

Abstract
Synaptic plasticity contributes to behavioral adaptations. As a key node in the reward pathway, the nucleus accumbens (NAc) is important for determining motivation-to-action outcomes. Across animal models of motivation including addiction, depression, anxiety, and hedonic feeding, selective recruitment of neuromodulatory signals and plasticity mechanisms have been a focus of physiologists and behaviorists alike. Experience-dependent plasticity mechanisms within the NAc vary depending on the distinct afferents and cell-types over time. A greater understanding of molecular mechanisms determining how these changes in synaptic strength track with behavioral adaptations will provide insight into the process of learning and memory along with identifying maladaptations underlying pathological behavior. Here, we summarize recent findings detailing how changes in NAc synaptic strength and mechanisms of plasticity manifest in various models of motivational disorders.
Keywords: Nucleus accumbens, plasticity, glia, glutamate, serotonin, opioids
Graphical Abstract
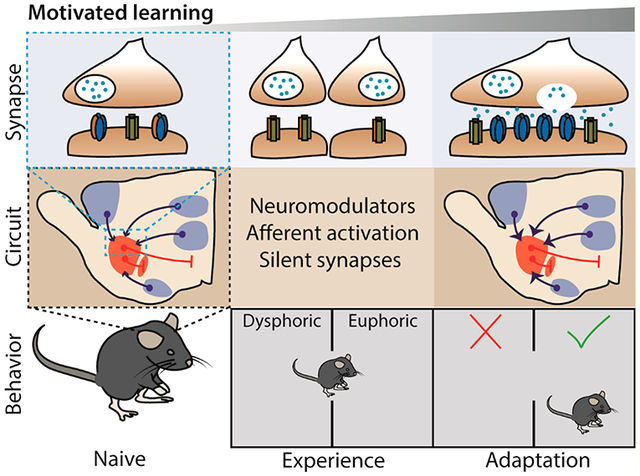
INTRODUCTION
The nucleus accumbens (NAc) is fundamental in driving goal-directed actions, integrating excitatory (glutamatergic) and neuromodulatory input along with local inhibitory control to optimize motivated behavioral outcomes. Long-term changes in synaptic strength within the NAc underlies experience-dependent neural plasticity.1,2 These synaptic adaptations include intricate molecular epigenetic, biochemical, electrophysiological, and morphological changes in individual neurons, ultimately reshaping synaptic function.3
Fast excitatory synaptic transmission occurs through postsynaptic α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and N-methyl-D-aspartate (NMDA) ionotropic glutamate receptors. AMPA receptors are the primary contributor to excitatory synaptic transmission. Their trafficking in and out of the membrane is paramount to the process of postsynaptic plasticity. NMDA receptors, as well as metabotropic glutamate (mGlu) and other G-protein-coupled receptor (GPCRs), can initiate signaling cascades, affecting AMPA receptor surface expression and subunit composition throughout reward learning in an experience-dependent and temporally dynamic manner.4–10 Many of these have been correlated or causally linked to motivational phenotypes in numerous models of developmental and psychiatric disorders. Maladaptive behaviors and the observed corresponding changes in NAc synaptic physiology are particularly well understood in models of addiction,4,11,12 stress and depression,13–19 but are also hallmarks of eating disorders,20 schizophrenia,21,22 pain perception23 and autism spectrum disorders.24,25 Because of the various contexts in which NAc synaptic plasticity is examined, creating a comprehensive model of the many plasticity mechanisms within this region has been elusive.
Here, we summarize mechanisms known to reshape NAc excitatory synaptic transmission and how they are altered in model systems of psychiatric disorders. These include glutamate-mediated synaptic plasticity, signaling via serotonin, opioids, and endocannabinoids, as well as glial and astrocytic synaptic interactions. This review highlights synaptic remodeling events that contribute to reward learning in healthy organisms and how these processes may serve as therapeutic targets for treatment of pathophysiologies underlying motivational disorders.
ANATOMY OF THE NAc
As a key component of the mesolimbic dopamine (DA) system, the NAc is a functional interface between the limbic and motor systems responsible for bringing motivation to action.26 The NAc is a part of the ventral striatum composed of shell and core subregions, which are thought to govern immediate responding to salient stimuli and conditioned reinforcement, respectively.1 The NAc is predominantly (~90%) made up of GABAergic medium spiny neurons (MSNs).27,28 MSNs, the output cells of the NAc, can be separated into one of two circuits distinguished by molecular, electrophysiological and anatomical properties.29,30 Herein, we will identify the MSN subtypes based on their expression of the type-1 or type-2 dopamine receptors (D1 and D2MSNs, respectively),31 in which D1MSNs largely project to the midbrain, while D2MSNs project to the ventral pallidum.32 However, it should be noted that this dichotomy is not as specific as the dorsal striatum as D1MSNs can also project to pallidal brain regions.30 Recruitment of D1 or D2MSNs has seemingly opposing effects on behavior: activation and activity of D1MSNs corresponds with an increase in reward seeking and locomotion while activation of D2MSNs promotes goal switching, catalepsy, and aversion.33–35 However, recent findings using in vivo calcium imaging in the NAc and dorsal striatum indicate that these cells act in concert to drive motivated behaviors.36,37 Importantly, NAc MSNs are quiescent cells that rely on concerted excitatory drive from multiple glutamatergic afferents to elicit action potential generation, propagating information flow through the NAc circuit. Therefore, the strength and activity of these glutamatergic synapses determines the likelihood of afferent information being transformed to postsynaptic action potential propagation, making them vital nodes in defining overall circuit function.
Alterations in glutamatergic transmission engenders the integrative role the NAc plays in directing behavior. Glutamatergic brain regions that project to the NAc, such as medial prefrontal cortex (PFC; cognitive processing in goal-directed behavior), basolateral amygdala (BLA; conditioning forms of learning including processing of positive and negative emotions), ventral subiculum of the hippocampus (Hipp; contextual learning), and the dorsomedial thalamus (DMT; aversion, attention shifting), as well as corelease of glutamate from midbrain dopamine regions,38–40 are thought to encode salient information pertaining to proprioceptive self-assessments and externally available stimuli.1,27,41–44 By adjusting the strength of inputs from these afferent regions, the NAc is able to transform emotional and environmental information into action.
NEUROMODULATORY SIGNALS DIRECT NAc CIRCUIT FUNCTION
Glutamate.
The strength of an afferent-MSN connection depends upon the number of release sites or synapses, the probability of vesicular release, and quantal size as determined by post synaptic receptor availability. In ex vivo electro-physiology studies, much of the observed changes are via modifications to quantal size by modifying the synaptic AMPA receptor population or by alteration in release probability. Comparisons of current amplitude fluxed through AMPA and NMDA receptors, referred to as an AMPA/NMDA ratio, is a common metric for examining differences in synaptic strength across slices and conditions. This metric is often accompanied by direct measurement of quantal AMPA currents in the presence of tetrodotoxin (miniature EPSCs; mEPSCs) or replacing Ca2+ with strontium to evoke asynchronous EPSCs as a means to examine synaptic AMPA receptor populations. Additional analyses of isolated AMPA or NMDA receptor currents, including decay kinetics, current−voltage relationships, and coefficient of variation also provide insight into receptor subunit expression.
Synaptic plasticity of glutamatergic synapses can be initiated by numerous neurotransmitters, including glutamate itself. NMDA receptors are both ligand- and voltage-gated channels that act as coincidence detectors in the synapse.45 Entry of the second messenger Ca2+ through these receptors directs synaptic remodeling to strengthen or weaken future synaptic events.46,47 In the NAc, NMDA signaling has been shown repeatedly to induce long-term depression (LTD) of synaptic transmission reducing postsynaptic AMPA surface expression and/or function (Figure 1C). Long-term potentiation (LTP) in the NAc is developmentally regulated48 and is sensitive to drug history.49 NMDA activation is known to trigger LTP and LTD via signaling through ERK, PKC, or coupling to CaMKII.4,5,47 In ex vivo slice preparations, LTP and LTD can be evoked in the NAc by stimulation of glutamate release at high (100 Hz)48–51 and low (1−13 Hz) frequencies, respectively.49,50,52–55 LTP/LTD induction is often mirrored by bidirectional postsynaptic trafficking of AMPA receptors mediated in part by changes in scaffolding protein association and phosphorylation state.7,51,56 Transport of AMPA receptors and other proteins into the post synaptic density following LTP results in a restructuring of the synaptic spines.57–60
Figure 1.

mGlu and NMDA plasticity mechanisms coordinate pre- and postsynaptic function of NAc glutamatergic synapses. (A) Extrasynaptic glutamate homeostasis couples to group II mGlu activation. Extra-synaptic glutamate is tightly regulated by astrocytic cysteine-glutamate antiporter (xCT). High extracellular glutamate activates group II mGlu receptors to decrease presynaptic release. (B) Postsynaptic activation of group-1 mGlu receptors is known to recruit PLC to generate IP3 and DAG. DAG can be further cleaved by DAGL to a free fatty acid and 2-arachidonyl glycerol (2AG), which can signal to presynaptic CB1Rs. Activation of CB1Rs can act via inhibition of VGCCs and/or activation of presynaptic potassium channels to decrease vesicular release. Additionally, activation group-1 mGlu receptors can also induce a calcium dependent synthesis of anandamide (AEA), which likewise acts on CB1Rs but also activates TRPV1 channels. Activation of TRPV1 at the membrane or on the ER induces a dynamin-dependent internalization of AMPA receptors. (C) NMDA dependent LTD and LTP. Endogenous glutamate/glycine binding and concurrent depolarization activates NMDA receptors allowing an influx of calcium which can couple to downstream phosphatase/kinase cascades. These likely include CamKII and calcineurin, which can phosphorylate/dephosphorylate AMPA receptors, respectively. This contributes to their insertion or removal from the postsynaptic density. However, this mechanism is not well-defined in the NAc.
