

Abstract
We report a series of glutamate and aspartate analogues designed using the hydroxy-1,2,3-triazole moiety as a bioisostere for the distal carboxylic acid. Compound 6b showed unprecedented selectivity among AMPA receptor subtypes, confirmed also by an unusual binding mode observed on the crystal structures in complex with the AMPA receptor GluA2 agonist binding domain. Here, a methionine (Met729) was highly disordered compared to previous agonist-bound structures. This observation provides a possible explanation for the pharmacological profile. In the structure with 7a, an unusual organization of water molecules around the bioisostere arises compared to previous structures of ligands with other bioisosteres. Aspartate analogue 8 with the hydroxy-1,2,3-triazole moiety directly attached to glycine was unexpectedly able to activate both the glutamate and glycine agonist binding sites of the NMDA receptor. These observations demonstrate novel features that arise when employing a hydroxyl-triazole moiety as bioisostere for the distal carboxylic acid in glutamate receptor agonists.
Keywords: Bioisosterism; Glutamic Acid; AMPA; Hydroxy-1,2,3-Triazole; NMDA
Graphical Abstract.
1. INTRODUCTION
(S)-Glutamic acid (glutamate, Glu, 1, Figure 1) is the major excitatory neurotransmitter in the central nervous system (CNS) and is crucial to normal brain functions. However, the neurotransmitter is also involved in a number of brain disorders.1-5 The effects of Glu are mediated by a highly heterogeneous receptor population comprising G protein-coupled metabotropic Glu receptors and ionotropic Glu receptors (iGluRs).1 The iGluRs are ligand-gated ion-channels that are divided into three major classes according to amino acid sequence identity and to their respective activation by the agonists N-methyl-D-aspartic acid (NMDA), (S)-2-amino-3-(3-hydroxy-5-methyl-isoxazol-4-yl)propionic acid ((S)-AMPA, 2) and kainic acid (kainate, KA).2 The iGluRs are homo- or heterotetrameric assemblies of various subunits; NMDA receptors being made up of the GluN1, GluN2A-D, and GluN3A-B subunits, AMPA receptors of the GluA1-4 subunits, and KA receptors of the GluK1-5 subunits. The majority of functional NMDA receptors in the CNS are heteromeric GluN1/GluN2 receptors composed by two GluN1 subunits that recognize the endogenous co-agonist glycine (or d-serine) and two GluN2 subunits that recognize Glu, whereas KA and AMPA receptors exist as both homo- and heteromeric combinations of their respective cloned subunits. X-ray crystallographic studies of full-length AMPA, KA and NMDA receptors6, 7 have established that the iGluRs are tetrameric assemblies 8-14 consisting of four structural subdomains: the intracellular C-terminal domain, the transmembrane domain forming the ion channel, the agonist binding domain (ABD), and the large extracellular N-terminal domain. A large number of structures of soluble iGluR ABDs constructs have revealed important information on the molecular basis for orthosteric ligand recognition, and the mechanisms underlying activation, desensitization, and allosteric modulation of the receptors. 15-17
Figure 1.

Structures of Glutamic acid, Kainic acid, NMDA, selective AMPA analogues and hydroxy-1,2,3-triazole analogues 6a-c, 7a-c and 8.
A large number of ligands showing selectivity among the three classes of iGluRs have been disclosed, but few ligands have been developed with specificity for the individual receptor subtypes within the classes, presumably due to high sequence identity among the subunits within each class. In particular, selectivity towards individual AMPA receptor subunits has been difficult to achieve. Nevertheless, some compounds (Figure 1) such as (RS)-2-amino-3-(4-chloro-3-hydroxyisoxazol-5-yl)propionic acid (Cl-HIBO, 3) and (RS)-3-hydroxy-4,5,6,7-tetrahydroisoxazolo[5,4-c]pyridine-7-carboxylic acid (7-HPCA, 4) are able to selectively activate GluA1/2,18, 19 exploiting the only difference between GluA1/2 and Glu3/4 among the amino acids in direct contact with Glu. Some preference towards GluA1 and GluA3 has been achieved by the continued development of MeTetAMPA (5a) through BnTetAMPA (5b) to aminomethyl and chloro substituted BnTetAMPA analogues 5c and 5d, respectively.20-22 The protrusion of the benzyl group in BnTetAMPA into a less conserved area is enabled by the movement of a conserved methionine (Met729 in GluA2) in the binding pocket, leading to the observed variation in the selectivity profiles of these analogues.
In recent years, we have been focusing on improving the strategy of scaffold hopping to replace the acidic moieties by acidic hydroxylated azoles in different lead compounds.23 Using this strategy, we successfully designed a new class of DHODH (dihydroorotate dehydrogenase) inhibitors,24-31 AKR1C3 (Aldo-Keto Reductase 1C3) inhibitors,32-34 inhibitors of the canonical NF-κB cascade 35-36 GABA (gamma-aminobutyric acid) analogues37-39 as well as Glu analogues.40 In light of the promising results obtained in this previous work, and in an attempt to explore the selectivity towards iGluRs, the hydroxy-1,2,3-triazole scaffold was used to bioisosterically replace the distal carboxylic group of Glu. For the AMPA analogues 6a and 7a 41 we previously reported that the hydroxy-1,2,3-triazole scaffold provides the opportunity to place a substituent oriented in two different directions as opposed to the isoxazolol moiety in AMPA. Here, we describe the synthesis and extensive pharmacological characterization at native and recombinant iGluRs of the Glu analogues 6a-c, 7a-c and the aspartate analogue 8 along with X-ray crystallographic studies of compounds 6b and 7a bound in the GluA2 ABD.
2. RESULTS
2.1. Chemistry: synthesis of target compounds 6a-c, 7a-c and 8.
The synthesis of racemic hydroxy-1,2,3-triazole Glu analogues 6a-c and 7a-c (Scheme 1) follows our previously published procedure, starting from the isomeric triazole building blocks 9a-c and 10a-c,41 which are obtained in a robust methodology for preparation of regio-substituted hydroxy-1,2,3-triazoles. Compounds 9a-c and 10a-c were first reduced to their corresponding alcohols 11a-c and 12a-c with NaBH4 in EtOH, subsequently reacted with N-bromosuccinimide (NBS) to yield 13a-c and 14a-c. Reaction with diethyl-2-acetamidomalonate in the presence of NaH followed by catalytic hydrogenation at atmospheric pressure afforded compounds 17a-c and 18a-c. Final deprotection of 17a-c and 18a-c in 6N HCl at reflux gave, after ion exchange chromatography, the zwitterion racemic mixture of compounds 6a-c and 7a-c in quantitative yields.
Scheme 1.

Synthesis of hydroxy-1,2,3-triazole Glu analogues 6a-c and 7a-c: i) NaBH4, EtOH abs; ii) NBS, (Ph)3P, dry DCM, −10°C; iii) diethyl-2-acetamidomalonate, NaH, dry THF; iv) H2, Pd/C 10% w/w, dry THF; v) a) 6N HCl, reflux; b) strong acidic cation exchange resin (elution with water until neutral pH. Finally the compound was eluted with 10% NH3 solution).
The synthesis of the zwitterion racemic mixture of triazole NMDA analogue 8 (Scheme 2), started from 14a treated with NaCN in ethanol to yield the nitrile 19. The latter subsequently reacted with trimethylchlorosilane (TMSCl) and ethanol to afford the ester 20. The NMDA analogues 8 was obtained by nitrosation of 20 and subsequent reduction of oxime 21 by catalytic hydrogenation at atmospheric pressure, followed by immediate hydrochloric acid hydrolysis of the intermediate ester 22.
Scheme 2.

Synthesis of compound 8: i) NaCN, EtOH / H2O 9:1 v/v; ii) TMSCl, abs EtOH, 50 °C; iii) NaH, dry ethanol, EtONO; iv) a) H2, Pd/C, dry ethanol; b) HCl gas, dry ethanol; v) a) 6N HCl, reflux; b) strong acidic cation exchange resin (elution with water until neutral pH, then recover of the desired compound by elution with 10% w/w NH3 solution).
2.2. Pharmacology.
Initially, compounds 6b-c, 7b-c, and 8 were characterized at the major iGluR subgroups in a radioligand binding assay using rat cortical synaptosomes (data for 6a and 7a were previously obtained and are reported in ref. 37 Table 1). Compounds like 6a-6c and 7a showed selective but moderate (μM) activity at AMPA receptors. 6a is a close analogue of AMPA and displayed an IC50 value of 1.4 μM in the [3H]AMPA binding assay, which is 100-fold higher than that displayed by AMPA itself. The ethyl and propyl substituted analogues 6b and 6c displayed similar potencies of 0.98 and 5.20 μM, respectively. In 7a, the movement of the methyl group to the neighboring nitrogen gives 7a similar potency as 6a at AMPA receptors (IC50 = 3.1 μM), although in this case, ethyl and propyl substitutions (7b and 7c) were not tolerated. Compound 8 displayed slightly weaker affinity to NMDA receptors (Ki = 34 μM) than NMDA itself (Ki = 6.2 μM).
Table 1.
Receptor binding at native iGluRs in rat brain synaptosomesa.
| Compound | [3H]AMPA IC50(μM) |
[3H]KA IC50(μM) |
[3H]CGP39653 Ki (μM) |
|---|---|---|---|
| (S)-Glu 42 | 0.34 | 0.38 | 0.20 |
| AMPA 20 | 0.039 | >100 | >100 |
| NMDA 42 | >100 | >100 | 6.2 |
| KA 43 | 4.0 | 0.007 | >100 |
| 6a 41 | 1.4 | > 100 | > 100 |
| 6b | 0.98 [6.03 ± 0.09] |
> 100 | > 100 |
| 6c | 5.2 [5.30 ± 0.09] |
> 100 | > 100 |
| 7a 41 | 3.1 | > 100 | > 100 |
| 7b | > 100 | > 100 | > 100 |
| 7c | > 100 | > 100 | > 100 |
| 8 | > 100 | > 100 | 34 [4.49 ± 0.09] |
AMPA, KA and NMDA receptors were studied using [3H]AMPA, [3H]KA and [3H]CGP 39653 as radioligands, respectively. Data are mean values of three individual experiments performed in triplicate. For 6b and 6c: pIC50 values with SEM in brackets. For 8: pKi values with SEM in brackets.
The AMPA receptor selectivity among compounds 6a-c, and 7a-c prompted an initial functional screening in a Ca2+/Fluo-4 assay using HEK293 cell lines expressing GluA1-, GluA2(Q)-, GluA3- and GluA4 (Table 2) to probe whether the compounds are agonists at the AMPA receptors. This demonstrated that all the compounds having affinity towards AMPA receptors displayed significant agonist activity (>5% of the maximal response induced by (S)-Glu) with an order of magnitude lower potency than Glu at GluA4. Full curves could not be generated at the other subtypes due to weaker agonist potencies in general using this assay, and therefore it was not possible determine any subtype selectivity of the compounds.
Table 2.
Functional properties of Glu and six hydroxy-1,2,3-triazole Glu analogues at GluA1-, GluA2(Q)-, GluA3- and GluA4-HEK293 cell lines in the Ca2+/Fluo-4 assay.
| Compound | GluA1i EC50 [pEC50 ± SEM] |
GluA2i(Q) EC50 [pEC50±SEM] |
GluA3i EC50 [pEC50±SEM] |
GluA4i EC50 [pEC50±SEM] |
|---|---|---|---|---|
| Glu | 33 [4.48 ± 0.07] |
57 [4.25 ± 0.03] |
24 [4.63 ± 0.05] |
4.8 [5.31 ± 0.02] |
| 6a | SAR@ 300 | SAR@ 300 | SAR@ 300 | 55 [4.25 ± 0.03] |
| 6b | SAR@ 100 | SAR@ 300 | SAR@ 100 | 35 [4.46 ± 0.07] |
| 6c | SAR@ 1000 | SAR@ 1000 | SAR@ 300 | 51 [4.29 ± 0.07] |
| 7a | SAR@ 1000 | SAR@ 1000 | SAR@ 1000 | ~100 [~4.0] |
| 7b | >1000a | >1000a | >1000a | >1000a |
| 7c | >1000a | >1000a | >1000a | SAR@ 1000 |
SAR@, significant agonist response >5% of the maximal response induced by (S)-Glu at concentrations indicated,
tested both as agonist and antagonist (using Glu EC80). All compounds were tested at concentrations up to 1000 μM, and an assay buffer containing 10 mM Ca2+ and 100 μM cyclothiazide was used in the experiments. EC50 values and concentrations for which significant agonist responses were observed (in those cases for which a complete concentration-response curves could not be obtained) are given in μM. pEC50 ± SEM are shown in brackets.
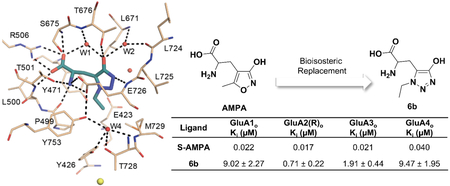
Following these results, we decided to investigate whether subtype selectivity could be observed among the AMPA receptor subtypes with these new bioisosteric analogues. We determined the binding affinities of selected compounds at cloned rat AMPA receptor subtypes expressed in Sf9 insect cells (Table 3). In general, the binding affinities were of the same magnitude (low micromolar) as in the synaptosome assay, but showing variation among subtypes. Some compounds (6a-c and 7a) showed an order of magnitude weaker affinity towards GluA1 than GluA2. The most potent compound 6b, with an affinity of 0.71 μM at GluA2 showed an order of magnitude weaker binding at both GluA1 and GluA4. This compound was further characterized functionally using two-electrode voltage-clamp electrophysiology with Xenopus oocytes expressing recombinant homomeric rat GluA2(Q)i. This assay demonstrated that the compound 6b was a partial agonist with EC50 of 65 ± 6 μM and Imax of 0.863 ± 0.015, where Glu Imax = 1.000 (Figure 2).
Table 3.
