

SUMMARY
Neuroligins, postsynaptic cell-adhesion molecules that are linked to neuropsychiatric disorders, are extensively studied, yet fundamental questions about their functions remain. Using in vivo replacement strategies in quadruple conditional knockout mice of all neuroligins to avoid heterodimerization artifacts, we show in hippocampal CA1 pyramidal neurons that neuroligin-1 performs two key functions in excitatory synapses by distinct molecular mechanisms. NMDA-receptor-dependent LTP requires trans-synaptic binding of postsynaptic neuroligin-1 to presynaptic β-neurexins but not the cytoplasmic sequences of neuroligins. In contrast, postsynaptic NMDA-receptor (NMDAR)-mediated responses involve a neurexin-independent mechanism that requires the neuroligin-1 cytoplasmic sequences. Strikingly, deletion of neuroligins blocked the spine expansion associated with LTP as monitored by two-photon imaging; this block involved a mechanism identical to that of the role of neuroligin-1 in NMDAR-dependent LTP. Our data suggest that neuroligin-1 performs two mechanistically distinct signalling functions, and that neurolign-1-mediated trans-synaptic cell-adhesion signalling critically regulates LTP.
In Brief
At excitatory synapses on CA1 pyramidal neurons lacking neuroligins, Nlgn1 rescues impairments in functional and structural LTP and in NMDA receptor-mediated synaptic transmission. Structure-function rescue experiments reveal that Nlgn1 plays mechanistically distinct roles in LTP and basal NMDA receptor-mediated transmission.
INTRODUCTION
Activity-dependent regulation of synaptic transmission determines the input-output relationships of neural circuits and critically contributes to the astounding information-processing capacity of the brain. However, the mechanisms of activity-dependent synaptic plasticity remain incompletely understood. At excitatory synapses, NMDA receptors (NMDARs) play a major role in shaping synaptic responses acutely and in the induction of long-term potentiation (LTP), a critically important form of synaptic plasticity (Bliss and Collingridge, 1993; Malenka and Bear, 2004; Malenka and Nicoll, 1999). How NMDARs are regulated and how LTP is controlled in an overall synaptic context, however, remains unclear. Decades of studies have elucidated the structure and function of NMDARs and the role of various postsynaptic proteins in LTP in some detail (Huganir and Nicoll, 2013; Luscher and Malenka, 2012; Nicoll, 2017), but the mechanisms that embed these key postsynaptic features into the function of the overall synapse are unexplored.
Over the past decade, it has become increasingly apparent that synaptic cell-adhesion molecules are critically involved in the specification of synaptic properties and in various forms of synaptic plasticity ( Südhof, 2008, 2017). A major post-synaptic family of synaptic cell-adhesion proteins are neuroligins, which are type I membrane proteins that interact with presynaptic neurexins. The mammalian brain expresses four neuroligins (Nlgn1 - Nlgn4) that exhibit distinct properties. Nlgn1 localizes to excitatory synapses, Nlgn2 to inhibitory and cholinergic synapses, Nlgn3 to both excitatory and inhibitory synapses, and Nlgn4 to glycinergic synapses (Budreck and Scheiffele, 2007; Graf et al., 2004; Hoon et al., 2011; Song et al., 1999; Varoqueaux et al., 2004). Stringent analyses of mouse neurons with constitutive or conditional genetic deletions of neuroligins revealed that neuroligins are not generally required for synaptogenesis, but confer critical properties onto their resident synapses (Chubykin et al., 2007; Foldy et al, 2013; Gibson et al., 2009; Poulopoulos et al., 2009; Varoqueaux et al., 2006; Zhang et al., 2015). This is particularly apparent for Nlgn1. Previous experiments showed that genetic deletion of Nlgn1 caused a decrease in NMDAR-mediated responses in cultured hippocampal neurons (Chubykin et al., 2007), and that conditional deletion of Nlgn1 alone or Nlgn123 together from hippocampal CA1 pyramidal neurons in vivo blocked NMDA receptor (NMDAR)-dependent LTP and simultaneously reduced the ratio of NMDAR-mediated excitatory postsynaptic currents (NMDAR EPSCs) to AMPA receptor-mediated EPSCs (AMPAR EPSCs) (Jiang et al., 2017).
However, the role of Nlgn1 in specifying synapse properties and enabling LTP has not been undisputed, and the mechanisms underlying the role of Nlgn1 in these processes remain controversial (Budreck et al., 2013; Hoy et al., 2013; Jiang et al., 2017; Shipman and Nicoll, 2012a, b). In considering these issues, a crucial variable is the approach used in a particular study. For example, one previous study (Shipman and Nicoll, 2012b) failed to observe a requirement for Nlgn1 in LTP in adult hippocampal CA1 pyramidal neurons but since this study used microRNAs, incomplete ablation of Nlgn1 expression and potential off-target effects of the microRNAs could have influenced their results. Another study (Budreck et al., 2013) employed constitutive Nlgn1 single knock-out (KO) mice for rescue experiments to explore the mechanism of Nlgn1 action, but the continued expression of other neuroligins in these mice raises the possibility of heterodimerization of rescue proteins with remaining neuroligins.
Considering the current state of the field, there are important implications in rigorously dissecting the role of Ngln1 in synaptic plasticity. First, Nlgn1 is required for the generation of LTP, which is one of the most important and extensively studied forms of neural plasticity. Second, Ngln1 mutations are associated with psychiatric disorders including autism and schizophrenia (Südhof, 2008; 2017). Therefore, a precise understanding of the function of Nlgn1 in LTP will advance understanding of the molecular mechanisms underlying a prototypic form of synaptic plasticity and in turn provide a molecular framework, which will be valuable for the identification of potential therapeutic targets for debilitating, common brain disorders.
Here, we perform a detailed structure-function analysis of the synaptic roles of Nlgn1 by performing in vivo rescue experiments in hippocampal CA1 pyramidal neurons in conditional single Nlgn1 and quadruple Nlgn1234 cKO mice in which expression of Cre-recombinase genetically deletes Nlgn1 or all four neuroligins, respectively. This approach enables the precisely timed replacement of endogenous Nlgn1 in identifiable neurons in vivo under conditions that exclude any possibility of heterodimerization between the rescue construct and remaining endogenous neuroligins. In mice lacking either only Nlgn1 or all neuroligins, expression of the Nlgn1 extracellular sequences anchored to the membrane by GPI-lipids was, surprisingly, sufficient to restore LTP as well as the growth of spines that accompanies LTP. These results indicate that the binding of Nlgn1 to PDZ domain-containing proteins such as PSD95 is dispensable for both functional and structural LTP. In contrast, Nlgn1 binding to presynaptic neurexins was critical for both functional and structural LTP, as LTP rescue by Nlgn1 was prevented by mutation of residues critical for neurexin binding. Consistent with a critical role of the Nlgn1-neurexin interactions in LTP, genetic deletion of presynaptic β-neurexins blocked LTP and spine growth. The rescue of basal NMDAR-mediated synaptic transmission due to Nlgn1234 deletion exhibited the opposite Nlgn1 domain-dependence: it required Nlgn1’s intracellular domain but not binding to neurexins. This dramatic molecular dissociation of the Nlgn1 domains required for LTP versus the maintenance of basal NMDAR-mediated synaptic transmission provides unexpected complexity to the molecular architecture responsible for regulating synaptic strength at excitatory synapses.
RESULTS
Nlgn1 Domain-Specific Rescue of NMDAR-dependent LTP in Nlgn1 cKO mice
Previous studies showed that Nlgn1 binds to the postsynaptic scaffolding protein PSD95 (Irie et al., 1997), that PSD95 may be involved in LTP (Ehrlich and Malinow, 2004; Stein et al., 2003), that Nlgn1 maintains postsynaptic NMDAR responses (Chubykin et al., 2007), and that conditional deletion of Nlgn1 in vivo abolished LTP (Jiang et al., 2017). Viewed together, these results suggest that Nlgn1 may transduce an extracellular signal by a PSD95-dependent mechanism, that this function of Nlgn1 may be related to its involvement with NMDARs, and that this process of signal transduction is essential for LTP. To test this hypothesis, we examined the mechanism of Nlgn1 action by performing rescue experiments in Nlgn1 cKO mice, a method that allowed testing of which domains of Nlgn1 are crucial for LTP.
We generated lentiviruses expressing a Cre-recombinase (Cre)-EGFP fusion protein and various mutant forms of Nlgn1 (Figure 1A). All Nlgn1 proteins contained an insert in splice sites A and B (Comoletti et al., 2007). Transmembrane Nlgn1 constructs were each tagged with a single N-terminal HA-epitope whereas GPI-anchored Nlgn1 constructs contained two consecutive HA-epitopes between the Nlgn-1 extracellular domain and the NCAM signal sequence (Gokce and Sudhof, 2013). Cre-EGFP and Nlgn1 were expressed from di-cistronic messages in which Nlgn1 sequences were translated via an IRES sequence (Figure 1A). We stereotactically injected a small amount of lentiviruses into the CA1 region of Nlgn1 cKO mice at three week of age (P21) to achieve sparse infection of CA1 pyramidal neurons (Figure 1B). Two to three weeks after stereotactic infections (at P35-P42), we prepared acute hippocampal slices from injected mice and performed whole-cell voltage-clamp recordings from CA1 pyramidal neurons (Figure 1C). NMDAR-dependent LTP was induced by applying two 100 Hz, 1 sec stimulus trains separated by 20 sec while holding the cells at 0 mV. Recordings from infected and uninfected cells were interleaved with one cell being recorded from each slice from the batch of slices prepared daily from the same animal.
Figure 1. Nlgn1 Domain-Specific Rescue of NMDAR-dependent LTP in Nlgn1 cKO mice.
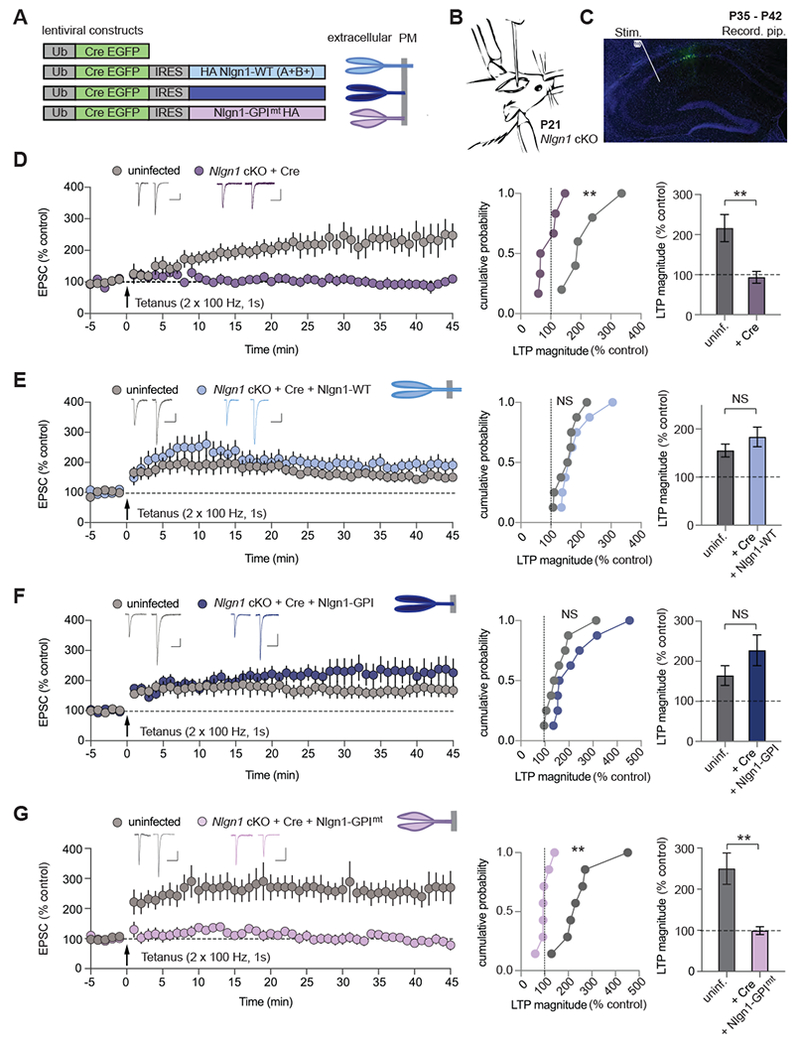
(A) Schematics of lentiviral constructs expressing Cre-EGFP fusion protein and various mutant forms of Nlgn1 (left) along with diagrams of Nlgn1 rescue proteins (right; PM: plasma membrane).