Both group-I (mGlu1/5) and group-II (mGlu2/3) metabotropic glutamate receptors are coupled to numerous signaling cascades that can exert pre- and postsynaptic effects.61–63 Postsynaptic group-I mGlu receptors are Gq-coupled GPCRs that can initiate AMPA internalization54,64,65 and/or an mGlu5 specific Ca2+-dependent endocannabinoid (eCB) production in NAc MSNs (Figure 1B). eCBs can signal to presynaptic cannabinoid type-1 receptors (CB1Rs)52,65 or postsynaptic TRPV1 receptors.54 Glutamate spillover following repeated vesicular fusion or glial-mediated release via cysteine-glutamate exchanger (xCT) and glial glutamate transporter (GLT-1) can also recruit presynaptic group-II receptors, which are Gi/o-coupled and decrease vesicular release probability (Figure 1A).66–68
Serotonin.
The NAc receives extensive inputs from the dorsal raphe nucleus (DRN), a mesencephalic structure rich in serotonin (5-HT)-containing perikarya.69 Consistent with the appositional relationship between 5-HT fibers and afferent synaptic inputs,70 5-HT has been shown to induce LTD of excitatory synaptic strength onto MSNs.71–74 This form of LTD (5-HT-LTD) is expressed at a presynaptic locus and mediated predominately via the 5-HT1B receptor, a Gi/o-coupled GPCR implicated in reward-related behavior (Figure 2). Low frequency stimulation (LFS) has also been shown to trigger 5-HT-LTD in a CB1R-dependent manner,71,75 indicating eCBs are downstream of 5-HT signaling and may function cooperatively to regulate NAc circuit dynamics.
Figure 2.

Oxytocin gates serotonergic LTD in the NAc. Oxytocin release from paraventricular nucleus (PVN) terminals drives release of 5-HT from dorsal raphe (DRN) afferents. 5-HT in the NAc may act on either pre or postsynaptic 5HT-1B receptors, which can either directly inhibit neurotransmitter release or indirectly through eCB signaling. Beyond several isolated studies, how 5-HT modifies NAc excitatory transmission is unknown.
The 5-HT1B receptor also mediates oxytocin (OT)-induced synaptic adaptations in the NAc. OT is a neuropeptide implicated in neuropsychiatric conditions featuring maladaptive social behavior, including autism and schizophrenia.73 Ex vivo bath-application of OT induces robust LTD of EPSCs onto D1 and D2MSNs in the NAc that is blocked by NAS-181, a selective 5-HT1B receptor antagonist.73 These data indicate that OT-mediated 5-HT release in the NAc triggers a form of presynaptic LTD that is required for social reward behavior.
Opioids.
Opioids are a widely expressed peptidergic modulatory system affecting neuronal function and circuity dynamics. The opioid system consists of four receptor subtypes (mu, delta, kappa, and opioid receptor like-1) and three endogenous ligands (endorphin, enkephalin, and dynorphin) with varying degrees of ligand specificity and expression patterns.76 Within the NAc, D1 and D2MSNs express endogenous opioids in a similarly dichotomous manner: D1MSNs primarily express dynorphin (Dyn) and D2MSNs express enkephalin (Enk), with striatal systems being largely described to lack beta-endorphin.77 However, opioid receptors (ORs) are broadly expressed and how they regulate excitatory transmission in the NAc remains obscure.78–80
Activation of mu, delta, or kappa ORs can drive “liking” or “wanting” behavioral outcomes in a manner dependent on NAc subregion.81 While in vivo studies have been abundant, less is known how these receptors control synaptic transmission. In the dorsal striatum, where MSNs are more discretely subdivided anatomically into a “patch” and “matrix” framework, mu ORs are found uniquely expressed in patches and are activated by enkephalin to decrease microcircuit inhibition and promote MSN activation.82 In the NAc, activation of mu ORs decreases NMDA and AMPA receptor currents with little effect on membrane properties83 with functional expression both preand postsynaptically (Figure 3).78 It has been demonstrated mu ORs exert strong control over thalamic but not motor cortex inputs into the dorsal striatum,84 but it is unclear whether NAc MSNs are under similar mu OR control. These findings suggest mu-OR signaling in the NAc may similarly be separable by afferent origin and should be investigated accordingly.
Figure 3.

Endogenous opioids regulate synaptic transmission in the NAc. Opioid receptors are expressed widely on glutamatergic terminals and cell bodies within the NAc. Mu-ORs likely function presynaptically and reduce release probability. Kappa-ORs are expressed on glutamatergic afferents (excluding the vHipp) and inhibit neurotransmitter release, particularly onto D1 (red) MSNs. Kappa-ORs are activated by Dyn which is produced locally by D1MSNs. Dyn can also inhibit dopamine release by acting on VTA terminals kappa-ORs. Activation of D1 receptors promotes prodynorphin expression, serving to inhibit glutamatergic drive onto these MSNs. While Dyn and Enk are produced by NAc MSNs, opioid peptides can also be released from hypothalamic projections from the Arcuate nucleus and the Lateral Hypothalamus.
A more recent report demonstrated that stimulation of Dyn+ NAc MSNs drives both reward and aversion via stimulation of the dorsal and ventral NAcSh, respectively, and is dependent on kappa OR signaling.85 Kappa ORs specifically decrease excitatory drive of BLA but not VH inputs onto D1MSNs and decrease inhibitory drive onto D2MSNs. Thus, kappa OR activation within the NAc results in increased transmission from the VH and BLA through D1MSN activation.86 It should be noted that kappa-OR signaling can also inhibit DA signaling by inhibiting VTA terminals.87 Beyond these few reports, mechanistic details of OR function in the NAc are largely unexplored.
Endocannabinoids.
The cannabinoid 1 receptor (CB1R) is implicated in substance abuse disorders.88–91 CB1Rs are the most abundant G-protein coupled receptor in the CNS and are localized mainly at presynaptic glutamate and GABA terminals in select neuronal populations.92 CB1Rs are activated by Delta-9-tetrahydrocannabinol, (Δ9-THC), the main psychoactive substance in Cannabis sativa. Endogenous cannabinoids are produced via postsynaptic de novo synthesis with subsequent release and retrograde activation of presynaptic CB1Rs. In the NAc, stimulation of mGlu5 receptors leads to a rise in postsynaptic calcium. This in turn leads to retrograde signaling through eCB release and activation of presynaptic CB1 receptors. Activation of CB1Rs reduces neurotransmitter release by decreasing release probability in a presynaptic K+ channel dependent manner (Figure 1).52,54,93–95 Contrary to the dorsal striatum, CB1Rs are not expressed in NAc MSNs but are expressed by NAc fast-spiking interneurons and on glutamatergic terminals.96
2-Arachidonylglycerol (2-AG) is the primary eCB mediating retrograde eCB signaling and is synthesized from diacylglycerol precursors by diacylglycerol lipaseα (DAGLα) in the adult brain. The eCB anandamide (AEA), in addition to CB1R activation, can also activate TRPV1 channels.97–100 TRPV1 is a nonselective cation channel that is highly permeable to calcium and is activated by acidic pH, high temperature and specific lipid species.97,98 TRPV1 function is commonly associated with presynaptic mechanisms including a form of LTD triggered by postsynaptic group I mGlu receptors at excitatory synapses on interneurons in the hippocampus.101 However, TRPV1 activation can also act postsynaptically to induce depression of excitatory synapses in the NAc core.54 This adds to the eCB system’s canonical role in regulating presynaptic release and positions it as a versatile modulator of NAc circuit function.
Glial Regulation of Drug-Reward Learning.
In addition to neuron-centric mechanisms of synaptic and behavioral plasticity, a growing body of research points to the importance of glia and the immune system. Specifically, microglia and astrocytes are increasingly found to play active roles in sculpting synaptic physiology and behavior. Microglia are the brain’s resident macrophage.102 These cells make up 10% of the brain parenchyma103 and play a key role in mediating immune responses in this region.104 Microglia play an important role in development, learning, and brain homeostasis105 by refining learning-induced spine formation as well as synaptic pruning (phagocytosis).106,107 In the context of drug-reward learning, the function of these cells appears complex and sometimes contradictory.
Importantly, microglia also influence synaptic function in the NAc. Microglia in the NAc express toll like receptor 4 (TLR4),108,109 a pattern-recognition receptor of the innate immune system that detects bacterial lipopolysaccharide110 and endogenous “danger signals” such as those produced during an inflammatory response.111 TLR4 knockout mice lack NMDA-dependent LTD in NAc core linking the immune system with synaptic plasticity.109 Beyond TLR4, microglia play a role NAc synaptic physiology and may mediate aspects of drug reward susceptibility. Tumor necrosis factor alpha (TNFα) is a proinflammatory cytokine upregulated in many conditions including after activation of TLR4.110 Microglial TNFα decreases synaptic strength as measured by AMPA/NMDA ratios the NAc D1MSNs to oppose synaptic and behavioral changes brought about with noncontingent cocaine exposure.112 These findings provide compelling evidence for the immune system facilitating and perhaps driving adaptations in NAc excitatory transmission.
Besides microglia, astrocytes play major roles in sculpting physiology and behavior.113 In the NAc, astrocytes are capable of regulating the concentration of extrasynaptic glutamate via the cysteine-glutamate exchanger (catalytic subunit = xCT) and GLT-1, which regulate extracellular glutamate levels. GLT- 1 is expressed on astrocytes and is responsible for glutamate uptake. Alterations in GLT-1 function can thus have profound impact on synaptic glutamate signaling.68,114 N-Acetylcysteine, which stimulates xCT, bidirectionally regulates EPSC amplitude in NAc MSNs; low doses (0.5 μM) decreases presynaptic release probability in a group-II mGlu dependent manner while high doses (50 μM) increase EPSC amplitude in via mGlu5 activation.115 The increase in extracellular glutamate acts on neuronal presynaptic mGlu2/3 to decrease vesicular release probability.10
EXPERIENCE RESHAPES NAc SYNAPSES AND PLASTICITY MECHANISMS
Experimentally, acute slice physiology has been instrumental in elucidating mechanisms of synaptic plasticity in the NAc. Importantly, in vivo experience can also drive new synapse formation, strengthen or weaken select afferent inputs, and impede or enhance molecular plasticity mechanisms. Such stimuli include those used in models of motivated appetitive behaviors, anxiety, and depression.116 From the seminal work of Thomas et al., which defined a correlational change in NAc MSN synaptic strength following cocaine exposure, investigating adaptations in synaptic function in acute slices following in vivo experience has led to developments in recent years showing a causal effect of synaptic plasticity and altered behavioral outcomes.49,55,117–121 As addressed below, this powerful approach has repeatedly demonstrated a functional relationship between glutamatergic synaptic strength and behavioral plasticity. As such, NAc synaptic plasticity has become nearly inseparable from questions interrogating reward and motivation. By focusing on the plasticity mechanisms within the NAc rather than the various psychiatric disease models, we aim to elucidate common mechanisms by which in vivo experiences drive change in the NAc circuit.
