

Summary
Small RNAs trigger the formation of epialleles that are silenced across generations. Consequently, RNA-directed epimutagenesis is associated with persistent gene repression. Here, we demonstrate that small interfering RNA-induced epimutations in fission yeast are still inherited even when the silenced gene is reactivated, and descendants can reinstate the silencing phenotype that only occurred in their ancestors. This process is mediated by the deposition of a phenotypically neutral molecular mark composed of tri-methylated histone H3 lysine 9 (H3K9me3). Its stable propagation is coupled to RNAi and requires maximal binding affinity of the Clr4/Suvar39 chromodomain to H3K9me3. In wild-type cells, this mark has no visible impact on transcription but causes gene silencing if RNA polymerase-associated factor 1 complex (Paf1C) activity is impaired. In sum, our results reveal a distinct form of epigenetic memory in which cells acquire heritable, transcriptionally active epialleles that confer gene silencing upon modulation of Paf1C.
Keywords: transgenerational inheritance, RNA interference, RNAe, heterochromatin, transcriptional gene silencing, epigenetic memory, phenotypic plasticity, Paf1C, H3K9me3, si3 mark
Graphical Abstract
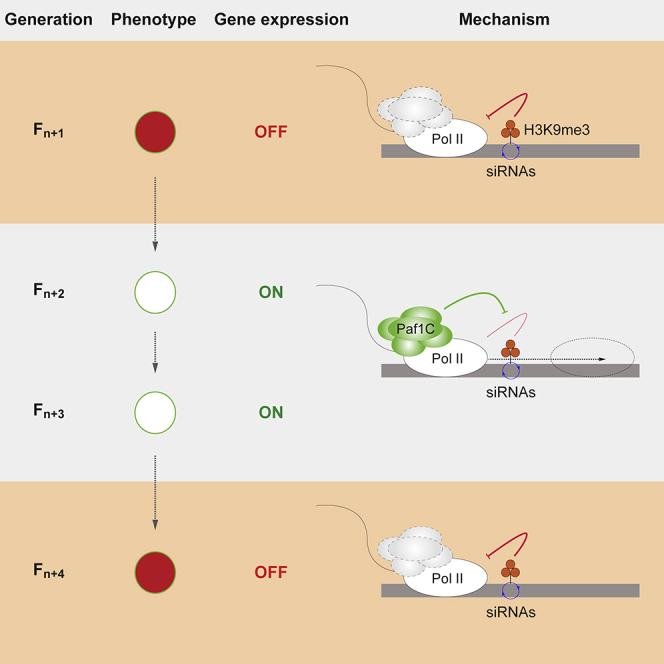
Highlights
-
•
S. pombe remembers and reinstates silencing that occurred in a previous generation
-
•
Paf1 modulation enables acquisition of a new trait that is plastic and inheritable
-
•
H3K9me3 functions as an epigenetic mark that is not repressive per se
-
•
Paf1C facilitates transcription elongation through H3K9-methylated nucleosomes
Duempelmann et al. describe an H3K9me3 and siRNA-dependent epimutation, which is installed during gene silencing and inherited across generations, even upon reactivation of the silenced gene. Yet silencing is reinstated in later generations if Paf1C is impaired, revealing H3K9me3 as a stable epigenetic mark that is not repressive per se.
Introduction
Phenotypic effects caused by epimutations rather than changes in DNA sequence have been described in various organisms. Epimutations are potentially adaptive if inherited across generations and might even respond to environmental challenges. Yet experimental evidence is scarce (Heard and Martienssen, 2014). Prominent examples of heritable phenotypic changes caused by epimutations are paramutation in plants and RNA-induced epigenetic silencing (RNAe) in nematodes, in which small RNAs trigger the formation of epialleles that are stably silenced across generations (Ashe et al., 2012, Chandler, 2007, Erhard and Hollick, 2011, Grentzinger et al., 2012, Luteijn et al., 2012, Shirayama et al., 2012). A similar phenomenon has recently been described in the fission yeast Schizosaccharomyces pombe (Yu et al., 2018), demonstrating that long-lasting gene silencing responses mediated by small RNAs are widespread. Consequently, RNA-directed epimutagenesis is associated with persistent gene repression.
Whereas it is now well established that RNA-induced repression of genes is heritable across generations, it has remained unknown whether mechanisms exist that robustly convey transgenerational memory of a silencing “experience,” without establishing a permanently repressed state. Here, we report the discovery of such a phenomenon in S. pombe: we show that RNAe, triggered by transient expression of hairpin-derived small interfering RNAs (siRNAs) coupled with mutation of the polymerase-associated factor 1 (Paf1), is stably propagated for at least 18 generations. Upon reintroduction of Paf1, the silent state is lost but resumes if Paf1 is again impaired in later generations, even in the absence of the original siRNA trigger. This process is mediated by the deposition of a phenotypically neutral molecular mark composed of tri-methylated histone H3 lysine 9 (H3K9me3), and its stable propagation is coupled to RNAi. Our results reveal that H3K9me3 functions as an epigenetic mark that is not repressive per se and imply that Paf1C’s function in promoting transcription elongation is particularly important within difficult-to-transcribe chromatin.
Results
A Point Mutation in Paf1 Enables Multi-generational Inheritance of Gene Silencing
In S. pombe, ectopic expression of primary siRNAs mediates de novo silencing of euchromatic genes through the formation of heterochromatin. These silent epialleles are only established upon the concurrent mutation of factors that negatively regulate this process (Flury et al., 2017, Kowalik et al., 2015, Yu et al., 2018), including mutations in subunits of the Paf1C, such as a nonsense mutation in the paf1+ gene (paf1-Q264Stop). We used this system to study mechanisms of epigenetic inheritance by expressing an RNA hairpin (ade6-hp) from a euchromatic locus (Figures S1A and S1B), which leads to the generation of primary siRNAs that induce strong heterochromatin-mediated silencing of ade6+ expression in paf1-Q264Stop cells (Kowalik et al., 2015). Because silencing of ade6+ causes a phenotypic switch from white to red cells when grown on limiting adenine plates, ade6+ ON (white) and ade6+ OFF (red) epialleles can be visually distinguished. The red phenotype of cells bearing an ade6+ OFF epiallele is stably propagated to the next generation in the same paf1-Q264Stop background, even in the absence of the primary siRNAs that induced the silent state, demonstrating epigenetic inheritance (Kowalik et al., 2015).
