

Abstract
Type 1 diabetes (T1D) is a T cell-mediated autoimmune disease in which insulin-producing β-cells are destroyed. Although butyrophilin-like 2 (BTNL2) has been shown to be a negative T cell regulator in vitro, its ability to inhibit T cell responses in vivo has not been determined. In this study, we determined the effect of a recombinant BTNL2-IgG2a Fc (rBTNL2-Ig) fusion protein on T1D development in vivo. We show here that in vivo administration of rBTNL2-Ig ameliorates T1D in NOD mice. This is associated with the ability of rBTNL2-Ig to inhibit the proliferation, activation, and inflammatory cytokine production from autoreactive T cells in vivo. In addition, rBTNL2-Ig treatment increases the generation of regulatory T cells. Our results suggest that targeting the BTNL2 pathway has the potential to be used in the prevention and treatment of patients with T1D.
Keywords: BTNL2, Type 1 Diabetes, autoreactive T cells, regulatory T cells
Introduction
Around 36 million people suffer from Type 1 Diabetes (T1D) worldwide according to a report from the International Diabetes Federation reported in 2015. T1D is an autoimmune disease characterized by the infiltration of lymphocytes into the islets of the pancreas followed by destruction of insulin-secreting beta-cells, leading to high blood-sugar concentrations that needs daily insulin injections. Exogenous insulin therapy is not a cure. If it is not treated properly, T1D can cause long-term complications, such as kidney disease, blindness, heart attacks, strokes and amputations.
The NOD mouse is the widely used animal model for human T1D [1]. NOD mice share many of the characteristics of this human disease. It is generally believed that T lymphocytes (T cells) play a major role in the pathogenesis of T1D. T cell activation requires at least two signals. The first signal is through the interaction between the T-cell receptor (TCR) and peptides presented on MHC molecules. The second signal requires the interaction between co-stimulatory and co-inhibitory molecules on antigen-presenting cells (APCs) and their receptors on T cells. Among the T cell co-stimulatory and co-inhibitory molecules, the B7 family is of central importance. B7 family members include B7–1, B7–2 [2], PD-L1 (B7-H1) [3], PD-L2 (B7-DC) [4], B7-H2 [also called inducible T cell co-stimulator ligand (ICOSL), B7h, B7RP-1, GL50] [5], B7-H3 [6], B7-H4 (B7x, B7S1) [7], B7-H5 (HHLA2) [8], and B7-H6 [9], etc.
Butyrophilin (BTN) and BTN-like (BTNL) molecules share sequence, structural, and functional similarity with the B7 family [10]. Therefore, BTN and BTNL molecules are considered as extended B7 family members [11]. It has been reported that BTNL2 can inhibit T cell proliferation and cytokine production in vitro [12, 13]. However, the ability of BTNL2 to inhibit T cell responses in vivo and to treat T1D has not been reported.
In this study, we determined the effect of rBTNL2-Ig fusion protein on T1D. We demonstrate here that in vivo administration of rBTNL2-Ig ameliorates T1D in NOD mice. This is related to its ability to inhibit T cell proliferation, activation and cytokine production in vivo. rBTNL2-Ig treatment also significantly increases the generation of regulatory T cells (Tregs) in NOD mice.
Methods
Mice
NOD mice were purchased from The Jackson Laboratory (Bar Harbor, ME). All mice were housed, treated, and handled according to a protocol approved by the University of Connecticut Animal Care and Use Committee.
Production of rBTNL2-Ig
rBTNL2-Ig fusion protein that contains the extracellular domain of the mouse BTNL2 (aa27–452) and the constant region of mouse IgG2a was produced by transfecting the gene containing expression vector into HEK-293 cells. The fusion protein was purified from the expression system using Protein G Sepharose 4 Fast Flow according to the manufacturer’s instructions (GE Healthcare). The endotoxin level was less than 0.01 EU/ml of 1 µg of purified rBTNL2-Ig as determined by a Limulus Amebocyte Lysate assay. Recombinant mouse IgG2a Fc protein (Control Ig) was purchased from BXCell (West Lebanon, NH).
In vitro T cell proliferation assay
T cells were purified from female NOD mice by an immunomagnetic system (Miltenyi Biote Inc., Auburn, CA), and the purity of the cells was usually >95%. T cells were stimulated with anti-CD3 antibody (Biolegend) in the presence of rBTNL2-Ig or control Ig. The cells were pulsed with [3H] thymidine (1 µCi/well) (PerkinElmer, Inc., Downers Grove, IL) for12 hours before harvest, and then collected by a cell harvester (Brandel, Missouri City, Texas). [3H] thymidine incorporation was measured by liquid scintillation spectroscopy (PerkinElmer, Inc.).
