that pharmacological inhibition of Bax and Bak in neurons is feasible and may indeed provide thera-peutic benefit. However, off-target effects with these tool compounds may contribute to the morphological alterations observed, and the high concentrations required limit the use of these compounds to in vitro investigations only.
Evidence of the function of Bak in neuronal apoptosis has been controversial. Bak may promote neuronal survival under certain conditions and in development, while promoting neuronal cell death after stress such as in stroke (Fannjiang et al., 2003; Hardwick and Soane, 2013). In addition,althoughfull-length Bak isubiquitously expressed in mostcells, including central and peripheral nervous system neurons (Putcha et al., 2002; Sun et al., 2001), a neuron-specific splice variant of Bak, N-Bak, has been reported to have both anti- and pro-death functions (Sun et al., 2001; Uo et al., 2005). While evidence for the existence of this splice variant on the protein and functional level is inconclusive (Jakobson et al., 2012; Putcha et al., 2002), the inhibitors we have developed here offer the opportunity to investigate the role of Bax and Bak in the execution of MOMP in neurons and their role in the molecular pathophysiology of diseases of the nervous system.
To permeabilize membranes, Bax and Bak first homodimerize via the BH3-groove interface leading to the formation of heterogeneously sized higher-order oligomers via additional dimer interfaces (Czabotar et al., 2013; Dewson et al., 2012; Iyer etal., 2015; Maetal., 2013;Subburaj etal.,2015).Which ofthese species permeabilizes the membrane is controversial with recent evidence suggesting that relatively disordered oligomers of dimers are sufficient to permeabilize membranes (Uren et al., 2017). We have recently shown that the BH3-groove dimer nucleates the assembly of the Bax oligomeric pore, which is enlarged by dimerization at the helix 9 interface (Liao et al., 2016; Zhang et al., 2016). Using MSN-125 as a tool to dissect the molecular mechanism of Bax activation, we now show that blocking Bax at the dimer stage is sufficient to inhibit MOMP. Therefore, in contrast to results obtained with belted lipid nanodisks or MOM vesicles (Kushnareva et al., 2012; Xu et al., 2013) the minimal permeabilizing unit is a correctly formed dimer.
Crosslinking analysis revealed that MSN-125 disrupts helix 6 and helix 9 interactions, and some, but not all, interactions in the BH3-groove interface (Figures 5 and 6), suggesting that these interactions are necessary for Bax oligomerization. Consistent with this interpretation, the altered dimeric Bax structure is not recognized by the conformation-specific antibody 6A7 (Figure 3D). Therefore MSN-50 and MSN-125 can serve as important tools for identifying molecular interactions critical for forming functional Bax oligomers (Subburaj et al., 2015; Uren et al., 2017). We anticipate that these small molecules will allow further insight into the mechanism regulating formation, oligomerization, and the functional role of recently identified diverse oligomeric Bax-structures in forming an ordered “apoptotic pore” complex in the MOM (Grosse et al., 2016; Salvador-Gallego et al., 2016). Because MSN-125 inhibition of dimer interfaces is a downstream event and it inhibits heat-activated Bax (FigureS1I), we predict that the compounds will inhibit apoptosis induced by small molecules (Brahmbhatt et al., 2016) or p53 in addition to pro-apoptotic BH3-proteins (Follis et al., 2015).
In essence, three classes of functional Bax inhibitors have been reported (Bombrun et al., 2003; Hetz et al., 2005; Jiang et al., 2007; Peixoto et al., 2009, 2015; Polster et al., 2003), at least one class is sufficiently hydrophobic that it is expected to partition into membranes (Polster et al., 2003). Bax and Bak have to insert into and permeabilize membranes to lead to MOMP. Therefore, it is not surprising that other small molecules that alter the physical properties of membranes such as dibucaine, propranolol, and cholesterol have all been shown to partially inhibit the insertion of Bax into membranes, thereby reducing apoptosis (Christenson et al., 2008; Polster et al., 2003). However, gross perturbation of cellular membranes is expected to have wide-ranging effects on cellular physiology that are likely independent of a Bax/Bak-mediated pathway.
While access to some of the other molecules has been limited, it has been shown that most of them inhibit Bax, and they either do not inhibit Bak or the effect on Bak activity remains to be determined (Hetz et al., 2005). Furthermore, electrophysiology studies suggested that they act as channel blockers rather than inhibiting Bax oligomerization in the MOM or inhibiting Bak (Bombrun et al., 2003; Hetz et al., 2005; Peixoto et al., 2009; Polster et al., 2003). Using fluorescence spectroscopy and biochemical crosslinking, we show that MSN-50 and MSN-125 directly disrupt the correct formation of dimers and higher-order Bax oligomers, suggesting that they do not act as channel blockers.
The sole member of the third class, the MDM2 inhibitor Nutlin-3a, which we used as a control, has been reported to inhibit Bax and Bak oligomerization as an off-target effect, in addition to its intended inhibitory effect on the interaction of MDM2 and p53 (Jiang et al., 2007). Assessment of the molecular mechanism of inhibiting Bax and Bak by Nutlin-3a suggests it inhibits Bax dimerization (Jiang et al., 2007), indicating a different and uncharacterized molecular mechanism from the molecules described here that allow dimer formation but alter the interfaces. Moreover, it remains unclear how Nutlins confer protective effects in kidney tubular cells but also potentiate cancer cell death (Tovar et al., 2006). Finally, Nutlin-3a is much less potent in inhibiting MOMP than MSN-50 and MSN-125 (Figure S1B).
In aggregate our results demonstrate that small-molecule inhibition of Bax and Bak oligomerization via partial disruption of the canonical BH3 peptide-groove interface prevents cells from executing MOMP in response to diverse apoptotic stimuli. Our studies not only provide novel insight into the mechanism of Bax oligomerization in the MOM, but also provide crucial tools for future mechanistic studies of the molecular organization of Bax and Bak oligomeric structures in the MOM. These tool compounds may also facilitate the development of future compounds designed to inhibit apoptosis by restricting MOMP in a wide range of diseases, including acute neurodegeneration, or for mitigating dose-limiting side effects of some cancer treatments.