Using this approach, we have previously demonstrated that siRNA-mediated repression of ade6+ in the absence of Paf1C is mitotically stable and mediated by heterochromatic histone H3 lysine 9 (H3K9) modifications and secondary ade6+ siRNA production at the target locus (Kowalik et al., 2015). To test whether the repressed state of ade6+ is also stably maintained over multiple generations in the absence of the primary siRNA trigger, we repeatedly crossed red paf1-Q264Stop ade6+ OFF cells with white paf1-Q264Stop ade6+ ON cells (Figure 1A) and analyzed the degree of inheritance by tetrad dissection (Figures S1C and S1D). In total, we examined three independent pedigrees for 5 generations (F5), of which one we continued until F18. Spores of the 18th generation still formed red colonies (Figures 1A and S1C), indicating that the repressed phenotype is stably inherited. Segregation of the red phenotype was non-Mendelian (Figure S1C), excluding DNA sequence changes as the underlying cause for the observed heritability. Occasionally, we observed the red phenotype in more than 2 spores of a tetrad (Figure S1C), indicating that the ade6+ OFF allele can be paramutagenic (Chandler, 2010). Similar to RNAe in Caenorhabditis elegans (Luteijn and Ketting, 2013), we observed secondary ade6+ siRNAs in paf1-Q264Stop ade6+ OFF cells, but not in paf1-Q264Stop ade6+ ON cells derived from F10 spores (Figure 1B). These siRNAs spread up and downstream of the region initially targeted by the hairpin (Figure 1B). They also correlated well with enrichment of the heterochromatic histone modifications H3K9me2 and H3K9me3 (Figure 1C), which are associated with gene silencing (Jih et al., 2017). Consistent with a recent study (Yu et al., 2018), inheritance of the red silencing phenotype remained strictly dependent on the paf1-Q264Stop mutation, as all spores that inherited the paf1+ allele formed white colonies (Figures 1D and S1E). Thus, if primary siRNAs are transiently expressed from a euchromatic locus, acquisition and epigenetic inheritance of the ade6+ OFF silencing phenotype (red colonies) is only possible if Paf1C activity is impaired.
Figure 1.

Transgenerational Inheritance of siRNA-Directed Gene Silencing
(A) Silencing of the ade6+ gene was initiated in trans by a primary siRNA-producing ade6-hairpin (denoted by the symbol) in paf1-Q264Stop cells (F0). F0 cells with a silent epiallele (ade6+ OFF, red phenotype) were then crossed with paf1-Q264Stop cells lacking the ade6-hairpin (ade6+, white phenotype), followed by spore dissection on YE plates. Colonies forming from F1 spores that inherited the ade6+ OFF allele (red), but not the ade6-hairpin, were selected and again crossed with naive paf1-Q264Stop ade6+ cells (white). This was repeated until generation 18 (F18; see also Figure S1C).
(B) Small RNA (sRNA) sequencing performed with red (ade6+ OFF) or white (ade6+ ON) colonies from F10 spores. Read counts were normalized to library size. Black line denotes the part of ade6+ that is complementary to the primary siRNA-producing ade6-hairpin. One representative of many sequenced spores is shown.
(C) Chromatin immunoprecipitation (ChIP)-qPCR measuring H3K9me2 and H3K9me3 enrichments at ade6+ ON and ade6+ OFF epialleles, normalized to adh1+ and relative to a clr4Δ background control. n = 3 independent white or red colonies formed from F10 spores. Center values denote the mean; error bars denote SD.
(D) Tetrad dissection of F7 paf1-Q264Stop ade6+ OFF (red) cells crossed with naive paf1+ cells. F8 paf1+ spores never grew as red colonies (see also Figure S1E).
H3K9me3 and Secondary siRNAs Are Inherited with No Apparent Silencing Phenotype in Wild-Type Cells
H3K9 methylation is a well-established “repressive” histone modification. Hence, we concluded that the loss of the RNAe phenotype in all paf1+ progeny from heterozygous paf1-Q264Stop × paf1+ crosses must result from a failure to maintain methylated H3K9 in wild-type cells. However, we discovered that H3K9me2 and H3K9me3 marks were not erased from the reactivated ade6+ epiallele (Figures 2A–C). Furthermore, the secondary ade6+ siRNAs were also still produced in the paf1+ cells (Figures 2D, 2E, and S2). This demonstrates that H3K9 methylation marks and secondary siRNAs are stably propagated to the next generation, despite no apparent silencing phenotype. Therefore, we hypothesized that H3K9 methylation functions as a phenotypically neutral epimutation, conferring a cellular memory of the ancestral ade6+-silencing phenotype that might be reinstated by repeated Paf1C impairment. To test this, we crossed the paf1+ cells that had lost the silencing phenotype with paf1-Q264Stop cells and monitored gene silencing in the next generation (Figure S3A). Indeed, in almost half of the crosses, ade6+ silencing was re-established in paf1-Q264Stop cells, which was again stably maintained through mitosis (Figures S3B–S3D). Silencing occurred at the level of transcription, as H3K36me3 levels were strongly reduced on the ade6+ gene (Figures 2F and S3E). The paf1-Q264Stop allele did not affect transcription of other genes, and we only observed repression of ade6+ in paf1-Q264Stop cells if the ancestral strain had been exposed to ade6+ siRNAs (Figures 2G, 2H, and 5). These results demonstrate that RNAe installs a molecular mark—involving H3K9 methylation and secondary siRNA production—that is also compatible with active transcription. Because this mark resists erasure upon reactivation of the silent allele, it confers a cellular memory of gene silencing that had occurred in past generations. We hereafter refer to the marked ade6+ epiallele as ade6si3.
Figure 2.

H3K9 Methyl Marks Remain, even when the Silenced ade6+ Gene Is Reactivated
(A) Schematic illustrating that the ade6si3 OFF epiallele from a red paf1-Q264Stop parent retains a molecular mark (ade6si3) in paf1+ daughter cells (indicated by green color), although the silent state was lost (ade6si3 ON). If the silent state had already been lost in the paf1-Q264Stop parental generation (ade6+ ON), no mark was inherited to the next generation (ade6+).
(B and C) ChIP-qPCR measuring H3K9me2 (B) and H3K9me3 (C) enrichments at ade6+ genes in marked and non-marked wild-type cells, which show no silencing phenotype. Values are normalized to adh1+ and relative to a clr4Δ background control. n = 4 independent colonies growing from unrelated F11 paf1+ spores. Center values denote the mean, error bars denote SD, and p values were calculated with a two-tailed Student’s t test.
(D) sRNA sequencing performed with wild-type cells carrying the ade6si3 ON epiallele. Same cells as used in (B) and (C) are shown. See Figure S2 for all 4 biological replicates.