Treatment protocol
Groups of female NOD mice were injected with indicated doses of recombinant rBTNL2-Ig, or control Ig intraperitoneally (i.p.) twice weekly from 4 to 16 weeks of age. The mice were monitored for blood glucose levels by test strips (Advanced glucose meter, CVS health, USA) every week. Mice with a blood glucose measurement of greater than 250 mg/dL on two consecutive readings were considered diabetic.
Histological analysis
The pancreata were harvested from the mice, fixed with 4% paraformaldehyde and prepared for paraffin sections. The paraffin sections were stained with hematoxylin and eosin (H&E). At least 100 islets were counted and blindly scored. Double staining of insulin and CD45 was developed using primary antibodies followed by FITC- or Texas red-conjugated secondary antibody.
Isolation of islet-infiltrating cells (IICs)
Islets were isolated with collagenase IV (Gibco, USA) digestion of the pancreata, as described previously [14]. The IICs suspension was then subjected to flow cytometric analysis.
Flow cytometric analysis
Single-cell suspensions were generated from spleens as described previously [30]. Single-cell suspensions were stained with fluorochrome-conjugated antibodies against CD4, CD8, CD25, Foxp3, CD69, CD62L and CD44 (BioLegend, Inc, San Diego, CA, USA). Intracellular staining of cytokine γ-interferon (IFNγ) was performed after 4–5 hours of phorbol myristic acid (50 ng/ml, Sigma) plus ionomycin (1 µg/ml; Sigma) stimulation. Isotype-matched controls were included in all experiments. The samples were analyzed on a FACS Calibur or LSRFortessa X-20 Cell Analyzer (BD Biosciences). Data analysis was performed using FlowJo software (Tree Star, Ashland, OR)
Statistical analysis
P values were based on the two-sided Student’s t test. A confidence level above 95% (p <0.05) was determined to be significant.
Results
rBTNL2-Ig inhibits the proliferation and activation of T cells from NOD mice in vitro
It has been shown that rBTNL2-Ig can suppress T cell proliferation and cytokine production from non-diabetes-prone C57BL/6 mice [12, 13]. We determined whether rBTNL2-Ig could affect the autoreactive T-cell response. T cells were purified from female NOD mice, and stimulated with 1 µg/ml anti-CD3 antibody in the presence of graded doses of rBTNL2-Ig or control Ig protein. T cell proliferation was measured by [3H] thymidine incorporation. As shown in Figure 1A, rBTNL2-Ig inhibited anti-CD3-induced T cell proliferation in a dose-dependent manner. Furthermore, rBTNL2-Ig inhibited the T cell proliferation across a range of anti-CD3 concentrations (Figure 1B).
Figure 1.
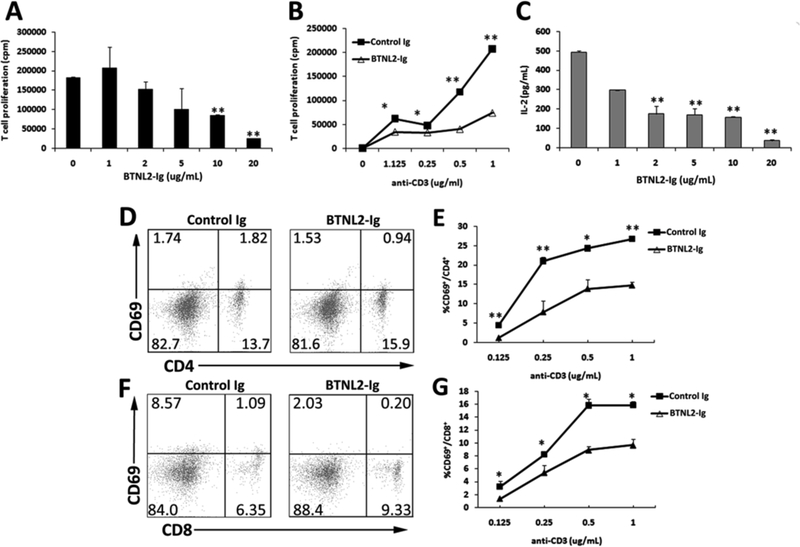
rBTNL2-Ig inhibits T cell function from NOD mice in vitro. Purified CD3+ T cells were stimulated with (A, C) 1 µg/ml plate bound anti-CD3, or (B, D-G) varying concentrations of anti-CD3 in the presence of indicated concentrations of rBTNL2-Ig or control Ig protein for (D-G) 12 h (for early T cell marker CD69 detection), or (A-C) 72 h (for T-cell proliferation and IL-2 production). (A, B) For the T-cell proliferation assay, the cells were pulsed with 1 µCi of [3H]-thymidine 12 hours before harvest. (C) The levels of IL-2 in the supernatant were measured by an ELISA kit. (D-G) T cells were examined for CD69 expression by CD4 and CD8 T cells by flow cytometry. (D, F) Representative flow cytometric profiles and (E, G) are statistical analysis of the percentages of CD69+ cells in CD4 or CD8 T cells. Data represent three independent experiments and are expressed as means ± SEM. *P < 0.05, ** P < 0.01 vs control Ig.