STAR✩METHODS
Detailed methods are provided in the online version of this paper and include the following:
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-Bax 6A7, 2D2 | Laboratory of Dr. Richard Youle; Hsu and Youle, 1997 | N/A |
| Sheep polyclonal anti-cytochrome c | Serum obtained from Caprilogics; antibody purified in our laboratory | N/A |
| Donkey anti- sheep - Alexa Fluor 488 | Invitrogen | Cat # A-11015 |
| Goat anti-mouse- Alexa Fluor 555 | Invitrogen | Cat # A-21422 |
| Bacterial and Virus Strains | ||
| Escherichia coli BL21-AI | New England Biolabs | Cat # C2528 |
| Escherichia coli DH5α | New England Biolabs | Cat # C2988J |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Nutlin-3a | Selleck | Cat # S8059; CAS: 675576-98-4 |
| Actinomycin D | Sigma | Cat # A9415; CAS: 50-76-0 |
| Staurosporine | Sigma | Cat # S5921; CAS: 62996-74-1 |
| Disuccinimidyl suberate | Thermo Scientific Pierce | Cat # 21655; CAS: 68528-80-3 |
| Crystal Violet | Sigma | Cat# HT901; CAS: 548-62-9 |
| 8-Aminonaphthalene-1,3,6-Trisulfonic Acid, Disodium Salt | Life Technologies | Cat # A350; CAS: 5398-34-5 |
| p-Xylene-Bis-Pyridinium Bromide | Life Technologies | Cat # X1525; CAS: 14208-10-7 |
| DAPI | Sigma | Cat # D9542; CAS: 28718-90-3 |
| IANBD | Life Technologies | Cat # D2004; CAS: 173485-12-6 |
| DACM | AnaSpec | Cat # AS-81403 |
| Alexa Fluor 568 | Life Technologies | Cat # A20341 |
| DiD | Life Technologies | Cat # D7757 |
| Caspase-8 | Enzo Life Sciences | Cat # BML-SE172-5000 |
| β-NADH | Sigma | Cat # N8129; CAS: 606-68-8 |
| l-lactic dehyrogenase | Sigma | Cat # L3916; CAS: 9001-60-9 |
| Experimental Models: Cell Lines | ||
| Mouse: Baby mouse kidney cells | Laboratory of Dr. Eileen White; Mathew et al., 2008 | N/A |
| Human: HCT116 cells | Wang and Youle, 2012 | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57bl/6J. E15 embryos to generate primary embryonic mouse brain cortical neuronal culture | The Jackson Laboratory, continuous
breeding in house |
JAX: 000664 |
| Mouse: B6.129-Bak1tm1Thsn/J | The Jackson Laboratory | JAX: 004183 |
| Recombinant DNA | ||
| pvitro1-blasti-smac-mCherry | Shamas-Din et al., 2014 | N/A |
| Software and Algorithms | ||
| GraphPad Prism | GraphPad Software, Inc., | https://www.graphpad.com/ |
| ImageJ | Natinal Institutes of Health | https://imagej.nih.gov/ij/ |
| SPSS | IBM | https://www.ibm.com/analytics/us/en/technology/spss/ |
| Other | ||
| TNT-coupled SP6 RNA polymerase/reticulocyte lysate system | Promega | Cat # L4600 |
| Cytochrome c ELISA | R&D systems | Cat # MCTC0 |
| L-α-phosphatidylcholine | Avanti Polar Lipids | Cat # 840051C |
| L-α-phosphatidylethanolamine | Avanti Polar Lipids | Cat # 841118C |
| L-α-phosphatidylinositol | Avanti Polar Lipids | Cat # 840042C |
| 1,2-dioleoyl-sn-glycero-3-phospho-Lserine(sodium salt) | Avanti Polar Lipids | Cat # 840035C |
| 1’,3’-bis[1,2-dioleoyl-sn-glycero-3-phospho]-sn-glycerol (sodium salt) | Avanti Polar Lipods | Cat # 710335C |
CONTACT FOR REAGENT AND RESOURCE SHARING
For reagents and resources other than MSN-50 and MSN-125 generated in this study or any other questions about the reagents please contact David W. Andrews (david.andrews@sri.utoronto.ca). For all questions and inquiries regarding BJ-1, BJ-1BP, MSN-50 and MSN-125 please contact Prabhat Arya (prabhata@drils.org) for questions and inquiries regarding DAN004 please contact Wibke Diederich (wibke.diederich@staff.uni-marburg.de).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
See Method Details below for details on cell cultures and culture conditions.
METHOD DETAILS
Protein Purification
Bax
Recombinant full length human Bax fused to a C- terminus intein tag containing a chitin binding domain was expressed in Escherichia coli BL21-AI and purified as described previously (Billen et al., 2008; Lovell et al., 2008). Briefly, cells were resuspended in lysis buffer (10 mM Hepes (pH 7.2), 100 mM NaCl, 0.2% CHAPS, 1 mM phenylmethylsulfonylfluoride (PMSF), 10 μg/ml DNase and 1 X protease inhibitor cocktail) and lysed by mechanical description in a French press. Bax was purified by affinity chromatography with a chitin column. Chitin beads were incubated with the lysate containing Bax for 2 hours at 4°C and washed with wash buffer (10 mM Hepes (pH 7.2), 500 mM NaCl, 0.5% CHAPS). Release of Bax from the intein tag was conducted by incubation of the chitin column in cleavage buffer (10mM Hepes (pH 7.2), 200 mM NaCl, 0.2 mM EDTA, 0.1% CHAPS and 100 mM β-mercaptoethanol) for 24–48 hrsat 4°C. Theeluate containing Bax was passed over a DEAE Sepharose column 2–3 X to remove nucleic acid contaminants, dialyzed in dialysis buffer (10 mM Hepes (pH 7), 200 mM NaCl, 0.2 mM EDTA, 10 % glycerol), and stored at −80°C.
Mutants of Bax with a single cysteine at position 126 or 134 intended for labelling with fluorescent dyes were purified as described above except Bax was released from the intein tag in buffer containing 100 mM hydroxylamine instead of β-mercaptoethanol to prevent leaving a free sulfhydryl at the carboxyl-terminus. The concentration of Bax was determined spectrophotometrically at 280 nm using the molar extinction coefficient 37,000 M−1cm−1. For labeling, N-N’-dimethyl-N-(iodoacetyl)-N’-(7-nitrobenzene-2-oxa-1,3-diazol-4-yl)ethylenediamine (IANBD; Life Technologies), N-(7-dimethylamino-4-methylcoumarin-3-yl) maleimide (DACM; Anaspec), or Alexa 568 C5-maleimide (Alexa568; Life Technologies) were prepared in DMSO and the concentrations were determined after dilution in methanol by absorbance using the dye molar extinction coefficients 25,000 M−1cm−1 at 479 nm, 27,000 M−1cm−1 at 380 nm, or 106,000 M−1cm−1 at 575 nm, respectively. Bax was labeled by incubation of the protein with a 10 fold molar excess of dye in labeling buffer (10 mM HEPES pH 7,0.2 M NaCl, 0.2 mM EDTA, 0.5% CHAPS w/v and 10% glycerol v/v) with rotation at room temperature for 2 hours in the dark. The reaction was quenched with 1 mM dithiothreitol. Free dye was removed from the sample by gel filtration chromatography (Sephadex G-25 Fine, GE Healthcare Life Sciences) in the labeling buffer without CHAPS.