AMPA Receptor Expression and Function Coincides with in Vivo Experience.
Expression and function of AMPA and NMDA receptors in the NAc are strongly associated with experience-dependent behavioral plasticity, particularly in drug abuse models.43,60 Thomas et al. demonstrated NAc shell MSNs have a reduced AMPA/NMDA ratio following repeated drug exposure and is concurrent with a reduction in NMDA-dependent LTD.122 This phenomenon was then shown to be mediated by the challenge dose of cocaine/saline administered prior to the recording.123 Thus, AMPA/NMDA ratios are decreased immediately following drug, but are strengthened following a short abstinence period and can be reduced again with re-exposure.124 These findings demonstrated a temporal restructuring of glutamatergic signaling within the NAc following salient experience. Similar results with calcium-permeable AMPA receptors (CP-AMPA) have been demonstrated following drug self-administration, leading AMPA- receptor expression and function to be thought of as a neural correlate of incubation of drug craving.119 However, it should be noted that the contingency of drug delivery determines the type of remodeling seen in the NAc with respect to AMPA subunit composition125 but both favor an increase in glutamatergic drive.
Synapse maturation is a developmental process underlying neural circuit formation and is considered a critical physiological substrate for learning and memory.126–128 In the NAc, the relative abundance of silent or AMPA receptor deficient synapses is increased following acute cocaine self-administration (Figure 4A). These nascent synapses are short-lived and mature over time via insertion GluA2-lacking CP-AMPA receptors (Figure 4B).119,125–127,129 Maturation occurs following several weeks after drug withdrawal and requires PSD95 and SAP102 MAGUK proteins.130 The generation of CPAMPA containing synapses is correlated with incubation of drug seeking in self-administration models131 and is not normally seen following noncontingent drug administration. However, recent work has demonstrated increases in AMPA rectification, a measurement CP-AMPA expression, following repeated noncontingent exposure. Short access to cocaine self-administration drove CP-AMPA expression at PFC-D1 synapses, while long-access, presumably resulting in enhanced negative withdrawal symptoms, drove CP-AMPA expression at D2MSNs specifically at BLA synapses.132 Additionally, some synapses, such as those from the DMT-NAc, are reported to contain a high density of CP-AMPA at baseline which is unaffected by drug history. However, the formation and maturation of silent synapses is seen at this input, suggesting maturation may proceed by a non CP-AMPA mechanism.133 Notably, increases in mature spine number are also seen in noncontingent exposure paradigms and are specific for D1MSNs.134,135
Figure 4.

Experience drives plasticity of NAc glutamatergic transmission in vivo. (A) Following salient experience, nascent “silent” synapses are formed in the NAc, lacking functional AMPA receptors but expressing high concentrations of GluN2B-NMDA receptors. (B) As these synapses mature, they are under the control of mGlu and NMDA-dependent plasticity mechanisms. These mechanisms are extensively observed in mature, experience-naïve animals. (C) Following extensive abstinence from the initial salient experience, such as chronic stress, drug self-administration, or after acute removal from highly palatable chow, GluA2-lacking AMPA receptors are more abundant in the postsynaptic density. In cocaine-specific contexts, GluN2C/D NMDA receptors are found in a higher concentration in a subset of synapses. Broadly, group-1 mGlu-dependent eCB signaling is decreased and lowered concentrations of extra-synaptic glutamate stemming from astrocytes leads to decreased group-II mGlu receptor inhibition of vesicular release. Additionally, NMDA and group-1 mGlu activation favor internalization of AMPA receptors.
While both NMDA and mGlu plasticity described above are initiated through local signals within the dendritic spine, transcription/translational changes are required to maintain the effect.17,136 Such changes include altered expression of Homer1a, CREB, and ΔfosB.29,126,137 Salient experience is also coupled to upregulation of transcription factors in the NAc that can alter AMPA/NMDA expression. Two well studied transcription factors, CREB and ΔfosB, are recruited following cocaine exposure and are sufficient to drive changes in synaptic transmission. CREB is expressed following salient experience and can drive behavioral responding to both aversive and rewarding stimuli.138 Cocaine-induced or viral-mediated over-expression of CREB alters membrane and synaptic properties of NAc MSNs.126,139 Likewise, ΔfosB is upregulated in the NAc following exposure to abused drugs140,141 and is associated with behavioral adaptations tied to addiction. Interestingly, over-expression ΔfosB in the NAc “silences” D1 synapses in the shell and core but may unsilence D2MSN synapses via AMPA insertion in the NAc shell. Notably, ΔfosB promotes the expression of GluA2 as well as CaMKII, and these effects are restricted to D1MSNs in the NAc.142–144 As such, ΔfosB is positioned as a critical transcriptional regulator of cocaine-induced synaptic adaptations in the NAc.
Alterations in NAc glutamatergic transmission are not limited to drug-contexts. Interestingly, appearance of mature CPAMPA containing synapses is also observed days after removing animals from a highly palatable “junk-food” diet (Figure 4C),145 suggesting palatable food and “natural” rewards may be a more potent driver of this adaptation. This may serve to increase appetitive drive for palatable food, as inhibiting glutamatergic transmission via intra-accumbens infusion of CNQX, an AMPA receptor antagonist, stimulates voracious feeding behavior.146
Additionally, models of depression and anxiety induced by stressors also drive remodeling of NAc glutamate synapses.17 Chronic restraint stress has been shown to impair the induction of LTD within the NAc core via an MC4R-dependent signaling cascade.13 This is mediated by a selective internalization of GluA2-containing receptors resulting in an unmasking of synaptic GluA2-lacking, Ca2+ permeable AMPA receptors selectively at D1MSNs. Chronic social defeat stress results in a decrease in mEPSC frequency at D1MSNs but an increase in synaptic events at D2MSNs. Chronic pain, which likewise induces an amotivational phenotype, caused a decrease in AMPA/NMDA ratios at D2MSNs. This is in part mediated by increased GluN2B-subunit expression but an abolition of NMDA mediated LTD at D2 synapses.23 Similarly, precipitated withdrawal from morphine, which induces conditioned place aversion, selectively strengthened DMT-NAc D2 synapses and coincided with an increase in AMPA rectification.121 Thus, the canonical model of increased synaptic connectivity at D1 and D2MSNs promoting reward and aversive behavior may be incomplete, as these adaptations coincide with both circumstances.
NMDA Function and Receptor-Dependent Plasticity Induction.
NMDA receptors are implicated in experience-dependent synaptic changes. Several studies have demonstrated that NMDA receptor activation correlates with drug-induced synaptic changes.61,147 GluN2B-containing NMDA receptors are of particular importance to experience-driven plasticity in the NAc. GluN2B receptors have much slower deactivation kinetics, resulting greater net ion flux and Ca2+ entry upon glutamate binding and depolarization. These large currents extend the temporal binding window that allows coupling of synaptic events to neuronal firing.45 GluN2B receptors are found in high concentrations throughout the developing brain and facilitate formation of new synaptic connections via their high concentration in silent synapses.128 The de novo generation of NAc silent synapses in adults occurs following acute withdrawal from drug self-administration and coincides with an increase in the relative expression of GluN2B.127 The formation of new synapses and their subsequent maturation (see above) suggests an increase in connectivity between glutamatergic afferent regions and the NAc following salient experience, increasing their influence on MSN activation. Importantly, the formation of these synapses has been demonstrated at specific afferent-NAc connections including the BLA,118 PFC,55 and DMT,133 demonstrating their prevalence in NAc circuit remodeling. Thus, GluN2B NMDA receptors are crucial for forming new synapses in response to in vivo experience.
NMDA receptors are also crucial for directing synaptic strength. Following noncontingent drug exposure, NMDA currents from the DMT are selectively enhanced via increase in GluN2C/D.53 The increase of NMDA function in cocaine treated animals also unmasked an NMDA-dependent LTD at D1MSN synapses. In line with this finding, resetting glutamatergic inputs from specific brain regions by inducing NMDA-dependent plasticity in vivo at specific inputs can diminish relapse like behavior in rodents. NMDA-dependent LTD of BLA-NAc synapses reduced cue-primed reinstatement to drug seeking.118 An LTD protocol at vHipp-NAc synapses, previously shown to be NMDA dependent, disrupted preference for the drug-paired lever in a cue-induced reinstatement task.117 Additionally, a NMDA-dependent LTD protocol of PFC-NAc synapses reduced locomotion in a cocaine-induced locomotor sensitization.49 However, the same in vivo NMDA-dependent LTD protocol was impaired selectively at PFC-D1 synapses in ex vivo slice preparations from mice withdrawn from cocaine self-administration and failed to reduce responding for the active lever in a cue induced reinstatement task.117 Yet others have shown the same LTD protocol of PFC-NAc synapses also required mGlu1, was only present in animals withdrawn from cocaine, and was able to reduce cue-induced reinstatement in rats.55 These findings suggest that NMDA-dependent LTD is able to ameliorate motivated behavior in experienced animals. The nuanced differences in the models and results indicate additional studies are necessary to clarify the impact of salient drug experience on NMDA signaling at specific synapses.