Binding of 4-hydroxy-1,2,3-triazole Glu analogues to AMPA receptor subtypes.
| Ligand | GluA1o | GluA2(R)o | GluA3o | GluA4o | GluA2o-ABD | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ki (μM) | nH | Ki (μM) | nH | Ki (μM) | nH | Ki (μM) | nH | Ki (μM) | nH | |
| (S)-2 | 0.022 | n.d. | 0.017 | n.d. | 0.021 | n.d. | 0.040 | n.d. | n.d. | n.d. |
| 5a | 0.0052 | n.d. | 0.0039 | n.d. | 0.0022 | n.d. | 0.0027 | n.d. | n.d. | n.d. |
| 5b | 7.7 | n.d. | 0.75 | n.d. | 0.39 | n.d. | 0.20 | n.d. | n.d. | n.d. |
| 6a | 15.7 ± 1.1# | 1.02 ± 0.17 | 1.87 ± 0.72# | 0.95 ± 0.09 | 4.98 ± 0.58 | 0.95 ± 0.25 | 4.08 ± 1.66 | 0.89 ± 0.07 | n.d. | n.d. |
| 6b | 9.02 ± 2.27## | 0.62 ± 0.04 | 0.71 ± 0.22**,## | 0.99 ± 0.04 | 1.91 ± 0.44 | 0.99 ± 0.03 | 9.47 ± 1.95 | 0.72 ± 0.03 | 0.32 ± 0.03** | 1.01 ± 0.15 |
| 6c | 54.1 ± 8.6### | 0.81 ± 0.07 | 3.25 ± 0.47### | 1.55 ± 0.65 | 11.9 ± 2.9 | 0.75 ± 0.09 | ≈ 100 | n.d. | n.d. | n.d. |
| 7a | 73 ± 34 | 0.97 ± 0.17 | 4.38 ± 2.18* | 0.80 ± 0.06 | 4.89 ± 0.51 | 0.82 ± 0.05 | 11.6 ± 2.1 | 0.91 ± 0.24 | 1.97 ± 0.17* | 0.92 ± 0.05 |
| 7b | > 100 | n.d. | > 100 | n.d. | ≈ 100 | n.d. | > 100 | n.d. | n.d. | n.d. |
| 7c | > 100 | n.d. | > 100 | n.d. | > 100 | n.d. | > 100 | n.d. | n.d. | n.d. |
Shown are means ± SEM from n ≥ 3 experiments conducted in triplicate at 12-15 ligand concentrations. n.d., not determined.
GluA2 not statistically significantly different from GluA2-ABD (p > 0.05, t-test).
GluA2 not statistically significantly different from GuA2-ABD (p > 0.05, t-test).
Ki at GluA1 statistically significantly different from GluA2 (p < 0.05, t-test).
Figure 2.

Normalized, averaged concentration-response data at homomeric rat GluA2(Q)i receptors expressed in Xenopus oocytes. Data are given as means ± SEM values of the pooled data. Responses from each oocyte were normalized to the maximum response of each oocyte before averaging. The top of the curve is fixed to 100% and the bottom to 0%. EC50 = 65 ± 6 μM, Hill slope = 1.07 ± 0.08 (n = 6 oocytes). Inset: Representative two-electrode voltage-clamp recording (Vh = −60 mV) with duplicate stimulations (0.3 – 1000 μM) followed by one stimulation at 3000 μM and one final stimulation of 1000 μM Glu.
We investigated the activity of compound 8 at recombinant NMDA receptors using two-electrode voltage-clamp electrophysiology. Initially, the compound was tested at 100 μM for the ability to activate GluN1/2A-D receptors in the absence of both Glu and Gly. Unexpectedly, the compound (100 μM) was able to activate several recombinant NMDA receptors in absence of both Glu and Gly, with the exception of the GluN1/2A subtype (Figure 3B).
Figure 3.

(A) Representative two-electrode voltage-clamp recording of responses from recombinant GluN1/2D receptors expressed in Xenopus oocytes. Reponses were activated by 100 μM compound 8 as indicated by the grey bar, and control responses were activated by co-application of 300 μM Glu plus 100 μM Gly. The horizontal scale bars indicate 30 sec and the vertical scale bars indicate 200 nA. (B) Summary of responses to 100 μM compound 8 alone as percentage of control at recombinant GluN1/2A-D receptors. Data are given as mean ± SEM values from 4-6 oocytes. (C, D) Concentration-response data for compound 8 at recombinant NMDA receptor subtypes measured using two-electrode voltage-clamp recordings in the continuous presence of either 300 μM Glu (C) or 100 μM Gly (D). Responses are normalized to maximal activation by 300 μM Glu plus 100 μM Gly. Data are given as mean ± SEM values from 4 oocytes.
We determined the activity of 8 in the presence of either Glu or Gly to probe the activity on GluN2 and GluN1 agonist binding sites, respectively (Figure 3C-D, Table 4). In the presence of 300 μM Glu (i.e. for activity at GluN1), compound 8 was a full agonist at GluN1 when expressed with GluN2C and GluN2D with potencies of 90 μM and 35 μM, respectively. The compound was a weaker agonist at GluN1/2A and GluN1/2B receptors (full curves could not be generated). We then tested the activity of compound 8 at GluN1/2A-D in the presence of 100 μM Gly (i.e. for activity at GluN2). Compound 8 was a partial agonist at GluN1/2B and GluN1/2D with agonist efficacies of 22% and 58% and potencies of 108 μM and 58 μM, respectively. No response was seen at GluN1/2A and a weak response at GluN1/2C was observed at 300 μM (full curve could not be generated).
Table 4.
Concentration-response data for compound 8 at recombinant NMDA receptors measured using two-electrode voltage-clamp electrophysiology.
| 8 + Glu | 8 + Gly | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| EC50 (μM) | Relative Imax (%) |
nH | n | EC50 (μM) |
Relative Imax (%) |
nH | n | |||
| GluN1/2A | - | 38 ± 1 at 300 μM |
- | 4 | GluN1/2A | - | No response at 300 μM |
- | 3 | |
| GluN1/2B | 52 ± 1 at 300 μM |
4 | GluN1/2B | 108 ± 13 | 22 ± 2 | 1.4 | 4 | |||
| GluN1/2C | 90 ± 4 | 107 ± 2 | 1.7 | 4 | GluN1/2C | - | 22 ± 2 at 300 μM |
- | 4 | |
| GluN1/2D | 35 ± 1 | 98 ± 1 | 1.6 | 4 | GluN1/2D | 58 ± 1 | 58 ± 1 | 1.4 | 3 |
Data for compound 8 + Glu were obtained in the presence of 300 μM Glu, and data for compound 8 + Gly were obtained in the presence of 300 μM Gly. Relative Imax is the fitted maximal response relative to control responses activated by 100-300 μM glutamate plus 100–300 μM glycine in the absence of compound 8. Low activity prevented determination of potencies in some cases (indicated by -). Data are mean ± SEM, nH is the Hill coefficient, and n is the number of oocytes.
2.3. Structure.
We crystallized 6b and 7a with the agonist binding domain of GluA2 (GluA2-ABD). Compound 6b displays the greatest affinity at GluA2 within this new series of compounds, whereas 7a allowed us to explore a novel substitution pattern on the five-membered ring. Both structures crystallized as a GluA2-ABD dimer (Figure 4), and X-ray diffraction data were collected to a resolution of 1.4 Å (6b) and 1.55 Å (7a) (Table 5). Unambiguous electron density was seen for both ligands (Figures 5A and 5D). Full GluA2-ABD domain closure around 6b (22° for chain A and 20° for chain B) and 7a (21° for chain A and B) were observed. The domain closure is similar to that induced by AMPA44 and supports that 6b and 7a function as agonists.
Figure 4.

Structure of GluA2-ABD with 6b (PDB ID: 6Q54). The GluA2-ABD dimer is shown in cartoon representation with chain A in beige and chain B in orange. A grey arrow indicates D1-D2 domain closure. Molecule 6b is shown in cyan sticks representation.
Table 5.
Crystal data, data collection, and refinement statistics of GluA2-ABD structures.
| Crystal Data | ||
|---|---|---|
| Ligand | Compound 6b | Compound 7a |
| Space group | P21212 | P21212 |
| Unit cell dimensions (Å) | 98.33, 121.74, 47.13 | 98.34, 122.25, 47.35 |
| Molecules in a.u.a | 2 | 2 |
| Data collection and processing | ||
| Resolution (Å) | 45.59-1.40 (1.48-1.40)b | 44.15-1.55 (1.63-1.5 |
| Unique reflections | 112,206 (16,195) | 83,759 (12,103) |
| Average multiplicity | 6.8 (6.7) | 6.8 (6.7) |
| Completeness (%) | 100 (99.9) | 100 (100) |
| Wilson B-factor (Å2) | 12.3 | 13.3 |
| Rmerge (%)c | 5.3 (45.8) | 8.7 (36.8) |
| I/σI | 8.3 (1.6) | 4.7 (1.6) |
| CC1/2 | 0.999 (0.935) | 0.997 (0.922) |
| Refinement | ||
| Numbers of: | ||
| Amino-acid residues (chain A/B) | 264 / 263 | 264 / 263 |
| Compound 6b/7a | 2 | 2 |
| Sulfate/glycerol/chloride/lithium/citrate/PEGd/PGEe/water | 10 / 4 / 3 / 3 / 1 / 1 / 2 / 703 | 4 / 12 / 3 / 2 / - / - / - / 751 |
| Rworkf(%) / Rfreeg (%) | 14.4 / 16.3 | 14.8 / 17.5 |
| Average B-values (Å2) for: | ||
| Amino-acid residues (chain A/B) | 21.6 / 20.9 | 24.1 / 21.9 |
| Compound 6b/7a | 15.0 | 15.1 |
| Sulfate/glycerol/chloride/lithium/citrate/PEG/PGE/water | 38.8 / 49.2 / 53.4 / 9.1 / 17.2 / 46.3 / 49.7 / 29.1 | 46.9 / 64.3 / 50.8 / 12.7 / - / - / - / 30.6 |
| RMS deviation bonds length (Å)/angles (deg) | 0.010 / 1.1 | 0.009 / 1.0 |
| Ramachandran outliers/ favored (%) h | 0 / 99.5 | 0 / 99.2 |
| Rotamer ouliers (%)/ Cβ outliers (%)/ Clash Score | 0.6 / 0 / 1.58 | 0.65 / 0 / 2.04 |
a.u.: Asymmetric unit of the crystal.
Values in parentheses correspond to the outermost resolution shell.
Rmerge = ΣhklΣi∣Ii,hkl-Ihkl ∣/ ΣhklΣi∣Ihkl∣, Ii,hkl is the intensity of an individual measurement of the reflection with Miller indices hkl, Ihkl is the intensity from multiple observations.
di(hydroxyethyl)ether.
triethylene glycol.
Rwork = Σhkl( ∥ Fo,hkl∣ - ∣Fc,hkl ∥ )/∣Fo,hkl∣, where ∣Fo,hkl∣ and ∣Fr,hkl∣ are the observed and calculated structure factor amplitudes, respectively.
Rfree is equivalent to Rwork, but calculated with 5 % of the reflections omitted from the refinement process.
The Ramachandran plot was calculated according to MolProbity.45
Figure 5.

Structures of GluA2-ABD with 6b (PDB ID: 6Q54) and 7a (PDB ID: 6Q60). (A) Simple PHENIX omit 2Fo–Fc map around 6b, Arg506 and Met729, carved at 1.6 Å and contoured at 1 sigma (chain A). GluA2-ABD residues within 4 Å of 6b are shown in beige sticks representation. Molecule 6b is shown in cyan sticks representation. (B) Hydrogen-bonding network (up to 3.2 Å; black dashed lines) among binding-site residues, 6b and water molecules (red spheres). (C) Comparison of binding mode of 6b (chain A), AMPA (grey; PDB code 1FTM, chain A) and Glu (yellow; PDB code 1FTJ, chain A). The structures were overlaid on lobe D1 residues. Only water molecules (within 4 Å of the ligands) are shown for clarity and colored according to the respective ligands. (D) Simple PHENIX omit 2Fo-Fc map around 7a, Arg506 and Met729, carved at 1.6 Å and contoured at 1 sigma (chain A). Molecule 7a is shown in salmon sticks representation. (E) Hydrogen bonding network among binding site residues, 7a and water molecules. (F) Zoom on the hydrogen bonding network of W3 and W4 in the GluA2-ABD structures with 6b (cyan), 7a (magenta) and AMPA (grey).
In the GluA2-ABD structure with 6b, the amino-acid moiety of 6b forms the same hydrogen bonds to protein residues as usually seen for agonists (Figure 5B)44 The 4-hydroxy-1,2,3-triazole moiety of 6b makes two direct hydrogen bonds to the receptor: one from the (a)-nitrogen atom to the backbone nitrogen atom of Glu726 and one from the 4-hydroxyl group to the side-chain hydroxyl group of Thr676 (Figure 5B). In addition, the 4-hydroxyl group of 6b forms two water-mediated contacts: one through W1 to Thr676 (backbone N) and the carboxylate group of 6b as well as one through W2 to Leu671 (backbone N) and Leu724 (backbone O). The 1-ethyl substituent is within van der Waals distance (4 Å cut-off) of Glu423, Tyr471, Pro499, Glu726, Met729 and Tyr753 (Figure 5B); a region in GluA2-ABD that has previously been denoted the hydrophobic pocket.44 The side chain of Met729 is poorly defined in the electron density, indicating a high degree of flexibility (Figure 5A). This is in contrast to other binding site residues that are well-defined (see Arg506 in Figure 5A). Met729 has previously been shown to undergo an induced fit depending of the ligand bound.44 In the structure with 6b, the side chain of Met729 bends away from the 1-ethyl substituent.
Interestingly, 6b binds in a mode similar to AMPA (Figure 5C), whereas other AMPA-related agonists containing a larger substituent than a methyl group in the 5-position (corresponding to the N(c)-position in 6b), such as t-butyl group (ATPA)46, 2-methyl-tetrazole (MeTetAMPA)47 or benzyl-tetrazole (BnTetAMPA),22 have been shown to switch to a binding mode similar to Glu.44 Therefore, compound 6a (N(c)-methyl) would most likely also bind in the AMPA binding mode, whereas 6c (N(c)-propyl) probably would switch into the glutamate binding mode due to otherwise steric clash with the residues forming the hydrophobic pocket.