Bcl-XL
Recombinant full length human Bcl-XL fused to a C- terminal intein tag was expressed in Escherichia coli DH5α cells as described previously (Billen et al., 2008). Bcl-XL was purified by affinity chromatography similar to Bax except the lysis and wash buffer contained 20mM Tris (pH 8), 500 mM NaCl, 0.5 mM EDTA, 1 % CHAPS, 1 mM PMSF, 10 μg/ml DNase and 1 X protease inhibitor cocktail and intein cleavage was conducted in cleavage buffer containing 20 mM Tris (pH 8), 200 mM NaCl, 20 % glycerol, 0.2 % CHAPS, 100 mM β-mercaptoethanol. The eluate containing Bcl- XL was further purified on a phenyl-Sepharose column by elution with cleavage buffer without any salt, dialyzed in dialysis buffer (Tris (pH 8), 20% glycerol) and stored at −80°C.
Bid
Recombinant full length murine Bid fused to a N-terminal histidine tag was expressed in Escherichia coli BL21-AI and purified as described previously (Shamas-Din et al., 2013). Briefly cells were resuspended in lysis buffer (10 mM Hepes (pH 7), 100mM NaCl, 10 mM imidazole, 1mM PMSF, 10 μg/ml DNase and 1 × protease inhibitor cocktail) and lysed using mechanical disruption. Nickel beads were incubated with the lysate for 2 hours at 4°C and washed with wash buffer (10 mM HEPES (pH 7), 300 mM NaCl, 1 % CHAPS, 10 mM imidazole). Fractions containing Bid were eluted in elution buffer (10 mM HEPES (pH 7), 100 mM NaCl, 200 mM imidazole, 0.1 % CHAPS, 10% glycerol). cBid was obtained by incubating fractions containing Bid with caspase-8 (Enzo Life Sciences) at room temperature for ~36–40 hrs. tBid was isolated by affinity chromatography using nickel heads and elution in elution buffer containing 2% (w/v) octylglucoside. cBid and tBid fractions were dialyzed in dialysis buffer (10 mM HEPES (pH 7), 100 mM NaCl, 0.1 mM EDTA, 10% glycerol) and stored at −80°C.
Bim
Recombinant full length murine BimL fused to a N-terminal histidine tag was expressed in Escherichia coli BL21-AI and purified as described previously (Sarosiek et al., 2013). Cells were lysed by mechanical disruption in lysis buffer (20 mM Tris (pH 8), 20 mM NaCl, 1% CHAPS, 5 mM imidazole, 20% glycerol). The lysate was passed through a nickel column and the column was washed with wash buffer (20 mM Tris (pH 8), 50 mM NaCl, 0.5 % CHAPS, 10 mM imidazole, 20% glycerol). Fractions containing Bim were obtained by eluting with elution buffer (20 mM Hepes (pH 7.2), 10 mM NaCl, 0.3% CHAPS, 300 mM imidazole, 20% glycerol). The concentration of NaCl in the eluate was adjusted to 150 mM and applied to a high performance phenyl sepharose column pre-equilibrated with buffer (20 mM Hepes (pH 7.2), 100 mM NaCl, 0.3% CHAPS, 20% glycerol). Fractions containing Bim were eluted in the equilibration buffer lacking NaCl, dialyzed in dialysis buffer (10 mM Hepes (pH 7), 1 mM NaCl, 20% glycerol), and stored at −80°C.
Liposome Permeabilization and Dye Release Assays
Mitochondria-like liposomes (48% phosphatidylcholine, 28% phosphatidylethanolamine, 10% phosphatidylinositol, 10% dioleoyl phophatidylserine and 4% tetraoleoyl cardiolipin (Avanti)) encapsulating aminonaphthalene1,3,6-trisulfonic acid (ANTS) and its quencher, 45 mM p-xylene-bis-pyridinium (DPX, Life Technologies) were prepared in buffer containing 10 mM HEPES (7.2), 200 mM potassium chloride, 5 mM magnesium chloride and 0.2 mM EDTA as described previously (Billen et al., 2008).
To assay inhibition of Bax, the fluorescence increase due to liposome permeabilization was measured (excitation at 355 nm and emission 520 nm) on a Tecan M1000 microplate reader. To ensure the background signal stabilized and to correct for compound fluorescence the initial fluorescence (F0) was recorded for 15–30 min at 37°C in the presence of compounds and liposomes before adding proteins. Bax and tBid were added (in that order) att0, and fluorescence changes (F) were measured for at least 2 h at 37°C.To estimate F100, Triton X-100 was added to a final concentration of 0.2% (w/v), and fluorescence was measured for 10 min at 37°C. The change in hydrophobicity due to the addition of detergent results in a slight overestimation of F100. The percentage release of ANTS/ DPX was calculated as [(F – F0)/(F100 – F0)]×100. For compound screening, % inhibition was calculated by [(% ReleaseBax + tBid-%Release Bax + tBid+ compound)/% Release Bax + tBid]*100. Z-scores were calculated by [(% inhibition - % inhibition population mean)/Standard deviation]. Unless indicated otherwise, all assays used 20 nM tBid and 100 nM Bax.
Small Molecule Bax Inhibitor Screen
A collection of benzofuran-based flavonoid-inspired, and several tetrahydroquinoline alkaloid-inspired compounds were screened for inhibition of tBid/Bax-mediated dye release in a MOMP-mimicking liposome permeabilization assay. In the first round 86 compounds were individually added to liposomes and then purified Bax and tBid were added to induce permeabilization of liposomes. Small molecules that inhibited liposome permeabilization with aZ-score >2 were re-assayed manually. The top two compounds were used to select additional compounds for a second round of screening.
Compounds and Synthesis
Diversity-oriented synthesis of the compounds screened including MSN-125, MSN-50 and the synthesis of DAN004 are described in the Methods S1. Nutlin-3a was obtained from Selleck.