(E) Length distribution and 5′ U bias of small RNAs shown in (D), which are characteristic of siRNAs. Read counts in (D) and (E) were normalized to library size.
(F and G) University of California Santa Cruz (UCSC) genome browser shots of ade6+ (F) and an unrelated euchromatic gene nap1+(G). ChIP sequencing (ChIP-seq) profiles for H3K36me3, total H3, and input for wild-type (with and without marked ade6+) and paf1-Q264Stop (with marked ade6+) cells are normalized to library size. All 3 biological replicates are shown in Figures S3E and 5.
(H) qRT-PCR measurement of ade6+ mRNA levels in wild-type and paf1-Q264Stop cells, with and without the molecular mark. mRNA levels were normalized to act1+ and shown relative to paf1+ade6+ ON. n = 4–9 biological replicates (individual data points). Center values denote the mean, error bars denote SD, and p values were calculated using two-tailed Student’s t tests.
(A–H) F10 paf1-Q264Stop ade6imp OFF cells were the same as shown in Figure 1.
Figure 5.

Paf1C Sustains Transcription when RNA Polymerase II Encounters H3K9me3 Marked Nucleosomes
(A) Schematic illustrating that Paf1C has an important role in sustaining transcription when RNA polymerase II encounters H3K9me3 marked nucleosomes.
(B) UCSC genome browser shots of naive and marked ade6+ genes. ChIP-seq profiles for H3K9me2, H3K9me3, RNA polymerase II, and H3K36me3 for wild-type and paf1-Q264Stop cells were normalized to library size.
(C) UCSC genome browser shots of an unrelated euchromatic gene nap1+. ChIP-seq profiles for RNA polymerase II for wild-type and paf1-Q264Stop cells were normalized to library size.
(D) Scatterplot quantifying RNA polymerase II (Pol II) redistribution between the 5′ end and the 3′ end of all expressed genes (n = 1,860; see STAR Methods). Shown are the log2 values of the ratio of read counts within the first 500 bp versus the last 500 bp of every gene. ade6+ (red) and nap1+ (blue) are highlighted. The left plot compares marked versus silenced cells, and the right plot compares marked with naive cells.
(E) Scatterplot comparing the levels of H3K36me3 over all genes. Shown are reads per million (RPM) values for each expressed gene (same gene list as in D). The left plot compares marked versus silenced cells, and the right plot compares marked with naive cells.
We assessed the mitotic stability of ade6si3 in Paf1C wild-type cells by exponentially growing paf1+ ade6si3 ON cells in liquid culture and crossing a sample every 3 days (30–40 mitotic divisions) with paf1-Q264Stop cells (Figure 3A). This revealed recurrence of the silent state in paf1-Q264Stop progeny even after 17 days of exponential growth (∼180 mitotic divisions) of the paf1+ ade6si3 ON parental cells, although this gradually declined with the number of cell divisions (Figure 3B). We next evaluated transgenerational inheritance of ade6si3 in the paf1+ background. We found that paf1+ grandparents were able to pass on the ade6si3 ON allele to a paf1+ parent, and silencing could still be re-established in their paf1-Q264Stop progeny (Figures 3C and 3D). Thus, the marked ade6si3 epiallele is stably maintained during both mitosis and meiosis.
Figure 3.

The ade6si3 Epiallele Is Stable across Generations in Wild-Type Cells
(A) Crossing scheme for testing the mitotic stability of the marked ade6si3 epiallele. F11 paf1+ade6si3 ON colonies were grown in liquid culture and crossed to paf1-Q264Stop ade6+ cells every 30–40 mitotic divisions. Restoration of silencing phenotype in F12 paf1-Q264Stop was assessed by random spore analysis.
(B) Analysis of silencing restoration as depicted in (A), which was performed eleven times. At least 1,000 F12 colonies (spores) per cross were inspected for the silencing phenotype.
(C) Crossing scheme for testing the meiotic stability of the marked ade6si3 epiallele. Colonies grown from F11 paf1+ade6si3 ON spores (A) were crossed to wild-type cells and subsequently to paf1-Q264Stop ade6+ cells.
(D) Restoration of the silencing phenotype in F13 paf1-Q264Stop cells was assessed by random spore analysis. At least 1,000 colonies grown from those spores per cross were inspected for the silencing phenotype. n = 11 different crosses. Centre values denote the mean; error bars denote SD.
(A–D) F10 paf1-Q264Stop ade6imp OFF cells were the same as shown in Figure 1.
In summary, impaired Paf1C activity enables primary siRNAs to install a molecular mark on the homologous protein-coding gene, which results in a strong silencing phenotype that is inherited over many generations. Upon reactivation of Paf1C, the silencing phenotype is lost, but the phenotypically neutral mark (secondary siRNA production and H3K9 methylation; si3) is still inherited by subsequent generations, even in the absence of the primary siRNAs that triggered marking in the ancestral strain. If Paf1C is again compromised, silencing is re-established and extremely stably maintained.
Epigenetic Memory of an Ancestral Gene-Silencing Episode Is Mediated by Coupling of Secondary siRNA Amplification and H3K9 Tri-methylation
Coupling of RNAi and histone H3K9 methylation is important to maintain the silent state of an RNAe response triggered by centromeric siRNAs (Yu et al., 2018). To investigate whether such coupling also mediates transgenerational inheritance of the marked ade6si3 epiallele, we crossed paf1-Q264Stop ade6si3 OFF cells with paf1+ cells lacking key RNAi factors (Ago1, Dcr1, or Rdp1) or the H3K9 methyltransferase Clr4 (Figure 4A). These were then crossed with paf1-Q264Stop cells expressing Clr4, Ago1, Dcr1, and Rdp1 to see whether the marked ade6si3 epiallele had been inherited and the silencing state could be re-established. However, in the next-generation paf1-Q264Stop cells, ade6+ could no longer be silenced (Figure 4B). Furthermore, secondary ade6+ siRNA production ceased in these mutants (Figures 4C, 4D, and S4). ade6+ siRNAs were also absent in the parental paf1+ clr4Δ cells (Figures 4C and S4B), and H3K9me3 and H3K9me2 were abolished in the parental paf1+ RNAi mutants (Figures 4E and 4F). This demonstrates coupling of siRNA production and histone H3K9 methylation in paf1+ cells for the transgenerational inheritance of the marked allele. To further dissect the requirement of specific H3K9 methylation states for stable marking, we employed a Clr4 mutant (Clr4-F449Y) that is deficient in catalyzing specifically H3K9me3 (Jih et al., 2017). We observed that ade6+ siRNA production was abolished in Clr4-F449Y-expressing cells (Figures 4C and S4B) and that the silencing phenotype was not re-established in the next generation (Figure 4B). In contrast, H3K9me2 was only lost in clr4-F449Y cells at the marked locus, but not at centromeric repeats (Figures 4F and 4H; Jih et al., 2017), where siRNA production was maintained (Figures 4D and S4C). Therefore, we conclude that epigenetic inheritance of the ade6si3 allele is mediated by coupling of secondary siRNA amplification and H3K9 tri-methylation. H3K9me2 catalysis alone (i.e., in the presence of Clr4-F449Y) is not sufficient to maintain the mark (Figures 4B, 4C, and 4F).