We next examined whether rBTNL2-Ig affects IL-2 production from T cells in vitro. As shown in Figure 1C, the production of IL-2 was reduced by 64.6%, 65.4%, 68.2% and 92.6% in the presence of 2, 5, 10 and μg/mL rBTNL2-Ig, respectively (Fig. 1C). We also determined whether rBTNL2-Ig affects T cell activation. CD69 is an early T-cell activation marker. We found that rBTNL2-Ig inhibited the expression of CD69 by CD4 and CD8 T cells, and the inhibition was across a range of anti-CD3 concentrations (Figure 1D-F). Taken together, these data suggest that rBTNL2-Ig inhibits the proliferation, IL-2 production, and activation of T cells from diabetes-prone NOD mice in vitro.
rBTNL2-Ig treatment ameliorates T1D in NOD mice
We then assessed the effect of rBTNL2-Ig on autoimmune diabetes. To this end, female NOD mice were injected with varying doses of rBTNL2-Ig (25 or 50 µg) or control Ig (25 µg) twice weekly from 4 to 16 weeks of age. The development of diabetes was monitored over time. As shown in Figure 2, 9% and 27% of NOD mice in the control-Ig-treated group developed diabetes at 12 and 16 weeks of age, respectively. In contrast, no NOD mice in the rBTNL2-Ig-treated groups developed diabetes during this period. The results suggest that rBTNL2-Ig treatment delayed diabetes onset in NOD mice.
Figure 2.
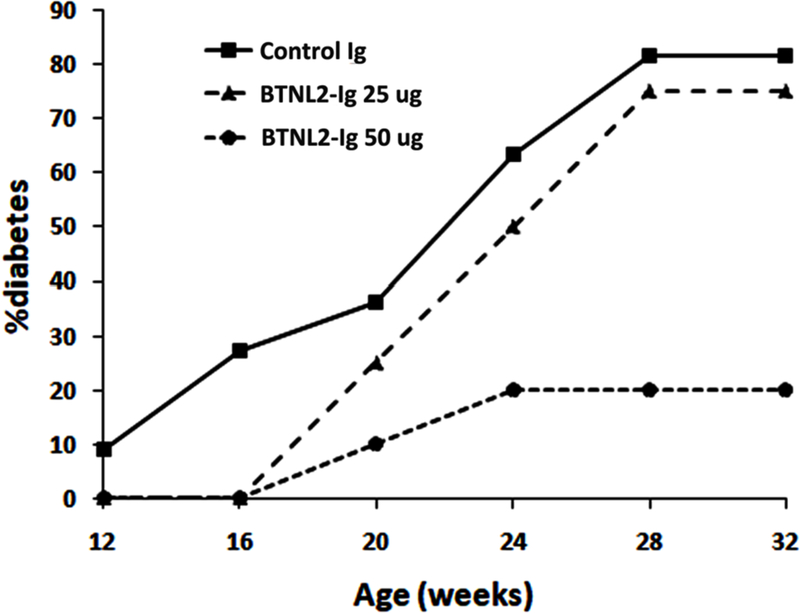
rBTNL2-Ig treatment delays the onset and reduces the incidence of autoimmune diabetes. Female NOD mice were injected i.p. with rBTNL2-Ig (25 or 50 µg) or control Ig (25 µg) twice weekly from 4 to 16 weeks of age. The mice were monitored for blood glucose levels each week. Mice with blood glucose of greater than 250 mg/dL on two consecutive readings were considered diabetic. The percentages of onset of diabetes are plotted at 12, 16, 20, 24, 28 and 32 weeks of age. The data are pooled from 3 independent experiments with 4–6 mice per group.
Beyond 16 weeks of age, the incidence of diabetes was slightly decreased in the 25 µg rBTNL2-Ig-treated group, with 83% incidence in control Ig-treated group, whereas 75% incidence in the rBTNL2-Ig-treated group at week 28. In contrast, the incidence of diabetes was significantly decreased in the 50 µg rBTNL2-Ig-treated group, with 80% vs 20% of incidence in the control Ig- vs rBTNL2-treated group at week 28. The data suggest that treatment with rBTNL2-Ig attenuates diabetes in NOD mice.
rBTNL2-Ig treatment increases the generation of Tregs and inhibits the activation of T cells in the pancreas
Studies have shown that CD4+Foxp3+ Tregs play an important role in the prevention of autoimmune diabetes. Since it has been reported that BTNL2 can increase the generation of Tregs in vitro[15], we analyzed the percentages of Tregs in IICs of the NOD mice. As shown in Figure 3A and B, rBTNL2-Ig treatment increased the percentages of CD4+Foxp3+ Tregs in IICs, as compared to control Ig treatment.