Compound 7a also binds in the AMPA binding mode, and the amino acid part and 1,2,3-triazole moiety of 7a form the same hydrogen bonds to protein residues as seen for 6b (Figure 5E). The introduction of a methyl group in the N(b)-position in 7a and absence of a substituent in the N(c)-position sequesters an additional water molecule (W3), forming contacts to the nitrogen atom in the N(c)-position of 7a, the hydroxyl group of Tyr471 and one water molecule (W4) (Figures 5E and 5F). Both W3 and W4 are absent in the structure of GluA2-ABD with AMPA (PDB code 1FTM48), whereas only W3 is absent in the structure with 6b. The major difference in the GluA2-ABD between the AMPA and 6b / 7a bound structures is the conformation of Met729 that moves further away from the ligand in the structures with 6b and 7a, also affecting the nearby surroundings (Figure 5F). These structural changes might lead to the sequestering of W4, forming contacts to the side chain hydroxyl groups of Tyr426, Thr728 and Tyr753, and also to Met729 (backbone N) in the structure with 6b. In the AMPA bound structure where W4 is not present the side chains of Thr728 and Tyr753 form a direct contact with each other as also seen in the structure with 7a.
Based on the binding affinities of 7b and 7c containing an ethyl and n-propyl group, respectively, in the N(b)-position it was possible to rule out that larger substituents than a methyl group at this position can be accommodated in the receptor (Table 3). This is in agreement with the crystal structure of GluA2-ABD with 7a, where the distance between the N(b)-methyl group and the Cβ atom of Met729 is only 3.4 Å. Therefore, introduction of a larger substituent in the 2-position would lead to steric clash with Met729 or require major structural changes in the receptor.
A 40-fold and 250-fold lower binding affinity of 6b and 7a at GluA2, respectively, compared to AMPA was observed (Table 3). Due to the very similar binding mode of 6b, 7a and AMPA, we find that the most likely explanation for this difference in binding affinity is the difference in pKa values for the 4-hydroxy-1,2,3-triazole moiety (6b: 6.42 and 7a: 6.11)41 compared to that of AMPA (4.8).49 Furthermore, the structural changes of Met729 in the GluA2-ABD accompanied by the difference in binding site water network might also contribute to the observed differences in binding affinities.
3. DISCUSSION.
We have reported the synthesis and pharmacological characterization of a series of glutamate and aspartate analogues with the hydroxy-1,2,3-triazole as bioisostere for the distal carboxylic acid. Four analogues showed selectivity among AMPA receptors, most pronounced with compound 6b that shows moderate binding affinity towards GluA2, but an order of magnitude lower affinity towards GluA1 and GluA4. This unprecedented selectivity profile is not mediated by differences in amino acid residues in direct contact with the ligand. The only sequence difference in the orthosteric site that mediates the selectivity of Cl-HIBO (3) is a tyrosine in GluA1/2 that is a phenylalanine in GluA3/4.50 However, 6b shows a different pharmacological profile. Previously, we have demonstrated that selectivity in BnTetAMPA analogues arise from movement of a methionine corresponding to Met729 in GluA2. This movement likely has varying energy costs in different AMPA subunits due to a subunit specific environment in this area, where the sequence differs among the subunits. It is therefore notable that the X-ray structure of 6b in GluA2-ABD shows an unprecedented weak electron density for Met729 in GluA2, indicating that 6b binding results in increased flexibility of this residue in GluA2-ABD compared to other ligands for which crystal structures exist. This could reduce the entropic cost of binding to GluA2-ABD and thereby explain the higher affinity towards GluA2-ABD compared to other subtypes. The structure of 7a shows that substituents larger than a methyl group cannot be accommodated which is in line with the observed lack of activity of compounds 7b and 7c.
The use of the hydroxy-1,2,3-triazole in aspartate analogues lead to compound 8 that shows activity among NMDA receptor subtypes. Most notable is the ability of compound 8 to activate both the GluN1 and GluN2 subunits that harbours the Gly and Glu binding sites, respectively, of the NMDA receptor. This is surprising, since the size requirement for agonists binding the Gly site is normally quite strict leaving little space for substituents on Gly. However, recently we have demonstrated that larger substituents can be accommodated.51 Since compound 8 also activates the Glu site, the hydroxy-1,2,3-triazole moiety is the first bioisostere capable of enabling the simultaneous activation of the Glu and Gly sites. It seems probable that the bioisostere of compound 8 is recognized in two different states: negatively charged in the Glu binding pocket and neutral in the Gly binding pocket. This would be enabled by the pKa of the bioisostere being close to 7 (6.4 for 6a).41
4. CONCLUSIONS.
This study provides structural insight into factors determining selectivity among iGluRs and identifies the hydroxy-1,2,3-triazole moiety as a novel bioisostere for a carboxylic acid, leading to important changes in the interaction patterns of the carboxylic acid binding pocket in iGluRs. This hydroxyazole system, beside providing the opportunity to place a substituent oriented in two different directions as opposed to the isoxazolol moiety in AMPA, because a relatively weak pKa (close to 7) was able to act in differently states depending from the different binding pockets requirements. The synthesis and the study of the properties investigation of enantiomerically pure hydroxy-1,2,3-traizole Glu analogues are in progress; the results will be presented in a following publication.
5. EXPERIMENTAL SECTION.
5.1. Chemistry.
5.1.1. General methods.
All chemical reagents were obtained from commercial sources (Sigma Aldrich, Alfa Aesar and FluoroChem) and used without further purification. Analytical grade solvents (acetonitrile, diisopropyl ether, diethyl ether, dichloromethane [DCM], dimethylformamide [DMF], ethanol 99.8% v/v [EtOH], ethyl acetate [EtOAc], methanol [MeOH], petroleum ether b.p. 40-60 °C [petroleum ether]) were used without further purification. When needed, solvents were dried on 4 Å molecular sieves. Tetrahydrofuran (THF) was distilled immediately prior to use from Na and benzophenone under N2. Thin layer chromatography (TLC) on silica gel was carried out on 5 × 20 cm plates with 0.25 mm layer thickness to monitor the process of reactions. Anhydrous Na2SO4 was used as a drying agent for the organic phases. Purification of compounds was achieved with flash column chromatography on silica gel (Merck Kieselgel 60, 230-400 mesh ASTM) using the eluents indicated or by CombiFlash Rf 200 (Teledyne Isco) with 5-200 mL/min, 200 psi (with automatic injection valve) using RediSep Rf Silica columns (Teledyne Isco) with the eluents indicated. Compounds synthesized in our laboratory generally varied between 90-99% purity. The biological experiments were only employed on compounds with a purity of at least 95%. Purity was checked using three analytical HPLC methods. HPLC analyses were performed on an UHPLC chromatographic system (Perkin Elmer, Flexar). The analytical columns were an HPLC Ascentis® Express RP-Amide (4.9 × 100 mm, 2.7 μm), HPLC Thermo Aquasil C18 (4.6 × 200 mm, 5 μm) and UHPLC Acquity CSH Fluoro-Phenyl (2.1 × 100 mm, 1.7 μm) (Waters). Compounds were dissolved in water with 0.1% trifluoroacetic acid and injected through a 20 μL loop. The mobile phase for the first method consisted of water / acetonitrile (ratio between 90/20 and 40/60, depending on the compound’s retention factor). UHPLC retention times were obtained at flow rates of 0.5 mL/min, and the column effluent was monitored at 262, 254 and 242 nm, referenced against a 360 nm wavelength. The mobile phase for the second method consisted of water / acetonitrile (ratio between 100/0 and 40/60, depending on the compound’s retention factor). UHPLC retention times were obtained at flow rates of 1.0 mL/min, and the column effluent was monitored at 262, 254 and 242 nm, referenced against a 360 nm wavelength. The mobile phase for the third method consisted of water / acetonitrile with 0.1% trifluoroacetic acid (ratio between 90/20 and 40/60, depending on the compound’s retention factor). UHPLC retention times were obtained at flow rates of 0.5 mL/min, and the column effluent was monitored at 266, 262, 254 and 242 nm, referenced against a 360 nm wavelength. Melting points (m.p.) were measured on a capillary apparatus (Büchi 540). The final m.p. determination was achieved by placing the sample at a temperature 10 °C below the m.p. and applying a heating rate of 1 °C/min. All compounds were routinely checked by 1H- and 13C-NMR and mass spectrometry. MS spectra were performed on Finnigan-Mat TSQ-700 (70 eV, direct inlet for chemical ionization [CI]) or Waters Micromass ZQ equipped with ESCi source for electrospray ionization mass spectra. 1H- and 13C-NMR spectra were performed on a Bruker Avance 300 instrument. For coupling patterns, the following abbreviations are used: br = broad, s = singlet, d = doublet, dd = doublet of doublets, t = triplet, q = quartet, h = sextet, m = multiplet. Chemical shifts (δ) are given in parts per million (ppm). Values marked with an asterisk are interchangeable. HRMS spectra were recorded on an LTQ Orbitrap mass spectrometer (Thermo Scientific, Bremen, Germany), equipped with an atmospheric pressure interface and an ESI ion source instrument. Compounds 9a-c and 10a-c were prepared following already described procedures.41, 52
General procedure for synthesis of compounds 6a-c and 7a-c.
A suspension of appropriate protected glutamate triazole derivatives (17a-c or 18a-c, 150 mg) in 6N HCl (25 mL) in a microwave vial, was stirring in a monomodal MW reactor at 100 °C for 4 h and then concentrated in vacuo. The crude solid material was then triturated with EtOAc, dissolved in water (10 mL) and the resulting solution concentrated in vacuo to afford the hydrochloric salt of the desired compound as a yellow solid. This latter was then converted to zwitterion using a Dowex 50W-X8 (200-400 mesh, capacity 1.7 meq/mL wet bed volume) ion-exchange resin affording the title compound. The resin activation was performed according to the following method. The resin was washed with water (three volumes of resin), 10 % w/w HCl (up to acidic pH), water (up to neutral pH), 10 % w/w NH3 (up to basic pH), water (up to neutral pH) and then 10% w/w HCl until acid pH was reached. The resin was then washed with water until neutrality of the eluate, then a solution of the hydrochloride salt, dissolved in slightly acid water to help solubility, was loaded on the top the column. The column was eluted with water until neutral pH, then with 10% w/w NH3 solution to recover the desired compound in zwitterionic form.
(RS) 2-Amino-3-(4-hydroxy-1-methyl-1H-1,2,3-triazol-5-yl)propanoic acid (6a).
White solid (m.p. = 220.2-221.4 °C; from trituration with diethyl ether). Yield 92% 1H-MMΈ. (300 MHz, D2O): δ, 3.31 (dd, J = 6.5, 1.3 Hz, 2H, −CH2CH−), 3.93 (s, 3H, -NCH3), 4.36 (t, J = 6.5 Hz, 1H −CH2CH−); 13C-NMR (75 MHz, D2O): δ, 23.3 (−CH2CH−), 37.3 (−NCH3), 51.2 (−CH2CH−), 115.9 (C-d), 155.8 (C-e), 170.7 (COOH). MS (ES+): 187 (M+1), (ES−): 185 (M-1). ESI-HRMS (m/z) [M + H]+ calcd. for C6H11N4O3 187.0826, obsd. 187.0825.
(RS) 2-Amino-3-(1-ethyl-4-hydroxy-1H-1,2,3-triazol-5-yl)propanoic acid (6b).
White solid (m.p. = 233.0-239.7 °C (dec.), from trituration with diethyl ether). Yield 72% 1H-NMR (300 MHz, D2O + MeOH): δ 1.25 (t, J = 7.1 Hz, 3H, −NCH2CH3), 2.60 (dd, J = 15.0, 8.7 Hz, 1H, −CH2CH−), 2.81 (dd, J = 15.0, 5.1 Hz, 1H, −CH2CH−), 3.26-3.39 (m, 1H, −CH2CH−), 4.04 (q, J = 7.1 Hz, 2H, −NCH2CH3); 13C-NMR (75 MHz, D2O + MeOH): δ 15.3 (−NCH2CH3), 28.6 (−CH2CH−), 44.6 (−NCH2CH3), 56.2 (−CH2CH−) 115.9 (C-d), 165.0 (C-e), 182.5 (−COOH). MS (ES+): 201 (M+1), (ES−): 199 (M-1). ESI-HRMS (m/z) [M + H]+ calcd. for C7H13N4O3 201.0982, obsd. 201.0981.
(RS) 2-Amino-3-(4-hydroxy-1-propyl-1H-1,2,3-triazol-5-yl)propanoic acid (6c).
White solid, (m.p. = 233.0-241.7 °C (dec.), from trituration with diethyl ether). Yield 88%. 1H-NMR (300 MHz, D2O + MeOH): δ 0.85 (t, J = 7.4 Hz, 3H, −NCH2CH2CH3), 1.83 (h, J = 7.3 Hz, 2H, −NCH2CH2CH3), 3.23 (dd, J = 6.1, 1.4 Hz, 2H, −CH2CH−), 4.05 (dd, J = 6.4, 5.6 Hz, 1H, −CH2CH−), 4.21 (t, J = 7.2 Hz, 2H, −NCH2CH2CH3); 13C-NMR (75 MHz, D2O + MeOH): δ 10.7 (−NCH2CH2CH3), 23.0 (−NCH2CH2CH3) 24.3 (−CH2CH−), 52.2 (−NCH2CH2CH3), 53.8 (−CH2CH−), 116.1 (C-d), 157.2 (C-e), 173.3 (−COOH). MS (ES+): 215 (M+1), (ES−) 213 (M-1). ESI-HRMS (m/z) [M + H]+ calcd. for C8H15N4O4 215.1139, obsd. 215.1138.
(RS) 2-Amino-3-(5-hydroxy-2-methyl-2H-1,2,3-triazol-4-yl)propanoic acid (7a).