Figure 4.

H3K9me3 and siRNAs Preserve the ade6si3 Epiallele in paf1+ Cells without Affecting Transcription
(A) Crossing scheme for testing the requirement of H3K9me3 and RNAi for the heritability of the molecular mark (B–H) or for testing heritability of the mark if the affinity of Chp1 or Clr4 for H3K9me3 is reduced (Chp1-F61A and Clr4-W31G) or abolished (Chp1-V24R and Clr4-W41G) in otherwise wild-type cells (I).
(B) Restoration of the silencing phenotype in F12 paf1-Q264Stop cells (otherwise wild-type), assessed by random spore analysis. If the F11 paf1+ parent was mutant for any of the factors depicted (mutantX), silencing was never re-established. Number of colonies grown from F12 spores that were inspected for the silencing phenotype is as follows: 442, 1,161, and 1,167 (wild-type, ade6si3); 4,892, 5,070, and 4,982 (wild-type, ade6+); 2,549, 1,586, and 1,906 (clr4Δ); 2,743, 651, and 631 (clr4-F449Y); 1,368, 2,019, and 2,219 (ago1Δ); 1,980, 1,444, and 1,447 (dcr1Δ); and 2,063, 1,775, and 1,918 (rdp1Δ). Center values denote the mean; error bars denote SD.
(C and D) sRNA sequencing was performed with F11 wild-type ade6si3 and paf1+mutantX cells. Enrichments are shown at ade6+ (C) and dg (D). Counts were normalized to library size. All biological triplicates of wild-type ade6si3 and mutantX are shown in Figure S4.
(E–H) ChIP-qPCR measuring H3K9me2 and H3K9me3 enrichments at ade6+ genes (E and F) and the centromeric dg repeat (G and H), normalized to adh1+ and relative to a clr4Δ background control in F11 wild-type ade6si3 and paf1+mutantX cells that produced F12 spores analyzed in (B). Center values denote the mean; error bars denote SD.
(B–H) n ≥ 3 different crosses (pedigrees) for each genetic background.
(I) Restoration of the silencing phenotype in F12, in which Chp1 and Clr4 had again normal affinities for H3K9me3, was assessed by random spore analysis (see A for crossing scheme). Number of F12 paf1-Q264Stop colonies (otherwise wild-type) that were inspected for the silencing phenotype is as follows: 1,113, 1,049, 1,372, and 1,294 (wild-type); 2,308, 1,113, 2,200, 1,077, and 1,317 (clr4-W31G); 1,615, 708, and 2,155 (clr4-W41G); 1,196, 1,249, 1,035, and 1,292 (chp1-V24R); and 2,146, 1,012, 1,014, 1,015, and 652 (chp1-F61A). Center values denote the mean; error bars denote SD; n, number of crosses (pedigrees) for each genetic background.
(J) Model depicting positive feedback loops that preserve the marked epiallele. Clr4 recognizes H3K9me3 and catalyzes H3K9 methylation on newly deposited histones. H3K9me2 is sufficient to stabilize RITS binding to chromatin. RITS together with H3K9me3 retain Clr4 activity at the marked locus. Amplification of secondary siRNAs further stabilizes RITS association with the marked locus, promoting recruitment of Clr4 activity.
Inheritance of the Phenotypically Neutral Epimutation Depends on Maximal Binding Affinity of the Clr4/Suvar39 Chromodomain to H3K9me3
We next investigated how H3K9 tri-methylation maintains inheritance of the ade6si3 epiallele. Methylation of H3K9 creates a binding site for chromodomain-containing proteins. The chromodomains of Chp1 and Clr4, both essential factors for RNAi-directed heterochromatin silencing, bind H3K9me3 more tightly than H3K9me2, and the chromodomain of Chp1 has the highest binding affinity for H3K9me (Schalch et al., 2009). Chp1 forms the RNA-induced transcriptional silencing complex (RITS) together with Ago1 and Tas3 (Verdel et al., 2004). Such high-affinity binding between Chp1 and H3K9me3 is critical for de novo formation of heterochromatin at centromeric repeats, but it is not required for the maintenance of heterochromatin once it is established (Schalch et al., 2009). In agreement with these observations, we observed that ade6si3 was maintained in cells expressing a mutant of Chp1, Chp1-F61A (Figure 4A and 4I). This mutant binds H3K9me3 with lower affinity, which is similar to the affinity of wild-type Chp1 binding to H3K9me2 (Schalch et al., 2009). Thus, high-affinity binding of the RITS complex to H3K9me3 is not a prerequisite for inheritance of the ade6si3 epiallele (Figures 4A and 4I). However, in cells expressing Chp1-V24R, which cannot bind methylated H3K9 at all, the mark was lost (Figure 4I). To test whether the requirement of H3K9me3 for stable marking is also linked to Clr4, we reduced the high H3K9me3 binding affinity of the Clr4 chromodomain to the lower affinity for H3K9me2 by mutating W41 to G (Schalch et al., 2009). In contrast to Chp1-F61A, the mark was lost and the silencing phenotype could not be re-established. This also occurred with the complete H3K9me3 binding mutant Clr4-W31G (Figure 4I). Therefore, maximal binding affinity of the Clr4, but not the Chp1, chromodomain to H3K9me3 is required for the inheritance of the marked ade6si3 epiallele. These results support a model in which Clr4 recognizes H3K9me3, which had been deposited during the initial primary siRNA-mediated silencing episode, and catalyzes the same modification on newly deposited histones. This positive feedback loop is not self-sufficient but further depends on RITS binding to methylated H3K9 and subsequent amplification of secondary siRNAs, which promote further recruitment of Clr4 activity to the marked locus (Figure 4J).