Mitochondria Isolation and Permeabilization Assay
BMK Mitochondria
Bax–/–lBak–/–or Bax–/–baby mouse kidney (BMK) cells stably expressing a fusion protein comprised of the mitochondrial import peptide of Smac (amino acids 1–58) fused to the amino-terminus of the red fluorescence protein, mCherry were maintained in 3 μg/ml Blasticidin (Bioshop) in DMEM containing 10% FBS (Life Technologies). Heavy membranes containing mitochondria were isolated and assayed as described previously (Shamas-Din et al., 2014). Briefly, cells were lysed by nitrogen cavitation at 150psi for 10 min on ice in buffer containing 20mM HEPES (pH 7.2), 250 mM sucrose, 150 mM potassium chloride, 1 mM EDTA, 1 X protease inhibitor cocktail. Nuclei and cell debris were removed by centrifugation of the lystate at 2000g for 4 min at 4°C. Heavy membranes containing mitochondria were obtained by centrifuging the supernatant at 13000g for 10 min at 4°C and washing the pellet once in lysis buffer. Membrane fractions (0.2 mg/ml protein) were incubated with the compound, followed by the addition of 2 nM tBid and 20 nM Bax for the Bax–/–lBak–/–mitochondria, or 0.5 nM tBid for the Bax–/– mitochondria for 30 min at 37°C. Mitochondria were then pelleted by centrifugation at 13000 g for 10 min. The release of Smac-mCherry was assessed by fluorometric analysis of the supernatant and pellet fractions at excitation of 580 nm and emission of 610 nm in a Tecan M1000 microplate reader. The percentage release of Smac-mCherry was calculated as [Fsupernatant / (Fsupernatant + Fpellet )] x 100.
Mouse Liver Mitochondria
Mitochondria were isolated from livers of, Bak–/– or wt mice and frozen as described (Yamaguchi et al., 2007). Briefly, mouse livers were lysed by dounce homogenization in AT buffer containing 300 mM Trehalose, 10 mM HEPES-KOH (pH 7.5), 10 mM potassium chloride, 1 mM EGTA, 1 mM EDTA and 0.1% (w/v) BSA. Tissue components were removed by centrifugation at 600g for 10 min. To obtain heavy membranes containing mitochondria, the supernatant was centrifuged at 3500g for 15 min. The pellet was resuspended in AT buffer and centrifuged at 1500g for 5 min. The resulting supernatant was centrifuged at 5500g for 10 min (step performed twice). Mitochondrial fractions (50 mg/ml) were frozen at −80°C. When required, mitochondrial samples were thawed rapidly, washed in AT buffer containing 80 mM potassium chloride and resuspended in wash buffer supplemented with 5 mM succinate, 2 mM ATP, 10 μM phosphocreatine and 10 μg/ml creatine kinase.
To test inhibition of Bak by DAN004, mitochondria were isolated from wt mouse liver to remove endogenous Bax. After thawing, membrane fractions (1 mg/ml protein) were incubated with compound and 5 nM c Bid and pelleted as described above. The supernatant and pellet fractions were analyzed by immunoblotting using a cytochrome c antibody affinity purified in our laboratory. Immunoblots were developed using chemiluminescence and intensities were recorded using a CCD camera (DNR). Image analysis was conducted using ImageJ to quantify the release of cytochrome c.
To measure mitochondrial permeabilization for constitutively active Bak protein synthesized in SP6 RNA polymerase/reticulocyte lysate-based transcription and translation coupled system, 40 μM MSN-50 was added to 2 μl Bak prior to the addition of mitochondria and cytochrome c release was assayed by ELISA (Zhang et al., 2016). As a control, inhibition was measured after addition of 2 μl Bcl-XL synthesized in vitro instead of MSN-50.
Long Term Survival after Replating
For experiments with MSN-125 and MSN-50 male HCT-116 (Wang and Youle, 2012) and BMK cells (Mathew et al., 2008) of unspecified sex were seeded at a cell density of 3000 cells/well in a 96 well plate (Sarstedt). For experiments with DAN004 BMK cells were seeded at 750 cells/well in 384 well plates. Cells were plated in in DMEM (Life Technologies) containing 10% FBS (Sigma) and after adhering, cells were treated with the indicated concentration of the inhibitors for 3–4 hours followed by addition of actinomycin D (ActD) (Sigma) or staurosporine (STS) for another 4 hours. The ActD/STS (+ compound) containing media was then replaced with drug-free media (containing inhibitor) and cells were grown for 4 days (to confluency for DMSO controls) and re-plated into 24 or 96 well plates (Sarstedt) for MSN and DAN004 inhibitors, respectively. Surviving cells were allowed to grow until the DMSO controls on the same plate just reached confluency and then the plate was stained with crystal violet (Sigma). Images of the plates were obtained using a flat-bed scanner. For 96 well plates, quantification of surviving cells was done by absorbance measurements at 600 nm as described previously (Brahmbhatt et al., 2016).
Immunofluorescence
HCT-116 cells were pre-treated with 10 μM MSN-125 for three hours, followed by the addition of actinomycin D (50 ng/ml final) for 24 hours. Cells were fixed using paraformaldehyde and analyzed by immunofluorescence by double staining with primary sheep anti-cytochrome c antibody (Caprilogics) and mouse anti-Bax 6A7 monoclonal antibodies. Secondary donkey anti-sheep-Alexa Fluor 488 and goat anti-mouse-Alexa Fluor 555 (Invitrogen) antibodies were used for microscopy. The nuclei of HCT-116 cells were stained with DAPI according to the manufacturer’s instructions (Sigma). Cells were imaged using a Zeiss LSM710 confocal microscope and associated software.