(B) Illustration of mouse lentiviral stereotactic injection into the CA1 region of the hippocampus at P21.
(C) Annotated confocal image of a hippocampal slice from a mouse (P35-P42) showing lentiviral Cre-EGFP + AAV-DJ-CMV-DIO-EGFP (cytoplasmic) infection in the CA1. The stimulation electrode (Stim.) is placed in stratum radiatum and the recording pipette (Recording pip.) is sealed onto a CA1 pyramidal neuron. DAPI (blue), GFP (green). Scale bar: 500 μm.
(D – G) NMDAR-dependent LTP in Nlgn1 cKO mice following Cre-EGFP expression and simultaneous expression of Nlgn1 rescue constructs. Summary of LTP time course (left panels) in uninfected and lentiviral infected CA1 pyramidal neurons showing sample average EPSCs pre- and post-tetanus (35 – 40 min); cumulative frequency plots of LTP magnitudes from individual experiments (middle panels); and quantification of LTP magnitude (right panels). The lentiviruses expressed (D) Cre-EGFP, (E) Cre-EGFP IRES wild-type Nlgn1 with splice sites A and B in (Nlgn1-WT), (F) Cre-EGFP IRES GPI membrane anchored extracellular domain of Nlgn1 (Nlgn1-GPI), (G) GPI anchored Nlgn1, as in F, mutated at neurexin binding sites (Nlgn1-GPImt). Scale bars: 50 pA, 100 ms. Numbers in bar graphs correspond to experimental ‘n’ of cells/animals. All values plotted with error bars are mean ± SEM. Mann-Whitney test, *p<0.05, **p<0.01, NS = not significant.
Consistent with previous results, LTP was blocked by deletion of Nlgn1 alone (Jiang et al., 2017) (Figure 1D). Simultaneous expression of Nlgn1-WT rescued LTP (Figure 1E), as did expression of the extracellular domain of Nlgn1 anchored to the plasma membrane by a GPI-lipid modification (Nlgn1-GPI) (Gokce and Sudhof, 2013), which thus lacks the transmembrane region and cytoplasmic sequences (Figure 1F). However, mutating the neurexin-binding site of Nlgn1-GPI (Nlgn1-GPImt, Arac et al., 2007) completely prevented rescue of NMDAR-dependent LTP by Nlgn1-GPI (Figure 1G). When expressed in cultured neurons, all of these constructs trafficked to the plasma membrane and showed levels of expression similar to or less than that of endogenous Nlgn1 (Figure S1).
Nlgn1 Domain-Specific Rescue of NMDAR-dependent LTP in Nlgn1234 cKO mice
These results suggest that, surprisingly, the binding of Nlgn1 to presynaptic neurexins but not the transduction of a neurexin binding signal to the postsynaptic cytoplasmic Nlgn1 sequences is critical for the role of Nlgn1 in LTP. However, other neuroligins are expressed in Nlgn1 cKO neurons and could heterodimerize with the rescue constructs (Poulopoulos et al., 2012). Because of this possibility, our results in Nlgn1 cKO neurons are not conclusive because it is possible that mutant Nlgn1-GPI used for rescues could have reversed the block of LTP by heteromerizing with endogenous Nlgn3, and thereby couple the extracellular Nlgn1-GPI domains to the intracellular Nlgn3 effector sequences. To exclude this possibility, we generated quadruple Nlgn1234 cKO mice in which Nlgn1, Nlgn2, and Nlgn3 are expressed normally but deleted by Cre-recombinase, whereas Nlgn4 is constitutively ablated (Jamain et al., 2008) (Figure S2A). Consistent with the results of genetically deleting Nlgn1 (Figure 1), Cre-mediated deletion of all neuroligins in vivo using lentivirus injections at P21 blocked NMDAR-dependent LTP (Figure 2A). Strikingly, LTP was again rescued by expression of either Nlgn1-WT (Figure S2B) or of Nlgn1-GPI (Figure 2B). Since Nlgn1-GPI proteins can no longer heterodimerize with an endogenous neuroligin in the quadruple KO neurons, this result establishes that the transmembrane region and cytoplasmic sequences of Nlgn1 are not essential for LTP. To assess whether Nlgn1-GPI mediated LTP is indeed NMDAR-dependent, we bath applied APV (50 μM) before tetanic stimulation (Figure S2C). The presence of APV prevented the generation of Nlgn1-GPI mediated LTP, providing classic evidence that it is NMDAR-dependent. Moreover, again as in Nlgn1 cKO mice, mutation of the neurexin-binding site in Nlgn1-GPI (Nlgn1-GPImt) blocked rescue of LTP (Figure 2C). As an additional test of the critical role of Nlgn1 binding to neurexins in LTP, we made the neurexin binding mutations in full-length Nlgn1, which also failed to rescue LTP upon ablation of Nlgn1234 (Figure S2B).
Figure 2. Nlgn1 Domain-Specific Rescue of NMDAR-dependent LTP in Nlgn1234 cKO mice.
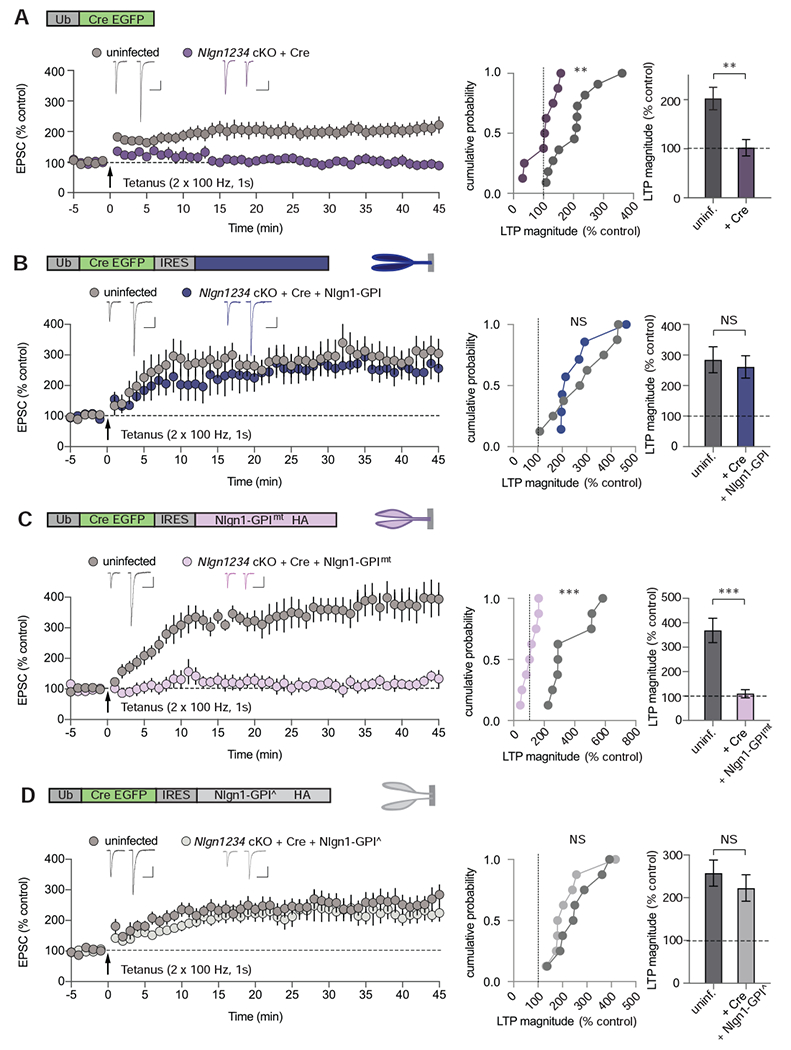
(A – D) NMDAR-dependent LTP in Nlgn1234 cKO mice following Cre-EGFP expression and simultaneous expression of Nlgn1 rescue constructs. Schematics of lentiviral constructs and Nlgn1 rescue proteins are shown above each set of summary graphs. Summary of LTP time course (left panels) in uninfected and lentiviral infected CA1 pyramidal neurons showing sample average EPSCs pre- and post-tetanus (35 – 40 min); cumulative frequency plots of LTP magnitudes from individual experiments (middle panels); and quantification of LTP magnitude (right panels). The lentiviruses expressed: (A) Cre-EGFP, (B) Cre-EGFP IRES GPI membrane anchored extracellular domain of Nlgn1 (Nlgn1-GPI), (C) Cre-EGFP IRES GPI anchored Nlgn1, mutated at neurexin binding sites (Nlgn1-GPImt), (D) Cre-EGFP IRES GPI anchored Nlgn1 with mutated dimerization sites (Nlgn1-GPI^), Scale bars: 50 pA, 100 ms. Numbers in bars correspond to experimental ‘n’ of cells/animals. All values plotted with error bars are mean ± SEM. Mann-Whitney test,*p<0.05, **p<0.01, ***p<0.001, NS = not significant.
Neuroligin dimerization has been suggested to be critical for Nlgn1 function as monitored with a mutation that was not tested for its effect on Nlgn1 folding but based on its sequence was assumed to be disruptive (Shipman and Nicoll, 2012a). However, neuroligin dimerization is not required for the synapse-inducing activity of Nlgn1 as analysed with a mutation that was shown by biophysical measurement to not significantly impair Nlgn1 folding (Ko et al., 2009). To test whether Nlgn1 homodimerization is required for LTP, we generated a mutant version of Nlgn1-GPI that still folds correctly and binds neurexins but does not dimerize (Arac et al., 2007; Ko et al., 2009). Expression of this mutant, which also lacks all cytoplasmic and transmembrane sequences of Nlgn1, in Nlgn1234 quadruple cKO neurons robustly rescued LTP (Figure 2D). Thus, the intracellular and transmembrane sequences of Nlgn1 and the dimerization of Nlgn1 are unexpectedly dispensable for LTP, but binding of Nlgn1 to neurexins is essential for LTP. These results imply that Nlgn1 transduces a trans-synaptic neurexin signal that is required for LTP via a mechanism that is independent of Nlgn1 binding to PSD95.