RNA Polymerase II Accumulates at the Beginning of an H3K9 Methylated Gene if Paf1C Activity Is Impaired
Our results reveal that such coupling of siRNA- and H3K9me3-positive feedback regulation is still not sufficient to silence transcription of a protein-coding gene in paf1+ cells. This attributes an important activity to Paf1C that facilitates transcription through H3K9me3 marked nucleosomes (Figure 5A). Supporting this, we observed a buildup of RNA polymerase II at the 5′ end of the marked ade6si3 OFF, but not on other genes in paf1-Q264Stop cells (Figures 5B–5D). Because H3K36 on ade6si3 OFF is not methylated (Figures 5B and 5E), we conclude that RNA polymerase II frequently stalls on H3K9 methylated nucleosomes if Paf1C activity is impaired. This implies that Paf1C’s established function in promoting transcription elongation (Vos et al., 2018) is particularly important within difficult-to-transcribe chromatin. This is different from its less well-understood function in RNA 3′ end formation (Van Oss et al., 2017), which is important to prevent de novo deposition of H3K9 methyl marks (Kowalik et al., 2015). Thus, persistent impairment of Paf1C activity is required for manifestation of a silencing phenotype, but not for sustainable coupling of RNAi and H3K9me3 once initiated.
Discussion
Our findings demonstrate that fission yeast has the ability to remember gene-silencing episodes that have occurred in previous generations. Unlike other examples of heritable epimutations (Baulcombe and Dean, 2014, Heard and Martienssen, 2014, Iwasaki and Paszkowski, 2014, Klosin and Lehner, 2016, Luteijn and Ketting, 2013, Yu et al., 2018), the phenomenon described here is unique in that the phenotypic change only manifests under the same conditions that had enabled acquisition of the epiallele. This shows that S. pombe cells can acquire a new trait that is plastic and can be passed down to its offspring. In C. elegans, environmental stresses give rise to changes in heritable small RNAs, which have been proposed to silence specific genes that could in turn improve the progeny’s chance to cope with similar stresses (Rechavi and Lev, 2017). Because initiation of epigenetic gene silencing has only been observed in the presence of an enabling genetic mutation in S. pombe (Flury et al., 2017, Kowalik et al., 2015, Yu et al., 2018), modulation of repressors, such as Paf1C, is imperative for such an RNAe response to occur in the wild. Importantly, we noticed impaired recovery of spores after tetrad dissection of homozygous paf1-Q264Stop crosses (Figures S1C and S1E). This shows that functional Paf1C is vital for the reproductive success of S. pombe, and hence, RNAe-enabling mutations in Paf1C subunits are unlikely to become fixed during evolution. Yet our results raise the intriguing hypothesis that external factors, such as chemicals or metabolites present in the yeast’s immediate environment, could transiently impair Paf1C activity and thereby enable the acquisition of a si3-marked epiallele. In light of this, we find it interesting that impaired Paf1C activity leads to seemingly stochastic production of siRNAs using endogenous mRNAs as templates (Kowalik et al., 2015), which continue to be amplified also in subsequent generations (Figure S5). Because these siRNAs have no homology to constitutive heterochromatin, they are not expected to trigger silencing that remains in wild-type cells (Yu et al., 2018) but rather leave, as we show here, a phenotypically neutral molecular mark. Therefore, transient inactivation of Paf1C could trigger random marking of protein-coding genes, some of which, or combinations thereof, may lead to an increased population fitness under adverse conditions. Such an epigenetic bet-hedging strategy would be ideal for a unicellular fungus to survive in a dynamic environment and could explain why this extremely potent form of cellular memory has evolved in this organism. Although challenging, it will be an exciting task to discover natural conditions that would lead to such a response.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-H3, rabbit polyclonal | Abcam | Cat# ab1791, RRID:AB_302613 |
| anti-H3K9me2, mouse monoclonal | MBL International | Cat# MABI0307, RRID:AB_11124951 |
| anti-H3K9me3, rabbit polyclonal | Active Motif | Cat# 39161, RRID:AB_2532132 |
| anti-H3K14ac, rabbit monoclonal | Novus | Cat# NB110-57051, RRID:AB_843953 |
| anti-H3K36me3, rabbit polyclonal | Abcam | Cat# ab9050, RRID:AB_306966 |
| anti-RNA polymerase II, mouse monoclonal | Covance Research Products Inc | Cat# MMS-126R, RRID:AB_10013665 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Glusulase | PerkinElmer | NEE154001EA |
| nourseothricin dihydrogen sulfate (NAT) | Fisher or WERNER BioAgents GmbH | Cat# 5029426 or Cat# 5.0 |
| G418 sulfate (Geneticin) | Roche or Invitrogen/Life Technologies | Cat# 04727878001-2 or Cat# 10131027 |
| Hygromycin | Sigma or Invitrogen/Life Technologies | Cat# H7772 or Cat# 10687010 |
| NEBNext High-Fidelity 2X PCR Master Mix | NEB | Cat# M0541S |
| PrimeScript RT Master Mix | Takara | Cat# RR036A |
| Formaldehyde | Sigma | Cat# F8775 |
| Tween 20 | Sigma | Cat# P9416 |
| PMSF | Sigma | Cat# P7626 |
| cOmplete Protease Inhibitor Cocktail, EDTA-free | Roche | Cat# 05056489001 |
| Dynabeads M-280 Sheep anti-Mouse IgG | Thermo Fisher | Cat# 11202D |
| Dynabeads M-280 Sheep anti-Rabbit IgG | Thermo Fisher | Cat# 11203D |
| Proteinase K | Roche | Cat# 3115879001 |
| RNase A | Roche | Cat# 10109169001 |
| AMPure XP Beads | Beckman Coulter | Cat# A63881 |
| 5-Fluoroorotic Acid (FOA) | US biological, Thermo Fisher | Cat# 207291-8-4 |
| Critical Commercial Assays | ||
| MasterPure Yeast RNA Purification Kit | Epicenter | Cat# MCR85102 |
| Protein Assay Dye Reagent Concentrate | Bio-Rad | Cat# 500-0006 |
| QIAseq miRNA Library Kit | QIAGEN | Cat# 331505 |
| SsoAdvanced Universal SYBR Green Supermix | Bio-Rad | Cat# 172-5274 |
| NEBNext Ultra II DNA Library Prep Kit for Illumina | NEB | Cat# E7645L |
| NEBNext Multiplex Oligos for Illumina | NEB | Cat# E7600S |
| Deposited Data | ||
| smallRNA and ChIP-seq data | This paper | GEO: GSE120352 |
| Experimental Models: Organisms/Strains | ||
| S. pombe: Strain background: 972, see Table S1 | This paper | N/A |
| Oligonucleotides | ||
| Primers, see Table S2 | This paper | N/A |
| Recombinant DNA | ||
| pJET1.2 - clr4-F449Y | This paper | pMB1858 |
| pJET1.2 - clr4-W31G::hph | This paper | pMB1928 |
| pJET1.2 - clr4-W41G::hph | This paper | pMB1929 |
| pJET1.2 - chp1-V24R::hph | This paper | pMB1931 |
| pJET1.2 - chp1-F61A::hph | This paper | pMB1932 |
| Software and Algorithms | ||
| Plugin Cell Counter 2.2.2 | Fiji | https://imagej.nih.gov/ij/plugins/cell-counter.html |
| bcl2fastq2 v2.17 | Illumina | https://support.illumina.com/sequencing/sequencing_software/bcl2fastq-conversion-software.html |
| STAR | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| bedtools (version 2.26.0) | Quinlan, 2014 | https://github.com/arq5x/bedtools2/ |
| bedGraphToBigWig (from UCSC binary utilities) | Kent et al., 2010 | http://genome.ucsc.edu/ |
| R | Team, 2014 | https://www.r-project.org/ |
| QuasR | Gaidatzis et al., 2015 | https://bioconductor.org/packages/release/bioc/html/QuasR.html |
| Other | ||
| Illumina HiSeq2500 | Illumina | N/A |
| Agilent 1100 system | Agilent | N/A |
| MSM System 400 | Singer Instruments | Serial number 686-400-43911 |
| Bioruptor Plus sonication device | Diagenode | N/A |
Contact for Reagent and Resource Sharing
Published research reagents from the FMI are shared with the academic community under a Material Transfer Agreement (MTA) having terms and conditions corresponding to those of the UBMTA (Uniform Biological Material Transfer Agreement). All custom codes used to analyze data and generate figures are available upon reasonable request. Genome-wide datasets are deposited at GEO under the accession number GSE120352. Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Marc Bühler (marc.buehler@fmi.ch).