Figure 3.
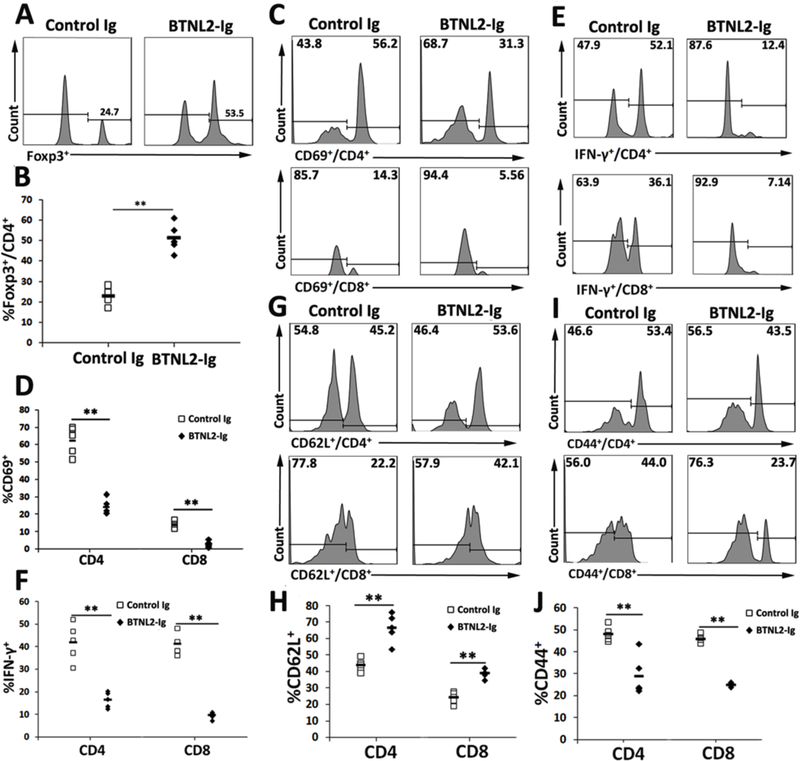
rBTNL2-Ig increases the percentage of Tregs and reduces the activation of T cells in IICs. IICs were harvested from rBTNL2-Ig- or control Ig-treated NOD mice at week 32 of age. The IICs were analyzed for the percentages of (A, B) CD4+Foxp3+ Tregs, (C, D) CD69+ cells in CD4 and CD8 T cells, (E, F) IFNγ-producing CD4 and CD8 T cells, (G, H) CD62L+ cells and (I, J) CD44+ in CD4 and CD8 T cells. (A, C, E, G, I) Representative flow cytometric profiles and (B, D, F, H, J) statistical analysis of means ± SEM. *P < 0.05, ** P < 0.01 vs control Ig. The data are representative of 3 independent experiments with 4–6 mice per group.
We next evaluated the activation of T cells in IICs, and found that rBTNL2-Ig treatment reduced CD69 expression on both CD4 and CD8 T cells (Figure 3C, D). The results suggest that rBTNL2-Ig inhibited the activation of CD4 and CD8 T cells in IICs. We also performed intracellular staining of IFN-γ. As shown in Figure 3E and F, rBTNL2-Ig treatment decreased the percentages of IFNγ–producing CD4 and CD8T cells, suggesting an inhibition of T helper 1 (Th1) and cytotoxic CD8 cell function after rBTNL2-Ig treatment.
To further confirm that rBTNL2-Ig treatment inhibits T cell activation, we analyzed the expression of CD62L and CD44 by CD4 and CD8 T cells in IICs. It has been reported that naïve T cells express CD62L, whereas effective T cells express CD44. As shown in Figure 3G–J, rBTNL2-Ig treatment increased the percentages of CD62L+ cells, but decreased the percentages of CD44+ cells in both CD4 and CD8 T cells. Collectively, our data suggest that rBTNL2-Ig treatment increases the generation of Tregs and reduces the activation of both CD4 and CD8 T cells in IICs.
rBTNL2-Ig also increases Treg generation and suppresses T cell activation in the spleen
We next determined whether the enhanced Treg generation and reduced T cell activation also occurred in the spleen of rBTNL2-Ig-treated NOD mice. As shown in Figure 4A and B, rBTNL2-Ig treatment increased the percentage of CD4+CD25+Foxp3+ Tregs in the spleen. In addition, rBTNL2-Ig reduced the percentages of CD69+ cells (Figure 4C, D) and IFNγ-producing cells (Figure 4E, F) in splenic CD4 and CD8 T cells.
