

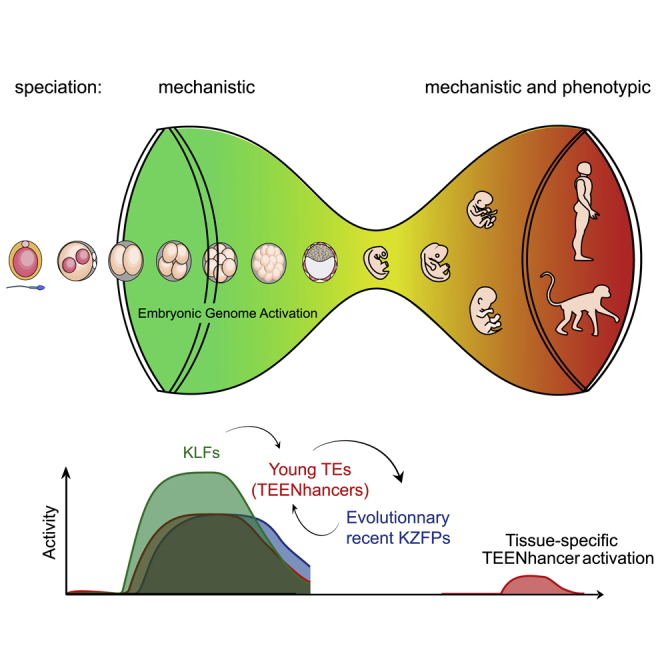
Summary
Expansion of transposable elements (TEs) coincides with evolutionary shifts in gene expression. TEs frequently harbor binding sites for transcriptional regulators, thus enabling coordinated genome-wide activation of species- and context-specific gene expression programs, but such regulation must be balanced against their genotoxic potential. Here, we show that Krüppel-associated box (KRAB)-containing zinc finger proteins (KZFPs) control the timely and pleiotropic activation of TE-derived transcriptional cis regulators during early embryogenesis. Evolutionarily recent SVA, HERVK, and HERVH TE subgroups contribute significantly to chromatin opening during human embryonic genome activation and are KLF-stimulated enhancers in naive human embryonic stem cells (hESCs). KZFPs of corresponding evolutionary ages are simultaneously induced and repress the transcriptional activity of these TEs. Finally, the same KZFP-controlled TE-based enhancers later serve as developmental and tissue-specific enhancers. Thus, by controlling the transcriptional impact of TEs during embryogenesis, KZFPs facilitate their genome-wide incorporation into transcriptional networks, thereby contributing to human genome regulation.
Keywords: Transposable elements, SVA, HERVK, HERVH, KRAB-zinc finger proteins, Krüppel-like factors, embryonic genome activation, morula, cis-regulatory elements, human genome evolution
Graphical Abstract
Highlights
-
•
KLFs foster EGA by activating enhancers embedded in young TEs (TEENhancers)
-
•
TEENhancers confer a degree of species specificity to early genome activation
-
•
TEENhancers stimulate the expression of KZFPs responsible for their repression
-
•
These KZFPs in turn facilitate TEENhancers’ exaptation as tissue-specific regulators
Transposable elements (TEs) are key to the evolutionary turnover of regulatory sequences but potentially toxic to the host. Trono and colleagues demonstrate that KRAB zinc-finger proteins tame the activity of TEs during human early embryogenesis, thus allowing for their genome-wide incorporation into species-specific transcriptional networks.
Introduction
In the human genome, more than 4.5 million sequences can be readily identified as derived from transposable elements (TEs), accounting for at least 50% of its DNA content. Most of these TEs are endogenous retroelements (EREs), replicating through a copy-and-paste mechanism based on reverse transcription of an RNA intermediate and integration of its DNA product into the genome, whether they are ERVs (endogenous retroviruses), LINEs (long interspersed nuclear elements), SINEs (short interspersed nuclear elements) (which include the primate-specific Alu repeats), or the Hominidae-restricted SVAs (SINE-VNTR-Alu). Long discarded as junk DNA, TEs are increasingly recognized as major motors of genome evolution. They notably act as insertional mutagens and constitute recombination hotspots, owing to their repetitive nature. Only one in about ten thousand human TEs is still capable of transposition, but waves of TE expansion have coincided with major phenotypic shifts during evolution, for instance, mammalian radiation or emergence of the primate lineage (Chalopin et al., 2015, Cordaux and Batzer, 2009).