Experimental Model and Subject Details
Schizosaccharomyces pombe strains used in this study are derivates of the standard laboratory strain 972 and are listed in Table S1. Strains were grown at 30°C either in liquid YES media (160rpm) or on solid agarose plates (YE or YES plates).
Method Details
Strains and plasmids
S. pombe strains were generated by genetic crossing or by homologous recombination with transformed DNA (Bähler et al., 1998, Forsburg and Rhind, 2006). For the generation of the point mutants clr4-F449Y, clr4-W31G, clr4-W41G, chp1-V24R, chp1-F61A, first the entire clr4 ORF or the first 90bp of chp1 were deleted with URA3 from Candida albicans and then replaced by the mutated ORF at the same locus by FOA counter selection. ago1Δ, clr4Δ, dcr1Δ and rdp1Δ mutants were generated by deletion of the entire ORF with a hphMX or natMX cassette. All strains used in this study are listed in Table S1.
Mating and spore analysis
For a genetic cross, cells of opposite mating type were mixed in 10 μL H2O, plated on SPAS plates (Forsburg and Rhind, 2006) and incubated at room temperature for 2-3 days. Cells were suspended in 1 mL H2O and asci formation was assessed by microscopy. For random spore analysis (RSA), 2 μL glusulase were added and incubated either at 30°C for 8 hours or at room temperature overnight to kill vegetative cells. Different dilutions were plated to receive 20-500 colonies/plate. For tetrad dissection (Figures 1D, S1C, and S1E), 10 μL of a 1:100 tetrad asci dilution were dispersed on a yeast extract (YE) plate (Forsburg and Rhind, 2006) and tetrads were separated with a Singer Instruments MSM System. After 6 hours at room temperature, spores were dissected and plates were incubated at 30°C to allow colony formation (4-5 days).
Analysis of epigenetic inheritance
Inheritance of the marked ade6+ epiallele
F10 h- paf1-Q264Stop ade6si3 OFF cells (SPB3850) were crossed with h+ paf1+ ade6+ ON cells (SPB3728). After 2 days, RSA was performed and spores were plated on YE plates. After 4-5 days colonies were genotyped. h- paf1+ colonies were selected and grown in 55ml YES (Forsburg and Rhind, 2006). Cells were harvested at OD = 1 for ChIP, small RNA sequencing, and further mating. F11 h- paf1+ were crossed to h+ paf1-Q264Stop::kanR ade6+ ON (SPB2063). After 2 days, RSA was performed and cells were plated on YE + geneticin (100ug/ml) plates to select for paf1-Q264Stop::kanR cells. After 5-7 days, images were acquired and the phenotype of the F12 paf1-Q264Stop colonies was manually quantified with the Fiji plugin Cell Counter.
Mitotic stability of the marked ade6+ epiallele
For Figures 3A and 3B, F10 h- paf1-Q264Stop ade6si3 OFF cells (SPB3850) were crossed with h+ paf1+ ade6+ ON cells (SPB3728). After 2 days, RSA was performed. 4 days later, F11 h- paf1+ ade6si3 ON colonies were transferred to YES media and grown at 30°C at 200 rpm. The liquid cultures were kept in an exponential growth phase by diluting with YES media twice a day. Every 72 hours (30-40 mitotic cell divisions) a sample was crossed to h+ paf1-Q264Stop::kanR ade6+ ON cells (SPB2063). After 2 days, RSA was performed and cells were plated on YE geneticin (100ug/ml) plates. 5-7 days later images were acquired and the phenotype of the F12 paf1-Q264Stop colonies was quantified.
Meiotic stability of the marked ade6+ epiallele
For Figures 3C and 3D, F10 h- paf1-Q264Stop ade6si3 OFF cells (SPB3850) were crossed to h+ paf1+ ade6+ ON cells (SPB3728). After 2 days, RSA was performed. After 4 days F11 h- paf+ ade6si3 ON colonies were crossed to h+ paf+ ade6+ ON cells. After 2 days, F12 tetrad asci were inoculated in 1ml H2O + 2 μL glusulase and incubated at 30°C for 10 hours with occasional vortexing. Spore pellets were resuspended in YES media and incubated at 30°C for 3 days for recovery. Then cells were crossed with h+ paf1-Q264Stop::kanR ade6+ ON cells (SPB2063). After 2 days, RSA was performed and cells were plated on YE geneticin (100ug/ml) plates. 5-7 days later images were acquired and the phenotype of the F12 paf1-Q264Stop colonies was determined.