Conditional Deletion of all Neuroligins in Adult Neurons Selectively Impairs NMDAR- But Not AMPAR-mediated Synaptic Responses
Knockout or knockdown of Nlgn1 has consistently been found to reduce NMDAR-mediated synaptic currents in CA1 pyramidal neurons with varying effects on AMPAR-mediated synaptic currents, depending on the manipulation performed and preparation being studied (Budreck et al., 2013; Hoy et al., 2013; Jiang et al., 2017). Moreover, triple deletion of Nlgn1, Nlgn2, and Nlgn3 has been shown to generally decrease synaptic responses in cultured neurons, although no loss of synapses was observed (Chanda et al., 2017; Varoqueaux et al., 2006). However, the effect of the deletions of all neuroligins on basal excitatory synaptic strength in vivo in mature synapses on adult neurons has not been examined. This is an important topic since impairment in diverse synaptic properties might influence the induction of LTP. Furthermore, genetic deletion of neuroligins in cultured neurons or young mice may influence synaptic responses via effects on synaptogenesis or synapse maturation, not because neuroligins play a critical role in maintaining basal synaptic transmission at mature synapses. To address this question, we again injected lentiviruses expressing Cre-recombinase into the CA1 region of P21 Nlgn1234 cKO mice, and performed simultaneous whole-cell voltage-clamp recordings 2 – 3 weeks later from infected and adjacent non-infected CA1 pyramidal neurons in acute slices (Figure 3A). Surprisingly, we found that AMPAR EPSCs were not significantly altered by deletion of all neuroligins (Figure 3B, C). NMDAR EPSCs, however, were on average reduced by ~40% (Figure 3B, C). These results are consistent with previous work using the Nlgn123 cKO and Nlgn1 cKO mouse lines in which the ratio of NMDAR EPSCs to AMPAR EPSCs (the NMDAR/AMPAR ratio) was reduced following genetic ablation of Nlgn123 or Nlgn1 alone while AMPAR miniature EPSCs were unaffected (Jiang et al., 2017). To test further whether AMPAR-mediated synaptic currents are affected by knock-out of neuroligins, we recorded miniature EPSCs first in Nlgn1 cKO mice and then in the Nlgn1234 cKO mice. Consistent with previous findings (Jiang et al., 2017), conditional deletion of Nlgn1 had no effect on AMPAR miniature EPSCs (Figure S3A). Similarly, conditional deletion of Nlgn1234 did not change the frequency and amplitude of miniature EPSCs (Figure S3B). These findings further strengthen the hypothesis that the numbers of postsynaptic AMPARs are not affected by neuroligin deletion and also suggest that neither are the numbers of functional synapses on CA1 pyramidal neurons.
Figure 3. Nlgn1’s Intracellular Domain is Required to Maintain Basal NMDAR-Mediated Synaptic Transmission.
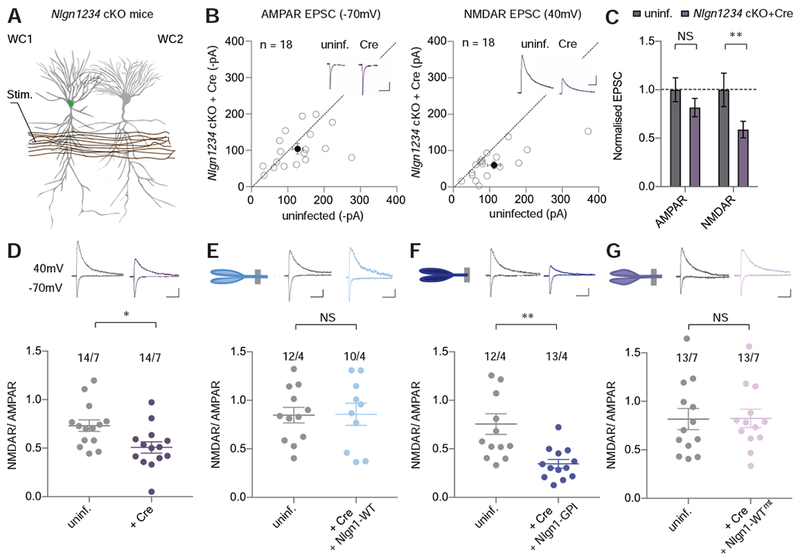
(A) Schematic of simultaneous dual whole-cell (WC) recording from adjacent lentiviral infected (left) and uninfected CA1 pyramidal cells (right). The electrode (Stim.) evokes EPSCs by stimulating axons in stratum radiatum.
(B) Scatter plot of AMPAR EPSCs (left) and NMDAR EPSCs (right) of lentiviral Cre infected (y-axis) and uninfected (x-axis) neuron pairs (unfilled circles) from Nlgn1234 cKO mice (n=10). Filled circles represent means ± SEMs. Insets show representative traces (scale bars: 50 pA, 100 ms).
(C) Summary graphs of normalized AMPAR- (left) and NMDAR EPSCs (right) from data shown in B. Bars represent mean ± SEM. Wilcoxon signed rank test, *p<0.05, **p<0.01, NS = not significant.
(D – G) Summary plots of ratio of NMDAR- to AMPAR-mediated EPSCs in CA1 pyramidal neurons of Nlgn1234 cKO mice. Representative traces are shown above each graph (scale bars: 50 pA, 100 ms). (D) Uninfected and lentiviral Cre infected cells, (E) Uninfected and lentiviral Cre + Nlgn1-WT infected cells, (F) Uninfected and lentiviral Cre + Nlgn1-GPI infected cells, (G) Uninfected and lentiviral Cre + Nlgn1-WTmt infected cells. Numbers above each data set are cells/animals. Lines illustrate mean ± SEM. Two-tailed Student’s t-test, *p<0.05, **p<0.01, ***p<0.001, NS = not significant.
NMDAR-mediated Synaptic Transmission Requires Nlgn1’s Intracellular Domain
Because the dual recordings showed that the deletion of neuroligins produced a specific reduction in NMDAR EPSCs that is most likely due to the loss of Nlgn1, we examined whether this reduction could be rescued by re-expression of wild-type Nlgn1. For this purpose, we compared the ratio of NMDAR-versus AMPAR-mediated synaptic responses between infected and uninfected cells from the same set of acute slices. The decrease in NMDAR/AMPAR ratios caused by genetic deletion of Nlgn1234 (Figure 3D) was fully rescued by expression of Nlgn1-WT (Figure 3E), confirming that Nlgn1 mediates this phenotype. We then asked whether the Nlgn1 sequences required for LTP match those required for NMDAR-mediated responses. Unexpectedly, we found that Nlgn1-GPI, which lacks Nlgn1’s intracellular domain (Figure 3F), was unable to rescue the decrease in NMDAR/AMPAR ratio in neuroligin-deficient neurons, whereas full-length Nlgn1 with the neurexin-binding mutation fully rescued the NMDAR/AMPAR ratios similarly to Nlgn1-WT (Figure 3G). To further test the reproducibility of these results, we also performed simultaneous paired recordings from adjacent control and molecularly manipulated CA1 pyramidal neurons. Consistent with the AMPAR/NMDAR ratio results, expression of Nlgn1-WT or Nlgn1-WT with the neurexin-binding mutation in neurons in which Nlgn1234 was genetically deleted rescued NMDAR EPSCs but Nlgn1-GPI did not; AMPAR EPSCs were unaffected by any of these manipulations (Figure S4). Thus, the critical roles of Nlgn1 in LTP and NMDAR-mediated synaptic transmission require distinct sequences, implying different transduction mechanisms.
Conditional Deletion of Presynaptic β-neurexins Inhibits LTP
The lack of LTP rescue by mutant Nlgn1 that is unable to bind presynaptic neurexins (i.e., by Nlgn1 with the neurexin-binding mutation) strongly suggests that presynaptic neurexins are required for LTP, as previously suggested for LTP at excitatory synapses formed by CA1 pyramidal neurons on excitatory regular-firing neurons in the subiculum (Aoto et al., 2013). To test the critical role of neurexins at Schaffer collateral synapses on CA1 pyramidal neurons, we specifically focused on β-neurexins using the triple conditional KO mice that allow deletion of all three β-neurexins (Nrxn123β cKO mice; Anderson et al., 2015). This focus on β-neurexins was motivated by our current observation that Nlgn1 containing inserts in both sites of alternative splicing (SSA and SSB) fully rescues LTP in Nlgn1-deficient neurons and previous findings that this splice variant exclusively binds to β-neurexins (Boucard et al., 2005; Ichtchenko et al., 1995). To be able to stimulate only axonal inputs onto CA1 pyramidal neurons that lack β-neurexins, we co-infected the CA3 region of Nrxn123β cKO mice or BL6 control mice at P21 with adeno-associated viruses (AAVs) expressing Cre-recombinase (AAV-DJ-Syn-Cre-GFP) and with AAVs expressing ChETA under Cre control (AAV-DJ-EF1-DIO ChETA-eYFP; Figure 4A). We prepared slices from these mice 3 – 4 weeks later, and analysed AMPAR EPSCs evoked by full-field light pulses applied at 0.1 Hz for establishing baseline synaptic strength (Figure 4B). LTP was elicited by optogenetic activation of ChETA-expressing axons using 10 tetani of 50 stimuli delivered at 50 Hz, separated by 10 seconds while holding the CA1 pyramidal neurons at 0 mV. This stimulation protocol produced an LTP of greater than 120% in 6 of 8 cells recorded from control slices prepared from BL6 WT mice, but in only 1 of 8 cells from the Nrxn123β cKO mice (Figure 4C-E), indicating that presynaptic β-neurexins are crucial for LTP at excitatory synapses on CA1 pyramidal neurons. Measurement of light-evoked NMDAR EPSCs and AMPAR EPSCs revealed that NMDAR/AMPAR ratios were unaffected by the β-neurexin deletion (Figure 4F). Furthermore, paired pulse ratios of AMPAR EPSCs (50 msec interstimulus interval) were also not changed by the β-neurexin deletion (Figure 4G) suggesting that changes in basal presynaptic release probability cannot account for the block of LTP. It is surprising that deletion of β-neurexins alone inhibited CA3 to CA1 LTP since β-neurexins are much less abundant in the hippocampus than α-neurexins (Fuccillo et al., 2015). These results suggest that the Nlgn1-neurexin interaction required for LTP at these synapses might be β-neurexin and Nlgn1 splice site B specific.
Figure 4. Conditional Deletion of Presynaptic β-neurexins Inhibits LTP.