Primary Neuronal Cultures
All animal procedures were performed in accordance with the local standards for animal care and were approved by the animal care committee at Sunnybrook Research Institute. To establish primary neuron cultures of mixed sex, cerebral cortices from male and female mouse embryos (embryonic day E14.5–15) were dissected and cultured for up to 10 days to ensure maturation of neurons as described (Mergenthaler et al., 2012). Neurons from one embryo were considered as one independent “n”. A maximum of 3 embryos of the same litter were used. Briefly, neurons were cultured in Neurobasal-A medium (Life Technologies) supplemented with B-27 (Life Technolgies), 0.5 mM L-glutamine and 25 μM glutamate. The medium was partially replaced on day 6 in culture with Neuro- basal-A supplemented with B-27 and L-glutamine. On day 9, neurons were treated with either 25 μM or 100 μM glutamate in BSS0 (116 mM NaCl, 5.4 mM KCl, 0.8 mM MgSO4, 1 mM NaH2PO4, 26.2 mM NaHCO3, 10 μM glycine, 1.8 mM CaCl2, 10 mM HEPES pH 7.4) for 30 minutes (37°C, 5% CO2) after washing the cultures with PBS. Medium was pooled and added to cultures after the incubation period with or without addition of 5 μM MSN-125. Cell death was analyzed 20–24 hours after glutamate treatment by measuring lactate dehydrogenase release as previously described (Mergenthaler et al., 2012). Briefly, lactate dehydrogenase concentration in medium was analyzed by measuring NADH to NAD+ turnover (absorbance at 340 nm) in a coupled spectrophotometric assay on aTecan M1000 microplate reader at 37°C and normalized to total lactate dehydrogenase levels after volume correction. For each measurement, 200 μl LDH buffer (33 mM KH2PO4, 66 mM K2HPO4) containing 210 μM β-NADH (Sigma), pH 7.4 were added to 50 μl media supernatant in a 96 well plate. Immediately before starting the measurements, 25 μl LDH buffer containing 22.7 mM sodium pyruvate were added. Total lactate dehydrogenase release was measured after incubating neuronal cultures with 0.5% TritonX-100 for 30 minutes (37° C). All measurements were normalized to control reactions containing 500 units/l l-lactic dehydrogenase (Sigma). Statistical analysis was performed in Prism 5.0 (GraphPad) or SPSS 23 (IBM). ANOVA was performed after normality testing.
Bax:Bax Bax:Membrane FRET Assay
Bax:Bax or Bax:Membrane binding was assessed in a PTI Quantamaster fluorimeter using the FRET assay described previously (Lovell et al., 2008). For Bax:Bax FRET measurements, compounds dissolved in DMSO or as controls equivalent volumes of DMSO were added after liposomes but prior to the proteins, thereby allowing correction for compound fluorescence. Liposomes were then incubated with DAC-134C-Bax and Initial fluorescence (F0) was recorded (excitation, 380 nm and emission, 460 nm). Decreased DAC fluorescence (F) due to FRET was measured after the addition of NBD-126C-Bax and tBid.
For Bax:membrane FRET measurements, 10 μM DAN004 was incubated with 20 nM of the donor-labeled protein, Alexa568–126C-Bax at 37°C for 10 minutes before adding 20 nM BIM and liposomes that were reconstituted in the presence of the acceptor fluorophore DiD (1,1’-Dioctadecyl-3,3,3’,3’-tetramethylindodicarbocyanine perchlorate; Life Technologies). DiD is incorporated into the reconstituted bilayer, and can be used as the acceptor for proteins binding membranes as previously described (Shamas-Din et al., 2013). FRET was measured by recording the initial fluorescence intensity (F0; excitation, 580 nm and emission, 595 nm) of Alexa568-Bax incubated with DAN004. The fluorescence intensity of Alexa568-Bax decreased upon addition of the acceptor, DiD incorporated liposomes (F) due to FRET.
Disuccinimidyl Suberate (DSS) Crosslinking
Samples containing liposomes (125 μM total lipids) were incubated with the compounds, 5 nM tBid and 100 nM Bax at 30°C for 2 hours in a low protein binding microtiter plate (Corning). DSS (Pierce) was added (2 mM final concentration) for 30min at room temperature followed by quenching with 20 mM Tris-Cl (pH 8) as described (Billen et al., 2008). Proteins were separated by SDS-PAGE and Bax was detected by immunoblotting with the monoclonal antibody 2D2 at a dilution of 1:10,000. Immunoblots were digitized by imaging with a CCD camera and the intensity of the dimer and larger oligomer bands was analyzed using ImageJ. To combine the data from multiple independent experiments the blots were normalized using the total intensity of dimer and larger oligomers in the “no compound” lane. Oneway ANOVA statistical test was performed in GraphPad Prism.
Disulfide Crosslinking
The single-cysteine Bax mutants were constructed and designated as described before (Zhang et al., 2016). The [35S]Met-labeled Bax or Bak mutant proteins were synthesized in either a wheat germ-based in vitro translation (IVT) system or a transcription/translation (TNT) coupled SP6 RNA polymerase/reticulocyte lysate system (Promega) as described (Zhang et al., 2016). After adding 20 μg/ml of cycloheximide to terminate translation, 15 or 7 μl of the resultant Bax or Bak proteins from the IVT or TNT system, respectively, were mixed with mitochondria containing 0.5 mg/ml total protein. Mitochondria purified from liver of mice lack Bax as it is a cytosolic protein in liver. Mitochondria were treated with 18 mM N-ethylmaleimide (NEM) to block the sulfhydryl groups of mitochondrial proteins, thereby preventing them from crosslinking with the cysteine(s) in Bax or Bak mutants via disulphides in the following oxidation reactions. In addition, 40 μM MSN-50 or MSN-125,1 mM DTT and for reactions containing Bax, 16 μM of a peptide containing residues 53–86 of Bax including the BH3-region (BH3-peptide) that activates Bax (Tan et al., 2006), were added and the samples were incubated at 37°C for 1 h. The resulting mitochondria and bound Bax or Bak were pelleted by centrifugation at 4°C and 13,000g for 5 min, and then resuspended in 100 μl buffer A (110 mM KOAc, 1 mM Mg(OAc)2and 25 mM Hepes, pH 7.5). The resulting samples were treated with 1 mM NaAsO4 to eliminate the residual DTT, and then divided into two aliquots. One aliquot was treated with redox catalyst copper (II) (1,10-phenanthroline)3 (CuPhe; consisting of 0.3 mM CuSO4and 1 mM 1,10-phenanthroline) to induce disulfide crosslinking. After incubation on ice for 30 min, the oxidation reactions were quenched with 18 mM NEM, 100 mM ethyl-enediaminetetraacetic acid (EDTA) and 1 mM NaAsO4. As the “0 min” control, the other aliquot was treated with NEM, EDTA and NaAsO4 to block the disulfide crosslinking prior to the addition of CuPhe. The resulting samples were precipitated with Cl3CCOOH, and then analyzed with non-reducing and as controls (not shown) reducing SDS-PAGE (15%), followed by phosphor-imaging to detect the radioactive Bax proteins and their disulphide-linked dimers. Intensities of both dimer and monomer bands in the phosphor-imaging data were measured and used to calculate the fraction of dimer. The fraction of dimer for each Bax or Bak sample in the absence or presence of a MSN compound was normalized to the mean fraction of dimer in the absence of the MSN compound from two to three independent experiments.