Recent findings demonstrate that global TLR4 knockout mice lack NMDA-LTD in the NAc core and display reduced drug-induced locomotion and place preference.109 Importantly, there is evidence suggesting that pharmacologic antagonism of TLR4 attenuates drug reward learning to both opioids148 and cocaine.149 Such findings led to the idea that drugs of abuse directly interact with TLR4 to induce cellular changes.150 Also implied is that TLR4 is necessary for drug-reward learning. However, there is controversy surrounding some of these points.151 These findings suggest a link between TLR4 expressing microglia and drug-induced adaptations in NAc NMDA plasticity.
mGlu Plasticity Shifts Postsynaptically Following Drug Experience.
mGlu function is negatively impacted by both acute and chronic exposure to cocaine (Figure 4B, C). Many studies have demonstrated the induction of mGlu5-dependent LTD is blunted following a single or repeated cocaine experience [for examples, see refs 52, 54, 63–65, 152, and 153]. Cocaine-induced abolition of mGlu LTD is thought to be mediated by changes in structural protein Homer isoforms. Homer1a expression induced by acute cocaine exposure sequesters mGlu5 from the membrane surface but increases mGlu1.137,154 This switch in synaptic control from mGlu5 to mGlu1 is proposed to change downstream plasticity targets to favor CP-AMPA internalization over eCB production.65,137,154 This is consistent with the finding that mGlu dependent eCB production is altered in rodents exposed to cocaine, but this is not due to changes in CB1R expression/function.52,152
Much like NMDA-LTD, putative mGlu dependent plasticity is also modified in a synapse specific manner. Withdrawal from drug self-administration unmasked an mGlu1 and NMDA dependent LTD at PFC-NAc shell MSNs.55 Likewise, a separate LTD protocol previously shown to be mGlu-dependent was capable of evoking LTD at PFC-NAc shell synapses and was enhanced in mice that administered cocaine.117 In vivo induction of this putative mGlu-LTD at PFC synapses also ameliorated cue-induced drug seeking. Notably, mGlu1 PAMs infused into the NAc are able to achieve a similar effect in a cue-induced reinstatement task.120 Additionally, recent reports have demonstrated an mGlu1 positive allosteric modulator (PAM) can “reset” CP-AMPA containing synapses in the NAc while also reducing drug induced place preference.130 Given the expression of mGlu1 LTD at PFC synapses in cocaine exposed mice and the efficacy of in vivo PFC mGlu-LTD, it is possible that the ability of intra-NAc PAM infusion to also reduce drug seeking is mediated via action on PFC terminals. Because of this, it may be of interest for future studies to focus on experience-induced alterations in group-I mGlu-dependent plasticity in a synapse-specific manner.
In addition to group-I mGlu function, signaling through group-II mGlu receptors are also heavily tied to drug experience and are coupled to glutamate homeostatic regulation by astrocytes.67,155 Notably, extracellular glutamate is elevated following drug exposure in self-administering animals.156 These changes in extracellular glutamate concentrations arise via downregulation of NAc astrocytic xCT (Figure 4C). Withdrawal from cocaine and nicotine downregulates xCT.68,157 In similar studies, multiple drugs of abuse have been shown to downregulate GLT-1, which can be pharmacologically rescued by Ceftriaxone.114 This results in increased neuronal presynaptic release probability promoting increased glutamatergic transmission for drug-related signals/cues leading to relapse.10 Increasing astrocyte activity using Gq-coupled designer-receptor exclusively activated by designer drugs (DREADD) resulted in increased extracellular glutamate and was associated with decreased cue-induced reinstatement in rats.158
mGlu and NMDA signaling seem to be differentially recruited throughout reward learning in animal drug-exposure models. However, it is yet unclear how these changes result in altered circuit function. For one, the multitude of experimental paradigms, including rodent model, behavioral setup, and cell-type/input specificity, obfuscate comparisons across studies. Additionally, it is apparent that contingent and spatial recognition are more efficient at driving change in the NAc circuitry than context association or home cage experiences in rodents. However, there is an emerging trend suggesting salient experience induces an increased susceptibility to NMDA- and mGlu1-dependent AMPA internalization and a reduction presynaptic control by group-II mGlu and mGlu5-dependent eCB signaling.
Neuromodulatory Regulation of NAc Synapses Following Salient Experience.
The majority of studies investigating changes in glutamatergic NAc signaling have focused on AMPA, NMDA, and mGlu receptors. However, the various modulatory signals that interact with these receptors can also be impacted by salient experience. While the role of 5-HT in drug-related behavior remains enigmatic, 5-HT1B activity has been shown to contribute to the reinforcing properties of psychostimulants, including cocaine and amphetamine.159,160 5-HT-LTD in the NAc is impaired for up to 72 h following a single in vivo administration of cocaine, an effect rescued by a membrane-permeable PKA inhibitor.71
Similarly, opioids and ORs within the NAc are impacted by salient experience and can drive behavior. Repeated force swim stress induces a kappa-opioid dependent ERK1/2 phosphor-ylation within the NAc.161 Additionally, stress induces phosphorylation of kappa ORs162 consistent with theirs and dynorphin’s role in stress-induced behaviors.87 Dyn signaling is also implicated in models of drug abuse163 but only recently has a synaptic phenotype been demonstrated. Cocaine exposure can selectively impair dynorphin-A induced LTD of glutamatergic synapses with no effect on inhibitory transmission.164 Additionally, these authors found that dynorphin-B exhibited non kappa-OR dependent effects that were unaffected by cocaine. As previous work has focused extensively on Dyn and kappa OR signaling, future studies should focus on delta and mu ORs synaptic function and how they are impacted by salient experience.
Endocannabinoid signaling is also tied to salient experience, although this is in part due to its known dependence on mGlu signaling in the NAc. While CB1R activation is important for the expression of these behavioral phenotypes, few studies have identified changes in the receptor or eCB synthetic enzymes. Of note, stimulant exposure impacts mGlu-dependent eCB production but leaves CB1R function intact.152 However, acute exposure to the CB1R agonist THC results in a desensitization of the receptor and blunts eCB-LTD.165 Chronic exposure similarly blunts eCB-LTD but plasticity of the synapse is rescued by group-II mGlu receptors,166 suggesting that CB1R-dependent plasticity mechanisms may be replaced by alternative signaling cascades. Following extinction training from cocaine self-administration, 2-AG concentration is greatly increased in the NAc.167 This increase in 2-AG may be compensatory for the increases in glutamatergic signaling normally seen following drug withdrawal.
While drugs of abuse seem to have limited immediate effect on CB1R control of synaptic transmission, there is evidence tying NAc eCB signaling to hedonic feeding and motivated behaviors. Notably, Cnr1−/− null mice exhibit phenotypes that coincide with reduced motivation to obtain hedonic stimuli. This is somewhat unsurprising given the known effects of ingesting Cannabis sativa.168 In rodents, 2-AG and anandamide concentrations are increased within the NAc following fasting, and intra-NAc administration of 2-AG drives voracious feeding behavior in sated rats.169 Additionally, long-term exposure to a palatable diet decreases CB1R availability in the NAc.170 It is worth noting that Oginsky et al. observed an increase in CPAMPA expression following acute removal from palatable chow while the animals examined by Harrold et al. were still on the diet. Taken together, it is possible that eCB signaling within the NAc is functioning reactively to changes in glutamate transmission rather than acting as a driving force for synaptic remodeling in and of itself. Future studies should focus on examining expression and function of eCB synthetic and degradative enzymes following salient challenge.
CONCLUSION
While there is an ever increasing body of knowledge describing the synaptic machinery within the NAc, how synaptic plasticity influences MSN recruitment to direct neuronal circuit function remains a lofty goal for physiologists and behaviorists alike. It is also worth noting that many secondary signaling proteins recruited by the plasticity mechanisms described above are well characterized in other brain regions but have not been validated within the NAc. Given the heterogeneity of plasticity mechanisms available to individual synaptic connections within the NAc itself, these signaling cascades likewise may be unique to the NAc, differ between cell type and projecting brain region, and thus warrant additional studies.
It remains unclear how synaptic signaling mechanisms observed ex vivo are utilized in vivo, or how an animal’s experiences are transduced into plasticity of accumbens synapses. It should be noted that the majority of findings summarized above pertain entirely to observations in monosynaptic connectivity and does not describe the great deal of integrative power the NAc has when considering its variety of inputs and modulatory systems function in tandem.27 However, with the advent of in vivo imaging of neuronal activity in awake behaving animals, targeted pharmacology and optogenetics, the field is equipped to answer these questions. While these techniques have been employed to map the brain’s reward circuitry, future studies should clarify how plasticity mechanisms gate synaptic function and behavior in real time. It is through these studies that we may gain insight as to how the interaction of pharmacology and physiology drive behavior in a synapse specific manner.
ACKNOWLEDGMENTS
The authors would like to thank the editors of this special edition for inviting us to write this review and to previous members of the Grueter lab who contributed to the initial outlining process.