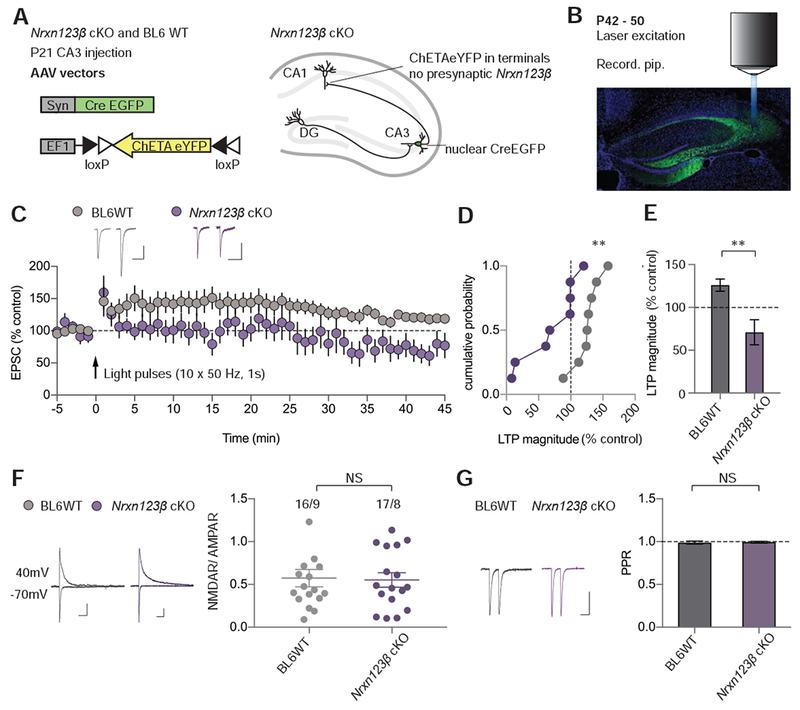
(A) Schematics of AAV constructs used for injection into the CA3 at P21 (left). Schematic of Nrxn123β cKO mouse hippocampal slice infected with AAV-DJ-Syn-Cre-GFP (nuclear) + AAV-DJ-EF1-DIO-ChETA-eYFP and thus expressing nuclear Cre-GFP, ChETA-eYFP and no Nrxn123β (right).
(B) Annotated confocal image of hippocampal slice from a mouse (P42 – 50), showing AAV-DJ-Syn-Cre-GFP + AAV-DJ-EF1-DIO-ChETA-eYFP infection in the CA3 region. The recording pipette (Recording pip.) is sealed onto a CA1 pyramidal neuron and the laser is projected onto the slice via the epifluorescent microscope objective (not drawn to scale). DAPI (blue), GFP (green). Scale bar: 500 μm.
(C) Summary of LTP time course with sample EPSCs pre- and post-induction (35 – 40 min).
(D) Cumulative frequency plot of LTP magnitude from individual experiments.
(E) Quantification of LTP magnitude for AAV infected cells in BL6WT and Nrxn123β cKO animals (numbers = cells/animals).
(F) Ratio of NMDAR- to AMPAR-mediated EPSCs in AAV infected neurons in BL6WT and Nrxn123β cKO mice. Representative traces are shown on left (scale bars: 50 pA, 50 ms). Summary plots of mean NMDAR/AMPAR ratios from individual neurons are shown on right (numbers = cells/animals). Lines are mean ± SEM.
(G) Paired pulse ratios from AAV infected neurons in BL6WT and Nrxn123β cKO mice.
Representative traces are shown on left (scale bars: 50 pA, 50 ms). Summary bar graph of PPRs (mean ± SEM) are shown on right (numbers = cells/animals). Mann Whitney test,**p<0.01, NS = not significant.
Neuroligin Deletion Blocks LTP-induced Growth of Dendritic Spines
NMDAR-dependent LTP, defined as an increase in the amplitude of AMPAR EPSCs, is accompanied by a morphological restructuring of synapses and spines that exhibits as a hallmark an increase in dendritic spine volume (Hedrick and Yasuda, 2017; Kasai et al., 2010). Studies on the molecular mechanisms of spine growth during LTP have revealed a central role of the actin cytoskeleton and Rho GTPase proteins (Hedrick and Yasuda, 2017; Kasai et al., 2010), but how these changes are embedded in the overall organization of the synapse via trans-synaptic signalling by cell-adhesion molecules remains unexplored. Although structural LTP is commonly monitored at the single spine level using glutamate uncaging, we reasoned that induction of LTP by repetitive activation of voltage-gated Ca2+-channels (referred to as voltage-pulse LTP) should cause spine growth throughout the dendritic tree, thus making it relatively easy to detect. Voltage-pulse LTP shares physiological and molecular mechanisms with NMDAR-dependent LTP, including a requirement for Nlgn1 and synaptotagmin-1, but differs from NMDAR-dependent LTP in that it is NMDAR-independent and converges with classical NMDAR-dependent LTP at the level of Ca2+-influx (Jiang et al., 2017; Kato et al., 2009; Wu et al., 2017; Wyllie et al., 1994). To image spines from wildtype or neuroligin-deficient CA1 pyramidal neurons, we injected BL6 control mice or Nlgn1234 quadruple cKO mice at P21 with lentiviruses expressing a nuclear Cre-EGFP fusion protein (Kaeser et al., 2011) and with AAVs expressing a Cre-dependent, cytoplasmic EGFP (AAV-DJ-CMV-DIO-EGFP). This procedure resulted in sparse expression of cytoplasmic EGFP in CA1 pyramidal neurons, allowing us to image their dendritic spines in acute slices using 2-photon microscopy (Figures 5A and 5B).
Figure 5. Neuroligin1234 Deletion Impairs LTP-induced Growth of Dendritic Spines.
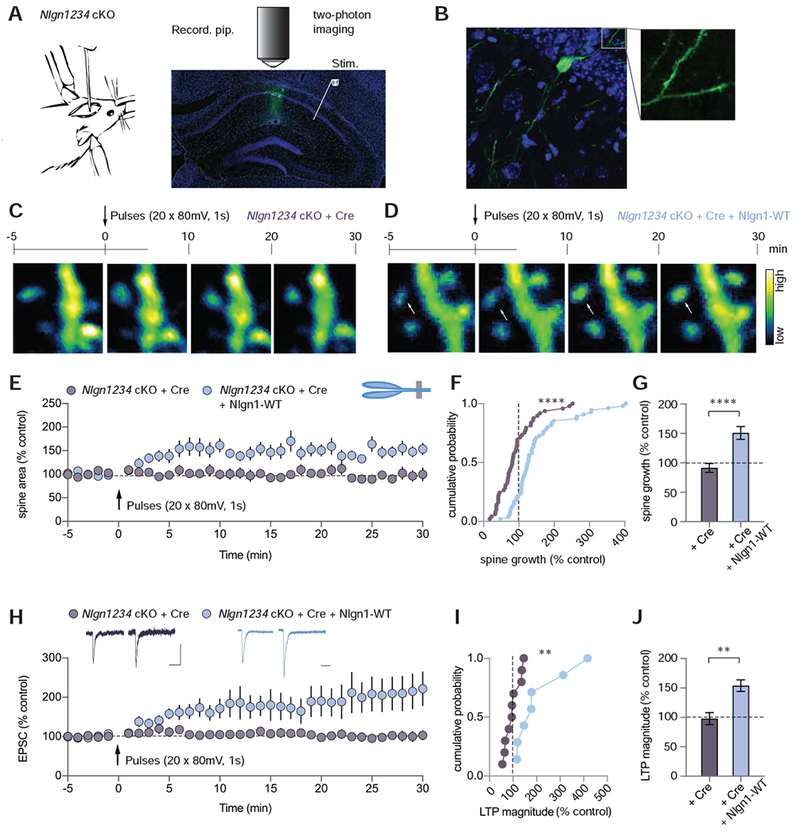
(A) Illustration of mouse lentiviral stereotactic injection into the CA1 region of the hippocampus (left). Annotated confocal image of hippocampal slice (right) from a mouse showing lentiviral Cre-EGFP + AAV-DJ-CMV-DIO-EGFP (cytoplasmic) infection of CA1 pyramidal neurons. The recording pipette (Recording pip.), sealed onto a CA1 pyramidal neuron, was used to elicit LTP. While recording, a dendrite segment of the patched cell was imaged with a two-photon microscope at a frequency of 96 frames per minute. DAPI (blue), GFP (green). Scale bar: 500 μm.
(B) Confocal image of EGFP-positive cell in slice imaged with 60X objective. DAPI (blue), GFP (green). Scale bar: 10 μm.
(C, D) The time course of experiments with representative two-photon images of dendritic segments from Nlgn1234 cKO slices infected with AAV-DJ-CMV-DIO-EGFP (cytoplasmic) and (C) lentiviral Cre-EGFP or (D) Cre-EGFP + Nlgn1-WT. White arrow shows example spine that grew relative to baseline. Bar on right displays the color coding reflecting the degree of fluorescence intensity. Scale bar: 2.5 μm
(E) Summary time course of spine growth during VGCC-dependent LTP in Nlgn1234 cKO mice expressing Cre-EGFP or Cre-EGFP + Nlgn1-WT.
(F) Cumulative frequency plot of changes in spine area for all imaged spines in the two conditions.
(G) Quantification of spine growth magnitude (mean ± SEM; numbers = spines/cells).
(H) Summary time course of EPSCs following induction of VGCC-dependent LTP. Insets show sample traces pre- and post-pulses (Scale bars: 50 pA, 50 ms).
(I) Cumulative frequency plot of LTP magnitude for individual cells.
(J) Quantification of LTP magnitude (mean ± SEM; numbers = cells/animals). Mann Whitney test,**p<0.01, ****p<0.0001.
To elicit voltage-pulse LTP while imaging dendritic spines on secondary dendritic branches, we performed whole-cell voltage-clamp recordings from cytoplasmic EGFP-expressing neurons 3 – 4 weeks after viral injections (Figure S5A). After obtaining a stable baseline of AMPAR EPSCs, we applied 20 depolarizing voltage pulses (80 mV, 1 sec separated by 6 s) to cells clamped at −70 mV. In wildtype cells expressing Cre and Cre-dependent EGFP, robust functional and structural LTP was elicited (Figures S5B-H). Approximately 70% of imaged spines grew by >20% while ~30% of spines were stagnant or shrank (Figure S5E). In marked contrast, neither structural nor functional LTP were elicited in neuroligin-deficient CA1 pyramidal neurons (Figures 5C, 5E-J). Only ~20% of spines grew in these cells following the voltage pulses, whereas >70% of spines were stagnant or decreased in volume (Figure S6A). The loss of structural and functional LTP due to the deletion of all neuroligins in Nlgn1234 quadruple cKO mice was completely rescued by expression of wildtype Nlgn1 (Figures 5D-J and S6A), indicating that similar to its role in functional LTP, Nlgn1 is critically required for the growth of spines that defines structural LTP.
Nlgn1 Binding to Neurexins is Crucial for Structural and Functional Voltage-Pulse LTP
To mechanistically dissect Nlgn1’s role in structural LTP, we performed Nlgn1 rescue experiments using Nlgn1-GPI, which lacks the transmembrane region and cytoplasmic sequences of Nlgn1, and mutant full-length Nlgn1 that is unable to bind to neurexins. Analogous to the results from experiments on functional NMDAR-dependent LTP, the extracellular domain of Nlgn1 (Nlgn1-GPI) alone rescued functional and structural voltage-pulse LTP on a Nlgn1234 null background, while the mutant Nlgn1 with the neurexin-binding site inactivation failed to rescue either (Figure 6). Similar to the findings from control cells or from Nlgn1234-deficient cells expressing wild-type Nlgn1, ~60% of spines in Nlgn1-GPI expressing cells grew following induction of voltage-pulse LTP, whereas only ~20% of spines grew in the cells expressing the mutant Nlgn1 unable to bind to neurexins (Figure S6B). Although the growth of spines accompanying LTP was diminished by neuroligin deletion and differentially affected by the Nlgn1 replacement constructs, baseline spine size and spine density were not influenced by any of the genetic manipulations (Figure S6C, D). Furthermore, wild-type Nlgn1 and Nlgn1-GPI also rescued functional voltage-pulse LTP in Nlgn1 single cKO mice (Figure S7). Together, these results demonstrate that the domain requirements of Nlgn1 for supporting structural LTP are identical to those for functional LTP, with the Nlgn1 intracellular domain being dispensable, but the binding of the extracellular domain to neurexins being essential (Figure 7). They also provide further evidence that the number of synapses on CA1 pyramidal neurons and basal synaptic strength are not affected by neuroligin deletion.