White solid (m.p. = 152.2-155.8 °C; from trituration with diethyl ether). Yield 98%. 1H-NMR (300 MHz, D2O): δ 3.22 (d, J = 5.9 Hz, 2H, −CH2CH−), 3.90 (s, 3H, −NCH3), 4.29 (t, J = 5.9 Hz, 1H −CH2CH-); 13C-NMR (75 MHz, D2O): δ 24.3 (−CH2CH−), 41.3 (−NCH3), 52.6 (−CH2CH−), 125.8 (C-d), 156.2 (C-e), 171.5 (−COOH). MS (ES+): 187 (M+1), (ES−): 185 (M-1). ESI-HRMS (m/z) [M + H]+ calcd. for C6H11N4O3 187.0826, obsd. 187.0825.
(RS) 2-Amino-3-(2-ethyl-5-hydroxy-2H-1,2,3-triazol-4-yl)propanoic acid (7b).
White solid, (m.p. = 196.0-199.3°C (dec.), from trituration with diethyl ether). Yield 93%. 1H-NMR (300 MHz, D2O + MeOH): δ 1.32 (t, J = 7.3 Hz, 3H, −NCH2CH3), 3.04 (dd, J = 15.9, 7.7 Hz, 1H, −CH2CH−), 3.14 (dd, J = 15.9, 4.7 Hz, 1H, −CH2CH−), 3.94 (dd, J = 7.7, 4.7 Hz, 1H, −CH2CH−), 4.12 (q, J = 7.3 Hz, 2H, −NCH2CH3); 13C-NMR (75 MHz, D2O + MeOH): δ 14.0 (−NCH2CH3), 25.2 (−CH2CH−), 48.9 (−NCH2CH3), 54.1 (−CH2CH−) 126.9 (C-d), 158.1 (C-e), 173.6 (−COOH). MS (ES+): 201 (M+1), (ES−): 199 (M-1). ESI-HRMS (m/z) [M + H]+ calcd. for C7H13N4O3 201.0982, obsd. 201.0980.
(RS) 2-Amino-3-(5-hydroxy-2-propyl-2H-1,2,3-triazol-4-yl)propanoic acid (7c).
White solid, (m.p. = 233.2-235.4 °C; from trituration with diethyl ether). Yield 93% 1H-NMR (300 MHz, D2O + MeOH): δ 0.77 (t, J = 7.4 Hz, 3H, −NCH2CH2CH3), 1.77 (h, J = 7.2 Hz, 2H, −NCH2CH2CH3), 3.06 (dd, J = 15.9, 7.6 Hz, 1H, −CH2CH−), 3.16 (dd, J = 15.9, 4.8 Hz, 1H, −CH2CH−), 3.95 (dd, J = 7.6, 4.8 Hz, 1H, −CH2CH−), 4.08 (t, J = 6.8 Hz, 2H, −NCH2CH2CH3); 13C-NMR (75 MHz, D2O + MeOH): δ 10.8 (−NCH2CH2CH3), 23.2 (−NCH2CH2CH3) 25.8 (−CH2CH−), 54.7, 56.6 (−CH2CH−, −NCH2CH2CH3), 127.4 (C-d), 158.4 (C-e), 174.1 (−COOH). MS (ES+): 215 (M+1), (ES−): 213 (M-1). ESI-HRMS (m/z) [M + H]+ calcd. for C8H15N4O4 215.1139, obsd. 215.1137.
(RS) 2-Amino-2-(5-hydroxy-2-methyl-2H-1,2,3-triazol-4-yl)acetic acid (8).
A suspension of 22 (0.507 mmol; 0.120 g) in 6N hydrochloric acid (10 mL) was heated at reflux for 6 h and then concentrated in vacuo. The crude solid material was then triturated with ethyl acetate, dissolved in water (10 mL) and the resulting solution concentrated in vacuo to afford the hydrochloride of the title compound as a yellow solid. The hydrochloride of compound 8 was then converted to zwitterion using a Dowex 50W-X8 (200-400 mesh, capacity 1.7 meq/mL wet bed volume) ion-exchange resin to afford the title compound as a white solid. (m.p. = 125.3-127.0 °C. from trituration with diethyl ether). Yield 90 %. 1H-NMR (300 MHz, D2O + MeOH): δ 3.53 (s, 3H, −NCH3), 4.10 (s, 1H, -CH); 13C-NMR (75 MHz, D2O + MeOH): δ 40.9 (−NCH3), 51.3 (−CH), 134.0 (C-d), 164.4 (C-e), 180.8 (−COOH). MS (ES+): 173 (M+1), (ES−): 171 (M-1). ESI-HRMS (m/z) [M + H]+ calcd. for C5H9N4O3 173.0669, obsd. 173.0669.
General procedure for ester reduction to alcohol (11a-c and 12a-c).
Sodium borohydride (20.00 mmol) was added portion wise over 5 min to a solution of corresponding ester (9a-c and 10a-c) (1.00 mmol) in dry EtOH (60 mL) and the mixture was stirred under inert atmosphere at 30 °C overnight. The reaction mixture was poured into iced water (100 mL) and the ethanol evaporated in vacuo. The aqueous layer was extracted with diethyl ether (3 × 50 mL). The organic layers were collected, dried and concentrated in vacuo. The crude was purified by flash chromatography (eluent: DCM / MeOH 95:5 v/v).
(4-(Benzyloxy)-1-methyl-1H-1,2,3-triazol-5-yl)methanol (11a).
White solid (m.p. = 74.5-75.1 °C; from trituration with diisopropyl ether). Yield 98%. 1H-NMR (300 MHz, CDCl3): δ 2.33 (br s, 1H, −CH2OH), 4.01 (s, 3H, −NCH3), 4.58 (s, 2H, −CH2OH), 5.35 (s, 2H, −OCH2Ph), 7.30-7.48 (m, 5H, aromatic protons); 13C-NMR (75 MHz, CDCl3): δ 35.8 (−NCH3), 51.4 (−CH2OH), 72.3 (−OCH2Ph), 119.3 (C-d), 128.3, 128.4, 128.5 (aromatic carbons), 136.5 (Cipso), 157.5 (C-e). MS (CI): 220 (M+1).
(4-(Benzyloxy)-1-ethyl-1H-1,2,3-triazol-5-yl)methanol (11b).
Colorless oil. Yield 92%. 1H-NMR (300 MHz, CDCl3): δ 1.49 (t, J = 7.3 Hz, 3H, −NCH2CH3), 2.66 (br s, 1H, −CH2OH), 4.32 (q, J = 7.3 Hz, 2H, −NCH2CH3), 4.55 (s, 2H, −CH2OH), 5.31 (s, 2H, −OCH2Ph) 7.28-7.42 (m, 5H, aromatic protons); 13C-NMR (75 MHz, CDCl3): δ 15.4 (−NCH2CH3), 44.6 (−NCH2CH3), 51.4 (−CH2OH), 72.4 (−OCH2Ph), 118.7 (C-d), 128.4, 128.5, 128.6 (aromatic carbons), 136.6 (Cipso), 157.5 (C-e). MS (ES+): 234 (M+1).
(4-(Benzyloxy)-1-propyl-1H-1,2,3-triazol-5-yl)methanol (11c).
White solid (m.p. = 74.5-75.1 °C; from trituration with diisopropyl ether). Yield 90%. 1H-NMR (300 MHz, CDCl3): δ 0.96 (t, J = 7.4 Hz, 3H, −NCH2CH2CH3), 1.78-2.11 (m, 2H, −NCH2CH2CH3), 2.20 (s, 1H, −OH), 4.26 (t, 2H, J = 7.4 Hz, −NCH2CH2CH3), 4.56 (s, 2H, −CH2OH), 5.37 (s, 2H, −OCH2Ph), 7.28 - 7.50 (m, 5H, aromatic protons); 13C-NMR (75 MHz, CDCl3): δ 11.2 (−NCH2CH2CH3), 23.5 (−NCH2CH2CH3), 51.1, 51.7 (−NCH2CH2CH3, −CH2OH), 72.7 (−OCH2Ph), 119.0 (C-d), 128.4, 128,5, 128.6 (aromatic carbons), 136.7 (Cipso), 157.5 (C-e). MS (ES+): 248 (M+1).
(5-(Benzyloxy)-2-methyl-2H-1,2,3-triazol-4-yl)methanol (12a).
White solid (m.p. = 54.9-55.6 °C; from trituration with diisopropyl ether). Yield 95%. 1H-NMR (300 MHz, CDCl3): δ 2.19 (br s, 1H, −CH2OH), 3.99 (s, 3H, −NCH3), 4.63 (s, 2H, −CH2OH), 5.24 (s, 2H, −OCH2Ph), 7.28 - 7.47 (m, 5H, aromatic protons); 13C-NMR (75 MHz, CDCl3): δ 41.7 (−NCH3), 54.5 (−CH2OH), 72.2 (−OCH2Ph), 128.0, 128.3, 128.5 (aromatic carbons), 131.6 (C-d), 136.3 (Cipso), 157.9 (C-e). MS (CI): 220 (M+1).
(5-(Benzyloxy)-2-ethyl-2H-1,2,3-triazol-4-yl)methanol (12b).
Colorless oil. Yield 94 %. 1H-NMR (300 MHz, CDCl3): δ 1.48 (t, J = 7.3 Hz, 3H, −NCH2CH3), 2.26 (br s, 1H, −CH2OH), 4.26 (q, J = 7.3 Hz, 2H, −NCH2CH3), 4.63 (s, 2H, −CH2OH), 5.24 (s, 2H, −OCH2Ph) 7.28-7.49 (m, 5H, aromatic protons); 13C-NMR (75 MHz, CDCl3): δ 14.7 (−NCH2CH3), 50.1 (−NCH2CH3), 54.7 (−CH2OH), 72.2 (−OCH2Ph), 128.2, 128.4, 128.6 (aromatic carbons), 131.3 (C-d), 136.5 (Cipso), 157.8 (C-e). MS (ES+): 234 (M+1).
(5-(Benzyloxy)-2-propyl-2H-1,2,3-triazol-4-yl)methanol (12c).
Colorless oil. Yield 93%. 1H-NMR (300 MHz, CDCl3): δ 0.91 (t, J = 7.3 Hz, 3H, −NCH2CH2CH3), 1.84-1.98 (m, 2H, −NCH2CH2CH3), 1.98 (s, 1H, −CH2OH), 4.17 (t, 2H, J = 7.1 Hz, −NCH2CH2CH3), 4.64 (s, 2H, −CH2OH), 5.25 (s, 2H, −OCH2Ph), 7.28-7.48 (m, 5H, aromatic protons); 13C-NMR (75 MHz, CDCl3): δ 11.2 (−NCH2CH2CH3), 23.2 (−NCH2CH2CH3), 54.8, 56.7 (−CH2OH, −NCH2CH2CH3), 72.3 (−OCH2Ph), 128,4, 128.7 (aromatic carbons), 131.3 (C-d), 136.5 (Cipso), 157.8 (C-e). MS (ES+): 248 (M+1).
General procedure for synthesis bromide derivatives (13a-c and 14a-c).
NBS (1.50 mmol) was added portion wise over 30 min under inert atmosphere to a cooled (−10 °C) solution of appropriate alcohol (11a-c or 12a-c, 1.00 mmol) and triphenylphosphine (1.10 mmol) in dry DCM (30 mL). The reaction mixture was stirred at −10 °C for 1 h and then directly loaded into a flash chromatography column (eluent: DCM) to afford compound as a colorless oil.
4-(Benzyloxy)-5-(bromomethyl)-1-methyl-1H-1,2,3-triazole (13a).
Colorless oil. Yield 81%. 1H-NMR (300 MHz, CDCl3): δ 3.91 (s, 3H, −NCH3), 4.36 (s, 2H, −CH2Br), 5.32 (s, 2H, −OCH2Ph), 7.19-7.43 (m, 5H, aromatic protons); 13C-NMR (75 MHz, CDCl3): δ 16.8 (−CH2Br), 35.6 (−NCH3), 71.8 (−OCH2Ph), 115.9 (C-d), 128.1, 128.2, 128.4 (aromatic carbons), 136.2 (Cipso), 157.5 (C-e). MS (CI): 282/284 (M+1).
4-(Benzyloxy)-5-(bromomethyl)-1-ethyl-1H-1,2,3-triazole (13b).
Colorless oil. Yield 65%. 1H-NMR (300 MHz, CDCl3): δ 1.58 (t, J = 7.3 Hz, 3H, −NCH2CH3) 4.32 (q, J = 7.3 Hz, 2H, −NCH2CH3), 4.43 (s, 2H, −CH2Br), 5.42 (s, 2H, −OCH2Ph) 7.27-7.52 (m, 5H, aromatic protons); 13C-NMR (75 MHz, CDCl3): δ 15.0 (−NCH2CH3), 16.9 (−CH2Br), 44.4 (−NCH2CH3), 72.0 (−OCH2Ph), 115.4 (C-d), 128.3, 128.4, 128.6 (aromatic carbons), 136.6 (Cipso), 157.9 (C-e). MS (ES+): 296/298 (M+1).
4-(Benzyloxy)-5-(bromomethyl)-1-propyl-1H-1,2,3-triazole (13c).
Colorless oil. Yield 60%. 1H NMR (300 MHz, CDCl3): δ 1.0 (t, J = 7.4 Hz, 3H, −NCH2CH2CH3), 1.91-2.07 (m, 2H, -NCH2CH2CH3), 4.24 (t, J = 7.4 Hz, 2H, −NCH2CH2CH3), 4.43 (s, 2H, −CH2Br), 5.42 (s, 2H, −OCH2Ph), 7.28-7.53 (m, 5H, aromatic protons); 13C NMR (75 MHz, CDCl3): δ 11.3 (−NCH2CH2CH3), 17.0 (−CH2Br), 23.2 (−NCH2CH2CH3), 50.1 (−NCH2CH2CH3), 72.1 (−OCH2Ph), 115.6 (C-d), 128.4, 128.5, 128.7 (aromatic carbons), 136.6 (Cipso), 157.9 (C-e). MS (ES+): 310 / 312 (M+1).
4-(Benzyloxy)-5-(bromomethyl)-2-methyl-2H-1,2,3-triazole (14a).