Funding
National Institute of Drug Abuse DA031699 and DA040630. NARSAD Young Investigator Award (to B.A.G)
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).Everitt BJ, and Robbins TW (2005) Neural systems of reinforcement for drug addiction: from actions to habits to compulsion. Nat. Neurosci 8, 1481–1489. [DOI] [PubMed] [Google Scholar]
- (2).Voorn P, Vanderschuren LJ, Groenewegen HJ, Robbins TW, and Pennartz CM (2004) Putting a spin on the dorsal-ventral divide of the striatum. Trends Neurosci 27, 468–474. [DOI] [PubMed] [Google Scholar]
- (3).Volkow ND, Wang GJ, Fowler JS, Tomasi D, and Telang F (2011) Addiction: beyond dopamine reward circuitry. Proc. Natl. Acad. Sci. U. S. A 108, 15037–15042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Joffe ME, Grueter CA, and Grueter BA (2014) Biological substrates of addiction. Wiley Interdiscip Rev. Cogn Sci 5, 151–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Grueter BA, Rothwell PE, and Malenka RC (2012) Integrating synaptic plasticity and striatal circuit function in addiction. Curr. Opin. Neurobiol 22, 545–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Huang YH, Schluter OM, and Dong Y (2011) Cocaine-induced homeostatic regulation and dysregulation of nucleus accumbens neurons. Behav. Brain Res 216, 9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Wolf ME (2010) Regulation of AMPA receptor trafficking in the nucleus accumbens by dopamine and cocaine. Neurotoxic. Res 18, 393–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Luscher C, and Huber KM (2010) Group 1 mGluR-dependent synaptic long-term depression: mechanisms and implications for circuitry and disease. Neuron 65, 445–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Kalivas PW, Volkow N, and Seamans J (2005) Unmanageable motivation in addiction: a pathology in prefrontalaccumbens glutamate transmission. Neuron 45, 647–650. [DOI] [PubMed] [Google Scholar]
- (10).Kalivas PW (2009) The glutamate homeostasis hypothesis of addiction. Nat. Rev. Neurosci 10, 561–572. [DOI] [PubMed] [Google Scholar]
- (11).Koob GF (2011) Neurobiology of Addiction. Focus 9, 55–65. [Google Scholar]
- (12).Koob GF (2008) A role for brain stress systems in addiction. Neuron 59, 11–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Lim BK, Huang KW, Grueter BA, Rothwell PE, and Malenka RC (2012) Anhedonia requires MC4R-mediated synaptic adaptations in nucleus accumbens. Nature 487, 183–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Christoffel DJ, Golden SA, Walsh JJ, Guise KG, Heshmati M, Friedman AK, Dey A, Smith M, Rebusi N, Pfau M, Ables JL, Aleyasin H, Khibnik LA, Hodes GE, Ben-Dor GA, Deisseroth K, Shapiro ML, Malenka RC, Ibanez-Tallon I, Han MH, and Russo SJ (2015) Excitatory transmission at thalamo-striatal synapses mediates susceptibility to social stress. Nat. Neurosci 18, 962–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Heshmati M, and Russo SJ (2015) Anhedonia and the brain reward circuitry in depression. Curr. Behav Neurosci Rep 2, 146–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Khibnik LA, Beaumont M, Doyle M, Heshmati M, Slesinger PA, Nestler EJ, and Russo SJ (2016) Stress and Cocaine Trigger Divergent and Cell Type-Specific Regulation of Synaptic Transmission at Single Spines in Nucleus Accumbens. Biol. Psychiatry 79, 898–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Russo SJ, and Nestler EJ (2013) The brain reward circuitry in mood disorders. Nat. Rev. Neurosci 14, 609–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Bagot RC, Parise EM, Pena CJ, Zhang HX, Maze I, Chaudhury D, Persaud B, Cachope R, Bolanos-Guzman CA, Cheer JF, Deisseroth K, Han MH, and Nestler EJ (2015) Ventral hippocampal afferents to the nucleus accumbens regulate susceptibility to depression. Nat. Commun 6, 7062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Francis TC, Chandra R, Friend DM, Finkel E, Dayrit G, Miranda J, Brooks JM, Iniguez SD, O’Donnell P, Kravitz A, and Lobo MK (2015) Nucleus accumbens medium spiny neuron subtypes mediate depression-related outcomes to social defeat stress. Biol. Psychiatry 77, 212–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).DiLeone RJ, Taylor JR, and Picciotto MR (2012) The drive to eat: comparisons and distinctions between mechanisms of food reward and drug addiction. Nat. Neurosci 15, 1330–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Lee JS, Jung S, Park IH, and Kim JJ (2015) Neural Basis of Anhedonia and Amotivation in Patients with Schizophrenia: The Role of Reward System. Curr. Neuropharmacol 13, 750–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).McCollum LA, and Roberts RC (2015) Uncovering the role of the nucleus accumbens in schizophrenia: A postmortem analysis of tyrosine hydroxylase and vesicular glutamate transporters. Schizophr Res 169, 369–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Schwartz N, Temkin P, Jurado S, Lim BK, Heifets BD, Polepalli JS, and Malenka RC (2014) Decreased motivation during chronic pain requires long-term depression in the nucleus accumbens. Science 345, 535–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Fuccillo MV (2016) Striatal Circuits as a Common Node for Autism Pathophysiology. Front. Neurosci 10, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Rothwell PE (2016) Autism Spectrum Disorders and Drug Addiction: Common Pathways, Common Molecules, Distinct Disorders? Front. Neurosci 10, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Mogenson GJ, Jones DL, and Yim CY (1980) From motivation to action: functional interface between the limbic system and the motor system. Prog. Neurobiol 14, 69–97. [DOI] [PubMed] [Google Scholar]
- (27).Sesack SR, and Grace AA (2010) Cortico-Basal Ganglia reward network: microcircuitry. Neuropsychopharmacology 35, 27–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Meredith GE (1999) The synaptic framework for chemical signaling in nucleus accumbens. Ann. N. Y. Acad. Sci 877, 140–156. [DOI] [PubMed] [Google Scholar]
- (29).Grueter BA, Robison AJ, Neve RL, Nestler EJ, and Malenka RC (2013) FosB differentially modulates nucleus accumbens direct and indirect pathway function. Proc. Natl. Acad. Sci. U. S. A 110, 1923–1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Kupchik YM, Brown RM, Heinsbroek JA, Lobo MK, Schwartz DJ, and Kalivas PW (2015) Coding the direct/indirect pathways by D1 and D2 receptors is not valid for accumbens projections. Nat. Neurosci 18, 1230–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Lobo MK, and Nestler EJ (2011) The striatal balancing act in drug addiction: distinct roles of direct and indirect pathway medium spiny neurons. Front. Neuroanat 5, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Smith RJ, Lobo MK, Spencer S, and Kalivas PW (2013) Cocaine-induced adaptations in D1 and D2 accumbens projection neurons (a dichotomy not necessarily synonymous with direct and indirect pathways). Curr. Opin. Neurobiol 23, 546–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Lobo MK, Covington HE 3rd, Chaudhury D, Friedman AK, Sun H, Damez-Werno D, Dietz DM, Zaman S, Koo JW, Kennedy PJ, Mouzon E, Mogri M, Neve RL, Deisseroth K, Han MH, and Nestler EJ (2010) Cell type-specific loss of BDNF signaling mimics optogenetic control of cocaine reward. Science 330, 385–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Bock R, Shin JH, Kaplan AR, Dobi A, Markey E, Kramer PF, Gremel CM, Christensen CH, Adrover MF, and Alvarez VA (2013) Strengthening the accumbal indirect pathway promotes resilience to compulsive cocaine use. Nat. Neurosci 16, 632–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Pascoli V, Terrier J, Hiver A, and Luscher C (2015) Sufficiency of Mesolimbic Dopamine Neuron Stimulation for the Progression to Addiction. Neuron 88, 1054–1066. [DOI] [PubMed] [Google Scholar]
- (36).Natsubori A, Tsutsui-Kimura I, Nishida H, Bouchekioua Y, Sekiya H, Uchigashima M, Watanabe M, de Kerchove d’Exaerde A, Mimura M, Takata N, and Tanaka KF (2017) Ventrolateral Striatal Medium Spiny Neurons Positively Regulate Food-Incentive, Goal-Directed Behavior Independently of D1 and D2 Selectivity. J. Neurosci 37, 2723–2733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Cui G, Jun SB, Jin X, Pham MD, Vogel SS, Lovinger DM, and Costa RM (2013) Concurrent activation of striatal direct and indirect pathways during action initiation. Nature 494, 238–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Morales M, and Root DH (2014) Glutamate neurons within the midbrain dopamine regions. Neuroscience 282, 60–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Adrover MF, Shin JH, and Alvarez VA (2014) Glutamate and dopamine transmission from midbrain dopamine neurons share similar release properties but are differentially affected by cocaine. J. Neurosci 34, 3183–3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Qi J, Zhang S, Wang HL, Barker DJ, Miranda-Barrientos J, and Morales M (2016) VTA glutamatergic inputs to nucleus accumbens drive aversion by acting on GABAergic interneurons. Nat. Neurosci 19, 725–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Britt JP, Benaliouad F, McDevitt RA, Stuber GD, Wise RA, and Bonci A (2012) Synaptic and behavioral profile of multiple glutamatergic inputs to the nucleus accumbens. Neuron 76, 790–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Stuber GD, Britt JP, and Bonci A (2012) Optogenetic modulation of neural circuits that underlie reward seeking. Biol. Psychiatry 71, 1061–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Luscher C, and Malenka RC (2011) Drug-evoked synaptic plasticity in addiction: from molecular changes to circuit remodeling. Neuron 69, 650–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Do-Monte FH, Minier-Toribio A, Quinones-Laracuente K, Medina-Colon EM, and Quirk GJ (2017) Thalamic Regulation of Sucrose Seeking during Unexpected Reward Omission. Neuron 94, 388–400 e384.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Paoletti P, Bellone C, and Zhou Q (2013) NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci 14, 383–400. [DOI] [PubMed] [Google Scholar]
- (46).Winder DG, and Sweatt JD (2001) Roles of serine/threonine phosphatases in hippocampal synaptic plasticity. Nat. Rev. Neurosci 2, 461–474. [DOI] [PubMed] [Google Scholar]