Small RNA sequencing and analysis
Total RNA from exponentially growing cultures was extracted with the MasterPure Yeast RNA Purification Kit (Epicenter). Small RNA libraries were prepared with the QIAseq miRNA Library Kit (QIAGEN) according to manufacturer’s instructions and sequenced with an Illumina HiSeq2500. 3′ adaptor sequences were removed from the raw reads using Cutadapt (Cutadapt –a “adapter”–discard-untrimmed -m 18) (Martin, 2011). Reads < 18 nt and untrimmed reads were removed. Trimmed reads were aligned to the S. pombe genome (ASM294 version 2.24) using Bowtie (bowtie -f -M 10000 -v 0 -S–best–strata) (Langmead et al., 2009). All analysis was done with uniquely mapping reads, which were extracted using mapping quality (mapq = 255) since bowtie assigns multi-mapping reads mapq = 0.
Chromatin immunoprecipitation
50 mL of exponentially growing cells were cross-linked at OD = 1/1.2 with 0.9% Formaldehyde for 15 min at room temperature. The crosslinking reaction was quenched with 2.6 mL Glycine (2.5 M) for 5 min at room temperature. After 2 washing steps with ice cold PBS Tween 20 (0.02%), cell pellets were lysed in ChIP lysis buffer (50 mM HEPES KOH pH 7.5, 140 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% Na-deoxycholate, 1 mM PMSF, 1x Roche cOMPLETE protease inhibitor cocktail) using a bead-beater (three times 1 min). Lysates were sonicated with a Bioruptor three times 10 × 30 s (30 s off) with 5 min pause in between. After centrifugation for 1x 5 min and 1x 15min, the protein concentration of the supernatant was determined using the Bio-Rad Assay. Equal protein amounts (1ug-2ug) were incubated with antibody overnight (12-16 hr) at 4°C (2.5 μL histone H3-specific antibody, 2.5 μL histone H3K9me2-specific antibody, 3 μL histone H3K9me3-specific antibody, 2.5 μL H3K36me3-specific antibody, and 2.5 μL RNA polymerase II antibody). Then supernatant + antibody were incubated with 30/40 μL Dynabeads. Washes were performed three times with lysis buffer, once with wash buffer (10 mM Tris/HCl pH 8, 250 mM LiCl, 0.5% NP40, 0.5% sodium deoxycholate and 1 mM EDTA) and once with TE buffer. Eluates were de-crosslinked in TE and 1% SDS over night at 65°C and subsequently treated with RNase A (0.2 mg/mL) for 1 hr at 37°C and 0.1 mg protease K for 1 hr at 65°C. DNA was purified using AMPure XP Beads and eluted in 20 μL 10mM Tris/HCl pH 8.
For ChIP-qPCR, SsoAdvanced Universal SYBR Green Supermix (Bio-Rad) was used. Enrichment was calculated by normalization to the adh1+ or act1+ locus over clr4Δ, which lacks H3K9me2 and H3K9me3. Primers used in this study are listed in Table S2.
For multiplex ChIP sequencing, samples were prepared with NEBNext Ultra II DNA Library Prep Kit for Illumina (NEB) and NEBNext Multiplex Oligos for Illumina (NEB).
ChIP-seq analysis
ChIP libraries were sequenced 50bp single-end on an Illumina HiSeq 2500 instrument. Raw data were demultiplexed and converted to fastq format using bcl2fastq2 v1.17 and mapped using STAR with the following parameters: –alignIntronMax 1–alignEndsType EndToEnd–outFilterType Normal–seedSearchStartLmax 30–outFilterMultimapNmax 10000–outSAMattributes NH HI NM MD AS nM–outMultimapperOrder Random–outSAMmultNmax 1–outSAMunmapped Within. For bigwig track generation by bedtools (version 2.26.0) and bedGraphToBigWig (from UCSC binary utilities), non-aligned reads and muli-mapping reads were discarded and read coverage was normalized to 1 million genome mapping reads (RPM).
H3K36me3 and RNA Pol II quantification was performed as follows: Overlapping genes, genes smaller than 1 kb and genes having another gene closer than 10 bp were excluded to avoid wrong assignment of reads. Then analysis was restricted to expressed genes only (i.e., genes with an RPM > 15), which yielded a final list of 1860 non-overlapping and expressed genes that could be unambiguously quantified. For H3K36me3, RPM values per gene were displayed as a scatterplot. For RNA polymerase II, we quantified the relocalization of Pol II relative to the gene ends. This was done by calculating the ratio of reads mapping to the first 500bp of every gene divided by the number of reads mapping to the last 500bp of the same gene. The log2 values of this ratio (log2(read count5′end / read count 3′end)) were then plotted as scatterplot. Due to the insertion of a resistance cassette after the paf1-Q264Stop allele, we removed this gene from the analysis as the modification of the genomic locus gave rise to ChIP and mapping artifacts that precluded quantification.
Quantification and Statistical Analysis
The number of n biological replicates is given in each figure legend. p values were calculated using two-tailed Student’s t tests with two-sample assuming unequal variances. The error bars in the plots denote SD, the center values denote the mean. No data or subjects were excluded.
Data and Software Availability
Genome-wide datasets are deposited at GEO under the accession number GEO: GSE120352 or were previously described in Kowalik et al., 2015 (GEO: GSE59170). All custom codes used to analyze data and generate figures are available upon reasonable request.
Acknowledgments
We thank members of the Bühler lab for discussions, L. Kaaji for comments on the manuscript, and H. Pickersgill (Life Science Editors) for editing services. We thank N. Laschet for technical assistance and K. Hirschfeld and S. Smallwood for library construction and next-generation sequencing. This work has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (grant agreement no. 681213 - REpiReg) and the Novartis Research Foundation.
Author Contributions
L.D. and M.B. conceived ideas, designed and performed experiments, analyzed data, generated yeast strains, prepared figures, and wrote the manuscript. Y.S., S.L., A.A., and D.O. performed experiments and generated strains. F.M. and L.D. performed computational analyses. M.B. supervised the study and secured funding. All authors discussed results and commented on the manuscript.
Declaration of Interests
The Friedrich Miescher Institute for Biomedical Research (FMI) receives significant financial contributions from the Novartis Research Foundation.
Published: March 18, 2019
Footnotes
Supplemental Information can be found with this article online at https://doi.org/10.1016/j.molcel.2019.02.009.