TEs were named “controlling elements” by their discoverer Barbara McClintock, because their moves within the genome of maize correlated with phenotypic changes (McClintock, 1956). Britten and Davidson (1971) subsequently proposed that TEs contribute to the genome-wide distribution of regulatory sequences that allow a cell to respond to a single stimulus by changing the expression of many of its genes, for instance, when a signaling pathway is triggered following activation of a cell surface receptor. Modern genomics validated this model by revealing that sequences recognized by many transcription factors reside within TEs, explaining why only a minority of TF-binding regions are conserved between human and mouse, and by demonstrating that TE-embedded regulatory sequences influence gene expression by acting as promoters, enhancers, repressors, terminators, or insulators as well as through a variety of post-transcriptional effects (reviewed in Chuong et al., 2017).
Thus, TEs play a prominent role in renewing the pool of TF binding sites collectively engaged in multiple aspects of gene regulation and disseminated over extensive regions of the genome. This poses a conundrum, because in order to be inherited, transposition events must occur during early embryogenesis and in the germline. On the one hand, the widely opened chromatin state that characterizes these periods is favorable to a broad distribution of new TF-binding-sites-bearing TE insertions. On the other hand, this requires that transposition-competent TEs be activated at these stages, and it implies that transcriptionally active sequences will be newly introduced in regions of the genome where they could be profoundly disruptive, hence rapidly eliminated by negative selection.
The present work solves this conundrum by unveiling the role of KRAB (Krüppel-associated box)-containing zinc finger proteins (KZFPs) as key facilitators of the domestication of TE-embedded regulatory sequences. Encoded in the hundreds by most higher vertebrates, including humans, KZFPs are characterized by an N-terminal KRAB domain and a C-terminal array of DNA-binding zinc fingers (ZFs). The ZF regions of a majority of KZFPs recognize TEs in a sequence-specific manner, and their KRAB domain can recruit KAP1 (KRAB-associated protein 1) (also known as TRIM28 or tripartite motif protein 28), which serves as a scaffold for a heterochromatin-inducing machinery comprising the histone methyltransferase SETDB1, the histone-deacetylase-containing NurD complex, heterochromatin protein 1 (HP1), and DNA methyltransferases (Ecco et al., 2017). Correspondingly, the KZFP/KAP1 system represses many TEs expressed in mouse, human embryonic stem cells (ESCs), and early embryo (Yang et al., 2017, Guo et al., 2017, Theunissen et al., 2016, Wolf et al., 2015, Göke et al., 2015, Guo et al., 2014, Smith et al., 2014, Turelli et al., 2014, Castro-Diaz et al., 2014, Matsui et al., 2010, Rowe et al., 2010, Wolf and Goff, 2009). This was initially interpreted as primarily responsible for preventing the spread of TEs, and rare TE/KZFPs pairs indeed display signs of mutational escape supporting such an arms race mode (Jacobs et al., 2014). However, a recent characterization of human KZFPs indicated that these proteins partner up with their targets to establish largely species-specific transcriptional networks (Imbeault et al., 2017), suggesting that KZFPs promote the domestication of TEs. Here, we validate this hypothesis by revealing that young TE-based enhancers broadly induced during human embryonic genome activation (EGA) are rapidly tamed by KZFPs of approximately similar evolutionary ages before serving later as lineage- or tissue-specific regulators of gene expression. Thus, rather than primarily involved in limiting the spread of TEs, KZFPs act as tolerogenic agents that facilitate the genome-wide exaptation and pleiotropic engagement of TE-based regulatory sequences, thus playing a critical role in the evolutionary turnover of transcriptional networks.
Results
Evolutionarily Recent TEs Are Activated during Human EGA and in Naive Human ESCs
Upon re-analyzing chromatin accessibility and single-cell transcriptome data from human pre-implantation embryos (Gao et al., 2018, Yan et al., 2013), we found first that at least one-third of the genomic sites opened during this period were embedded in TEs (Figure S1A) and second that the expression of these TEs increased between 4-cell (4C) and morula stages to drop in blastocyst and be similarly low in embryo-derived pluripotent stem cells (Figure S1B). Most of these TEs belonged to primate- and notably Hominoidea (ape)-restricted families, including many human-specific integrants from the LTR5Hs/HERVK, LTR7/HERVH, and SVA subgroups (Figures 1A and S1A–S1D). We further noted that these TE integrants tended to be close to genes also transcribed during EGA (Figure 1B).
Figure 1.