Figure 4.
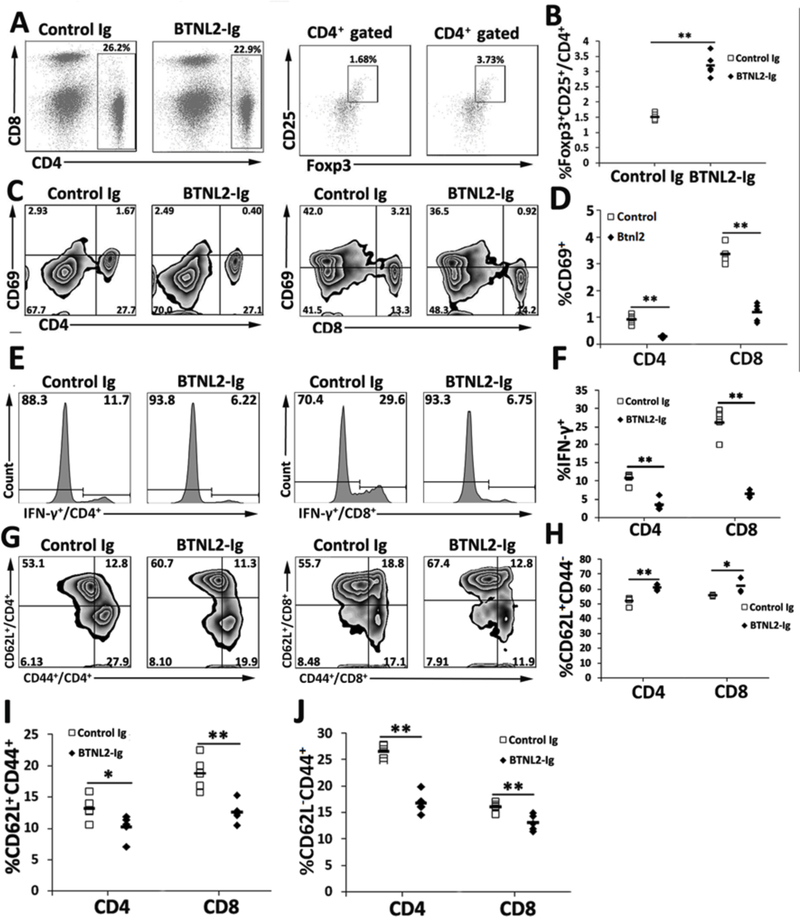
rBTNL2-Ig increases the generation of Tregs and decreases the activation of T cells in the spleen. The spleens were harvested from rBTNL2-Ig or control Ig-treated NOD mice at week 32 of age. The splenocytes were analyzed for the percentages of (A, B) CD4+ CD25+Foxp3+ Tregs, (C, D) CD69+ cells in CD4 and CD8 T cells, (E, F) IFNγ-producing CD4 and CD8 T cells, and (G-J) CD44−CD62L+ naïve, CD44+CD62L− effective memory, and CD44+CD62L+ central memory CD4 and CD8 T cells. (A, C, E, G, I) Representative flow cytometric profiles and (B, D, F, H, J) statistical analysis of means ± SEM. *P < 0.05, ** P < 0.01 vs control Ig. The data are representative of 3 independent experiments with 4–6 mice per group.
T cells can be divided into CD44−CD62L+ naïve, CD44+CD62L− effective memory, and CD44+CD62L+ central memory T cells based on the expression of CD44 and CD62L. We examined these T cell subsets in the NOD mice. As shown in Figure 4G–J, rBTNL2-Ig treatment increased the percentages of naïve T cells and decreased the percentages of effective memory and central memory CD4 and CD8 T cells. Taken together, our results suggest that rBTNL2-Ig treatment enhances the generation of Tregs and inhibits the activation and IFNγ cytokine production from T cells in the spleen.
rBTNL2-Ig reduced the inflammation infiltration of islet
We also performed histological examination with H&E and immunofluorescent staining to determine the degree of insulitis in the NOD mice at 32 weeks of age. In control Ig-treated mice, the patterns of insulitis were visible and the islets were infiltrated seriously. In contrast, in rBTNL2-Ig-treated mice, the degree of inflammation was decreased and the islets maintained nearly intact morphology (Fig. 5A). We counted the islets under a microscope and evaluated the scores. We found that the percentages of islets with low scores increased and islets with high scores decreased after rBTNL2-Ig treatment (Fig. 5B).
Figure 5.