QUANTIFICATION AND STATISTICAL ANALYSIS
See individual sections above for details on the statistics used for analysis.
Supplementary Material
SIGNIFICANCE
Mitochondrial outer membrane permeabilization and subsequent cell death underlies several degenerative diseases including acute neurodegeneration in stroke and can be an undesired side effect of cancer therapy in normal tissues. MOMP is executed by the pro-apoptotic Bcl-2 protein family members Bax and Bak in the MOM, and is an irreversible step in committing a cell to die. We report the discovery of small-molecule inhibitors of both Bax and Bak oligomerization that promote cell survival after apoptotic stimuli and protect primary neurons from death after excitotoxicity. We demonstrate that these compounds prevent Bax and Bak oligomerization by partially disrupting the canonical BH3 peptide-groove interface and preventing interactions at subsequent interfaces and thereby establish that neither the Bax/Bak monomer nor the partially defective dimer is sufficient to permeabilize membranes and thus to execute MOMP. Our work thus provides novel insight into the Bax/ Bak-mediated regulation of MOMP as well as novel tools to dissect the mechanisms of Bax/Bak oligomerization leading to MOMP.
Highlights
Discovery of novel small-molecule inhibitors of Bax and Bak Xin Niu, Hetal Brahmbhatt, oligomerization
Compounds provide evidence that monomeric Bax/Bak is insufficient for MOMP
Compounds prevent Bax/Bak oligomerization at specific dimer sites
Compounds prevent cell death and mediate neuroprotection after excitotoxicity
ACKNOWLEDGMENTS
We thank M. Falcone for conducting the compound partitioning experiments and C. Griffiths for assistance in the screen. This work was supported by the Ontario Institute for Cancer Research through funding provided by theGovernment of Ontario, the Terry Fox Research Institute, the W. Garfield Weston Foundation – Brain Canada Foundation Multi-Investigator Research Initiative (MIRI), and Canadian Institutes of Health Research, Foundation Grant FDN143312 (D.W.A.), the National Cancer Institute of Canada and Department of Science and Technology (P.A.), the Shanghai Municipal Science and Technology Commission 10410704000 (J.Y. and D.W.A.), the Shanghai Municipal Education Commission J50201 (J.Y.), the European Union’s Seventh Framework Program (FP7/2008–2013) under Grant Agreement 627951 (Marie Curie IOF to P.M.), and the German Academic Exchange Service DAAD PPP Canada program with funds of the German Federal Ministry of Education and Research (grant 57212163 to P.M.), and the United States NIH grant GM062964 and the Oklahoma Center for the Advancement of Science and Technology grant HR16–026, and a Presbyterian Health Foundation seed grant GRF00000125 (J.L.). D.W.A. is a Tier 1 Canada Research Chair. P.M. is a fellow of the BIH Charité Clinician Scientist Program funded by the Charité Universitätsmedizin Berlin and the Berlin Institute of Health, and R.J. received a PhD fellowship from the Council of Scientific and Industrial Research.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes four figures, one table, and supplemental text and can be found with this article online at http://dx.doi.org/10.1016/j.chembiol.2017.03.011.
REFERENCES
- Andrews DW (2014). Pores of no return. Mol. Cell 56, 465–466. [DOI] [PubMed] [Google Scholar]
- Annis MG, Soucie EL, Dlugosz PJ, Cruz-Aguado JA, Penn LZ, Leber B, and Andrews DW (2005). Bax forms multispanning monomers that oligomerize to permeabilize membranes during apoptosis. EMBO J. 24, 2096–2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billen LP, Kokoski CL, Lovell JF, Leber B, and Andrews DW (2008). Bcl-XL inhibits membrane permeabilization by competing with Bax. PLoS Biol. 6, e147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogner C, Leber B, and Andrews DW (2010). Apoptosis: embedded in membranes. Curr. Opin. Cell Biol. 22, 845–851. [DOI] [PubMed] [Google Scholar]
- Bombrun A, Gerber P, Casi G, Terradillos O, Antonsson B, and Halazy S (2003). 3,6-Dibromocarbazole piperazine derivatives of 2-propanol as first inhibitors of cytochrome c release via Bax channel modulation. J. Med. Chem. 46, 4365–4368. [DOI] [PubMed] [Google Scholar]
- Brahmbhatt H, Uehling D, Al-Awar R, Leber B, and Andrews D (2016). Small molecules reveal an alternative mechanism of Bax activation. Biochem. J. 473, 1073–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christenson E, Merlin S, Saito M, and Schlesinger P (2008). Cholesterol effects on BAX pore activation. J. Mol. Biol. 381, 1168–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czabotar PE, Westphal D, Dewson G, Ma S, Hockings C, Fairlie WD, Lee EF, Yao S, Robin AY, Smith BJ, et al. (2013). Bax crystal structures reveal how BH3 domains activate Bax and nucleate its oligomerization to induce apoptosis. Cell 152, 519–531. [DOI] [PubMed] [Google Scholar]
- D’Orsi B, Bonner H, Tuffy LP, Dussmann H, Woods I, Courtney MJ, Ward MW, and Prehn JH (2012). Calpains are downstream effectors of bax-dependent excitotoxic apoptosis. J. Neurosci. 32, 1847–1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Orsi B, Kilbride SM, Chen G, Perez Alvarez S, Bonner HP, Pfeiffer S, Plesnila N, Engel T, Henshall DC, Dussmann H, et al. (2015). Bax regulates neuronal Ca2+ homeostasis. J. Neurosci. 35, 1706–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danial NN, and Korsmeyer SJ (2004). Cell death: critical control points. Cell 116, 205–219. [DOI] [PubMed] [Google Scholar]
- Degenhardt K, Sundararajan R, Lindsten T, Thompson C, and White E(2002). Bax and Bak independently promote cytochrome C release from mitochondria. J. Biol. Chem. 277, 14127–14134. [DOI] [PubMed] [Google Scholar]