Colorless oil. Yield 93%. 1H-NMR (300 MHz, CDCl3): δ 4.02 (s, 3H, −NCH3), 4.46 (s, 2H, −CH2Br), 5.27 (s, 2H, −OCH2Ph), 7.30-7.52 (m, 5H, aromatic protons); 13C-NMR (75 MHz, CDCl3): δ 20.4 (−CH2Br), 42.1 (−NCH3), 72.3 (−OCH2Ph), 128.0, 128.4, 128.7 (aromatic carbons), 128.8 (C-d), 136.3 (Cipso), 158.0 (C-e). MS (CI): 282/284 (M+1).
4-(Benzyloxy)-5-(bromomethyl)-2-ethyl-2H-1,2,3-triazole (14b).
Colorless oil. Yield 66%. 1H-NMR (300 MHz, CDCl3): δ 1.50 (t, J = 7.3Hz, 3H, −NCH2CH3), 4.27 (q, J = 7.3 Hz, 2H, −NCH2CH3), 4.48 (s, 2H, −CH2Br), 5.28 (s, 2H, −OCH2Ph) 7.32-7.48 (m, 5H, aromatic protons); 13C-NMR (75 MHz, CDCl3): δ 14.6 (−NCH2CH3), 20.6 (−CH2Br), 50.3 (−NCH2CH3), 72.2 (−OCH2Ph), 128.1, 128.3, 128.4, (C-d), 128.6 (aromatic carbons), 136.3 (Cipso), 157.9 (C-e). MS (ES+): 296 / 2989 (M+1).
4-(Benzyloxy)-5-(bromomethyl)-2-propyl-2H-1,2,3-triazole (14c).
Colorless oil. Yield 66%. 1H NMR (300 MHz, CDCl3): δ 0.84 (t, J = 7.4 Hz, 3H, −NCH2CH2CH3), 1.77-1.92 (m, 2H, - NCH2CH2CH3), 4.11 (t, J = 7.4 Hz, 2H, −NCH2CH2CH3), 4.40 (s, 2H, −CH2Br), 5.20 (s, 2H, −OCH2Ph), 7.24-7.43 (m, 5H, aromatic protons); 13C NMR (75 MHz, CDCl3): δ 11.4 (−NCH2CH2CH3), 20.8 (−CH2Br), 23.4 (−NCH2CH2CH3), 57.2 (−NCH2CH2CH3), 72.6 (−OCH2Ph), 128.4, 128.7, 128.8 (C-d), 128.9 (aromatic carbons), 136.7 (Cipso), 158.1 (C-e). MS (ES+): 310 / 312 (M+1).
General procedure for synthesis of compounds 17a-c and 18a-c.
Sodium hydride (60% w/w, 1.20 mmol) was added portion wise to an ice cooled solution of diethyl-2-acetamidomalonate (1.10 mmol) in dry THF (35 mL) kept under inert atmosphere. The reaction mixture was stirred at rt for 1 h, then a solution of appropriate bromide derivatives (13a-c or 14a-c, 1.00 mmol) in dry THF (10 mL) was added over 15 min. The resulting mixture was stirred at rt for 36 h, and then poured into a saturated solution of NH4Cl (80 mL). The THF was evaporated under reduced pressure and the aqueous layer was extracted with diethyl ether (3 × 50 mL). The organic layers were collected, dried and concentrated in vacuo to afford the compound as a white solid (15a-c and 16a-c). This latter was used in the next step without any further purification and characterized via mass spectroscopy.
Pd/C (10% w/w, 90 mg) was added to a solution of the solid material rising from the previously step (15a-c or 16a-c, 0.900 g) in dry THF (30 mL). The resulting mixture was vigorously stirred under hydrogen (1 atm) at rt for 1 h. The resulting mixture was then filtered over celite and the filtrate was concentrated in vacuo. The crude material was purified by flash chromatography (eluent: DCM / 2-propanol 95:5 v/v) to afford compounds 17a-c and 18a-c.
Diethyl 2-acetamido-2-((4-(benzyloxy)-1-methyl-1H-1,2,3-triazol-5-yl)methyl)malonate (15a).
The crude material was purified by flash chromatography (first petroleum ether / EtOAc 7:3 v/v next 6:4 v/v) to afford the compound as a white solid. MS (CI): 419 (M+1).
Diethyl 2-acetamido-2-((4-(benzyloxy)-1-ethyl-1H-1,2,3-triazol-5-yl)methyl)malonate (15b).
The crude material was purified by flash chromatography (first petroleum ether / EtOAc 7:3 v/v next 6:4 v/v) to afford the compound as a white solid. MS (CI): 433 (M+1).
Diethyl 2-acetamido-2-((4-(benzyloxy)-1-propyl-1H-1,2,3-triazol-5-yl)methyl)malonate (15c).
The crude material was purified by flash chromatography (eluent starting from petroleum ether / EtOAc 9:1 v/v to 7:3) to afford the compound as a white solid. MS (CI): 447 (M+1).
Diethyl 2-acetamido-2-((5-(benzyloxy)-2-methyl-2H-1,2,3-triazol-4-yl)methyl)malonate (16a).
The crude material was purified by flash chromatography (first petroleum ether / EtOAc 7:3 v/v next 6:4 v/v) to afford the compound as a white solid. MS (CI): 419 (M+1).
Diethyl 2-acetamido-2-((5-(benzyloxy)-2-ethyl-2H-1,2,3-triazol-4-yl)methyl)malonate (16b).
The crude material was purified by flash chromatography (first petroleum ether / EtOAc 7:3 v/v next 6:4 v/v) to afford the compound as a white solid. MS (CI): 433 (M+1).
Diethyl 2-acetamido-2-((5-(benzyloxy)-2-propyl-2H-1,2,3-triazol-4-yl)methyl)malonate (16c).
The crude material was purified by flash chromatography (eluent petroleum ether / acetone 8:2 v/v) to afford the compound as a colorless oil. MS (CI): 447 (M+1).
Diethyl 2-acetamido-2-((4-hydroxy-1-methyl-1H-1,2,3-triazol-5-yl)methyl)malonate (17a).
White solid (m.p. = 181.4-182.4 °C; from trituration with diisopropyl ether). Yield 69% over two steps. 1H-NMR (300 MHz, DMSO): δ 1.16 (t, J = 7.1 Hz, 6H −OCH2CH3), 193 (s, 3H, −NHCOCH3) 3.46 (s 2H, −CH2C−), 3.68 (s, 3H, −NCH3), 4.04-4.23 (m, 4H −OCH2CH3), 8.34 (s, 1H, −NHCOCH3), 9.95 (br s, 1H, −OH), 13C-NMR (75 MHz, DMSO): δ 13.7 (−OCH2CH3), 22.1 (−NHCOCH3), 25.6 (-CH2C−), 35.1 (−NCH3), 62.0 (−OCH2CH3), 65.3 (−CH2C−), 112.7 (C-d), 157.1 (C-e), 166.8 (−COOEt), 170.1 (−NHCOCH3). MS (CI): 329 (M+1).
Diethyl 2-acetamido-2-((1-ethyl-4-hydroxy-1H-1,2,3-triazol-5-yl)methyl)malonate (17b).
Colourless oil. Yield 67% over two steps. 1H-NMR (300 MHz, CDCl3): δ 1.27 (t, J = 6.8 Hz, 6H, −OCH2CH3), 1.44 (t, J = 7.0 Hz, 3H, −NCH2CH3), 2.01 (s, 3H, −NHCOCH3), 3.72 (s, 2H, −CH2C−), 4.15 (q, J = 7.0 Hz, 2H, −NCH2CH3), 4.21-437 (m, 4H, −OCH2CH3), 6.91 (s, 1H, −NHCOCH3), 7.81 (vvbr s, 1H, −OH), 13C-NMR (75 MHz, CDCl3): δ 14.0 (−OCH2CH3), 15.4 (−NCH2CH3), 23.1 (−NHCOCH3), 26.2 (−CH2C−), 44.2 (−NCH2CH3), 63.3 (−OCH2CH3), 65.3 (−CH2C−), 112.9 (C-d), 157.3 (C-e), 167.1 (−COOEt), 170.2 (−NHCOCH3). MS (ES+): 343 (M+1).
Diethyl 2-acetamido-2-((4-hydroxy-1-propyl-1H-1,2,3-triazol-5-yl)methyl)malonate (17c).
White solid (m.p. = 167.3 - 169.7 °C; from trituration with diisopropyl ether). Yield 69% over two steps. 1H-NMR (300 MHz, CDCl3): δ 0.90 (t, J = 7.4 Hz, 3H, −NCH2CH2CH3), 1.27 (t, J = 7.1 Hz, 6H, −OCH2CH3), 1.75-1.91 (m, 2H, −NCH2CH2CH3), 2.01 (s, 3H, −NHCOCH3), 3.72 (s, 2H, −CH2C−), 4.07 (t, J = 7.3 Hz, 2H, −NCH2CH2CH3), 4.23-4.35 (m, 4H, −OCH2CH3), 6.89 (s, 1H, −NHCOCH3); 13C-NMR (75 MHz, CDCl3): δ 11.1 (−NCH2CH2CH3), 14.0 (−OCH2CH3), 23.1 (−NCH2CH2CH3), 23.6 (−NHCOCH3), 26.2 (−CH2C−), 50.5 (−NCH2CH2CH3), 63.3 (−OCH2CH3), 65.3 (−CH2C−), 113.0 (C-d), 157.5 (C-e), 167.1 (−COOEt), 170.1 (−NHCOCH3). MS (ES+): 357 (M+1).
Diethyl 2-acetamido-2-((5-(benzyloxy)-2-methyl-2H-1,2,3-triazol-4-yl)methyl)malonate (18a).
White solid (m.p. = 124.5-125.4 °C; from trituration with diisopropyl ether). Yield 70% over two steps. 1H-NMR (300 MHz, DMSO): δ 1.16 (t, J = 7.0 Hz, 6H −OCH2CH3), 186 (s, 3H, −NHCOCH3), 3.34 (s 2H, −CH2C−), 3.81 (s, 3H, −NCH3), 4.14 (q, J = 7.0 Hz, 4H −OCH2CH3), 7.96 (s, 1H, −NHCOCH3), 10.17 (s, 1H, −OH), 13C-NMR (75 MHz, DMSO): δ 14.6 (−OCH2CH3), 23.0 (−NHCOCH3), 27.5 (−CH2C−), 42.0 (−NCH3), 62.5 (−OCH2CH3), 65.6 (−CH2C−), 126.2 (C-d), 158.3 (C-e), 167.9 (−COOEt), 170.2 (−NHCOCH3). MS (CI): 329 (M+1).
Diethyl 2-acetamido-2-((5-(benzyloxy)-2-ethyl-2H-1,2,3-triazol-4-yl)methyl)malonate (18b).
White solid (m.p. = 110.7-111.9 °C; from trituration with diisopropyl ether). Yield 33% over two steps. 1H-NMR (300 MHz, CDCl3): δ 1.27 (t, J = 7.1 Hz, 6H, −OCH2CH3), 1.41 (t, J = 7.3 Hz, 3H, −NCH2CH3), 2.02 (s, 3H, −NHCOCH3), 3.65 (s, 2H, −CH2C−), 4.14 (q, J = 7.3 Hz, 2H, −NCH2CH3), 4.25 (q, J = 7.1 Hz, 4H, −OCH2CH3), 6.67 (s, 1H, −NHCOCH3), 9.84 (br s, 1H, −OH), 13C-NMR (75 MHz, CDCl3): δ 14.1 (−OCH2CH3), 14.6 (−NCH2CH3), 23.1 (−NHCOCH3), 27.6 (−CH2C−), 49.8 (−NCH2CH3), 62.9 (−OCH2CH3), 66.2 (−CH2C−), 126.1 (C-d), 157.7 (C-e), 167.6 (−COOEt), 169.9 (−NHCOCH3). MS (ES+): 343 (M+1).
Diethyl 2-acetamido-2-((5-(benzyloxy)-2-propyl-2H-1,2,3-triazol-4-yl)methyl)malonate (18c).
White solid (m.p. = 147.1-149.7 °C; from trituration with diisopropyl ether). Yield 60% over two steps. 1H-NMR (300 MHz, CDCl3): δ 0.86 (t, J = 7.4 Hz, 3H, −NCH2CH2CH3), 1.27 (t, J = 7.1 Hz, 6H, −OCH2CH3), 1.83 (h, J = 7.2 Hz, 2H, −NCH2CH2CH3), 2.02 (s, 3H, −NHCOCH3), 3.65 (s, 2H, −CH2C−), 4.06 (t, J = 7.0 Hz, 2H, −NCH2CH2CH3), 4.25 (q, J = 7.1 Hz, 4H, −OCH2CH3), 6.67 (s, 1H, −NHCOCH3), 8.70 (vv br s, 1H, −OH), 13C-NMR (75 MHz, CDCl3): δ 11.1 (−NCH2CH2CH3), 14.0 (−OCH2CH3), 23.0 (−NCH2CH2CH3), 23.1 (−NHCOCH3), 27.5 (−CH2C−), 56.4 (−NCH2CH2CH3), 62.9 (−OCH2CH3), 66.2 (−CH2C−), 126.0 (C-d), 157.7 (C-e), 167.6 (−COOEt), 169.9 (−NHCOCH3). MS (ES+): 357 (M+1).
2-(5-(Benzyloxy)-2-methyl-2H-1,2,3-triazol-4-yl)acetonitrile (19).
Compound 14a (5.73 mmol; 1.61 g) was added to a solution of NaCN (11.46 mmol; 0.562 g) in EtOH / water 9:1 v/v (50 mL). The reaction was stirred at rt for 24 h, and then poured into water (60 mL). EtOH was evaporated in vacuo and the aqueous layer was extracted with DCM (3 × 50 mL). The organic layers were collected, dried and concentrated in vacuo. The crude material was purified by flash chromatography (eluent: from petroleum ether / EtOAc 9:1 v/v to 7:3) to afford the title compound as a colorless oil. Yield 53%. 1H-NMR (300 MHz, DMSO): δ 3.99 (s, 5H, −NCH3 overlapped -CH2CN), 5.25 (s, 2H, −OCH2Ph), 7.28-7.51 (m, 5H, aromatic protons); 13C-NMR (75 MHz, DMSO): δ 12.4 (−CH2CN), 41.8 (−NCH3), 71.8 (−OCH2Ph), 117.2 (−CH2CN), 121.9 (C-d), 127.9, 128.2, 128.4 (aromatic carbons), 136.2 (Cipso), 156.9 (C-e). MS (CI): 229 (M+1).