- (47).Luscher C, and Malenka RC (2012) NMDA receptor-dependent long-term potentiation and long-term depression (LTP/LTD). Cold Spring Harbor Perspect. Biol 4, a005710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Schramm NL, Egli RE, and Winder DG (2002) LTP in the mouse nucleus accumbens is developmentally regulated. Synapse 45, 213–219. [DOI] [PubMed] [Google Scholar]
- (49).Pascoli V, Turiault M, and Luscher C (2012) Reversal of cocaine-evoked synaptic potentiation resets drug-induced adaptive behaviour. Nature 481, 71–75. [DOI] [PubMed] [Google Scholar]
- (50).Kombian SB, and Malenka RC (1994) Simultaneous LTP of non-NMDA- and LTD of NMDA-receptor-mediated responses in the nucleus accumbens. Nature 368, 242–246. [DOI] [PubMed] [Google Scholar]
- (51).Yao WD, Gainetdinov RR, Arbuckle MI, Sotnikova TD, Cyr M, Beaulieu JM, Torres GE, Grant SG, and Caron MG (2004) Identification of PSD-95 as a regulator of dopamine-mediated synaptic and behavioral plasticity. Neuron 41, 625–638. [DOI] [PubMed] [Google Scholar]
- (52).Robbe D, Kopf M, Remaury A, Bockaert J, and Manzoni OJ (2002) Endogenous cannabinoids mediate long-term synaptic depression in the nucleus accumbens. Proc. Natl. Acad. Sci. U. S. A 99, 8384–8388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Joffe ME, and Grueter BA (2016) Cocaine Experience Enhances Thalamo-Accumbens N-Methyl-D-Aspartate Receptor Function. Biol. Psychiatry 80, 671–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Grueter BA, Brasnjo G, and Malenka RC (2010) Postsynaptic TRPV1 triggers cell type-specific long-term depression in the nucleus accumbens. Nat. Neurosci 13, 1519–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Ma YY, Lee BR, Wang X, Guo C, Liu L, Cui R, Lan Y, Balcita-Pedicino JJ, Wolf ME, Sesack SR, Shaham Y, Schluter OM, Huang YH, and Dong Y (2014) Bidirectional modulation of incubation of cocaine craving by silent synapse-based remodeling of prefrontal cortex to accumbens projections. Neuron 83, 1453–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Malinow R, and Malenka RC (2002) AMPA receptor trafficking and synaptic plasticity. Annu. Rev. Neurosci 25, 103–126. [DOI] [PubMed] [Google Scholar]
- (57).Meyer D, Bonhoeffer T, and Scheuss V (2014) Balance and stability of synaptic structures during synaptic plasticity. Neuron 82, 430–443. [DOI] [PubMed] [Google Scholar]
- (58).Nestler EJ (2013) Cellular basis of memory for addiction. Dialogues Clin. Neurosci 15, 431–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Bosch M, Castro J, Saneyoshi T, Matsuno H, Sur M, and Hayashi Y (2014) Structural and molecular remodeling of dendritic spine substructures during long-term potentiation. Neuron 82, 444–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Russo SJ, Dietz DM, Dumitriu D, Morrison JH, Malenka RC, and Nestler EJ (2010) The addicted synapse: mechanisms of synaptic and structural plasticity in nucleus accumbens. Trends Neurosci 33, 267–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Cahill E, Pascoli V, Trifilieff P, Savoldi D, Kappes V, Luscher C, Caboche J, and Vanhoutte P (2014) D1R/GluN1 complexes in the striatum integrate dopamine and glutamate signalling to control synaptic plasticity and cocaine-induced responses. Mol. Psychiatry 19, 1295–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Niswender CM, and Conn PJ (2010) Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu. Rev. Pharmacol. Toxicol 50, 295–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Grueter BA, McElligott ZA, and Winder DG (2007) Group I mGluRs and long-term depression: potential roles in addiction? Mol. Neurobiol 36, 232–244. [DOI] [PubMed] [Google Scholar]
- (64).Loweth JA, Tseng KY, and Wolf ME (2013) Using metabotropic glutamate receptors to modulate cocaine’s synaptic and behavioral effects: mGluR1 finds a niche. Curr. Opin. Neurobiol 23, 500–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).McCutcheon JE, Loweth JA, Ford KA, Marinelli M, Wolf ME, and Tseng KY (2011) Group I mGluR activation reverses cocaine-induced accumulation of calcium-permeable AMPA receptors in nucleus accumbens synapses via a protein kinase C-dependent mechanism. J. Neurosci 31, 14536–14541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Moussawi K, Pacchioni A, Moran M, Olive MF, Gass JT, Lavin A, and Kalivas PW (2009) N-Acetylcysteine reverses cocaine-induced metaplasticity. Nat. Neurosci 12, 182–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Moussawi K, Riegel A, Nair S, and Kalivas PW (2011) Extracellular glutamate: functional compartments operate in different concentration ranges. Front. Syst. Neurosci 5, 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Knackstedt LA, LaRowe S, Mardikian P, Malcolm R, Upadhyaya H, Hedden S, Markou A, and Kalivas PW (2009) The role of cystine-glutamate exchange in nicotine dependence in rats and humans. Biol. Psychiatry 65, 841–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Brown P, and Molliver ME (2000) Dual serotonin (5-HT) projections to the nucleus accumbens core and shell: relation of the 5-HT transporter to amphetamine-induced neurotoxicity. J. Neurosci 20, 1952–1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Soghomonian JJ, Descarries L, and Watkins KC (1989) Serotonin innervation in adult rat neostriatum. II. Ultrastructural features: a radioautographic and immunocytochemical study. Brain Res 481, 67–86. [DOI] [PubMed] [Google Scholar]
- (71).Burattini C, Battistini G, Tamagnini F, and Aicardi G (2014) Low-frequency stimulation evokes serotonin release in the nucleus accumbens and induces long-term depression via production of endocannabinoid. J. Neurophysiol 111, 1046–1055. [DOI] [PubMed] [Google Scholar]
- (72).Muramatsu M, Lapiz MD, Tanaka E, and Grenhoff J (1998) Serotonin inhibits synaptic glutamate currents in rat nucleus accumbens neurons via presynaptic 5-HT1B receptors. Eur. J. Neurosci 10, 2371–2379. [DOI] [PubMed] [Google Scholar]
- (73).Dolen G, Darvishzadeh A, Huang KW, and Malenka RC (2013) Social reward requires coordinated activity of nucleus accumbens oxytocin and serotonin. Nature 501, 179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Mathur BN, Capik NA, Alvarez VA, and Lovinger DM (2011) Serotonin induces long-term depression at corticostriatal synapses. J. Neurosci 31, 7402–7411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Best AR, and Regehr WG (2008) Serotonin evokes endocannabinoid release and retrogradely suppresses excitatory synapses. J. Neurosci 28, 6508–6515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Al-Hasani R, and Bruchas MR (2011) Molecular mechanisms of opioid receptor-dependent signaling and behavior. Anesthesiology 115, 1363–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Khachaturian H, Lewis ME, Haber SN, Houghten RA, Akil H, and Watson SJ (1985) Prodynorphin peptide immunocytochemistry in rhesus monkey brain. Peptides 6 (Suppl 2), 155–166. [DOI] [PubMed] [Google Scholar]
- (78).Chartoff EH, and Connery HS (2014) It’s MORe exciting than mu: crosstalk between mu opioid receptors and glutamatergic transmission in the mesolimbic dopamine system. Front. Pharmacol 5, 116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Bruchas MR, Land BB, and Chavkin C (2010) The dynorphin/kappa opioid system as a modulator of stress-induced and pro-addictive behaviors. Brain Res 1314, 44–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Lutz PE, and Kieffer BL (2013) The multiple facets of opioid receptor function: implications for addiction. Curr. Opin. Neurobiol 23, 473–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Castro DC, and Berridge KC (2014) Opioid hedonic hotspot in nucleus accumbens shell: mu, delta, and kappa maps for enhancement of sweetness ″liking″ and ″wanting″. J. Neurosci 34, 4239–4250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Banghart MR, Neufeld SQ, Wong NC, and Sabatini BL (2015) Enkephalin Disinhibits Mu Opioid Receptor-Rich Striatal Patches via Delta Opioid Receptors. Neuron 88, 1227–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Martin G, Nie Z, and Siggins GR (1997) mu-Opioid receptors modulate NMDA receptor-mediated responses in nucleus accumbens neurons. J. Neurosci 17, 11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Atwood BK, Kupferschmidt DA, and Lovinger DM (2014) Opioids induce dissociable forms of long-term depression of excitatory inputs to the dorsal striatum. Nat. Neurosci 17, 540–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Al-Hasani R, McCall JG, Shin G, Gomez AM, Schmitz GP, Bernardi JM, Pyo CO, Park SI, Marcinkiewcz CM, Crowley NA, Krashes MJ, Lowell BB, Kash TL, Rogers JA, and Bruchas MR (2015) Distinct Subpopulations of Nucleus Accumbens Dynorphin Neurons Drive Aversion and Reward. Neuron 87, 1063–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Tejeda HA, Wu J, Kornspun AR, Pignatelli M, Kashtelyan V, Krashes MJ, Lowell BB, Carlezon WA Jr., and Bonci A (2017) Pathway- and Cell-Specific Kappa-Opioid Receptor Modulation of Excitation-Inhibition Balance Differentially Gates D1 and D2 Accumbens Neuron Activity. Neuron 93, 147–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Muschamp JW, and Carlezon WA Jr. (2013) Roles of nucleus accumbens CREB and dynorphin in dysregulation of motivation. Cold Spring Harbor Perspect. Med 3, a012005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Clarke TK, Bloch PJ, Ambrose-Lanci LM, Ferraro TN, Berrettini WH, Kampman KM, Dackis CA, Pettinati HM, O’Brien CP, Oslin DW, and Lohoff FW (2013) Further evidence for association of polymorphisms in the CNR1 gene with cocaine addiction: confirmation in an independent sample and meta-analysis. Addiction biology 18, 702–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Zuo L, Kranzler HR, Luo X, Yang BZ, Weiss R, Brady K, Poling J, Farrer L, and Gelernter J (2009) Interaction between two independent CNR1 variants increases risk for cocaine dependence in European Americans: a replication study in family-based sample and population-based sample. Neuropsychopharmacology 34, 1504–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Hirvonen J, Goodwin RS, Li CT, Terry GE, Zoghbi SS, Morse C, Pike VW, Volkow ND, Huestis MA, and Innis RB (2012) Reversible and regionally selective downregulation of brain cannabinoid CB1 receptors in chronic daily cannabis smokers. Mol. Psychiatry 17, 642–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Schacht JP, Hutchison KE, and Filbey FM (2012) Associations between cannabinoid receptor-1 (CNR1) variation and hippocampus and amygdala volumes in heavy cannabis users. Neuropsychopharmacology 37, 2368–2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Kano M, Ohno-Shosaku T, Hashimotodani Y, Uchigashima M, and Watanabe M (2009) Endocannabinoid-mediated control of synaptic transmission. Physiol. Rev 89, 309–380. [DOI] [PubMed] [Google Scholar]