Evolutionarily Recent TEs Are Activated during Human EGA and in Naive hESCs
(A) Age-related chromatin accessibility of TE loci in human embryo (inferred from DNase-seq data re-analyzed from Gao et al., 2018) stratified according to the age of sequences obtained through coordinates conversion of human sequences using liftOver (indicated species are the farthest with an orthologous sequence). Red dots represent percent of observed accessible TE loci. Black dots are similarly analyzed from 100 random shuffling of all accessibility sites. p values (top) are represented in −log10(pval).
(B) Proximity between accessible human-specific TEs and EGA-induced genes. Volcano plot where, for each TE subfamily, the distance of its accessible integrants in human embryo to EGA genes is compared to the distance to all other genes is shown. Fold change is computed from the median distance to EGA genes versus the median distance to other genes (y axis). p values were computed with a Wilcoxon test (x axis) and adjusted for multiple testing with the Benjamini and Hochberg method. Single-cell RNA-seq was from Yan et al. (2013). Fraction of accessible TE integrants for each subfamily is indicated by dot size.
(C) Comparative chromatin accessibility of naive and primed hESCs with early human embryos. ATAC-seq was performed on naive and primed hESCs. Loci more accessible in either setting (2-fold differences; p < 0.05) were intersected with similar data from pre-implantation embryo (black dots, re-analyzing data from Gao et al., 2018) and roadmap DNase-seq (red dots for the placental tissues and gray dots for other tissues). Numbers of common accessible loci were plotted (log2).
(D) Comparative chromatin status of naive and primed hESCs. Merged ATAC-seq peaks from naive and primed hESCs were used as a reference to plot ATAC-seq status of these loci in 4C and morula (re-analyzing data from Gao et al., 2018) and their H3K27ac enrichment in naive or primed hESCs. Loci were ordered from top to bottom based on their enrichment in chromatin accessibility in naive compared to primed hESCs.
(E) H3K27ac status of age-stratified human TEs in naive compared to primed hESCs using subfamily add-up of normalized read counts. ∗∗∗p ≤ 0.001 for the comparisons of each age category being different than 0 using t test. Green dots represent LTR7/HERVH integrants, with C, B, and Y indicating the corresponding LTR7 subclasses, with and without their internal part. Cyan and orange dots represent LTR5Hs/HERVK and SVA subfamilies, respectively, with A, B, C, D, E, and F designating SVA subclasses.
(F) Young TEs of the LTR7/HERVH, LTR5/HERVK, and SVA subgroups are enriched in naive-specific super-enhancers. Relative representation of naive versus primed TE-based enhancers in super-enhancers is shown (defined as in Ji et al., 2016, corresponding to a merged list of large and distal H3K27ac regions). y axis represents Log2 fold enrichment (compared to random) differences for a specific TE subfamily between naive and primed super-enhancers; x axis represents integrants number of a TE subfamily belonging to naive and primed specific super-enhancers.
See also Figure S1.
To ask how the epigenetic state of these TEs might impact on human early development, we took advantage of embryo-derived human ESCs (hESCs). In their original primed state, these cells roughly correspond to the post-implantation epiblast, and they can be converted to a more naive state by overexpression of the KLF2 and NANOG transcription factors (KN) and/or by exposure to an inhibitory cocktail (KN+2i/L or 4-5i/LA; Takashima et al., 2014, Theunissen et al., 2014). Based on their transcriptome and on their chromatin status, characterized by assay for transposase-accessible chromatin with highthroughput sequencing (ATAC-seq) as a corollary for transcription factor (TF) accessibility of the underlying DNA, we determined that naive hESCs closely resemble pre-implantation embryo (Figures 1C, 1D, and S1E). We then profiled histone acetylation (by deep DNA sequencing of chromatin immunoprecipitated with an antibody specific for histone 3 acetylated on lysine 27 [H3K27ac ChIP-seq]) in naive and primed hESCs to map regulatory elements active in either setting and could correlate H3K27ac levels with naive-specific accessible genomic loci (Figure 1D), including many TE integrants of the SVA, LTR5Hs-HERVK, and LTR7-HERVH subfamilies, level of which decreased in primed cells (Figures 1E and S1F). We additionally observed that several SVAs and ERV long terminal repeats (LTRs) provided transcription start sites (TSSs) for coding genes or long non-coding RNAs (lncRNAs), although some intronic SVAs were sites of alternative splicing (Figure S1H). However, far more frequent were the hundreds of LTR5Hs, LTR7Y/B, and SVA loci strongly marked by H3K27ac without direct link to gene transcripts (Figure S1I). This suggested that these elements functioned as enhancers, a view further supported by their frequent clustering in regions previously defined as super-enhancers in naive hESCs (Figure 1F).