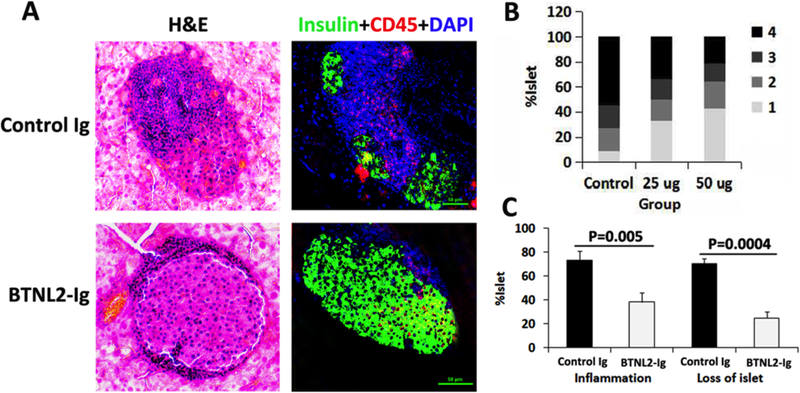
rBTNL2-Ig treatment attenuates insulitis in NOD mice. Pancreata were harvested from control Ig- and rBTNL2-Ig-treated NOD mice at 32 weeks of age. Sections of pancreata were subjected to H&E staining, or immunofluorescent staining with anti-insulin and anti-CD45 antibodies. (A) Representative sections of H&E and immunofluorescent staining. (B) Insulitis scores based on H&E staining. Scores were given as follows: score 1, free of infiltrates; score 2, <25%; score 3, 25–50%, and score 4, >50% infiltrates per islet. (C) Percentages of inflammation and loss of islets calculated according to the severity of intra islet infiltrates and insulin staining, respectively. The data are representative of 3 independent experiments with 4–6 mice per group.
We then calculated the percentages of islets with inflammation infiltration and loss. As shown in Figure 5C, rBTNL2-Ig treatment significantly reduced both inflammation and loss of islets, as compared to control Ig treatment.
Discussion
We show here that in vivo administration of rBTNL2-Ig attenuates autoimmune diabetes in NOD mice. To the best of our knowledge, this is the first report that rBTNL2-Ig fusion protein is able to ameliorate T1D in vivo. The NOD mouse is the best available animal model for human T1D [1]. The development of diabetes in NOD mice is the result of an imbalance of pathogenic/activated T cells and Tregs. Our data suggest that rBTNL2-Ig inhibits pathogenic/activated T cells by affecting their proliferation, activation, and inflammatory cytokine production.
It has been reported that the putative receptor for BTNL2 is expressed on activated T cells [12, 13], suggesting that BTNL2 can directly acts on T cells. This notion is supported by our data and those from others showing that rBTNL2-Ig inhibits the proliferation of purified T cells [12, 13]. The BTNL2 putative receptor seems to be distinct from the receptors for the known B7 family molecules because the BTNL2 protein failed to bind to these receptors [12, 13]. The nature of the BTNL2 receptor remains to be determined. It has also been shown that rBTNL2-Ig inhibits proximal TCR signals including AP-1, NFAT and NFκB in DO11.10 hybridoma cells [13]. The BTNL2-induced signal pathways in T cells also need to be further investigated.
We have, in addition, demonstrated that rBTNL2-Ig treatment significantly increased the generation of Tregs in NOD mice. Our results are consistent with previous report that BTNL2 can induce the production of Tregs in vitro [15]. Tregs typically express high levels of CD25 and the transcription factor Foxp3. The thymus supports the development of Tregs, generating so-called natural Tregs that populate the periphery. Since the phenotypes of the BTNL2-induced Tregs are similar to thymus-derived Tregs [15], it is possible that rBTNL2-Ig treatment enhances the development of natural Tregs in the thymus. However, the possibility that the increased production of Tregs in BTNL2-Ig-treated NOD mice is caused by enhanced production of induced Tregs in the periphery cannot be excluded.
The BTNL2 mutation has been associated with many autoimmune diseases, such as sarcoidosis, ulcerative colitis, Kawasaki disease, osteoarthritis, systemic lupus erythematosus, multiple sclerosis, Graves’ disease, Crohn’s disease, and rheumatoid arthritis [16]. BTNL2 is thought to play a role in limiting unwanted inflammation and enhancing the generation of Tregs. Our results showing that rBTNL2-Ig treatment attenuates T1D in mice are consistent with these results.
In conclusion, we have shown that in vivo administration of rBTNL2-Ig ameliorates T1D in NOD mice. Both the inhibition of pathogenic/activated T cells and generation of Tregs likely play a critical role in attenuating T1D. The ability of rBTNL2-Ig to negatively regulate autoimmune diabetes in NOD mice provides proof of principle for further investigation. More research is needed to understand the mechanisms by which BTNL2 attenuates autoimmune diabetes. Our results suggest that rBTNL2-Ig has the potential to be used in the clinical treatment of T1D.