- (93).Ohno-Shosaku T, and Kano M (2014) Endocannabinoid-mediated retrograde modulation of synaptic transmission. Curr. Opin. Neurobiol 29, 1–8. [DOI] [PubMed] [Google Scholar]
- (94).Hoffman AF, and Lupica CR (2001) Direct actions of cannabinoids on synaptic transmission in the nucleus accumbens: a comparison with opioids. J. Neurophysiol 85, 72–83. [DOI] [PubMed] [Google Scholar]
- (95).Robbe D, Alonso G, and Manzoni OJ (2003) Exogenous and endogenous cannabinoids control synaptic transmission in mice nucleus accumbens. Ann. N. Y. Acad. Sci 1003, 212–225. [DOI] [PubMed] [Google Scholar]
- (96).Winters BD, Kruger JM, Huang X, Gallaher ZR, Ishikawa M, Czaja K, Krueger JM, Huang YH, Schluter OM, and Dong Y (2012) Cannabinoid receptor 1-expressing neurons in the nucleus accumbens. Proc. Natl. Acad. Sci. U. S. A 109, E2717–2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Kauer JA, and Gibson HE (2009) Hot flash: TRPV channels in the brain. Trends Neurosci 32, 215–224. [DOI] [PubMed] [Google Scholar]
- (98).Ramsey IS, Delling M, and Clapham DE (2006) An introduction to TRP channels. Annu. Rev. Physiol 68, 619–647. [DOI] [PubMed] [Google Scholar]
- (99).Zygmunt PM, Petersson J, Andersson DA, Chuang H, Sorgard M, Di Marzo V, Julius D, and Hogestatt ED (1999) Vanilloid receptors on sensory nerves mediate the vasodilator action of anandamide. Nature 400, 452–457. [DOI] [PubMed] [Google Scholar]
- (100).Renteria R, Jeanes ZM, and Morrisett RA (2014) Ethanol attenuation of long-term depression in the nucleus accumbens can be overcome by activation of TRPV1 receptors. Alcohol.: Clin. Exp. Res 38, 2763–2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (101).Gibson HE, Edwards JG, Page RS, Van Hook MJ, and Kauer JA (2008) TRPV1 channels mediate long-term depression at synapses on hippocampal interneurons. Neuron 57, 746–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).Prinz M, and Priller J (2014) Microglia and brain macrophages in the molecular age: from origin to neuropsychiatric disease. Nat. Rev. Neurosci 15, 300–312. [DOI] [PubMed] [Google Scholar]
- (103).Kettenmann H, Kirchhoff F, and Verkhratsky A (2013) Microglia: new roles for the synaptic stripper. Neuron 77, 10–18. [DOI] [PubMed] [Google Scholar]
- (104).Joseph B, and Venero JL (2013) A brief overview of multitalented microglia. Methods Mol. Biol 1041, 3–8. [DOI] [PubMed] [Google Scholar]
- (105).Bilbo SD, and Schwarz JM (2012) The immune system and developmental programming of brain and behavior. Front. Neuroendocrinol 33, 267–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (106).Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, Ransohoff RM, Greenberg ME, Barres BA, and Stevens B (2012) Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 74, 691–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (107).Parkhurst CN, Yang G, Ninan I, Savas JN, Yates JR 3rd, Lafaille JJ, Hempstead BL, Littman DR, and Gan WB (2013) Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 155, 1596–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (108).Schwarz JM, Smith SH, and Bilbo SD (2013) FACS analysis of neuronal-glial interactions in the nucleus accumbens following morphine administration. Psychopharmacology (Berl) 230, 525–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (109).Kashima DT, and Grueter BA (2017) Toll-like receptor 4 deficiency alters nucleus accumbens synaptic physiology and drug reward behavior. Proc. Natl. Acad. Sci. U. S. A 114, 8865–8870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (110).Bohannon JK, Hernandez A, Enkhbaatar P, Adams WL, and Sherwood ER (2013) The immunobiology of toll-like receptor 4 agonists: from endotoxin tolerance to immunoadjuvants. Shock 40, 451–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (111).O’Neill LA (2008) Primer: Toll-like receptor signaling pathways–what do rheumatologists need to know? Nat. Clin. Pract. Rheumatol 4, 319–327. [DOI] [PubMed] [Google Scholar]
- (112).Lewitus GM, Konefal SC, Greenhalgh AD, Pribiag H, Augereau K, and Stellwagen D (2016) Microglial TNF-alpha Suppresses Cocaine-Induced Plasticity and Behavioral Sensitization. Neuron 90, 483–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (113).Clarke LE, and Barres BA (2013) Emerging roles of astrocytes in neural circuit development. Nat. Rev. Neurosci 14, 311–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (114).Knackstedt LA, Melendez RI, and Kalivas PW (2010) Ceftriaxone restores glutamate homeostasis and prevents relapse to cocaine seeking. Biol. Psychiatry 67, 81–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (115).Kupchik YM, Moussawi K, Tang XC, Wang X, Kalivas BC, Kolokithas R, Ogburn KB, and Kalivas PW (2012) The effect of N-acetylcysteine in the nucleus accumbens on neuro-transmission and relapse to cocaine. Biol. Psychiatry 71, 978–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (116).Richard JM, Castro DC, Difeliceantonio AG, Robinson MJ, and Berridge KC (2013) Mapping brain circuits of reward and motivation: in the footsteps of Ann Kelley. Neurosci. Biobehav. Rev 37, 1919–1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (117).Pascoli V, Terrier J, Espallergues J, Valjent E, O’Connor EC, and Luscher C (2014) Contrasting forms of cocaine-evoked plasticity control components of relapse. Nature 509, 459–464. [DOI] [PubMed] [Google Scholar]
- (118).Lee BR, Ma YY, Huang YH, Wang X, Otaka M, Ishikawa M, Neumann PA, Graziane NM, Brown TE, Suska A, Guo C, Lobo MK, Sesack SR, Wolf ME, Nestler EJ, Shaham Y, Schluter OM, and Dong Y (2013) Maturation of silent synapses in amygdala-accumbens projection contributes to incubation of cocaine craving. Nat. Neurosci 16, 1644–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (119).Conrad KL, Tseng KY, Uejima JL, Reimers JM, Heng LJ, Shaham Y, Marinelli M, and Wolf ME (2008) Formation of accumbens GluR2-lacking AMPA receptors mediates incubation of cocaine craving. Nature 454, 118–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (120).Loweth JA, Scheyer AF, Milovanovic M, LaCrosse AL, Flores-Barrera E, Werner CT, Li X, Ford KA, Le T, Olive MF, Szumlinski KK, Tseng KY, and Wolf ME (2014) Synaptic depression via mGluR1 positive allosteric modulation suppresses cue-induced cocaine craving. Nat. Neurosci 17, 73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (121).Zhu Y, Wienecke CF, Nachtrab G, and Chen X (2016) A thalamic input to the nucleus accumbens mediates opiate dependence. Nature 530, 219–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (122).Thomas MJ, Beurrier C, Bonci A, and Malenka RC (2001) Long-term depression in the nucleus accumbens: a neural correlate of behavioral sensitization to cocaine. Nat. Neurosci 4, 1217–1223. [DOI] [PubMed] [Google Scholar]
- (123).Boudreau AC, Reimers JM, Milovanovic M, and Wolf ME (2007) Cell surface AMPA receptors in the rat nucleus accumbens increase during cocaine withdrawal but internalize after cocaine challenge in association with altered activation of mitogen-activated protein kinases. J. Neurosci 27, 10621–10635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (124).Kourrich S, Rothwell PE, Klug JR, and Thomas MJ (2007) Cocaine experience controls bidirectional synaptic plasticity in the nucleus accumbens. J. Neurosci 27, 7921–7928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (125).McCutcheon JE, Wang X, Tseng KY, Wolf ME, and Marinelli M (2011) Calcium-permeable AMPA receptors are present in nucleus accumbens synapses after prolonged withdrawal from cocaine self-administration but not experimenter-administered cocaine.J. Neurosci 31, 5737–5743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (126).Brown TE, Lee BR, Mu P, Ferguson D, Dietz D, Ohnishi YN, Lin Y, Suska A, Ishikawa M, Huang YH, Shen H, Kalivas PW, Sorg BA, Zukin RS, Nestler EJ, Dong Y, and Schluter OM (2011) A silent synapse-based mechanism for cocaine-induced locomotor sensitization. J. Neurosci 31, 8163–8174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (127).Huang YH, Lin Y, Mu P, Lee BR, Brown TE, Wayman G, Marie H, Liu W, Yan Z, Sorg BA, Schluter OM, Zukin RS, and Dong Y (2009) In vivo cocaine experience generates silent synapses. Neuron 63, 40–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (128).Kerchner GA, and Nicoll RA (2008) Silent synapses and the emergence of a postsynaptic mechanism for LTP. Nat. Rev. Neurosci 9, 813–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (129).Lee BR, and Dong Y (2011) Cocaine-induced metaplasticity in the nucleus accumbens: silent synapse and beyond. Neuropharmacology 61, 1060–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (130).Shukla A, Beroun A, Panopoulou M, Neumann PA, Grant SG, Olive MF, Dong Y, and Schluter OM (2017) Calcium-permeable AMPA receptors and silent synapses in cocaine-conditioned place preference. EMBO J 36, 458–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (131).Loweth JA, Tseng KY, and Wolf ME (2014) Adaptations in AMPA receptor transmission in the nucleus accumbens contributing to incubation of cocaine craving. Neuropharmacology 76 Pt B, 287–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (132).Terrier J, Luscher C, and Pascoli V (2016) Cell-Type Specific Insertion of GluA2-Lacking AMPARs with Cocaine Exposure Leading to Sensitization, Cue-Induced Seeking, and Incubation of Craving. Neuropsychopharmacology 41, 1779–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (133).Neumann PA, Wang Y, Yan Y, Wang Y, Ishikawa M, Cui R, Huang YH, Sesack SR, Schluter OM, and Dong Y (2016) Cocaine-Induced Synaptic Alterations in Thalamus to Nucleus Accumbens Projection. Neuropsychopharmacology 41, 2399–2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (134).MacAskill AF, Cassel JM, and Carter AG (2014) Cocaine exposure reorganizes cell type- and input-specific connectivity in the nucleus accumbens. Nat. Neurosci 17, 1198–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (135).Kim J, Park BH, Lee JH, Park SK, and Kim JH (2011) Cell type-specific alterations in the nucleus accumbens by repeated exposures to cocaine. Biol. Psychiatry 69, 1026–1034. [DOI] [PubMed] [Google Scholar]