Krüppel-like Factors Are Major Early Embryonic Activators of the Human Genome
A search for transcription factor motifs in regions with naive-specific DNA accessibility revealed enrichment in binding sites for the pluripotency-associated KLF family members and the trophectoderm-associated factor AP-2 (TFAP2) (Figure S1G). This was consistent with a dual potential for these cells toward both embryonic and extra-embryonic differentiation and with the recent finding that TFAP2C participates in opening enhancers in this setting (Pastor et al., 2018). KLF4 and its homolog KLF17 stood out among 30 genes, the levels of which were at least 50-fold higher in morula and naive ESCs, compared to, respectively, 4C embryos and primed ESCs (Figure 2A; Table S1). Interestingly, hKLF17 was recently found capable of rescuing KLF2/4/5 triple knockout (KO) mouse ESCs (Yamane et al., 2018). ChIP-seq analyses in naive hESCs with an antibody against endogenous KLF4 further revealed that this factor was enriched at numerous pre-implantation and naive-specific accessible sites also adorned with H3K27ac (Figure S2A). KLF4 was notably associated with LTR7/HERVH, LTR5Hs/HERVK, and SVA, the old world monkey-, ape-, and human-specific TEs active in this setting (Figure S2A), as well as with some young LINEs from the L1Hs, L1PA2, and L1PA3 subgroups (data not shown). OCT4 was highly expressed in both naive and primed hESCs (Table S1), but it was bound to pre-implantation and naive-specific opened chromatin loci only in naive cells, suggesting that its recruitment to these sites required KLF4 (Figure S2A). This hypothesis was confirmed in the setting of reprogramming experiments of skin fibroblasts, where OCT4 bound these sequences only when KLF4 was also expressed, as well as in primed hESCs overexpressing KLF4 or KLF17 (Figures S2A and S2B). In this latter setting, H3K27ac deposition was further induced over a similar set of genomic sites (Figure S2C) that partly recapitulated the patterns observed in naive hESCs (Figures 2B and S2A) with hundreds of TSS, many naive-specific, and thousands of TEs, most belonging to the HERVH, SVA, LTR5Hs/HERVK, and L1Hs subfamilies (Figures 2C and S2D). HERVH and HERVK transcription was stimulated in this setting, but SVA transcripts remained low, possibly due to countering influences guarding their promoter from the influence of the enhancer located at their 3′ end (Figures 2C and 2D). We could verify that the KLF4-binding sequence present in LTR5Hs and SVA conferred KLF4 and KLF17 responsiveness to a GFP reporter system, as did a dCAS9 activator fusion protein (CRISPRa) targeted to this DNA sequence (Figure 2D). Finally, we could document the activation of genes situated in the vicinity of both activated HERVs and SVAs in primed hESCs overexpressing KLFs (Figure 2E). We conclude that KLF4 and KLF17 act as main drivers of the human pre-implantation transcription program notably by activating young transposable element-based enhancers.
Figure 2.

Krüppel-like Factors Are Major Inducers of EGA and Naive-Specific TE Enhancers
(A) KLF4 and KLF17 expression during early human embryonic development and stem cells, determined by re-analyzing single-cell RNA-seq data from Yan et al. (2013). Single-cell data points are grouped according to developmental stage: oocytes (Oo); zygotes (Zy); 2-cell (2C); 4-cell (4C); 8-cell (8C); morula (Mo); with late blastocyst split into epiblast (Epi), primitive endoderm (PE), and trophectoderm (TE); p0 and p10 representing passage numbers of ESCs derived from blastocyst and naive (N) and primed (P) hESCs (Theunissen et al., 2016).
(B) Pairwise comparison of KLF4/17-induced acetylation loci. H3K27ac ChIP was performed 5 days after transducing primed hESCs with GFP-, KLF4-, or KLF17-expressing lentiviral vectors, thus identifying H3K27ac loci with increased signal (n = 4,446; adjusted p value < 0.05); pie charts represent its proportion (black part; in % of the 4,446 loci) overlapping with chromatin accessibility in hESCs (determined by ATAC-seq, with naive versus primed specific loci defined by ≥2-fold difference with adjusted p < 0.05), human embryo DNase-seq, H3K27ac (K27ac), and OCT4- or KLF4-enrichment (p < 10e−5) in indicated cells. Color scale corresponds to Spearman correlation of pairwise loci comparisons.