Figure 6. Nlgn1 Binding to Neurexins is Essential for Structural and Functional VGCC-dependent LTP.
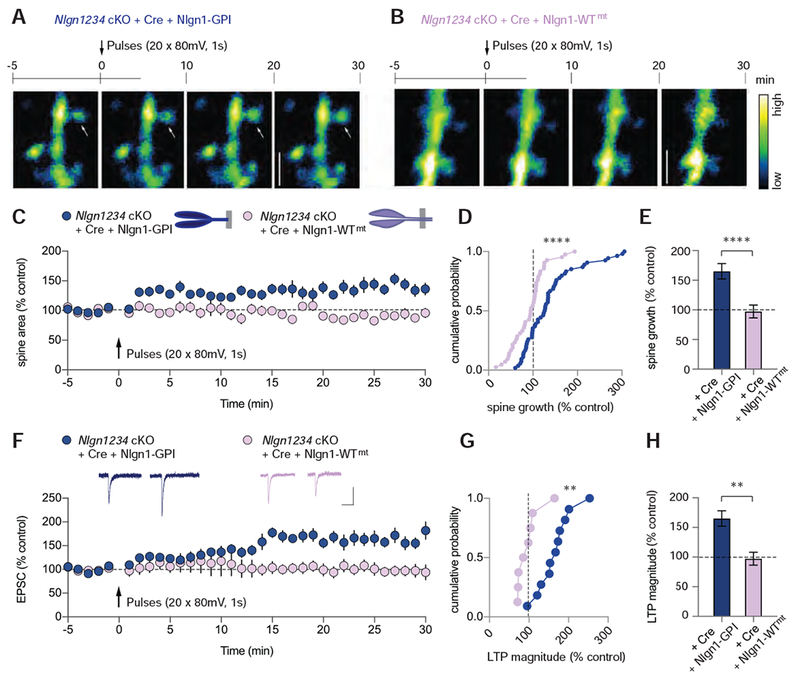
(A) (A, B) The time course of experiments with representative two-photon images of dendritic segments from Nlgn1234 cKO slices infected with AAV-DJ-CMV-DIO-EGFP (cytoplasmic) and (A) lentiviral Cre-EGFP + Nlgn1-GPI or (B) Cre-EGFP + Nlgn1-WTmt. White arrow indicates an example spine that grew relative to baseline. Bar on right displays the color coding reflecting the degree of fluorescence intensity. Scale bar: 2.5 μm
(C) Summary time course of spine growth comparing Nlgn1-GPI rescue with Nlgn1-WTmt rescue.
(D) Cumulative frequency plot of changes in spine area for all imaged spines in the two conditions.
(E) Quantification of spine growth magnitude (mean ± SEM; numbers = spines/cells).
(F) Summary time course of EPSCs following induction of VGCC-dependent LTP. Insets show sample traces pre- and post-pulses (Scale bars: 50 pA, 50 ms).
(G) Cumulative frequency plot of LTP magnitude for individual cells.
(H) Quantification of LTP magnitude (mean ± SEM; numbers = cells/animals). Mann Whitney test,**p<0.01, ****p<0.0001.
Figure 7. Summary Diagrams of Nlgn1 Domain-specific Regulation of LTP and Basal Synaptic Transmission.
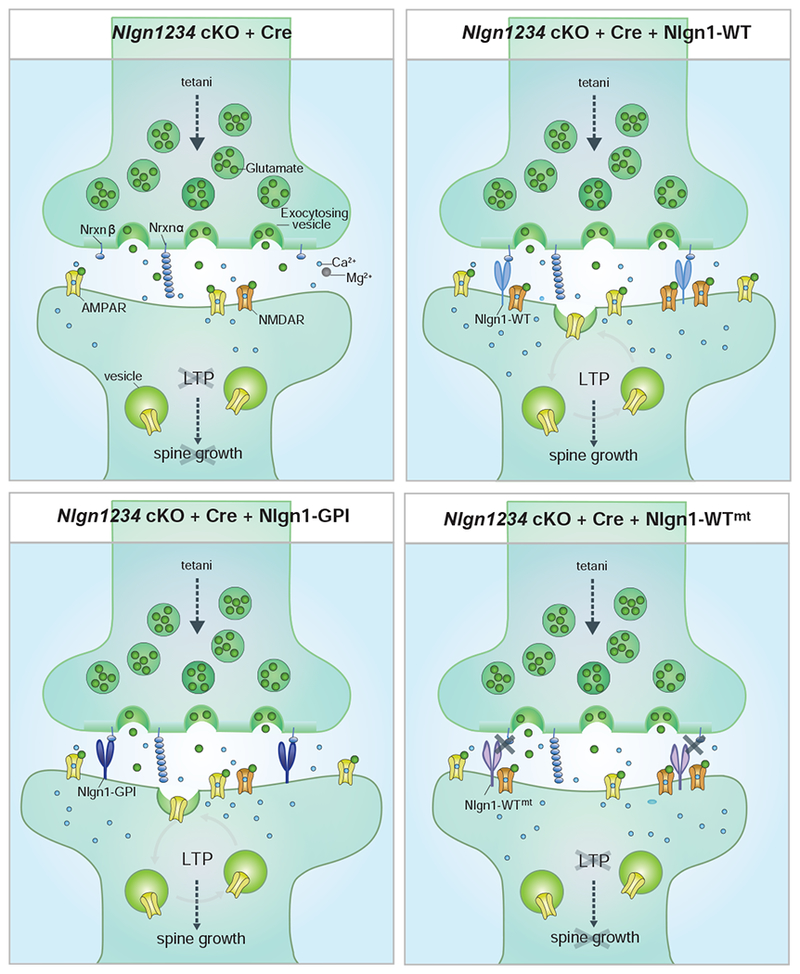
When all four Nlgns are deleted (upper left), NMDAR-mediated transmission is reduced due to loss of synaptic NMDARs and functional LTP and spine growth are blocked. Replacing Nlgn1234 with Nlgn1-WT (upper right) restores NMDARs as well as LTP and spine growth (upper right). Replacing Nlgn1234 with extracellular Nlgn1 lacking its transmembrane and cytoplasmic domains (Nlgn1-GPI; lower left)) rescues LTP ad spine growth, but not NMDARs. In contrast rescue with Nlgn1 that cannot bind to presynaptic Nrxn123β but contains its transmembrane and intracellular domains (lower right) rescues NMDARs, but not LTP and spine growth.
DISCUSSION
Despite decades of work, the role of neuroligins in synaptic transmission and synaptic plasticity is not well understood. Based on the homology between different neuroligins (Ichtchenko et al., 1995), their common localization to synapses (Graf et al., 2004; Song et al., 1999), and their powerful activity in artificial synapse-formation assays (Scheiffele et al., 2000), it was initially thought that all neuroligins perform similar functions in regulating synapse formation. More recent results, however, have led to a very different conclusion, namely that different neuroligins perform distinct functions – often functions that seem to have little relation to each other – and that these functions are unrelated to the initial formation of synapses and more important for the specific organization of synapse properties (Chubykin et al., 2007; Foldy et al., 2013; Jiang et al., 2017; Polepalli et al., 2017; Varoqueaux et al., 2006; Zhang et al., 2015). All of these functions, as defined by genetic manipulations, operate in mature synapses, but have been challenged by work performed primarily using knockdown approaches that appeared to argue for a role of neuroligins in glia instead of neurons or in determining synapse numbers instead of synapse properties (Kwon et al., 2012; Shipman and Nicoll, 2012a,b; Stogsdill et al., 2017). Further uncertainty was injected into the debate on neuroligin functions, moreover, by our recent finding that the deletion of Nlgn1 in adult neurons causes both a loss of NMDAR-dependent LTP and a decrease in NMDAR-mediated synaptic responses, but that these two phenotypes may potentially be unrelated to each other because LTP was also impaired when induced in an NMDAR-independent manner (Jiang et al., 2017). Finally, additional ambiguity was provided by the fact that in all of the studies to date, a given neuroligin was investigated in cells in which at least one other neuroligin continued to be expressed, which creates the confounding possibility of heterodimerization between any recombinant, virally expressed neuroligin and remaining endogenous neuroligins (Poulopoulos et al., 2012).
To address these issues, in the present study we examined the precise role of Nlgn1 in mature excitatory synapses on CA1 pyramidal neurons, arguably the best studied synapses and neurons in the mammalian brain, both in Nlgn1 single and Nlgn1234 quadruple cKO mice, allowing us to unequivocally assign functions to specific Nlgn1 phenotypes. Furthermore, our experiments were performed under conditions that exclude a role for neuroligins in glia because we sparsely infected neurons as visualized directly by microscopy, and that also minimize influences of on-going, large-scale synapse restructuring as observed in dissociated neuronal cultures and cultured slices since we performed all studies in acute slices after in vivo manipulations in young adult mice.
Our study allows several conclusions. First, deletion of all neuroligins had minimal effect on AMPAR-mediated synaptic responses and miniature AMPAR EPSCs, as well as no detectable effect on spine density and spine size. Together these results provide strong evidence that the number of synapses, number of postsynaptic AMPARs and basal neurotransmitter release were unchanged following neuroligin deletion. Thus, neuroligins are not essential building blocks of synapses as such. Second, the deletion of either Nlgn1 alone or of all neuroligins caused a block of NMDAR-dependent LTP and a decrease in NMDAR-mediated synaptic responses, showing that these previously characterized phenotypes (Jiang et al., 2017) are truly indicative of functions of Nlgn1 and are not influenced by a possible interplay between neuroligin isoforms. This conclusion was further strengthened by the rescue of these phenotypes in quadruple Nlgn1234 KO neurons by Nlgn1 alone. Third, the function of Nlgn1 in maintaining NMDAR-mediated responses does not involve neurexin-binding by Nlgn1 since it was fully rescued by mutant Nlgn1 that is unable to bind to neurexins, but does necessitate interactions of the cytoplasmic sequences of Nlgn1 since deletion of these sequences abolished this Nlgn1 function. The most plausible interaction for this function is binding of the Nlgn1 C-terminus to PSD95 (Irie et al., 1997).
Fourth, and maybe most surprisingly, the extracellular domain of Nlgn1 when attached to the membrane by a lipid GPI-anchor was fully able to rescue LTP in Nlgn-deficient synapses even when the extracellular domain was rendered monomeric by a mutation. This result is unexpected given that it mechanistically dissociates the role of Nlgn1 in LTP from its role in regulating NMDAR-mediated synaptic transmission and suggests that Nlgn1 is essential for LTP by a purely extracellular interaction. It may also seem surprising that a ~40% reduction in NMDAR-mediated synaptic transmission is not sufficient to prevent LTP. However, this result is entirely consistent with the previous finding that reducing NMDAR-mediated synaptic transmission by ~40% using APV, rather than neuroligin deletion, was also not sufficient to prevent LTP (Jiang et al., 2017). These findings can all be readily explained by the long-standing hypothesis that the quantitative level of the rise in postsynaptic spine Ca2+ triggered by activation of NMDARs determines the direction of the subsequent synaptic strength change and that LTP requires a rise in postsynaptic spine Ca2+ concentration beyond some critical threshold (Lisman, 1989; Malenka, 1991; Malenka and Nicoll, 1993: Cummings et al., 1996). Based on this hypothesis, we assume that our strong LTP induction protocol activated the synaptic NMDARs remaining after neuroligin deletion to a degree that was sufficient to raise postsynaptic Ca2+ concentration beyond the LTP threshold. Furthermore, the fact that we can generate structural and functional LTP via activation of voltage-dependent Ca2+ channels is consistent with the ability to generate LTD via activation of these channels (Cummings et al., 1996) and suggests that the triggering of these forms of Ca2+-dependent synaptic plasticity do not depend on the source of the rise in postsynaptic Ca2+.