- Delbridge AR, Valente LJ, and Strasser A (2012). The role of theapoptotic machinery in tumor suppression. Cold Spring Harb Perspect. Biol. 4, 10.1101/cshperspect.a008789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewson G, Kratina T, Sim HW, Puthalakath H, Adams JM, Colman PM, and Kluck RM (2008). To trigger apoptosis, Bak exposes its BH3 domain and homodimerizes via BH3:groove interactions. Mol. Cell 30, 369–380. [DOI] [PubMed] [Google Scholar]
- Dewson G, Ma S, Frederick P, Hockings C, Tan I, Kratina T, and Kluck RM (2012). Bax dimerizes via a symmetric BH3:groove interface during apoptosis. Cell Death Differ. 19, 661–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel T, Plesnila N, Prehn JH, and Henshall DC (2011). In vivo contributions of BH3-only proteins to neuronal death following seizures, ischemia, and traumatic brain injury. J. Cereb. Blood Flow Metab. 31, 1196–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fannjiang Y, Kim CH, Huganir RL, Zou S, Lindsten T, Thompson CB, Mito T, Traystman RJ, Larsen T, Griffin DE, et al. (2003). BAK alters neuronal excitability and can switch from anti- to pro-death function during postnatal development. Dev. Cell 4, 575–585. [DOI] [PubMed] [Google Scholar]
- Follis AV, Llambi F, Merritt P, Chipuk JE, Green DR, and Kriwacki RW (2015). Pin1-induced proline isomerization in cytosolic p53 mediates BAX activation and apoptosis. Mol. Cell 59, 677–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosse L, Wurm CA, Bruser C, Neumann D, Jans DC, and Jakobs S (2016). Bax assembles into large ring-like structures remodeling the mitochondrial outer membrane in apoptosis. EMBO J. 35, 402–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardwick JM, and Soane L (2013). Multiple functions of BCL-2 family proteins. Cold Spring Harb Perspect. Biol. 5, 10.1101/cshper-spect.a008722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz C, Vitte PA, Bombrun A, Rostovtseva TK, Montessuit S, Hiver A, Schwarz MK, Church DJ, Korsmeyer SJ, Martinou JC, et al. (2005). Bax channel inhibitors prevent mitochondrion-mediated apoptosis and protect neurons in a model of global brain ischemia. J. Biol. Chem. 280,42960–42970. [DOI] [PubMed] [Google Scholar]
- Hsu YT, and Youle RJ (1997). Nonionic detergents induce dimerization among members of the Bcl-2 family. J. Biol. Chem. 272, 13829–13834. [DOI] [PubMed] [Google Scholar]
- Hsu YT, and Youle RJ (1998). Bax in murinethymus isasoluble monomeric protein that displays differential detergent-induced conformations. J. Biol. Chem. 273, 10777–10783. [DOI] [PubMed] [Google Scholar]
- Iyer S, Bell F, Westphal D, Anwari K, Gulbis J, Smith BJ, Dewson G, and Kluck RM (2015). Bak apoptotic pores involve a flexible C-terminal region and juxtaposition of the C-terminal transmembrane domains. Cell Death Differ. 22, 1665–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakobson M, Lintulahti A, and Arumae U (2012). mRNA for N-Bak, a neuron-specific BH3-only splice isoform of Bak, escapes nonsense-mediated decay and is translationally repressed in the neurons. Cell Death Dis. 3, e269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang M, Pabla N, Murphy RF, Yang T, Yin XM, Degenhardt K, White E, and Dong Z (2007). Nutlin-3 protects kidney cells during cisplatin therapy by suppressing Bax/Bak activation. J. Biol. Chem. 282, 2636–2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushnareva Y, Andreyev AY, Kuwana T, and Newmeyer DD (2012). Bax activation initiates the assembly of a multimeric catalyst that facilitates Bax pore formation in mitochondrial outer membranes. PLoS Biol. 70, e1001394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leber B, Geng F, Kale J, and Andrews DW (2010). Drugstargeting Bcl-2 family members as an emerging strategy in cancer. Expert Rev. Mol. Med. 72, e28. [DOI] [PubMed] [Google Scholar]
- Liao C, Zhang Z, Kale J, Andrews DW, Lin J, and Li J (2016). Conformational heterogeneity of Bax helix 9 dimer for apoptotic pore formation. Sci. Rep. 6, 29502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovell JF, Billen LP, Bindner S, Shamas-Din A, Fradin C, Leber B, and Andrews DW (2008). Membrane binding by tBid initiates an ordered series of events culminating in membrane permeabilization by Bax. Cell 735, 1074–1084. [DOI] [PubMed] [Google Scholar]
- Ma S, Hockings C, Anwari K, Kratina T, Fennell S, Lazarou M, Ryan MT, Kluck RM, and Dewson G (2013). Assembly of the Bak apoptotic pore: a critical role for the Bak protein alpha6 helix in the multimerization of homodimers during apoptosis. J. Biol. Chem. 288, 26027–26038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew R, Degenhardt K, Haramaty L, Karp CM, and White E (2008). Immortalized mouse epithelial cell models to study the role of apoptosis in cancer. Methods Enzymol. 446, 77–106. [DOI] [PubMed] [Google Scholar]
- Mergenthaler P, Dirnagl U, and Meisel A (2004). Pathophysiology ofstroke: lessons from animal models. Metab. Brain Dis. 79, 151–167. [DOI] [PubMed] [Google Scholar]
- Mergenthaler P, Kahl A, Kamitz A, van Laak V, Stohlmann K, Thomsen S, Klawitter H, Przesdzing I, Neeb L, Freyer D, et al. (2012). Mitochondrial hexokinase II (HKII) and phosphoprotein enriched in astrocytes(PEA15) form a molecular switch governing cellular fate depending on the metabolic state. Proc. Natl. Acad. Sci. USA 709, 1518–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikhailov V, Mikhailova M, Degenhardt K, Venkatachalam MA, White E, and Saikumar P (2003). Association of Bax and Bak homo-oligomers in mitochondria. Bax requirement for Bak reorganization and cytochrome c release. J. Biol. Chem. 278, 5367–5376. [DOI] [PubMed] [Google Scholar]
- Moldoveanu T, Follis AV, Kriwacki RW, and Green DR (2014). Many players in BCL-2 family affairs. Trends Biochem. Sci. 39, 101–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peixoto PM, Ryu SY, Bombrun A, Antonsson B, and Kinnally KW (2009). MAC inhibitors suppress mitochondrial apoptosis. Biochem. J. 423, 381–387. [DOI] [PubMed] [Google Scholar]