Supplemental Information
References
- Ashe A., Sapetschnig A., Weick E.-M., Mitchell J., Bagijn M.P., Cording A.C., Doebley A.-L., Goldstein L.D., Lehrbach N.J., Le Pen J. piRNAs can trigger a multigenerational epigenetic memory in the germline of C. elegans. Cell. 2012;150:88–99. doi: 10.1016/j.cell.2012.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bähler J., Wu J.Q., Longtine M.S., Shah N.G., McKenzie A., 3rd, Steever A.B., Wach A., Philippsen P., Pringle J.R. Heterologous modules for efficient and versatile PCR-based gene targeting in Schizosaccharomyces pombe. Yeast. 1998;14:943–951. doi: 10.1002/(SICI)1097-0061(199807)14:10<943::AID-YEA292>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Baulcombe D.C., Dean C. Epigenetic regulation in plant responses to the environment. Cold Spring Harb. Perspect. Biol. 2014;6:a019471. doi: 10.1101/cshperspect.a019471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler V.L. Paramutation: from maize to mice. Cell. 2007;128:641–645. doi: 10.1016/j.cell.2007.02.007. [DOI] [PubMed] [Google Scholar]
- Chandler V.L. Paramutation’s properties and puzzles. Science. 2010;330:628–629. doi: 10.1126/science.1191044. [DOI] [PubMed] [Google Scholar]
- Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erhard K.F., Jr., Hollick J.B. Paramutation: a process for acquiring trans-generational regulatory states. Curr. Opin. Plant Biol. 2011;14:210–216. doi: 10.1016/j.pbi.2011.02.005. [DOI] [PubMed] [Google Scholar]
- Flury V., Georgescu P.R., Iesmantavicius V., Shimada Y., Kuzdere T., Braun S., Bühler M. The histone acetyltransferase Mst2 protects active chromatin from epigenetic silencing by acetylating the ubiquitin ligase Brl1. Mol. Cell. 2017;67:294–307.e9. doi: 10.1016/j.molcel.2017.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsburg S.L., Rhind N. Basic methods for fission yeast. Yeast. 2006;23:173–183. doi: 10.1002/yea.1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaidatzis D., Lerch A., Hahne F., Stadler M.B. QuasR: quantification and annotation of short reads in R. Bioinformatics. 2015;31:1130–1132. doi: 10.1093/bioinformatics/btu781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grentzinger T., Armenise C., Brun C., Mugat B., Serrano V., Pelisson A., Chambeyron S. piRNA-mediated transgenerational inheritance of an acquired trait. Genome Res. 2012;22:1877–1888. doi: 10.1101/gr.136614.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heard E., Martienssen R.A. Transgenerational epigenetic inheritance: myths and mechanisms. Cell. 2014;157:95–109. doi: 10.1016/j.cell.2014.02.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki M., Paszkowski J. Epigenetic memory in plants. EMBO J. 2014;33:1987–1998. doi: 10.15252/embj.201488883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jih G., Iglesias N., Currie M.A., Bhanu N.V., Paulo J.A., Gygi S.P., Garcia B.A., Moazed D. Unique roles for histone H3K9me states in RNAi and heritable silencing of transcription. Nature. 2017;547:463–467. doi: 10.1038/nature23267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent W.J., Zweig A.S., Barber G., Hinrichs A.S., Karolchik D. BigWig and BigBed: enabling browsing of large distributed datasets. Bioinformatics. 2010;26:2204–2207. doi: 10.1093/bioinformatics/btq351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klosin A., Lehner B. Mechanisms, timescales and principles of trans-generational epigenetic inheritance in animals. Curr. Opin. Genet. Dev. 2016;36:41–49. doi: 10.1016/j.gde.2016.04.001. [DOI] [PubMed] [Google Scholar]
- Kowalik K.M., Shimada Y., Flury V., Stadler M.B., Batki J., Bühler M. The Paf1 complex represses small-RNA-mediated epigenetic gene silencing. Nature. 2015;520:248–252. doi: 10.1038/nature14337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., Trapnell C., Pop M., Salzberg S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luteijn M.J., Ketting R.F. PIWI-interacting RNAs: from generation to transgenerational epigenetics. Nat. Rev. Genet. 2013;14:523–534. doi: 10.1038/nrg3495. [DOI] [PubMed] [Google Scholar]
- Luteijn M.J., van Bergeijk P., Kaaij L.J.T., Almeida M.V., Roovers E.F., Berezikov E., Ketting R.F. Extremely stable Piwi-induced gene silencing in Caenorhabditis elegans. EMBO J. 2012;31:3422–3430. doi: 10.1038/emboj.2012.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal. 2011;17:10–12. [Google Scholar]
- Quinlan A.R. BEDTools: the swiss-army tool for genome feature analysis. Curr. Protoc. Bioinformatics. 2014;47:11.12.1–11.12.34. doi: 10.1002/0471250953.bi1112s47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rechavi O., Lev I. Principles of transgenerational small RNA inheritance in Caenorhabditis elegans. Curr. Biol. 2017;27:R720–R730. doi: 10.1016/j.cub.2017.05.043. [DOI] [PubMed] [Google Scholar]
- Schalch T., Job G., Noffsinger V.J., Shanker S., Kuscu C., Joshua-Tor L., Partridge J.F. High-affinity binding of Chp1 chromodomain to K9 methylated histone H3 is required to establish centromeric heterochromatin. Mol. Cell. 2009;34:36–46. doi: 10.1016/j.molcel.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirayama M., Seth M., Lee H.-C., Gu W., Ishidate T., Conte D., Jr., Mello C.C. piRNAs initiate an epigenetic memory of nonself RNA in the C. elegans germline. Cell. 2012;150:65–77. doi: 10.1016/j.cell.2012.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Team, R.C. (2014). R: a language and environment for statistical computing. http://www.R-project.org/
- Van Oss S.B., Cucinotta C.E., Arndt K.M. Emerging insights into the roles of the Paf1 complex in gene regulation. Trends Biochem. Sci. 2017;42:788–798. doi: 10.1016/j.tibs.2017.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdel A., Jia S., Gerber S., Sugiyama T., Gygi S., Grewal S.I.S., Moazed D. RNAi-mediated targeting of heterochromatin by the RITS complex. Science. 2004;303:672–676. doi: 10.1126/science.1093686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos S.M., Farnung L., Boehning M., Wigge C., Linden A., Urlaub H., Cramer P. Structure of activated transcription complex Pol II-DSIF-PAF-SPT6. Nature. 2018;560:607–612. doi: 10.1038/s41586-018-0440-4. [DOI] [PubMed] [Google Scholar]
- Yu R., Wang X., Moazed D. Epigenetic inheritance mediated by coupling of RNAi and histone H3K9 methylation. Nature. 2018;558:615–619. doi: 10.1038/s41586-018-0239-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Genome-wide datasets are deposited at GEO under the accession number GEO: GSE120352 or were previously described in Kowalik et al., 2015 (GEO: GSE59170). All custom codes used to analyze data and generate figures are available upon reasonable request.