Fifth, Nlgn1 function in LTP involves trans-synaptic binding of β-neurexins, suggesting that although Nlgn1 acts only via extracellular interactions in this function, it must be signalling in a neurexin-dependent manner. Sixth and finally, Nlgn1 function in LTP extends to the structural rearrangement associated with LTP as monitored by the LTP-induced spine enlargement that was blocked by the Nlgn1 deletion but rescued by the same Nlgn1 mutations which also rescued functional LTP. This result, viewed together with the other LTP findings, suggests that Nlgn1-mediated signalling is essential for the expression of LTP, i.e. the long-lasting synaptic restructuring that enables chronic changes in synaptic strength that are characteristic for LTP. However, formally, it remains possible that somehow the Nlgn1-β-neurexins trans-synaptic interaction is critically involved in the initial postsynaptic triggering of LTP, which requires a postsynaptic rise in calcium via NMDARs or voltage-dependent Ca2+ channels.
In considering these observations, it should be noted that the phenotypes of the Nlgn1 deletions are by no means incremental. LTP was blocked essentially completely, suggesting that Nlgn1 signalling effects a gating function on LTP induction or expression, and the NMDAR levels at synapses were likely decreased significantly upon the in vivo deletion of Nlgn1 (Budreck et al., 2013). These phenotypes reflect truly neuronal mechanisms since in our experiments we selectively and sparsely manipulate neurons, not glia. Thus, although our data clearly support the conclusion that neuroligins are not fundamental building blocks of synapses, they can be considered almost as fundamental in constructing the dynamic properties of synapses that are controlled by NMDARs and are manifested among others by LTP. The overall picture that emerges from our study is that Nlgn1 is a multivalent signalling platform that does not act like a classical receptor with a unidirectional mechanism of action, but instead controls distinct effector pathways that are mediated by diverse molecular mechanisms.
Our results raise several questions, new and old. One issue regards differences between our current results and previous studies. RNAi of Nlgn1 in utero suggested that sparse deletion of Nlgn1 leads to a loss of synapses in layer 2/3 pyramidal neurons and the resulting decrease in synaptic strength (Kwon et al., 2012), which we did not observe in developing cultured neurons (Chanda et al., 2017), and do not observe here upon sparse genetic deletion of Nlgn1 in adult CA1 pyramidal neurons. It is possible that Nlgn1 has a developmental function in addition to its role in committed neurons or different functions in cortical versus hippocampal pyramidal neurons, but the Nlgn1 constitutive deletion also does not cause synapse loss (Kwon et al., 2012; Varoqueaux et al., 2006). Knockdown of Nlgn1 with microRNAs has also produced synapse loss (Shipman and Nicoll, 2012b), but again the phenotype is difficult to evaluate because it is different than that observed with genetic deletions. It also must be acknowledged that it is difficult to control for possible sequence-specific off-target effects. Overall, our current results are primarily at odds with knockdown studies, which have the same kinetics of protein loss but generally do not produce a complete ablation of protein expression. The possibility that the differences in phenotypes may be due to the fact that some neuroligin protein remains seems implausible since knockdown experiments appear to produce uniformly more severe phenotypes.
A second set of questions raised by our results regards the role of trans-synaptic signalling in LTP. We previously showed in subiculum synapses that NMDAR-dependent LTP was gated in an all-of-none fashion by a presynaptic neurexin signal that in turn was controlled by neurexin alternative splicing (Aoto et al., 2013). In the present paper, we extend this conclusion to LTP at Schaffer collateral/commissural synapses on CA1 pyramidal neurons by demonstrating that binding of postsynaptic Nlgn1 to presynaptic β-neurexins constitutes a similar all-or-none gate for NMDAR-dependent LTP. It has long been suggested that LTP must involve some sort of post-to-presynaptic trans-synaptic signalling (Luscher et al., 2000), since the postsynaptic morphological changes that accompany LTP are accompanied by presynaptic changes as evidenced in part by the fact that the postsynaptic density grows with LTP yet upon ultrastructural analysis always precisely aligns with the presynaptic active zone (Lisman and Harris, 1993; Südhof, 2012). That the binding of Nlgn1 to presynaptic neurexins is required for the spine growth accompanying LTP suggests that this specific neuroligin-neurexin trans-synaptic signalling may also be involved in the long-term morphological restructuring of the synapse following LTP. This trans-synaptic interaction also appears to dictate whether LTP expression is possible, greatly enlarging the computational plasticity of a given circuit.
Our results suggest that the maintenance of NMDARs at synapses does not require Nlgn1 interaction with Nrxns but does require the intracellular domain, whereas the very same domain is dispensable for LTP. This has major implications on the mechanism underlying Nlgn1’s role in LTP and basal transmission and contrasts with previous conclusions that Nlgn1 mediates LTP and basal NMDAR-mediated synaptic transmission through the same mechanism (Budreck et al., 2013). These previous conclusions were based on the finding that the extracellular domain of Nlgn1 alone can rescue both a reduction of NMDAR-mediated current as well as LTP. The discrepancy in results is most likely due to heterodimerization artefacts and developmental changes caused by the constitutive knock-out model. Finally, our results raise the question of mechanisms: how does Nlgn1 act as a multivalent signalling platform? What proteins besides neurexins bind to its extracellular sequences to mediate its functions, and how are binding signals of Nlgn1 and other neuroligins transduced into effector signals intracellularly? These questions point to a dearth of molecular information about synapses. They highlight how little we actually know about how synapses are constructed in a dynamic three-dimensional space and how much more we have to learn in order to understand these enigmatic computational units that govern information processing by the brain. The genetic deletion and rescue approaches used here to study the role of Nlgn1 in vivo at mature excitatory synapses should facilitate efforts to gain a more sophisticated understanding of the molecular basis of synaptic function and how mutations in synaptic proteins contribute to pathological brain function.
STAR*METHODS
Detailed methods are provided in the online version of this paper and include the following:
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resource and reagents should be directed to and will be fulfilled by the Lead Contact, Robert Malenka (malenka@stanford.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
The mouse lines Nlgn1 cKO and Nrxn123β cKO have been described previously (Anderson et al., 2015; Jiang et al., 2017). Nlgn1234 cKO mice were generated by breeding Nlgn123 cKO mice (Jiang et al., 2017) with Nlgn4 KO mice (Jamain et al., 2008). All mouse lines have been deposited at Jackson Labs with the exception of Nlgn1234 cKO. All procedures conformed to NIH Guidelines for the Care and Use of Laboratory Animals and were approved by the Stanford University Administrative Panel on Laboratory Animal Care. Male and female animals were used for all experiments. Three week-old mice were used for stereotactic injections and P0 pups for primary cultures. Animals were housed in the Research Animal Facility at Stanford University on a 12:12 light dark cycle with ad libitum access to food and water.
METHOD DETAILS
Plasmid Constructs and Viruses
Simplified diagrams of cloned constructs can be found in Figures 1 and 2. Lentiviruses were generated by the Stanford Gene Vector and Virus Core using transfected HEK293T cells producing lentiviral particles expressing Cre-recombinase and Nlgn1 from di-cistronic messages in which Nlgn1 sequences were translated via an IRES insert. All lentiviral vectors utilize the same backbone as in Kaeser et al. 2011. The wild-type neuroligin with splice sites A and B is described in Chanda et al. 2017. To facilitate trafficking of the GPI-anchored Nlgn1 protein to the cell surface, we designed Nlgn1-GPI according to the GPI-anchored constructs in Gökce et al. 2013. The Nlgn1 neurexin binding site mutant was generated by making previously described mutations L399A/N400A/D402N/E297A/K306A (Arac et al., 2007). The Nlgn1 dimerization mutant was generated by making previously described mutations F458A/M459A/W463A (Ko et al., 2009). Both these mutants were previously characterized and tested for correct folding, surface expression and binding affinities. The Stanford Gene Vector and Virus Core provided the stock viruses: lenti Ubiq-EGFP-cre, lenti Ubiq-EGFP-Δcre, AAV-DJ-Syn-Cre-GFP, AAV-DJ-EF1-DIO-ChETA-eYFP and AAV-DJ-CMV-DIO-EGFP (cytoplasmic).
Neuronal Cultures
Hippocampi were dissected from pups at P0 in ice cold HBSS buffer supplemented with 2 mM Ca2+ and 0.5 mM EGTA under a dissection hood and incubated in 10 U/ml papain solution in HBSS for 20 min, washed twice with HBSS and dissociated in serum media (MEM supplemented with 0.4% glucose, 5.4% FBS, 2 mM L-glutamine, 2.5% B-27) by tituration and plated on Matrigel (BD Biosciences) pre-coated coverslips in 24 well plates. After 24 hours 75% of the serum media was replaced with neurobasal media (Neurobasal media supplemented with 0.5 mM L-glutamine, 0.5% B-27). After another 3 days 50% of the old media was replaced with fresh media and additionally supplemented with 4 μM Ara-C (Sigma). On the same day, neurons were infected with lentiviruses.
Immunofluorescence
11 days after lentivirus infection cultured neurons were washed once with phosphate-buffered saline (PBS) and fixed with 4% paraformaldehyde (PFA) in PBS. After 3, 5 min washes in PBS neurons were blocked for 1 h in PBS containing 5% goat serum. To allow surface staining only, cells were not permeabilized by detergents. Primary antibody (mouse HA purified BioLegend MMS-101P) was applied with a dilution of 1:1000 overnight at 4°C. After 3 washes secondary goat anti-mouse Alexa 546 was applied with a dilution of 1:1000 for 1 h at room temperature and cells were subsequently washed 3 × 5 min before mounting.
Immunoblotting
Primary neurons were lysed at DIV15. Samples were run by SDS-PAGE and transferred to low autofluorescence polyvinylidene difluoride membrane (Immobilon, FL, Millipore) for Western blotting. Primary antibodies (mouse HA purified BioLegend MMS-101P 1:1000 and mouse β-actin antibody AC-15 SIGMA 1:2000) were applied overnight at 4 °C in blocking solution (1% milk powder in PBS). Secondary antibody (IR-Dye conjugation goat anti-mouse LI-COR Biosciences) was applied with a dilution of 1:10000 in blocking solution for 1 h. The membrane was subsequently scanned on the LI-COR Odyssey (LI-COR Biosciences) using the Image Studio software.
In vivo Stereotactic Injections
Female and male P21 mice (weight 10–12 g) were anesthetized by intraperitoneal injection using a mixture of ketamine (75 mg/kg body weight) and dexmedetomidine (0.375 mg/kg body weight). Heads were fixed on a Kopf stereotaxic apparatus and holes were drilled bilaterally into the skull at −1.8 mm posterior and −1.5 mm lateral to bregma for injection into the hippocampal CA1 region. The coordinates −1.8 mm posterior and −1.9 mm lateral were used for injections into the CA3. Glass cannulae filled with virus solution were lowered to a depth of 1.35 mm from the dura for CA1 or 1.8 mm for CA3, and 0.5 – 0.75 μl of virus solution was injected using a microinjection pump (Harvard Apparatus) at a flow rate of 0.1 μl/min. After surgery, atipamezole (10 mg/kg body weight) was injected by intraperitoneal injection and animals were monitored until fully awake.