Ethyl 2-(5-(benzyloxy)-2-methyl-2H-1,2,3-triazol-4-yl)acetate (20).
Dry EtOH (4.00 eq; 17.5 mmol; 1.02 mL), TMSC1 (4.00 eq; 17.5 mmol; 2.23 mL) and 19 (3.73 mmol; 0.850 g) were subsequently added to a dry flask kept under inert atmosphere at rt. The reaction mixture was stirred for 24 h, and then quenched with water (5 mL). Na2CO3 (14.9 mmol; 2.25 g) was then added portion wise to the reaction mixture. The obtained suspension was extracted with EtOAc (3 × 20 mL), the organic layers were collected, dried and concentrated in vacuo. The resulting crude material was purified by flash chromatography (eluent: petroleum ether / EtOAc 9:1 v/v) to afford the title compound as a colorless oil. Yield 84%. 1H-NMR (300 MHz, CDCl3): δ 1.23 (t, J = 7.1 Hz, 3H, −OCH2CH3), 3.61 (s, 2H, −CH2COOEt), 4.00 (s, 3H, −NCH3), 4.16 (q, J = 7.1 Hz, 2H, −OCH2CH3), 5.23 (s, 2H, −OCH2Ph), 7.28-7.46 (m, 5H, aromatic protons); 13C-NMR (75 MHz, CDCl3): δ 14.1 (−OCH2CH3), 29.7 (−CH2COOEt), 41.7 (−NCH3), 61.1 (−OCH2CH3), 72.0 (−OCH2Ph), 125.7 (C-d), 127.8, 128.1, 128.5 (aromatic carbons), 136.5 (Cipso), 158.4 (C-e), 169.9 (−CH2COOEt). MS (ES+): 276 (M+1).
(E/Z) Ethyl 2-(5-(benzyloxy)-2-methyl-2H-1,2,3-triazol-4-yl)-2-(hydroxyimino)acetate (21).
A solution of 20 (3.27 mmol; 0.900 g) in dry EtOH (15 mL) was slowly dropped into a solution of sodium hydride (60% w/w in mineral oil, 4.00 eq, 13.1 mmol, 0.503 g) in dry EtOH, stirred and kept under inert atmosphere. The reaction mixture was stirred at 0 °C for 30 min and then EtONO (4.00 eq; 13.1 mmol; 12.4 mL) was added. Then the reaction mixture was stirred rt for 20 h, and then glacial acetic acid was added to reach an acidic pH. The ethanol was evaporated in vacuo and the residue dissolved in water (50 mL) and the resulting solution was extracted with EtOAc (3 × 30 mL). The combined organic layers were collected, dried and concentrated in vacuo. The crude material was purified by flash chromatography (eluent: petroleum ether / EtOAc 7:3 v/v) to afford the title compound (mixture 1:1 of Z and E isomers) as a yellow solid. Yield 44%. 1H-NMR (300 MHz, CDCl3): δ 1.27, 1.32 (t, 3H, J = 7.1, −OCH2CH3), 4.02, 4.11 (s, 3H, −NCH3), 4.38, 4.29 (q, 2H, J = 7.1, −OCH2CH3), 5.23, 5.27 (s, 2H,−OCH2Ph), 7.27-7.47 (m, 5H, aromatic protons), 9.72 (s, 1H, −NOH); 13C-NMR (75 MHz, CDCl3): δ 14.1, 14.2 (−OCH2CH3), 42.4, 42.5 (−NCH3), 62.3, 62.4 (−OCH2CH3), 72.5, 72.6 (−OCH2Ph), 121.3, 124.1 (C-d), 127.8, 128.0, 128.3, 128.4, 128.5, 128.6 (aromatic carbons), 135.8, 136.2 (Cipso), 141.0, 143.9 (−C=NOH), 158.3, 159.0 (C-e), 162.4, 174.4 (−COOEt). MS (ES+): 305 (M+1).
Ethyl 2-amino-2-(5-(benzyloxy)-2-methyl-2H-1,2,3-triazol-4-yl)acetate (22).
Pd/C (10% w/w, 24 mg) and a saturated solution of hydrochloric acid in dry EtOH were added to a solution of 21 (0.789 mmol, 240 mg) in dry EtOH (35 mL) kept under inert atmosphere. The resulting mixture was vigorously stirred at rt under hydrogen for 40 min, then filtered over celite. The filtrate was concentrated in vacuo to give a crude solid material that was then triturated with diisopropyl ether, dissolved in water (10 mL) and the resulting solution concentrated in vacuo to afford the hydrochloric salt of the title compound as a yellow solid (m.p. = 63.2-64.1 °C; from trituration with diisopropyl ether). Yield 93%. 1H-NMR (300 MHz, D2O + MeOH): δ 1.21 (t, 3H, J = 7.1, −OCH2CH3), 3.97 (s, 3H, −NCH3), 4.29 (q, 2H, J = 7.1, −OCH2CH3), 5.38 (s, 1H, −CH); 13C-NMR (75 MHz, D2O + MeOH): δ 13.7 (−OCH2CH3), 42.2 (−NCH3), 47.8 (−CH−), 65.0 (−OCH2CH3), 123.2 (C-d), 156.8 (C-e), 168.0 (−COOEt). MS (ES+): 201 (M+1)
5.2. In vitro pharmacology.
5.2.1. Native receptor binding assays.
Affinities for native AMPA, KA and NMDA receptors in rat cortical synaptosomes were determined using 5 nM [3H]AMPA (57.5 Ci/mmol),53 5 nM [3H]KA (43.8 Ci/mmol),54 and 2 nM [3H]CGP 39653 (41.9 Ci/mmol),55 respectively, with minor modifications as previously described.56 Rat brain membrane preparations used in these receptor binding experiments were prepared according to a method previously described.57
5.2.2. Recombinant receptor binding assays.
Sf9 cells were cultured and infected with recombinant baculovirus of rat AMPA receptors (GluA1o-4o) and membranes prepared and used for binding as previously detailed.58, 59 The affinities of the compounds for GluA1o, GluA2(R)o, GluA3o and GluA4o were determined from competition experiments with 2-5 nM (RS)-[3H]AMPA. Italic letters in parentheses indicate the RNA-edited isoforms of the subunits. Ligand concentration-response curves were analyzed using GraphPad Prism v6 (GraphPad Software Inc., San Diego, CA) to determine the IC50, nH and Ki (n = 3 - 4 experiments at 12-15 ligand concentrations conducted in triplicate).
5.2.3. Two-electrode voltage-clamp electrophysiology for AMPA receptors.
Surgical procedures were conducted under the approval of the Danish Ministry of Justice Animal Experiments Inspectorate. Mature female Xenopus laevis were anaesthetized using 0.1% ethyl 3-aminobenzoate methanesulfonate (tricaine) and ovaries were surgically removed. The ovarian tissue was dissected and treated with 2 mg/mL collagenase in nominally Ca2+-free Barth’s medium (in mM: 88 NaCl, 1 KC1, 0.33 Ca(NO3)2, 0.41 CaCl2, 0.82 MgSO4, 2.4 NaHCO3, 10 HEPES, pH 7.4) for 2 h at rt. On the second day, oocytes were injected with 25-50 nL of (≈ 1 mg/mL) rat GluA2(Q)i cRNA and incubated in Barth’s medium with 0.10 mg/mL gentamicin (Sigma Chemical) and 1 % penicillin-streptomycin (Life Technologies, Paisley, UK) at 17 °C. Oocytes were typically used for recordings from 3 to 10 days post-injection and receptor responses were measured using two-electrode voltage-clamp electrophysiology (GeneClamp 500B, Axon Instruments, Union City, CA) with both microelectrodes filled with 3 M KCl. Recordings were made while the oocytes were continuously superfused with frog Ringer’s solution (in mM: 115 NaCl, 2 KCl, 1.8 BaCl2, 5 HEPES, pH 7.6). Compounds were dissolved in frog Ringer’s solution and added by bath application. Recordings were made at rt. Efficacy measurements were made in the presence of 100 μM cyclothiazide in order to block receptor desensitization. To determine the maximum response, oocytes were stimulated with 1 mM (S)-glutamate plus 100 μM cyclothiazide (n = 10 oocytes). Agonist concentration-response curves were analyzed using GraphPad Prism v6 (GraphPad Software Inc., San Diego, CA) to determine the EC50 using a four-parameter logistic equation (n = 6 oocytes).
5.2.4. Fluo-4/Ca2+ assay.
The functional properties of (S)-glutamate and compounds 6a-c, 7a-d and 8 were characterized at stable GluA1i-, GluA2(Q)i-, GluA3i- and GluA4i-HEK293 cell lines in the Fluo-4/Ca2+ assay essentially as previously described.60 The cells were split into poly-D-lysine-coated black 96-well plates with clear bottom (BD Biosciences, Bedford, MA). 16-24 hours later the medium was aspirated, and the cells were incubated in 50 μL assay buffer [Hanks Buffered Saline Solution (HBSS) containing 20 mM HEPES, 1 mM CaCl2, 1 mM MgCl2 and 2.5 mM probenecid, pH 7.4] supplemented with 6 μM Fluo4/AM (Molecular Probes, Eugene, OR) at 37 °C for 1 hour. The buffer was aspirated, the cells were washed with 100 μL assay buffer, and then 100 μL assay buffer supplemented with 100 μM cyclothiazide was added to the wells (in the antagonist experiments, this buffer was supplemented with various antagonist concentrations). Then the 96-well plate was assayed in a FlexStation3 Benchtop Multi-Mode Microplate Reader (Molecular Devices) measuring emission at 520 nm [in fluorescence units (FU)] caused by excitation at 485 nm a total of 90 sec before and after addition of 33 μL agonist solution (the agonist was dissolved in assay buffer). The experiments were performed in duplicate at least three times for each compound at each cell line.
5.2.5. Two-electrode voltage-clamp electrophysiology for NMDA receptors.
The cDNAs encoding rat NMDA receptor subunits GluN1-1a (Genbank accession number U11418 and U08261; hereafter GluN1), GluN2A (D13211), GluN2B (U11419), GluN2C (M91563), and GluN2D (L31611) were provided by Dr. S. Heinemann (Salk Institute, La Jolla, CA), Dr. S. Nakanishi (Osaka Bioscience Institute, Osaka, Japan), and Dr. P. Seeburg (University of Heidelberg). For expression in Xenopus laevis oocytes, the rat GluN2B open reading frame was edited without changing the amino acid sequence to remove a T7 RNA polymerase termination site located in the C-terminal domain.61 Xenopus oocytes were obtained from Rob Weymouth (Xenopus 1, Dexter, MI). Oocyte incubation, cRNA synthesis, and injections were performed essentially as previously described.62 Briefly, the oocytes were injected with cRNAs encoding GluN1 and GluN2 in a 1:2 ratio, and maintained at 17 °C in Barth’s solution containing (in mM) 88 NaCl, 1 KCl, 2.4 NaHCO3, 0.82 MgSO4, 0.33 mM Ca(NO3)2, 0.91 CaCl2, 10 HEPES (pH 7.5 with NaOH) supplemented with 100 IU/mL penicillin, 100 μg/mL streptomycin and 50 μg/mL gentamycin (Invitrogen, Life Technologies, Paisley, UK). Two-electrode voltage-clamp recordings were performed at rt 2-6 days post-injection with the extracellular recording solution comprised of (in mM) 90 NaCl, 1 KCl, 10 HEPES, 0.5 BaCl2 and 0.01 EDTA (pH 7.4 with NaOH) at a holding potential of −40 mV as previously described.62 NMDA receptor ligands were dissolved in extracellular recording solution and applied to the oocyte by gravity-driven perfusion. Concentration-response data measured using two-electrode voltage-clamp electrophysiology were fitted to the Hill equation to obtain EC50 values for individual oocytes as previously described.62
5.3. Crystal Structures.
5.3.1. Expression and purification.
The rat GluA2-ABD (segment S1 residues 413–527 and segment S2 residues 653–797, numbering according to UNP P19491, connected by a Gly-Thr linker) was expressed and purified essentially as previously described63 using 50 mM L-Asp in the buffers except for the final step. After removal of purification tag two expression remnants (Gly-Ala) residues are present in the N-terminus.
5.3.2. Crystallization and data collection.
Both ligands were crystallized in complex with GluA2-ABD using the hanging drop vapour diffusion method at 6 °C. The drops contained 1 μL of the protein-ligand solution and 1 μL of reservoir solution, and the reservoir volume was 0.5 mL. The protein-ligand solutions were as follows: GluA2-ABD in complex with 6b: 4.8 mg/mL GluA2-ABD, 4.8 mM 6b, 10 mM HEPES pH 7.0, 20 mM NaCl, 1 mM EDTA. GluA2-ABD in complex with 7a: 4.5 mg/mL GluA2-ABD, 7.5 mM 7a, 10 mM HEPES pH 7.0, 20 mM NaCl, 1 mM EDTA. Crystals for diffraction studies appeared within a couple of days, and were obtained with the following reservoir conditions: GluA2-ABD with 6b: 20% PEG4000, 0.3 M lithium sulfate, 0.1 M phosphate-citrate, pH 4.5. GluA2-ABD with 7a: 10% PEG4000, 0.1 M lithium sulfate, 0.1 M phosphate-citrate, pH 4.5 Crystals were flash cooled in liquid nitrogen after briefly soaking in cryo buffer consisting of the reservoir solution containing 10 % glycerol. X-ray data of the GluA2-ABD in complex with 6b and 7a were collected at the BioMAX beamline (MAXIV, Lund, Sweden) at a wavelength of 0.9800 Å.