(C) Genomic distribution of increased H3K27ac loci upon KLF4/17 overexpression in primed ESCs. (Left) Distribution between TSS (±500 bp) of coding genes and the non-TSS overlapping loci is shown: TEs (50% overlap from either TE or H3K27ac peak) or other regions; p values were computed with a permutation test (1,000 permutations); (right) distribution within indicated TE subfamilies with (blue) and without (gray) gain of H3K27ac enrichment is shown, further depicting loci with (light blue) or without (dark blue) increase expression in GFP versus both KLF4 and KLF17 (adjusted p value < 0.05). Numbers of integrants are indicated for each category.
(D) KLF4 binding on LTR5Hs and HERVK (SINE-R) region of SVA. KLF4 ChIP-seq signal in naive hESCs was superimposed on multiple sequence alignment of corresponding TEs. The rectangle highlights the common enhancer piece bound by KLF4, the one which was cloned from a SVA into a GFP vector activatable by CRISPRa, but not by KLF4 or KLF17 when KLF-motif is mutated. Error bars were established using SEM and p value using t test (∗∗∗ ≤ 0.001). See STAR Methods for details.
(E) KLF4-KLF17 overexpression in primed ESCs activates TE-close genes. Proportion of up- and downregulated genes upon KLF4-KLF17 overexpression is shown (y axes; p < 0.05), according to their distance to the closest TE (x axes). Upper and lower panels use TEs (SVA, LTR5Hs, and LTR7) with or without H3K27ac gains upon KLF4-KLF17 overexpression, respectively.
KLF-Activated, Young TE-Based Enhancers Regulate Naive hESC Transcription Networks
To test the functional impact of TE loci active in naive hESCs, we targeted these integrants with a dCAS9-KRAB fusion protein (CRISPRi), which can instate the repressive mark H3K9me3, hence, inactivate enhancers (Thakore et al., 2015). We established stable naive hESCs expressing CRISPRi together with guide RNAs (gRNAs) specific for either a sequence common to LTR5Hs and SVA or one found in LTR7B and LTR7Y, using in each case two gRNAs, each predicted to recognize a majority of the corresponding integrants (Figures 3A and S3A). We could document the loss of chromatin accessibility and the deposition of H3K9me3 at targeted loci, but not at TEs displaying more than one mismatch with the gRNAs (Figure S3B). Transcription from LTR5Hs-SVA integrants was decreased in cells transduced with CRISPRi and the corresponding gRNAs (Figure 3B), and a majority of genes located in the nearby vicinity (<100 kb) were secondarily repressed (Figure 3C) without significant increase in H3K9me3 or decrease in chromatin accessibility at their transcription start sites (Figure S3C). Interestingly, LTR7/HERVH integrants, which are typically transcribed in primed hESCs (Theunissen et al., 2016), were upregulated in this setting (Figure 3B). With the LTR7YB-specific gRNAs, changes were more global, with not only LTR7/HERVH but also LTR5Hs and SVA loci downregulated, and many genes were up- or downregulated irrespective of their proximity to LTR7 integrants (Figures S3D and S3E). This might be due either to the deregulation of genes affecting the general transcriptional program of the cells or to trans-acting influences of HERVH-derived lncRNAs as previously suggested (Lu et al., 2014). We then analyzed 3D nuclear architecture maps recently established by chromatin interaction analysis by paired-end tag sequencing (ChIA-PET) in naive and primed hESCs (Ji et al., 2016). This technique is based on the immunoprecipitation of a cohesin-containing complex protein (SMC1) followed by proximal DNA ligation, with sequencing of both ends of the DNA products to obtain a 3D map of DNA/DNA interactions, notably between promoters and enhancers. We first noted increased levels of reads over LTR5Hs and SVAs in naive compared to primed hESCs, suggesting higher rates of cohesin loading at these loci in the naive setting (Figure S3F). We could also document physical interactions between these TE and the promoters of genes that were downregulated when LTR5Hs and SVA were repressed (Figure S3G). For instance, there was a 40-kb distal interaction between the ST6GAL1 gene, the product of which is involved in the catalysis of the naive ESC and morula-specific glycoprotein CD75 (Collier et al., 2017) and a super-enhancer mostly composed of SVA-LTR5Hs (Figure 3D). Other genes involved in such interactions included PRODH, a neuron-specific gene that harbors an LTR5Hs-based enhancer 2 kb upstream of its promoter (Suntsova et al., 2013) as well as genes previously linked to hESC pluripotency, such as ZFP42 and C9ORF135, which encode, respectively, a naive-specific transcription factor and a pluripotency-linked membrane protein (Zhou et al., 2017). Ontology terms describing genes impacted by the LTR5Hs-SVA-targeting dCAS9-KRAB repressor and found to interact with SVA-LTR5Hs loci by ChIA-PET included transcription factors, notably KZFPs, and cellular processes likely to play important roles in early embryogenesis, such as mitochondrial functions and antiviral innate immunity (e.g., SAMHD1, which restricts Alu/LINE/SVA retro-transposition as well as exogenous viral infection) as well as WNT signaling pathway, cell cycle adhesion, and polarity (Table S2). Noteworthy, out of 275 genes recently documented as controlled by a broader set of putative LTR5-based enhancers in a human teratocarcinoma cell line (Fuentes et al., 2018), 87 were also downregulated in our CRISPRi experiment targeting LTR5Hs/SVA in naive hESCs (Figure S3H). Finally, many genes activated when KLF4-KLF17 or OKSM were overexpressed, respectively, in primed hESCs or fibroblasts were conversely downregulated when LTR5Hs-SVA-based enhancers were repressed by CRISPRi in these experimental settings (Figures S3I–S3K and 3E).