Electrophysiology
Mice were sacrificed at P35–42 and brains were rapidly placed into high sucrose cutting solution containing (in mM): 228 sucrose, 26 NaHCO3, 11 glucose, 2.5 KCl, 1 NaH2PO4, 7 MgSO4 and 0.5 CaCl2 and sliced on a Leica vibratome. The transverse hippocampal slices were then transferred into artificial cerebrospinal fluid (ACSF) containing (in mM): 119 NaCl, 26 NaHCO3, 11 glucose, 2.5 KCl, 1 NaH2PO4, 1.3 MgSO4 and 2.5 CaCl2 for recovery at 32°C for 30 min. After another 30 min of equilibrat ion at room temperature, slices were placed in a recording chamber perfused with 28–30°C ACSF infused with 95% O2 and 5% CO2. All slice recordings were performed with 50 μM picrotoxin in ACSF. Whole-cell recording pipettes (3–4 MΩ) were filled with an internal solution containing (in mM) 140 CsMeSO4, 8 CsCl, 10 HEPES, 0.25 EGTA, 2 Mg2ATP, 0.3 Na3GTP, 0.1 spermine, 7 phosphocreatine (pH 7.32–7.36; osmolarity 294–298). Recordings were made using a MultiClamp 700B amplifier (Molecular Devices), digitized at 10 kHz with the Digidata 1440A data acquisition system (Molecular Devices), and analyzed using AxoGraph. A theta glass pipette filled with ACSF and placed in CA1 stratum radiatum was used to stimulate Schaeffer collateral/commissural axons to evoke EPSCs. An Iso-Flex (A.M.P.I) stimulator was used to control the frequency, duration, and magnitude of the stimulus.
Prior to recordings, CA1 pyramidal neurons were visualized by infrared DIC, and GFP positive cells were identified by epifluorescence. After establishing whole cell recording configuration, a 5 – 10 min baseline was collected while holding cells at −70 mV and stimulating inputs at 0.1 Hz. To induce LTP, cells were depolarized to 0 mV and 2 trains of 100 Hz, 1 s separated by 20 s were delivered. To induce NMDAR-independent voltage-pulse LTP, 5 μM Bay K8644 (an L-type calcium channel activator) and 50 μM D-AP5 were added to ACSF. The induction protocol consisted of twenty, 1 sec postsynaptic depolarization pulses of 80 mV separated by 7 s. To generate summary graphs showing time course of LTP, individual experiments were normalized to the baseline and six consecutive traces were averaged to generate 1 min bins. These were then averaged across all cells in one condition. The magnitude of LTP was calculated on the basis of the averaged EPSC values from 35 to 40 min after the induction protocol. For all LTP experiments, one cell from each slice was recorded. Recordings from uninfected control cells were interleaved with those from virally infected cells and were made from the same set of slices prepared daily from a single animal. An independent experimenter (WM) performed the experiments in Figure S3, 4 and was blinded to the experimental manipulation that had been performed.
To examine basal synaptic transmission, neighboring Cre-recombinase infected and uninfected CA1 pyramidal cells were recorded simultaneously with Schaffer collateral /commissural stimulation at 0.1 Hz. AMPAR EPSCs were recorded as the peak current (2 msec window) at −70 mV and NMDAR EPSCs at +40 mV with their magnitude measured at 50 msec after the onset of the EPSC. For acquiring NMDAR/AMPAR ratios, infected and uninfected cells were recorded sequentially from the same slices. The ratio was calculated as NMDAR EPSCs (measured at 50 ms after the onset of EPSC at +40 mV) divided by AMPAR EPSCs (measured as the peak amplitude at −70 mV). For dual recordings shown in Figure S4, an independent experimenter (WM) performed the recordings blindly without knowledge of the manipulation that had been performed. mEPSCs were recorded in TTX (0.5 μM) and the concentration of Ca2+ in the ACSF was increased to 4 mM. For all electrophysiology experiments, cells were excluded if their series resistance was <20 MΩ or varied by >25% or if the required holding current was > 300pA. No statistical test was used to pre-determine sample size. Replication of key experiments are described in text.
ChETA-evoked LTP
For ChETA-evoked LTP experiments (Figure 4), recordings were made from uninfected CA1 pyramidal neurons while 473 nm light pulses (1–5 msec) from the Opto Duet Laser system (IkeCool Corporation) were delivered through the objective to the whole slice at 0.1 Hz, thereby stimulating ChETA infected neurons in the CA3. To induce LTP ten tetani (50 Hz, 1 sec) separated by 10 sec were given, while the cells were depolarized to 0 mV. Two light pulses at 50 ms intervals were delivered to measure paired pulse ratios (PPR).
Two-photon Imaging
Spines were imaged from secondary dendrites of CA1 pyramidal neurons while performing whole cell voltage clamp recordings using a water immersion 60x objective (Olympus) mounted on a custom built 2-photon microscope. A Ti:Sapphire laser (Chameleon) at 850 nm and a laser power < 100 mW was used to excite the fluorophores. 512 × 512 images with a scan time of 3 ms per line were acquired every minute with two slices of a z-stack (step size 1 micron) each containing 8 frames. MATLAB software synchronized the imaging with the recording, so that every minute 6 traces were collected at a frequency of 0.1 Hz and one set of images (2 z-slices, each 8 frames) were acquired. Electrophysiology recording data were collected and analyzed using pCLAMP 10 software. All recordings, imaging and analysis were done blindly without knowledge of the manipulation that the neuron had experienced.
Image analysis was performed using MetaMorph software. After averaging the frames, z-stacks were collapsed into maximum intensity projections. Background subtraction and threshold were set uniformly for all images in one experiment. ROIs were drawn for each spine and the total spine area detected was measured. For quantification spine areas were normalized to the mean of the baseline (−5 – 0 min). The summary plot contains all analysed spines from all cells. In the cumulative distribution, means of spine area from 25 – 30 min was plotted.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data are shown as mean ± SEM. Unless specifically mentioned, statistical significance was determined by the Mann Whitney test. NMDAR/AMPAR ratios in Figure 3 were distributed normally and therefore a two-tailed Student’s t test was used. For the experiments involving simultaneous dual recordings, significance was determined by the Wilcoxon’s signed rank test. Figure S6C and D required multiple comparisons and thus one-way Anova was used to assess significance. * = p < 0.05; ** = p < 0.01; *** = p < 0.001, **** = p < 0.0001. NS = not significant. Number of cells and animals (n/n) for each experiment are shown in each figure and described in each figure legend.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-HA | BioLegend | Ct# MMS-101P |
| Mouse anti-β-actin | Sigma-Aldrich | Ct# AC-15 |
| Mouse anti-Neuroligin 1 | Synaptic Systems | Ct# 129111 |
| IRDye® 800CW Goat anti-Mouse IgG | LI-COR Biosciences | Ct# 925-32210 |
| IRDye® 680LT Goat anti-Mouse IgG (H | LI-COR Biosciences | Ct# 925-68020 |
| Alexa 546, mouse | Invitrogen | Ct# A-21123; RRID: AB141592 |
| Bacterial and Virus Strains | ||
| AAV DJ-EF1-DIO ChETA-eYFP | Stanford Virus Core | GVVC-AAV-65 |
| AAV DJ-CMV DIO EGFP | Stanford Virus Core | GVVC-AAV-12 |
| AAV-DJ-Syn-Cre-GFP | Stanford Virus Core | GVVC-AAV-117 |
| (VSV-G) Ubiq-EGFP-cre | Stanford Virus Core | GVVC-lenti-11 |
| (VSV-G) Ubiq-EGFP-deleted cre | Stanford Virus Core | GVVC-lenti-12 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Picrotoxin | Sigma-Aldrich | Ct# P1675 |
| D-AP5 | Tocris | Ct# 0106 |
| (±)-Bay K 8644 | Tocris | Ct# 1544 |
| Cytosine β-D-arabinofuranoside (Ara C) | Sigma-Aldrich | Ct# 1768 |
| Experimental models: Organisms/Strains | ||
| Nlgn1 cKO (Nlgn1tm1Sud/J) | Jiang et al., 2017 | JAX: 023646 |
| Nlgn1234 cKO | This paper | |
| Nrxn123β cKO (B6;129-Nrxn3tm2Sud Nrxn1tm2Sud Nrxn2tm2Sud/J) | Anderson et al., 2013 | JAX: 008416 |
| Software and Algorithms | ||
| MATLAB | MathWorks | RRID:SCR_001622 |
| AxoGraph | AxoGraph | RRID:SCR_014284 |
| MetaMorph | Molecular Devices | RRID:SCR_002368 |
| pCLAMP 10 | Molecular Devices | RRID:SCR_011323 |
HIGHLIGHTS.