Acknowledgments
Funding
This work was supported by grants from NIH (1R01AI123131-01, LL) and Connecticut Regenerative Medicine Research Fund (16-RMB-UCONN-02, LL).
Footnotes
Conflict of Interest
The authors declare that they have no conflict of interest.
References
- [1].Anderson MS, Bluestone JA, Annu. Rev. Immunol 2005, 23, 447; [DOI] [PubMed] [Google Scholar]; Atkinson MA, Leiter EH, Nat. Med 1999, 5, 601; [DOI] [PubMed] [Google Scholar]; Roep BO, Atkinson M, von Herrath M, Nat. Rev. Immunol 2004, 4, 989. [DOI] [PubMed] [Google Scholar]
- [2].Freeman GJ, Gray GS, Gimmi CD, Lombard DB, Zhou LJ, White M, Fingeroth JD, Gribben JG, Nadler LM, J. Exp. Med 1991, 174, 625; [DOI] [PMC free article] [PubMed] [Google Scholar]; Freeman GJ, Gribben JG, Boussiotis VA, Ng JW, Restivo VA Jr., Lombard LA, Gray GS, Nadler LM, Science (New York, N.Y.) 1993, 262, 909. [DOI] [PubMed] [Google Scholar]
- [3].Dong H, Zhu G, Tamada K, Chen L, Nat. Med 1999, 5, 1365; [DOI] [PubMed] [Google Scholar]; Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, Fitz LJ, Malenkovich N, Okazaki T, Byrne MC, Horton HF, Fouser L, Carter L, Ling V, Bowman MR, Carreno BM, Collins M, Wood CR, Honjo T, J. Exp. Med 2000, 192, 1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, Iwai Y, Long AJ, Brown JA, Nunes R, Greenfield EA, Bourque K, Boussiotis VA, Carter LL, Carreno BM, Malenkovich N, Nishimura H, Okazaki T, Honjo T, Sharpe AH, Freeman GJ, Nat. Immunol 2001, 2, 261; [DOI] [PubMed] [Google Scholar]; Tseng SY, Otsuji M, Gorski K, Huang X, Slansky JE, Pai SI, Shalabi A, Shin T, Pardoll DM, Tsuchiya H, J. Exp. Med 2001, 193, 839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Wang S, Zhu G, Chapoval AI, Dong H, Tamada K, Ni J, Chen L, Blood 2000, 96, 2808; [PubMed] [Google Scholar]; Ling V, Wu PW, Finnerty HF, Bean KM, Spaulding V, Fouser LA, Leonard JP, Hunter SE, Zollner R, Thomas JL, Miyashiro JS, Jacobs KA, Collins M, Journal of immunology (Baltimore, Md. : 1950) 2000, 164, 1653; [DOI] [PubMed] [Google Scholar]; Swallow MM, Wallin JJ, Sha WC, Immunity 1999, 11, 423; [DOI] [PubMed] [Google Scholar]; Yoshinaga SK, Whoriskey JS, Khare SD, Sarmiento U, Guo J, Horan T, Shih G, Zhang M, Coccia MA, Kohno T, Tafuri-Bladt A, Brankow D, Campbell P, Chang D, Chiu L, Dai T, Duncan G, Elliott GS, Hui A, McCabe SM, Scully S, Shahinian A, Shaklee CL, Van G, Mak TW, Senaldi G, Nature 1999, 402, 827. [DOI] [PubMed] [Google Scholar]
- [6].Chapoval AI, Ni J, Lau JS, Wilcox RA, Flies DB, Liu D, Dong H, Sica GL, Zhu G, Tamada K, Chen L, Nat. Immunol 2001, 2, 269. [DOI] [PubMed] [Google Scholar]
- [7].Prasad DV, Richards S, Mai XM, Dong C, Immunity 2003, 18, 863; [DOI] [PubMed] [Google Scholar]; Sica GL, Choi IH, Zhu G, Tamada K, Wang SD, Tamura H, Chapoval AI, Flies DB, Bajorath J, Chen L, Immunity 2003, 18, 849; [DOI] [PubMed] [Google Scholar]; Zang X, Loke P, Kim J, Murphy K, Waitz R, Allison JP, Proceedings of the National Academy of Sciences of the United States of America 2003, 100, 10388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Zhao R, Chinai JM, Buhl S, Scandiuzzi L, Ray A, Jeon H, Ohaegbulam KC, Ghosh K, Zhao A, Scharff MD, Zang X, Proceedings of the National Academy of Sciences of the United States of America 2013, 110, 9879; [DOI] [PMC free article] [PubMed] [Google Scholar]; Zhu Y, Yao S, Iliopoulou BP, Han X, Augustine MM, Xu H, Phennicie RT, Flies SJ, Broadwater