- (136).Scheyer AF, Wolf ME, and Tseng KY (2014) A protein synthesis-dependent mechanism sustains calcium-permeable AMPA receptor transmission in nucleus accumbens synapses during withdrawal from cocaine self-administration. J. Neurosci 34, 3095–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (137).Szumlinski KK, Ary AW, and Lominac KD (2008) Homers regulate drug-induced neuroplasticity: implications for addiction. Biochem. Pharmacol 75, 112–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (138).Barrot M, Olivier JD, Perrotti LI, DiLeone RJ, Berton O, Eisch AJ, Impey S, Storm DR, Neve RL, Yin JC, Zachariou V, and Nestler EJ (2002) CREB activity in the nucleus accumbens shell controls gating of behavioral responses to emotional stimuli. Proc. Natl. Acad. Sci. U. S. A 99, 11435–11440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (139).Dong Y, Green T, Saal D, Marie H, Neve R, Nestler EJ, and Malenka RC (2006) CREB modulates excitability of nucleus accumbens neurons. Nat. Neurosci 9, 475–477. [DOI] [PubMed] [Google Scholar]
- (140).Nestler EJ (2008) Review. Transcriptional mechanisms of addiction: role of DeltaFosB. Philos. Trans. R. Soc., B 363, 3245–3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (141).Robison AJ, and Nestler EJ (2011) Transcriptional and epigenetic mechanisms of addiction. Nat. Rev. Neurosci 12, 623–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (142).Kelz MB, Chen J, Carlezon WA Jr., Whisler K, Gilden L, Beckmann AM, Steffen C, Zhang YJ, Marotti L, Self DW, Tkatch T, Baranauskas G, Surmeier DJ, Neve RL, Duman RS, Picciotto MR, and Nestler EJ (1999) Expression of the transcription factor deltaFosB in the brain controls sensitivity to cocaine. Nature 401, 272–276. [DOI] [PubMed] [Google Scholar]
- (143).Vialou V, Robison AJ, Laplant QC, Covington HE 3rd, Dietz DM, Ohnishi YN, Mouzon E, Rush AJ 3rd, Watts EL, Wallace DL, Iniguez SD, Ohnishi YH, Steiner MA, Warren BL, Krishnan V, Bolanos CA, Neve RL, Ghose S, Berton O, Tamminga CA, and Nestler EJ (2010) DeltaFosB in brain reward circuits mediates resilience to stress and antidepressant responses. Nat. Neurosci 13, 745–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (144).Robison AJ, Vialou V, Mazei-Robison M, Feng J, Kourrich S, Collins M, Wee S, Koob G, Turecki G, Neve R, Thomas M, and Nestler EJ (2013) Behavioral and structural responses to chronic cocaine require a feedforward loop involving DeltaFosB and calcium/calmodulin-dependent protein kinase II in the nucleus accumbens shell. J. Neurosci 33, 4295–4307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (145).Oginsky MF, Goforth PB, Nobile CW, Lopez-Santiago LF, and Ferrario CR (2016) Eating ‘Junk-Food’ Produces Rapid and Long-Lasting Increases in NAc CP-AMPA Receptors: Implications for Enhanced Cue-Induced Motivation and Food Addiction. Neuropsychopharmacology 41, 2977–2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (146).Maldonado-Irizarry CS, Swanson CJ, and Kelley AE (1995) Glutamate receptors in the nucleus accumbens shell control feeding behavior via the lateral hypothalamus. J. Neurosci 15, 6779–6788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (147).Beutler LR, Wanat MJ, Quintana A, Sanz E, Bamford NS, Zweifel LS, and Palmiter RD (2011) Balanced NMDA receptor activity in dopamine D1 receptor (D1R)- and D2R-expressing medium spiny neurons is required for amphetamine sensitization. Proc. Natl. Acad. Sci. U. S. A 108, 4206–4211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (148).Hutchinson MR, Northcutt AL, Hiranita T, Wang X, Lewis SS, Thomas J, van Steeg K, Kopajtic TA, Loram LC, Sfregola C, Galer E, Miles NE, Bland ST, Amat J, Rozeske RR, Maslanik T, Chapman TR, Strand KA, Fleshner M, Bachtell RK, Somogyi AA, Yin H, Katz JL, Rice KC, Maier SF, and Watkins LR (2012) Opioid activation of toll-like receptor 4 contributes to drug reinforcement. J. Neurosci 32, 11187–11200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (149).Northcutt AL, Hutchinson MR, Wang X, Baratta MV, Hiranita T, Cochran TA, Pomrenze MB, Galer EL, Kopajtic TA, Li CM, Amat J, Larson G, Cooper DC, Huang Y, O’Neill CE, Yin H, Zahniser NR, Katz JL, Rice KC, Maier SF, Bachtell RK, and Watkins LR (2015) DAT isn’t all that: cocaine reward and reinforcement require Toll-like receptor 4 signaling. Mol [DOI] [PMC free article] [PubMed] [Google Scholar]
- (150).Bachtell R, Hutchinson MR, Wang X, Rice KC, Maier SF, and Watkins LR (2015) Targeting the Toll of Drug Abuse: The Translational Potential of Toll-Like Receptor 4. CNS Neurol. Disord.: Drug Targets 14, 692–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (151).Tanda G, Mereu M, Hiranita T, Quarterman JC, Coggiano M, and Katz JL (2016) Lack of Specific Involvement of (+)-Naloxone and (+)-Naltrexone on the Reinforcing and Neuro-chemical Effects of Cocaine and Opioids. Neuropsychopharmacology 41, 2772–2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (152).Fourgeaud L, Mato S, Bouchet D, Hemar A, Worley PF, and Manzoni OJ (2004) A single in vivo exposure to cocaine abolishes endocannabinoid-mediated long-term depression in the nucleus accumbens. J. Neurosci 24, 6939–6945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (153).Shin S, Kwon O, Kang JI, Kwon S, Oh S, Choi J, Kim CH, and Kim DG (2015) mGluR5 in the nucleus accumbens is critical for promoting resilience to chronic stress. Nat. Neurosci 18, 1017–1024. [DOI] [PubMed] [Google Scholar]
- (154).Szumlinski KK, Abernathy KE, Oleson EB, Klugmann M, Lominac KD, He DY, Ron D, During M, and Kalivas PW (2006) Homer isoforms differentially regulate cocaine-induced neuroplasticity. Neuropsychopharmacology 31, 768–777. [DOI] [PubMed] [Google Scholar]
- (155).Kalivas PW, and Volkow ND (2011) New medications for drug addiction hiding in glutamatergic neuroplasticity. Mol. Psychiatry 16, 974–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (156).McFarland K, Lapish CC, and Kalivas PW (2003) Prefrontal glutamate release into the core of the nucleus accumbens mediates cocaine-induced reinstatement of drug-seeking behavior. J. Neurosci 23, 3531–3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (157).Scofield MD, and Kalivas PW (2014) Astrocytic dysfunction and addiction: consequences of impaired glutamate homeostasis. Neuroscientist 20, 610–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (158).Scofield MD, Boger HA, Smith RJ, Li H, Haydon PG, and Kalivas PW (2015) Gq-DREADD Selectively Initiates Glial Glutamate Release and Inhibits Cue-induced Cocaine Seeking. Biol. Psychiatry 78, 441–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (159).Fletcher PJ, Azampanah A, and Korth KM (2002) Activation of 5-HT(1B) receptors in the nucleus accumbens reduces self-administration of amphetamine on a progressive ratio schedule. Pharmacol., Biochem. Behav 71, 717–725. [DOI] [PubMed] [Google Scholar]
- (160).Barnes NM, and Sharp T (1999) A review of central 5-HT receptors and their function. Neuropharmacology 38, 1083–1152. [DOI] [PubMed] [Google Scholar]
- (161).Bruchas MR, Xu M, and Chavkin C (2008) Repeated swim stress induces kappa opioid-mediated activation of extracellular signal-regulated kinase 1/2. NeuroReport 19, 1417–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (162).Land BB, Bruchas MR, Lemos JC, Xu M, Melief EJ, and Chavkin C (2008) The dysphoric component of stress is encoded by activation of the dynorphin kappa-opioid system. J. Neurosci 28, 407–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (163).Shippenberg TS, Zapata A, and Chefer VI (2007) Dynorphin and the pathophysiology of drug addiction. Pharmacol. Ther 116, 306–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (164).Mu P, Neumann PA, Panksepp J, Schluter OM, and Dong Y (2011) Exposure to cocaine alters dynorphin-mediated regulation of excitatory synaptic transmission in nucleus accumbens neurons. Biol. Psychiatry 69, 228–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (165).Mato S, Chevaleyre V, Robbe D, Pazos A, Castillo PE, and Manzoni OJ (2004) A single in-vivo exposure to delta 9THC blocks endocannabinoid-mediated synaptic plasticity. Nat. Neurosci 7, 585–586. [DOI] [PubMed] [Google Scholar]
- (166).Mato S, Robbe D, Puente N, Grandes P, and Manzoni OJ (2005) Presynaptic homeostatic plasticity rescues long-term depression after chronic Delta 9-tetrahydrocannabinol exposure. J. Neurosci 25, 11619–11627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (167).Bystrowska B, Smaga I, Frankowska M, and Filip M (2014) Changes in endocannabinoid and N-acylethanolamine levels in rat brain structures following cocaine self-administration and extinction training. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 50, 1–10. [DOI] [PubMed] [Google Scholar]
- (168).Stice E, Figlewicz DP, Gosnell BA, Levine AS, and Pratt WE (2013) The contribution of brain reward circuits to the obesity epidemic. Neurosci. Biobehav. Rev 37, 2047–2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (169).Kirkham TC, Williams CM, Fezza F, and Di Marzo V (2002) Endocannabinoid levels in rat limbic forebrain and hypothalamus in relation to fasting, feeding and satiation: stimulation of eating by 2-arachidonoyl glycerol. Br. J. Pharmacol 136, 550–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (170).Harrold JA, Elliott JC, King PJ, Widdowson PS, and Williams G (2002) Down-regulation of cannabinoid-1 (CB-1) receptors in specific extrahypothalamic regions of rats with dietary obesity: a role for endogenous cannabinoids in driving appetite for palatable food? Brain Res 952, 232–238. [DOI] [PubMed] [Google Scholar]