Nlgn1 extracellular domain is sufficient to rescue LTP loss in cells lacking Nlgn’s
Nlgn1 intracellular domain is crucial for basal NMDAR-mediated transmission
Binding of Nlgn1 to presynaptic β-Nrxn’s is essential for LTP
Spine growth during LTP also requires Nlgn1 binding to β-Nrnx’s
ACKNOWLEDGMENTS
We thank Dr.’s Lu Chen, Jai Polepalli, Karthik Raju and Yu-Wei Wu for advice on electrophysiology, cloning and two-photon imaging. X.W. was supported by a postdoctoral fellowship from the Deutsche Forschungsgemeinschaft. The study was supported by NIH Grant P50 MH086403 to T.C.S. and R.C.M.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Anderson GR, Aoto J, Tabuchi K, Foldy C, Covy J, Yee AX, Wu D, Lee SJ, Chen L, Malenka RC, et al. (2015). β-Neurexins control neural circuits by regulating synaptic endocannabinoid signaling. Cell 162, 593–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoto J, Martinelli DC, Malenka RC, Tabuchi K, and Sudhof TC (2013). Presynaptic neurexin-3 alternative splicing trans-synaptically controls postsynaptic AMPA receptor trafficking. Cell 154, 75–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arac D, Boucard AA, Ozkan E, Strop P, Newell E, Sudhof TC, and Brunger AT (2007). Structures of neuroligin-1 and the neuroligin-1/neurexin-1 beta complex reveal specific protein-protein and protein-Ca2+ interactions. Neuron 56, 992–1003. [DOI] [PubMed] [Google Scholar]
- Bliss TV, and Collingridge GL (1993). A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361, 31–39. [DOI] [PubMed] [Google Scholar]
- Boucard AA, Chubykin AA, Comoletti D, Taylor P, and Sudhof TC (2005). A splice code for trans-synaptic cell adhesion mediated by binding of neuroligin 1 to alpha- and beta-neurexins. Neuron 48, 229–236. [DOI] [PubMed] [Google Scholar]
- Budreck EC, Kwon OB, Jung JH, Baudouin S, Thommen A, Kim HS, Fukazawa Y, Harada H, Tabuchi K, Shigemoto R, et al. (2013). Neuroligin-1 controls synaptic abundance of NMDA-type glutamate receptors through extracellular coupling. Proc. Natl. Acad. Sci. USA 110, 725–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budreck EC, and Scheiffele P (2007). Neuroligin-3 is a neuronal adhesion protein at GABAergic and glutamatergic synapses. Eur. J. Neurosci 26, 1738–1748. [DOI] [PubMed] [Google Scholar]
- Chanda S, Hale WD, Zhang B, Wernig M, and Sudhof TC (2017). Unique versus redundant functions of neuroligin genes in shaping excitatory and inhibitory synapse properties. J. Neurosci 37, 6816–6836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chubykin AA, Atasoy D, Etherton MR, Brose N, Kavalali ET, Gibson JR, and Sudhof TC (2007). Activity-dependent validation of excitatory versus inhibitory synapses by neuroligin-1 versus neuroligin-2. Neuron 54, 919–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comoletti D, Grishaev A, Whitten AE, Tsigelny I, Taylor P, and Trewhella J (2007). Synaptic arrangement of the neuroligin/beta-neurexin complex revealed by X-ray and neutron scattering. Structure 15, 693–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings JA, Mulkey RM, Nicoll RA and Malenka RC (1996). Ca2+ signlaing requirements for long-term depression in the hippocampus. Neuron 16, 825–833. [DOI] [PubMed] [Google Scholar]
- Ehrlich I, and Malinow R (2004). Postsynaptic density 95 controls AMPA receptor incorporation during long-term potentiation and experience-driven synaptic plasticity. J. Neurosci 24, 916–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Földy C, Malenka RC, Sudhof TC. (2013) Autism-associated neuroligin-3 mutations commonly disrupt tonic endocannabinoid signaling. Neuron 78, 498–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuccillo MV, Foldy C, Gokce O, Rothwell PE, Sun GL, Malenka RC, and Sudhof TC (2015). Single-Cell mRNA Profiling Reveals Cell-Type-Specific Expression of Neurexin Isoforms. Neuron 87, 326–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson JR, Huber KM, and Sudhof TC (2009). Neuroligin-2 deletion selectively decreases inhibitory synaptic transmission originating from fast-spiking but not from somatostatin-positive interneurons. J. Neurosci 29, 13883–13897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gokce O, and Sudhof TC (2013). Membrane-tethered monomeric neurexin LNS-domain triggers synapse formation. J. Neurosci 33, 14617–14628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graf ER, Zhang X, Jin SX, Linhoff MW, and Craig AM (2004). Neurexins induce differentiation of GABA and glutamate postsynaptic specializations via neuroligins. Cell 119, 1013–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedrick NG, and Yasuda R (2017). Regulation of Rho GTPase proteins during spine structural plasticity for the control of local dendritic plasticity. Curr. Opin. Neurobiol 45, 193–201. [DOI] [PubMed] [Google Scholar]
- Hoon M, Soykan T, Falkenburger B, Hammer M, Patrizi A, Schmidt KF, Sassoe-Pognetto M, Lowel S, Moser T, Taschenberger H, et al. (2011). Neuroligin-4 is localized to glycinergic postsynapses and regulates inhibition in the retina. Proc. Natl. Acad. Sci. USA 108, 3053–3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoy JL, Haeger PA, Constable JR, Arias RJ, McCallum R, Kyweriga M, Davis L, Schnell E, Wehr M, Castillo PE, et al. (2013). Neuroligin1 drives synaptic and behavioral maturation through intracellular interactions. J. Neurosci 33, 9364–9384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huganir RL, and Nicoll RA (2013). AMPARs and synaptic plasticity: the last 25 years. Neuron 80, 704–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichtchenko K, Hata Y, Nguyen T, Ullrich B, Missler M, Moomaw C, and Sudhof TC (1995). Neuroligin 1: a splice site-specific ligand for beta-neurexins. Cell 81, 435–443. [DOI] [PubMed] [Google Scholar]
- Irie M, Hata Y, Takeuchi M, Ichtchenko K, Toyoda A, Hirao K, Takai Y, Rosahl TW, and Sudhof TC (1997). Binding of neuroligins to PSD-95. Science 277, 1511–1515. [DOI] [PubMed] [Google Scholar]
- Jamain S, Radyushkin K, Hammerschmidt K, Granon S, Boretius S, Varoqueaux F, Ramanantsoa N, Gallego J, Ronnenberg A, Winter D, et al. (2008). Reduced social interaction and ultrasonic communication in a mouse model of monogenic heritable autism. Proc. Natl. Acad. Sci. USA 105, 1710–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang M, Polepalli J, Chen LY, Zhang B, Sudhof TC, and Malenka RC (2017). Conditional ablation of neuroligin-1 in CA1 pyramidal neurons blocks LTP by a cell-autonomous NMDA receptor-independent mechanism. Mol. Psychiatry 22, 375–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeser PS, Deng L, Wang Y, Dulubova I, Liu X, Rizo J, and Sudhof TC (2011). RIM proteins tether Ca2+ channels to presynaptic active zones via a direct PDZ-domain interaction. Cell 144, 282–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasai H, Fukuda M, Watanabe S, Hayashi-Takagi A, and Noguchi J (2010). Structural dynamics of dendritic spines in memory and cognition. Tr. Neurosci 33, 121–129. [DOI] [PubMed] [Google Scholar]
- Kato HK, Watabe AM, and Manabe T (2009). Non-Hebbian synaptic plasticity induced by repetitive postsynaptic action potentials. J. Neurosci 29, 11153–11160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko J, Zhang C, Arac D, Boucard AA, Brunger AT, and Sudhof TC (2009). Neuroligin-1 performs neurexin-dependent and neurexin-independent functions in synapse validation. EMBO J. 28, 3244–3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon HB, Kozorovitskiy Y, Oh WJ, Peixoto RT, Akhtar N, Saulnier JL, Gu C, and Sabatini BL (2012). Neuroligin-1-dependent competition regulates cortical synaptogenesis and synapse number. Nat. Neurosci 15, 1667–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman JE (1989). A mechanism for the Hebb and the anti-Hebb processes underlying learning and memory. Proc. Natl. Acad. Sci. USA 86, 9574–9578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman JE, and Harris KM (1993). Quantal analysis and synaptic anatomy--integrating two views of hippocampal plasticity. Tr. Neurosci 16, 141–147. [DOI] [PubMed] [Google Scholar]
- Lu W, Man H, Ju W, Trimble WS, MacDonald JF, and Wang YT (2001). Activation of synaptic NMDA receptors induces membrane insertion of new AMPA receptors and LTP in cultured hippocampal neurons. Neuron 29, 243–254. [DOI] [PubMed] [Google Scholar]
- Luscher C, and Malenka RC (2012). NMDA receptor-dependent long-term potentiation and long-term depression (LTP/LTD). Cold Spring Harb. Perspect. Biol 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luscher C, Nicoll RA, Malenka RC, and Muller D (2000). Synaptic plasticity and dynamic modulation of the postsynaptic membrane. Nat. Neurosci 3, 545–550. [DOI] [PubMed] [Google Scholar]
- Malenka RC (1991). Postsynaptic factors control the duration of synaptic enhancement in area CA1 of the hippocampus. Neuron 6, 53–60. [DOI] [PubMed] [Google Scholar]
- Malenka RC, and Bear MF (2004). LTP and LTD: an embarrassment of riches. Neuron 44, 5–21. [DOI] [PubMed] [Google Scholar]
- Malenka RC and Nicoll RA (1993). NMDA-receptor dependent synaptic plasticity: multiple forms and mechanisms. Tr. Neurosci 16, 521–527. [DOI] [PubMed] [Google Scholar]
- Nicoll RA (2017). A brief history of long-term potentiation. Neuron 93, 281–290. [DOI] [PubMed] [Google Scholar]
- Polepalli JS, Wu H, Goswami D, Halpern CH, Sudhof TC, and Malenka RC (2017). Modulation of excitation on parvalbumin interneurons by neuroligin-3 regulates the hippocampal network. Nat. Neurosci 20, 219–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulopoulos A, Aramuni G, Meyer G, Soykan T, Hoon M, Papadopoulos T, Zhang M, Paarmann I, Fuchs C, Harvey K, et al. (2009). Neuroligin 2 drives postsynaptic assembly at perisomatic inhibitory synapses through gephyrin and collybistin. Neuron 63, 628–642. [DOI] [PubMed] [Google Scholar]
- Poulopoulos A, Soykan T, Tuffy LP, Hammer M, Varoqueaux F, and Brose N (2012). Homodimerization and isoform-specific heterodimerization of neuroligins. Biochem. J 446, 321–330. [DOI] [PubMed] [Google Scholar]
- Scheiffele P, Fan J, Choih J, Fetter R, and Serafini T (2000). Neuroligin expressed in nonneuronal cells triggers presynaptic development in contacting axons. Cell 101, 657–669. [DOI] [PubMed] [Google Scholar]
- Shipman SL, and Nicoll RA (2012a). Dimerization of postsynaptic neuroligin drives synaptic assembly via trans-synaptic clustering of neurexin. Proc. Natl. Acad. Sci. USA 109, 19432–19437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shipman SL, and Nicoll RA (2012b). A subtype-specific function for the extracellular domain of neuroligin 1 in hippocampal LTP. Neuron 76, 309–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song JY, Ichtchenko K, Sudhof TC, and Brose N (1999). Neuroligin 1 is a postsynaptic cell-adhesion molecule of excitatory synapses. Proc. Natl. Acad. Sci. USA 96, 1100–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein V, House DR, Bredt DS, and Nicoll RA (2003). Postsynaptic density-95 mimics and occludes hippocampal long-term potentiation and enhances long-term depression. J. Neurosci 23, 5503–5506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stogsdill JA, Ramirez J, Liu D, Kim YH, Baldwin KT, Enustun E, Ejikeme T, Ji RR, and Eroglu C (2017). Astrocytic neuroligins control astrocyte morphogenesis and synaptogenesis. Nature 551, 192–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhof TC (2008). Neuroligins and neurexins link synaptic function to cognitive disease. Nature 455, 903–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhof TC (2012). The presynaptic active zone. Neuron 75, 11–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhof TC (2017). Synaptic neurexin complexes: a molecular code for the logic of neural circuits. Cell 171, 745–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varoqueaux F, Aramuni G, Rawson RL, Mohrmann R, Missler M, Gottmann K, Zhang W, Sudhof TC, and Brose N (2006). Neuroligins determine synapse maturation and function. Neuron 51, 741–754. [DOI] [PubMed] [Google Scholar]
- Varoqueaux F, Jamain S, and Brose N (2004). Neuroligin 2 is exclusively localized to inhibitory synapses. Eur. J. Cell Biol 83, 449–456. [DOI] [PubMed] [Google Scholar]
- Wu D, Bacaj T, Morishita W, Goswami D, Arendt KL, Xu W, Chen L, Malenka RC, and Sudhof TC (2017). Postsynaptic synaptotagmins mediate AMPA receptor exocytosis during LTP. Nature 544, 316–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyllie DJ, Manabe T, and Nicoll RA (1994). A rise in postsynaptic Ca2+ potentiates miniature excitatory postsynaptic currents and AMPA responses in hippocampal neurons. Neuron 12, 127–138. [DOI] [PubMed] [Google Scholar]
- Zhang B, Chen LY, Liu X, Maxeiner S, Lee SJ, Gokce O, and Sudhof TC (2015). Neuroligins sculpt cerebellar purkinje-cell circuits by differential control of distinct classes of synapses. Neuron 87, 781–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.