- Peixoto PM, Teijido O, Mirzalieva O, Dejean LM, Pavlov EV, Antonsson B, and Kinnally KW (2015). MAC inhibitors antagonize the pro- apoptotic effects of tBid and disassemble Bax/Bak oligomers. J. Bioenerg. Biomembr. 49, 65–74. [DOI] [PubMed] [Google Scholar]
- Polster BM, Basanez G, Young M, Suzuki M, and Fiskum G (2003). Inhibition of Bax-induced cytochrome c release from neural cell and brain mitochondria by dibucaine and propranolol. J. Neurosci. 23, 2735–2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakesch M, Denisov AY, Naim M, Gehring K, and Arya P (2008). The discovery of small molecule chemical probes of Bcl-X(L) and Mcl-1. Bioorg. Med. Chem. 76, 7443–7449. [DOI] [PubMed] [Google Scholar]
- Putcha GV, Harris CA, Moulder KL, Easton RM, Thompson CB, and Johnson EM Jr. (2002). Intrinsic and extrinsic pathway signaling during neuronal apoptosis: lessons from the analysis of mutant mice. J. Cell Biol. 757, 441–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvador-Gallego R, Mund M, Cosentino K, Schneider J, Unsay J, Schraermeyer U, Engelhardt J, Ries J, and Garcia-Saez AJ (2016). Bax assembly into rings and arcs in apoptotic mitochondria is linked to membrane pores. EMBO J. 35, 389–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarosiek KA, Chi X, Bachman JA, Sims JJ, Montero J, Patel L, Flanagan A,Andrews DW, Sorger P, and Letai A (2013). BID preferentially activates BAK while BIM preferentially activates BAX, affecting chemotherapy response. Mol. Cell 57, 751–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satsoura D, Kucerka N, Shivakumar S, Pencer J, Griffiths C, Leber B, Andrews DW, Katsaras J, and Fradin C (2012). Interactionofthefull-length Bax protein with biomimetic mitochondrial liposomes: a small-angle neutron scattering and fluorescence study. Biochim. Biophys. Acta 1818, 384–401. [DOI] [PubMed] [Google Scholar]
- Shamas-Din A, Bindner S, Zhu W, Zaltsman Y, Campbell C, Gross A, Leber B, Andrews DW, and Fradin C (2013). tBid undergoes multiple conformational changes at the membrane required for Bax activation. J. Biol. Chem. 288, 22111–22127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamas-Din A, Satsoura D, Khan O, Zhu W, Leber B, Fradin C, and Andrews DW (2014). Multiple partners can kiss-and-run: Bax transfers between multiple membranes and permeabilizes those primed by tBid. Cell Death Dis. 5, e1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subburaj Y, Cosentino K, Axmann M, Pedrueza-Villalmanzo E, Hermann E, Bleicken S, Spatz J, and Garcia-Saez AJ (2015). Bax monomers form dimer units inthe membranethat furtherself-assemble into multiple oligomeric species. Nat. Commun. 6, 8042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun YF, Yu LY, Saarma M, Timmusk T, and Arumae U (2001). Neuron- specific Bcl-2 homology 3 domain-only splice variant of Bak is anti-apoptotic in neurons, but pro-apoptotic in non-neuronal cells. J. Biol. Chem. 276, 16240–16247. [DOI] [PubMed] [Google Scholar]
- Szeto HH (2008). Mitochondria-targeted cytoprotective peptides for ischemia-reperfusion injury. Antioxid. Redox Signal. 70, 601–619. [DOI] [PubMed] [Google Scholar]
- Tan C, Dlugosz PJ, Peng J, Zhang Z, Lapolla SM, Plafker SM, Andrews DW, and Lin J (2006). Auto-activation of the apoptosis protein Bax increases mitochondrial membrane permeability and is inhibited by Bcl-2. J. Biol. Chem. 287, 14764–14775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovar C, Rosinski J, Filipovic Z, Higgins B, Kolinsky K, Hilton H, Zhao X, Vu BT, Qing W, Packman K, et al. (2006). Small-molecule MDM2 antagonists reveal aberrant p53 signaling in cancer: implications for therapy. Proc. Natl. Acad. Sci. USA 703, 1888–1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uo T, Kinoshita Y, and Morrison RS (2005). Neurons exclusively express N-Bak, a BH3 domain-only Bak isoform that promotes neuronal apoptosis. J. Biol. Chem. 280, 9065–9073. [DOI] [PubMed] [Google Scholar]
- Uren RT, O’Hely M, Iyer S, Bartolo R, Shi MX, Brouwer JM, Alsop AE, Dewson G, and Kluck RM (2017). Disordered clusters of Bak dimers rupture mitochondria during apoptosis. eLife 6, 10.7554/eLife.19944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, and Youle RJ (2012). Predominant requirement of Bax for apoptosis in HCT116 cells is determined by Mcl-1’s inhibitory effect on Bak. Oncogene 37, 3177–3189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis SN, Fletcher JI, Kaufmann T, van Delft MF, Chen L, Czabotar PE, Ierino H, Lee EF, Fairlie WD, Bouillet P, et al. (2007). Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science 375, 856–859. [DOI] [PubMed] [Google Scholar]
- Wilson-Annan J, O’Reilly LA, Crawford SA, Hausmann G, Beaumont JG, Parma LP, Chen L, Lackmann M, Lithgow T, Hinds MG, et al. (2003). Proapoptotic BH3-only proteins trigger membrane integration of pro-survival Bcl-w and neutralize its activity. J. Cell Biol. 762, 877–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu XP, Zhai D, Kim E, Swift M, Reed JC, Volkmann N, and Hanein D (2013). Three-dimensional structure ofBax-mediated pores in membrane bilayers. Cell Death Dis. 4, e683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi R, Andreyev A, Murphy AN, Perkins GA, Ellisman MH, and Newmeyer DD (2007). Mitochondria frozen with trehalose retain a number of biological functions and preserve outer membrane integrity. Cell Death Differ. 74, 616–624. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Zhu W, Lapolla SM, Miao Y, Shao Y, Falcone M, Boreham D, McFarlane N, Ding J, Johnson AE, et al. (2010). Baxforms an oligomer via separate, yet interdependent, surfaces. J. Biol. Chem. 285, 17614–17627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Subramaniam S, Kale J, Liao C, Huang B, Brahmbhatt H, Condon SG, Lapolla SM, Hays FA, Ding J, et al. (2016). BH3-in-groove dimerization initiates and helix 9 dimerization expands Bax pore assembly in membranes. EMBO J. 35, 208–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.