M, Ruff W, Taube JM, Zheng L, Luo L, Zhu G, Chen J, Chen L, Nature communications 2013, 4, 2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Brandt CS, Baratin M, Yi EC, Kennedy J, Gao Z, Fox B, Haldeman B, Ostrander CD, Kaifu T, Chabannon C, Moretta A, West R, Xu W, Vivier E, Levin SD, J. Exp. Med 2009, 206, 1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Abeler-Dorner L, Swamy M, Williams G, Hayday AC, Bas A, Trends Immunol 2012, 33, 34; [DOI] [PubMed] [Google Scholar]; Afrache H, Gouret P, Ainouche S, Pontarotti P, Olive D, Immunogenetics 2012, 64, 781; [DOI] [PubMed] [Google Scholar]; Arnett HA, Viney JL, Nat. Rev. Immunol 2014, 14, 559; [DOI] [PubMed] [Google Scholar]; Guo Y, Wang AY, Front Immunol 2015, 6, 421; [DOI] [PMC free article] [PubMed] [Google Scholar]; Rhodes DA, Reith W, Trowsdale J, Annu. Rev. Immunol 2016, 34, 151. [DOI] [PubMed] [Google Scholar]
- [11].Linsley PS, Peach R, Gladstone P, Bajorath J, Protein Sci 1994, 3, 1341; [DOI] [PMC free article] [PubMed] [Google Scholar]; Yang Y, Liu XK, Nguyen T, Bishop C, Graf D, Dong C, Journal of immunology (Baltimore, Md. : 1950) 2007, 178, 3661; [DOI] [PubMed] [Google Scholar]; Compte E, Pontarotti P, Collette Y, Lopez M, Olive D, European journal of immunology 2004, 34, 2089. [DOI] [PubMed] [Google Scholar]
- [12].Arnett HA, Escobar SS, Gonzalez-Suarez E, Budelsky AL, Steffen LA, Boiani N, Zhang M, Siu G, Brewer AW, Viney JL, J. Immunol 2007, 178, 1523. [DOI] [PubMed] [Google Scholar]
- [13].Nguyen T, Liu XK, Zhang Y, Dong C, J. Immunol 2006, 176, 7354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Holdcraft RW, Green ML, Breite AG, Circle L, Meyer ED, Adkins H, Harbeck SG, Smith BH, Gazda LS, Transplantation direct 2016, 2, e86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Swanson RM, Gavin MA, Escobar SS, Rottman JB, Lipsky BP, Dube S, Li L, Bigler J, Wolfson M, Arnett HA, Viney JL, J. Immunol 2013, 190, 2027. [DOI] [PubMed] [Google Scholar]
- [16].Valentonyte R, Hampe J, Huse K, Rosenstiel P, Albrecht M, Stenzel A, Nagy M, Gaede KI, Franke A, Haesler R, Koch A, Lengauer T, Seegert D, Reiling N, Ehlers S, Schwinger E, Platzer M, Krawczak M, Muller-Quernheim J, Schurmann M, Schreiber S, Nat. Genet 2005, 37, 357; [DOI] [PubMed] [Google Scholar]; Rybicki BA, Walewski JL, Maliarik MJ, Kian H, Iannuzzi MC, Am. J. Hum. Genet 2005, 77, 491; [DOI] [PMC free article] [PubMed] [Google Scholar]; Price P, Santoso L, Mastaglia F, Garlepp M, Kok CC, Allcock R, Laing N, Tissue Antigens 2004, 64, 575; [DOI] [PubMed] [Google Scholar]; Juyal G, Prasad P, Senapati S, Midha V, Sood A, Amre D, Juyal RC, Bk T, PloS one 2011, 6, e16565; [DOI] [PMC free article] [PubMed] [Google Scholar]; Mochida A, Kinouchi Y, Negoro K, Takahashi S, Takagi S, Nomura E, Kakuta Y, Tosa M, Shimosegawa T, Tissue Antigens 2007, 70, 128; [DOI] [PubMed] [Google Scholar]; Pathan S, Gowdy RE, Cooney R, Beckly JB, Hancock L, Guo C, Barrett JC, Morris A, Jewell DP, Tissue Antigens 2009, 74, 322; [DOI] [PubMed] [Google Scholar]; Moller M, Kwiatkowski R, Nebel A, van Helden PD, Hoal EG, Schreiber S, Microbes Infect 2007, 9, 522; [DOI] [PubMed] [Google Scholar]; Orozco G, Eerligh P, Sanchez E, Zhernakova S, Roep BO, Gonzalez-Gay MA, Lopez-Nevot MA, Callejas JL, Hidalgo C, Pascual-Salcedo D, Balsa A, Gonzalez-Escribano MF, Koeleman BP, Martin J, Hum. Immunol 2005, 66, 1235. [DOI] [PubMed] [Google Scholar]