5.3.3. Structure determination.
The diffraction data were processed with XDS64 and scaled using SCALA 65 within CCP4.66 The structures were solved with molecular replacement using PHASER67 in CCP4 and the structure of GluA2-ABD with 2-Me-Tet-AMPA as search model (PDB code 1M5B, chain A). The structures were first refined with AUTOBUILD68 and then in PHENIX.69 The program MAESTRO [version 10.1.013, Schrodinger, LLC, New York, NY, 2015] was used to generate coordinate files for 6b and 7a. The energy minimization and conformational search were performed in water. The parameter files for 6b and 7a for refinements in PHENIX were obtained using eLBOW,70 keeping the geometry obtained from MAESTRO. The structures were manually adjusted in COOT71 and refined in PHENIX with individual isotropic B-factors, NCS, TLS and riding H atoms. GluA2-ABD domain closures were calculated using the DynDom server72 relative to the apo structure of GluA2-ABD flop (PDB code 1FTO, chain A). Figures 4 and 5 were prepared in PyMOL [version 2.0.3, The PyMOL Molecular Graphics Systems, V.S., LLC].
Supplementary Material
ACKNOWLEDGEMENTS
This research was supported by funds from the University of Turin, Ricerca Locale 2016/2017 (grant nos. LOLM_RILO_17_01, BOSD_RILO_17_01), PRIN 2015 (LOLM_PRIN_2015_16_01), the Lundbeck Foundation. (P.T., S.M., J.S.K.), Danscatt (S.M., J.S.K., D.S.P.) and by the NIH (GM103546 and NS097536) to K.B.H. We wish to thank Dr. Livio Stevanato for performing all of the NMR experiments and for instrument maintenance and Heidi Peterson for help with expression, purification and crystallization. We wish to thank beamline scientists at BioMAX, MAX IV, Lund, Sweden for excellent support during data collections.
ABBREVIATION USED
- Glu
glutamate
- AMPA
(S)-2-amino-3-(3-hydroxy-5-methyl-4-isoxazolyl)propionic acid
- NMDA
N-methyl-D-aspartic acid
- KA
kainic acid
- GluRs
glutamate receptors
- iGluRs
ionotropic glutamate receptors
- mGluRs
metabotropic glutamate receptors
- ABD
agonist binding domain
- SAR@
significant agonist response
- MeTetAMPA
2-methyl-tetrazole-AMPA
- BnTetAMPA
benzyl-tetrazole-AMPA
Footnotes
CONFLICTS OF INTEREST
K.B.H. is a principal investigator on a research grant to University of Montana from Janssen Research & Development.
PDB CODES
PDB ID: 6Q54 (GluA2-ABD in complex with 6b), PDB ID: 6Q60 (GluA2-ABD in complex with 7a). Authors will release the atomic coordinates upon article publication.
ASSOCIATED CONTENT
Supporting info: a PDF containing 1H-NMR and 13C-NMR spectra of all compounds. Molecular formula strings and biological data (CSV file).
REFERENCES
- 1.Brauner-Osborne H; Egebjerg J; Nielsen EO; Madsen U; Krogsgaard-Larsen P Ligands for Glutamate Receptors: Design and Therapeutic Prospects. J Med Chem 2000, 43, 2609–2645. [DOI] [PubMed] [Google Scholar]
- 2.Traynelis SF; Wollmuth LP; McBain CJ; Menniti FS; Vance KM; Ogden KK; Hansen KB; Yuan H; Myers SJ; Dingledine R Glutamate Receptor Ion Channels: Structure, Regulation, and Function. Pharmacol Rev 2010, 62, 405–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Paoletti P; Bellone C; Zhou Q Nmda Receptor Subunit Diversity: Impact on Receptor Properties, Synaptic Plasticity and Disease. Nat Rev Neurosci 2013, 14, 383–400. [DOI] [PubMed] [Google Scholar]
- 4.Zhou Q; Sheng M Nmda Receptors in Nervous System Diseases. Neuropharmacology 2013, 74, 69–75. [DOI] [PubMed] [Google Scholar]
- 5.Lau CG; Zukin RS Nmda Receptor Trafficking in Synaptic Plasticity and Neuropsychiatric Disorders. Nat Rev Neurosci 2007, 8, 413–426. [DOI] [PubMed] [Google Scholar]
- 6.Meyerson JR; Kumar J; Chittori S; Rao P; Pierson J; Bartesaghi A; Mayer ML; Subramaniam S Structural Mechanism of Glutamate Receptor Activation and Desensitization. Nature 2014, 514, 328–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Meyerson JR; Chittori S; Merk A; Rao P; Han TH; Serpe M; Mayer ML; Subramaniam S Structural Basis of Kainate Subtype Glutamate Receptor Desensitization. Nature 2016, 537, 567–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen L; Dürr KL; Gouaux E X-Ray Structures of Ampa Receptor–Cone Snail Toxin Complexes Illuminate Activation Mechanism. Science 2014, 345, 1021–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karakas E; Furukawa H Crystal Structure of a Heterotetrameric Nmda Receptor Ion Channel. Science 2014, 344, 992–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee C-H; Lü W; Michel JC; Goehring A; Du J; Song X; Gouaux E Nmda Receptor Structures Reveal Subunit Arrangement and Pore Architecture. Nature 2014, 511, 191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sobolevsky AI; Rosconi MP; Gouaux E X-Ray Structure, Symmetry and Mechanism of an Ampa-Subtype Glutamate Receptor. Nature 2009, 462, 745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Twomey EC; Yelshanskaya MV; Grassucci RA; Frank J; Sobolevsky AI Structural Bases of Desensitization in Ampa Receptor-Auxiliary Subunit Complexes. Neuron 2017, 94, 569–580.e565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Twomey EC; Yelshanskaya MV; Grassucci RA; Frank J; Sobolevsky AI Channel Opening and Gating Mechanism in Ampa-Subtype Glutamate Receptors. Nature 2017, 549, 60–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yelshanskaya MV; Li M; Sobolevsky AI Structure of an Agonist-Bound Ionotropic Glutamate Receptor. Science 2014, 345, 1070–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stawski P; Janovjak H; Trauner D Pharmacology of Ionotropic Glutamate Receptors: A Structural Perspective. Bioorg Med Chem 2010, 18, 7759–7772. [DOI] [PubMed] [Google Scholar]
- 16.Hansen KB; Yi F; Perszyk RE; Furukawa H; Wollmuth LP; Gibb AJ; Traynelis SF Structure, Function, and Allosteric Modulation of Nmda Receptors. J Gen Physiol 2018, 150, 1081–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Karakas E; Regan MC; Furukawa H Emerging Structural Insights into the Function of Ionotropic Glutamate Receptors. Trends Biochem Sci 2015, 40, 328–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bjerrum EJ; Kristensen AS; Pickering DS; Greenwood JR; Nielsen B; Liljefors T; Schousboe A; Brauner-Osborne H; Madsen U Design, Synthesis, and Pharmacology of a Highly Subtype-Selective Glur1/2 Agonist, (Rs)-2-Amino-3-(4-Chloro-3-Hydroxy-5-Isoxazolyl)Propionic Acid (Cl-Hibo). J Med Chem 2003, 46, 2246–2249. [DOI] [PubMed] [Google Scholar]
- 19.Frydenvang K; Pickering DS; Greenwood JR; Krogsgaard-Larsen N; Brehm L; Nielsen B; Vogensen SB; Hald H; Kastrup JS; Krogsgaard-Larsen P; Clausen RP Biostructural and Pharmacological Studies of Bicyclic Analogues of the 3-Isoxazolol Glutamate Receptor Agonist Ibotenic Acid. J Med Chem 2010, 53, 8354–8361. [DOI] [PubMed] [Google Scholar]
- 20.Vogensen SB; Frydenvang K; Greenwood JR; Postorino G; Nielsen B; Pickering DS; Ebert B; Bolcho U; Egebjerg J; Gajhede M; Kastrup JS; Johansen TN; Clausen RP; Krogsgaard-Larsen P A Tetrazolyl-Substituted Subtype-Selective Ampa Receptor Agonist. J Med Chem 2007, 50, 2408–2414. [DOI] [PubMed] [Google Scholar]
- 21.Jensen AA; Christesen T; Bolcho U; Greenwood JR; Postorino G; Vogensen SB; Johansen TN; Egebjerg J; Brauner-Osborne H; Clausen RP Functional Characterization of Tet-Ampa [Tetrazolyl-2-Amino-3-(3-Hydroxy-5-Methyl- 4-Isoxazolyl)Propionic Acid] Analogues at Ionotropic Glutamate Receptors Glur1-Glur4. The Molecular Basis for the Functional Selectivity Profile of 2-Bn-Tet-Ampa. J Med Chem 2007, 50, 4177–4185. [DOI] [PubMed] [Google Scholar]
- 22.Wang SY; Larsen Y; Navarrete CV; Jensen AA; Nielsen B; Al-Musaed A; Frydenvang K; Kastrup JS; Pickering DS; Clausen RP Tweaking Subtype Selectivity and Agonist Efficacy at (S)-2-Amino-3-(3-Hydroxy-5-Methyl-Isoxazol-4-Yl)Propionic Acid (Ampa) Receptors in a Small Series of Bntetampa Analogues. J Med Chem 2016, 59, 2244–2254. [DOI] [PubMed] [Google Scholar]
- 23.Lolli M; Narramore S; Fishwick CW; Pors K Refining the Chemical Toolbox to Be Fit for Educational and Practical Purpose for Drug Discovery in the 21st Century. Drug Discov Today 2015, 20, 1018–1026. [DOI] [PubMed] [Google Scholar]
- 24.Giorgis M; Lolli ML; Rolando B; Rao A; Tosco P; Chaurasia S; Marabello D; Fruttero R; Gasco A 1,2,5-Oxadiazole Analogues of Leflunomide and Related Compounds. Eur J Med Chem 2011, 46, 383–392. [DOI] [PubMed] [Google Scholar]
- 25.Lolli ML; Giorgis M; Tosco P; Foti A; Fruttero R; Gasco A New Inhibitors of Dihydroorotate Dehydrogenase (Dhodh) Based on the 4-Hydroxy-1,2,5-Oxadiazol-3-Yl (Hydroxyfurazanyl) Scaffold. Eur J Med Chem 2012, 49, 102–109. [DOI] [PubMed] [Google Scholar]
- 26.Bonomo S; Tosco P; Giorgis M; Lolli M; Fruttero R The Role of Fluorine in Stabilizing the Bioactive Conformation of Dihydroorotate Dehydrogenase Inhibitors. J Mol Model 2013, 19, 1099–1107. [DOI] [PubMed] [Google Scholar]
- 27.Sainas S; Pippione AC; Giorgis M; Lupino E; Goyal P; Ramondetti C; Buccinna B; Piccinini M; Braga RC; Andrade CH; Andersson M; Moritzer AC; Friemann R; Mensa S; Al-Kadaraghi S; Boschi D; Lolli ML Design, Synthesis, Biological Evaluation and X-Ray Structural Studies of Potent Human Dihydroorotate Dehydrogenase Inhibitors Based on Hydroxylated Azole Scaffolds. Eur J Med Chem 2017, 129, 287–302. [DOI] [PubMed] [Google Scholar]
- 28.Lolli ML; Sainas S; Pippione AC; Giorgis M; Boschi D; Dosio F Use of Human Dihydroorotate Dehydrogenase (Hdhodh) Inhibitors in Autoimmune Diseases and New Perspectives in Cancer Therapy. Recent Pat Anticancer Drug Discov 2018, 13, 86–105. [DOI] [PubMed] [Google Scholar]
- 29.Sainas S; Pippione AC; Lupino E; Giorgis M; Circosta P; Gaidano V; Goyal P; Bonanni D; Rolando B; Cignetti A; Ducime A; Andersson M; Jarva M; Friemann R; Piccinini M; Ramondetti C; Buccinna B; Al-Karadaghi S; Boschi D; Saglio G; Lolli ML Targeting Myeloid Differentiation Using Potent 2-Hydroxypyrazolo[1,5- a]Pyridine Scaffold-Based Human Dihydroorotate Dehydrogenase Inhibitors. J Med Chem 2018, 61, 6034–6055. [DOI] [PubMed] [Google Scholar]
- 30.Pippione AC; Sainas S; Goyal P; Fritzson I; Cassiano GC; Giraudo A; Giorgis M; Tavella TA; Bagnati R; Rolando B; Caing-Carlsson R; Costa FTM; Andrade CH; Al-Karadaghi S; Boschi D; Friemann R; Lolli ML Hydroxyazole Scaffold-Based Plasmodium Falciparum Dihydroorotate Dehydrogenase Inhibitors: Synthesis, Biological Evaluation and X-Ray Structural Studies. Eur J Med Chem 2019, 163, 266–280. [DOI] [PubMed] [Google Scholar]
- 31.Sainas S; Pippione AC; Boschi D; Gaidano V; Circosta P; Cignetti A; Dosio F; Lolli ML Dhodh Inhibitors and Leukemia: An Emergent Interest for New Myeloid Differentiation Agents. Drugs of the Future 2018, 43, 823–834. [Google Scholar]
- 32.Pippione AC; Giraudo A; Bonanni D; Carnovale IM; Marini E; Cena C; Costale A; Zonari D; Pors K; Sadiq M; Boschi D; Oliaro-Bosso S; Lolli ML Hydroxytriazole Derivatives as Potent and Selective Aldo-Keto Reductase 1c3 (Akr1c3) Inhibitors Discovered by Bioisosteric Scaffold Hopping Approach. Eur J Med Chem 2017, 139, 936–946. [DOI] [PubMed] [Google Scholar]
- 33.Pippione AC; Carnovale IM; Bonanni D; Sini M; Goyal P; Marini E; Pors K; Adinolfi S; Zonari D; Festuccia C; Wahlgren WY; Friemann R; Bagnati R; Boschi D; Oliaro-Bosso S; Lolli ML Potent and Selective Aldo-Keto Reductase 1c3 (Akr1c3) Inhibitors Based on the Benzoisoxazole Moiety: Application of a Bioisosteric Scaffold Hopping Approach to Flufenamic Acid. Eur J Med Chem 2018, 150, 930–945. [DOI] [PubMed] [Google Scholar]
- 34.Lolli ML; Carnovale IM; Pippione AC; Wahlgren WY; Bonanni D; Marini E; Zonari D; Gallicchio M; Boscaro V; Goyal P; Friemann R; Rolando B; Bagnati R; Adinolfi S; Oliaro-Bosso S; Boschi D Bioisosteres of Indomethacin as Inhibitors of Aldo-Keto Reductase 1c3. ACS Medicinal Chemistry Letters 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pippione AC; Federico A; Ducime A; Sainas S; Boschi D; Barge A; Lupino E; Piccinini M; Kubbutat M; Contreras J-M; Morice C; Al-Karadaghi S; Lolli ML 4-Hydroxy-N-[3,5-Bis(Trifluoromethyl)Phenyl]-1,2,5-Thiadiazole-3-Carboxamide: A Novel Inhibitor of the Canonical Nf-Kb Cascade. MedChemComm 2017, 8, 1850–1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pippione AC; Sainas S; Federico A; Lupino E; Piccinini M; Kubbutat M; Contreras J-M; Morice C; Barge A; Ducime A; Boschi D; Al-Karadaghi S; Lolli ML N-Acetyl-3-Aminopyrazoles Block the Non-Canonical Nf-Kb Cascade by Selectively Inhibiting Nik. MedChemComm 2018, 9, 963–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lolli ML; Hansen SL; Rolando B; Nielsen B; Wellendorph P; Madsen K; Larsen OM; Kristiansen U; Fruttero R; Gasco A; Johansen TN Hydroxy-1,2,5-Oxadiazolyl Moiety as Bioisoster of the Carboxy Function. Synthesis, Ionization Constants, and Pharmacological Characterization of Gamma-Aminobutyric Acid (Gaba) Related Compounds. J Med Chem 2006, 49, 4442–4446. [DOI] [PubMed] [Google Scholar]
- 38.Tosco P; Lolli ML Hydroxy-1,2,5-Oxadiazolyl Moiety as Bioisoster of the Carboxy Function. A Computational Study on Gamma-Aminobutyric Acid (Gaba) Related Compounds. J Mol Model 2008, 14, 279–291. [DOI] [PubMed] [Google Scholar]
- 39.Giraudo A; Krall J; Nielsen B; Sorensen TE; Kongstad KT; Rolando B; Boschi D; Frolund B; Lolli ML 4-Hydroxy-1,2,3-Triazole Moiety as Bioisostere of the Carboxylic Acid Function: A Novel Scaffold to Probe the Orthosteric Gamma-Aminobutyric Acid Receptor Binding Site. Eur J Med Chem 2018, 158, 311–321. [DOI] [PubMed] [Google Scholar]
- 40.Lolli ML; Giordano C; Pickering DS; Rolando B; Hansen KB; Foti A; Contreras-Sanz A; Amir A; Fruttero R; Gasco A; Nielsen B; Johansen TN 4-Hydroxy-1,2,5-Oxadiazol-3-Yl Moiety as Bioisoster of the Carboxy Function. Synthesis, Ionization Constants, and Molecular Pharmacological Characterization at Ionotropic Glutamate Receptors of Compounds Related to Glutamate and Its Homologues. J Med Chem 2010, 53, 4110–4118. [DOI] [PubMed] [Google Scholar]
- 41.Pippione AC; Dosio F; Ducime A; Federico A; Martina K; Sainas S; Frolund B; Gooyit M; Janda KD; Boschi D; Lolli ML Substituted 4-Hydroxy-1,2,3-Triazoles: Synthesis, Characterization and First Drug Design Applications through Bioisosteric Modulation and Scaffold Hopping Approaches. MedChemComm 2015, 6, 1285–1292. [Google Scholar]
- 42.Clausen RP; Hansen KB; Cali P; Nielsen B; Greenwood JR; Begtrup M; Egebjerg J; Brauner-Osborne H The Respective N-Hydroxypyrazole Analogues of the Classical Glutamate Receptor Ligands Ibotenic Acid and (Rs)-2-Amino-2-(3-Hydroxy-5-Methyl-4-Isoxazolyl)Acetic Acid. Eur J Pharmacol 2004, 499, 35–44. [DOI] [PubMed] [Google Scholar]
- 43.Bunch L; Gefflaut T; Alaux S; Sagot E; Nielsen B; Pickering DS Pharmacological Characterization of (4r)-Alkyl Glutamate Analogues at the Ionotropic Glutamate Receptors--Focus on Subtypes Iglu(5-7). Eur J Pharmacol 2009, 609, 1–4. [DOI] [PubMed] [Google Scholar]
- 44.Pohlsgaard J; Frydenvang K; Madsen U; Kastrup JS Lessons from More Than 80 Structures of the Glua2 Ligand-Binding Domain in Complex with Agonists, Antagonists and Allosteric Modulators. Neuropharmacology 2011, 60, 135–150. [DOI] [PubMed] [Google Scholar]
- 45.Chen VB; Arendall WB 3rd; Headd JJ; Keedy DA; Immormino RM; Kapral GJ; Murray LW; Richardson JS; Richardson DC Molprobity: All-Atom Structure Validation for Macromolecular Crystallography. Acta Crystallogr D Biol Crystallogr 2010, 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lunn ML; Hogner A; Stensbol TB; Gouaux E; Egebjerg J; Kastrup JS Three-Dimensional Structure of the Ligand-Binding Core of Glur2 in Complex with the Agonist (S)-Atpa: Implications for Receptor Subunit Selectivity. J Med Chem 2003, 46, 872–875. [DOI] [PubMed] [Google Scholar]
- 47.Hogner A; Kastrup JS; Jin R; Liljefors T; Mayer ML; Egebjerg J; Larsen IK; Gouaux E Structural Basis for Ampa Receptor Activation and Ligand Selectivity: Crystal Structures of Five Agonist Complexes with the Glur2 Ligand-Binding Core. J Mol Biol 2002, 322, 93–109. [DOI] [PubMed] [Google Scholar]
- 48.Armstrong N; Gouaux E Mechanisms for Activation and Antagonism of an Ampa-Sensitive Glutamate Receptor: Crystal Structures of the Glur2 Ligand Binding Core. Neuron 2000, 28, 165–181. [DOI] [PubMed] [Google Scholar]
- 49.Madsen U; Schaumburg K; Brehm L; Curtis DR; Krogsgaard-Larsen P Ibotenic Acid Analogues. Synthesis and Biological Testing of Two Bicyclic 3-Isoxazolol Amino Acids. Acta Chem Scand B 1986, 40, 92–97. [DOI] [PubMed] [Google Scholar]
- 50.Banke TG; Greenwood JR; Christensen JK; Liljefors T; Traynelis SF; Schousboe A; Pickering DS Identification of Amino Acid Residues in Glur1 Responsible for Ligand Binding and Desensitization. The Journal of Neuroscience 2001, 21, 3052–3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Maolanon AR; Risgaard R; Wang SY; Snoep Y; Papangelis A; Yi F; Holley D; Barslund AF; Svenstrup N; Hansen KB; Clausen RP Subtype-Specific Agonists for Nmda Receptor Glycine Binding Sites. ACS Chem Neurosci 2017, 8, 1681–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sainas S; Pippione AC; Giraudo A; Martina K; Bosca F; Rolando B; Barge A; Ducime A; Federico A; Grossert SJ; White RL; Boschi D; Lolli ML Regioselective N-Alkylation of Ethyl 4-Benzyloxy-1,2,3-Triazolecarboxylate: A Useful Tool for the Synthesis of Carboxylic Acid Bioisosteres. J Heterocycl Chem 2018, 56, 501–519. [Google Scholar]
- 53.Honoré T; Nielsen M Complex Structure of Quisqualate-Sensitive Glutamate Receptors in Rat Cortex. Neuroscience Letters 1985, 54, 27–32. [DOI] [PubMed] [Google Scholar]
- 54.Braitman DJ; Coyle JT Inhibition of [3h]Kainic Acid Receptor Binding by Divalent Cations Correlates with Ion Affinity for the Calcium Channel. Neuropharmacology 1987, 26, 1247–1251. [DOI] [PubMed] [Google Scholar]
- 55.Sills MA; Fagg G; Pozza M; Angst C; Brundish DE; Hurt SD; Wilusz EJ; Williams M [3h]Cgp 39653: A New N-Methyl-D-Aspartate Antagonist Radioligand with Low Nanomolar Affinity in Rat Brain. Eur J Pharmacol 1991, 192, 19–24. [DOI] [PubMed] [Google Scholar]
- 56.Assaf Z; Larsen AP; Venskutonytė R; Han L; Abrahamsen B; Nielsen B; Gajhede M; Kastrup JS; Jensen AA; Pickering DS; Frydenvang K; Gefflaut T; Bunch L Chemoenzymatic Synthesis of New 2,4-Syn-Functionalized (S)-Glutamate Analogues and Structure-Activity Relationship Studies at Ionotropic Glutamate Receptors and Excitatory Amino Acid Transporters. Journal of Medicinal Chemistry 2013, 56, 1614–1628. [DOI] [PubMed] [Google Scholar]
- 57.Ransom RW; Stec NL Cooperative Modulation of [3h]Mk-801 Binding to the N-Methyl-D-Aspartate Receptor-Ion Channel Complex by L-Glutamate, Glycine, and Polyamines. J Neurochem 1988, 51, 830–836. [DOI] [PubMed] [Google Scholar]
- 58.Vogensen SB; Clausen RP; Greenwood JR; Johansen TN; Pickering DS; Nielsen B; Ebert B; Krogsgaard-Larsen P Convergent Synthesis and Pharmacology of Substituted Tetrazolyl-2-Amino-3-(3-Hydroxy-5-Methyl-4-Isoxazolyl)Propionic Acid Analogues. J Med Chem 2005, 48, 3438–3442. [DOI] [PubMed] [Google Scholar]
- 59.Sagot E; Pickering DS; Pu X; Umberti M; Stensbol TB; Nielsen B; Chapelet M; Bolte J; Gefflaut T; Bunch L Chemo-Enzymatic Synthesis of a Series of 2,4-Syn-Functionalized (S)-Glutamate Analogues: New Insight into the Structure-Activity Relation of Ionotropic Glutamate Receptor Subtypes 5, 6, and 7. J Med Chem 2008, 51, 4093–4103. [DOI] [PubMed] [Google Scholar]
- 60.Strange M; Brauner-Osborne H; Jensen AA Functional Characterisation of Homomeric Ionotropic Glutamate Receptors Glur1-Glur6 in a Fluorescence-Based High Throughput Screening Assay. Comb Chem High Throughput Screen 2006, 9, 147–158. [DOI] [PubMed] [Google Scholar]
- 61.Hansen KB; Ogden KK; Yuan H; Traynelis SF Distinct Functional and Pharmacological Properties of Triheteromeric Glun1/Glun2a/Glun2b Nmda Receptors. Neuron 2014, 81, 1084–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hansen KB; Tajima N; Risgaard R; Perszyk RE; Jorgensen L; Vance KM; Ogden KK; Clausen RP; Furukawa H; Traynelis SF Structural Determinants of Agonist Efficacy at the Glutamate Binding Site of N-Methyl-D-Aspartate Receptors. Mol Pharmacol 2013, 84, 114–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Krintel C; Frydenvang K; Ceravalls de Rabassa A; Kærn AM; Gajhede M; Pickering DS; Kastrup JS L-Asp Is a Useful Tool in the Purification of the Ionotropic Glutamate Receptor a2 Ligand-Binding Domain. The FEBS Journal 2014, 281, 2422–2430. [DOI] [PubMed] [Google Scholar]
- 64.Kabsch W Integration, Scaling, Space-Group Assignment and Post-Refinement. Acta Crystallogr D Biol Crystallogr 2010, 66, 133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Evans PR An Introduction to Data Reduction: Space-Group Determination, Scaling and Intensity Statistics. Acta Crystallogr D Biol Crystallogr 2011, 67, 282–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Winn MD; Ballard CC; Cowtan KD; Dodson EJ; Emsley P; Evans PR; Keegan RM; Krissinel EB; Leslie AG; McCoy A; McNicholas SJ; Murshudov GN; Pannu NS; Potterton EA; Powell HR; Read RJ; Vagin A; Wilson KS Overview of the Ccp4 Suite and Current Developments. Acta Crystallogr D Biol Crystallogr 2011, 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.McCoy AJ; Grosse-Kunstleve RW; Adams PD; Winn MD; Storoni LC; Read RJ Phaser Crystallographic Software. J Appl Crystallogr 2007, 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Terwilliger TC; Grosse-Kunstleve RW; Afonine PV; Moriarty NW; Zwart PH; Hung LW; Read RJ; Adams PD Iterative Model Building, Structure Refinement and Density Modification with the Phenix Autobuild Wizard. Acta Crystallogr D Biol Crystallogr 2008, 64, 61–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Adams PD; Afonine PV; Bunkoczi G; Chen VB; Davis IW; Echols N; Headd JJ; Hung LW; Kapral GJ; Grosse-Kunstleve RW; McCoy AJ; Moriarty NW; Oeffner R; Read RJ; Richardson DC; Richardson JS; Terwilliger TC; Zwart PH Phenix: A Comprehensive Python-Based System for Macromolecular Structure Solution. Acta Crystallogr D Biol Crystallogr 2010, 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Moriarty NW; Grosse-Kunstleve RW; Adams PD Electronic Ligand Builder and Optimization Workbench (Elbow): A Tool for Ligand Coordinate and Restraint Generation. Acta Crystallogr D Biol Crystallogr 2009, 65, 1074–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Emsley P; Lohkamp B; Scott WG; Cowtan K Features and Development of Coot. Acta Crystallogr D Biol Crystallogr 2010, 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hayward S; Berendsen HJ Systematic Analysis of Domain Motions in Proteins from Conformational Change: New Results on Citrate Synthase and T4 Lysozyme. Proteins 1998, 30, 144–154. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.