Figure 3.

TEENhancers Regulate Gene Expression in Naive hESCs
(A) Schematic representation of CRISPRi targeting of SVA-LTR5Hs common region. Venn diagram depicts the number of TEs predicted to be targeted by either gRNA, with bar plot indicating the percentage of the 2,808 commonly targeted integrants within each subfamily.
(B) Impact of CRISPRi on TEs expression. Naive hESCs were transduced with a dCAS9-KRAB lentiviral vector containing or not gRNAs against LTR5Hs-SVA (two different gRNAs each in quadruplicate). Expression of indicated TEs was compared between gRNA-expressing and control cells. One-sample t tests were computed to generate p values. ∗∗∗p ≤ 0.001.
(C) Impact of TE-targeting CRISPRi on gene expression. Number of up- and downregulated genes (p < 0.05 and fold change >10% between paired replicates) at indicated distance from closest CRISPRi-targeted TE is shown (in: TE within gene). (Top) Percentage of all up- or downregulated genes within 100 kb or farther away is shown.
(D) Long-range regulation of a gene by an early embryonic super-TEENhancer. (Top) H3K27ac, ATAC-seq, and ChIA-PET (Ji et al., 2016) profiles in naive and primed hESCs are shown. (Middle) RNA-seq in naive cells sorted for GFP-naive reporter in dCAS9-KRAB-transduced naive hESCs expressing (+) or not (−) the LTR5HS-SVA-targeting gRNA. (Bottom) Alignment of genomes of indicated species at homologous locus, revealing the human specificity of the TEENhancers. Then, TEs 1 through 6 (3 LTR5Hs and 3 SVAs) predicted to bind the gRNA are shaded in red.
(E) CRISPRi inhibits SVA-LTR5Hs-controlled gene activation during somatic reprogramming. Human primary fibroblasts containing dCAS9-KRAB with or without gRNA against LTR5Hs-SVA (gRNA+/−) were transduced or not (OSKM+/−) with OSKM-expressing Sendai virus for 6 days before measuring transcripts of HERVH, HERVK, and several genes found to interact with SVA-LTR5Hs by ChIA-PET and downregulated by CRISPRi in naive cells (normalized to RPLP0). Error bars represent SEM while the p value was established with a t test (∗∗∗ ≤ 0.001, ∗∗ ≤ 0.01, ∗ ≤ 0.01, and n.s. > 0.05).
In sum, these data reveal that recent TE colonizers of the human ancestral genome markedly influence transcription in naive hESCs and likely pre-implantation embryo, notably acting as stage-specific enhancers. To reflect their origin, young age, and transcriptional impact, we coined these elements TEENhancers.
Evolutionary Recent KZFPs Tame TEENhancers Active during Human Early Embryogenesis
We then asked whether KRAB zinc finger proteins, which are known TE repressors, were responsible for dampening the effect of TE-based enhancers activated at EGA and in naive hESCs. KZFP genes are often grouped in clusters, many on human chromosome 19, a consequence of their amplification by repeated episodes of gene and segment duplication (Huntley et al., 2006; Figures S4A and S4B). The approximate age of these genes can be assessed by examining the degree of conservation of the zinc fingerprints of their products, that is, the series of amino acids predicted to determine their DNA binding specificity (Imbeault et al., 2017, Liu et al., 2014). Applying this principle, we noticed that clusters of evolutionarily recent human KZFPs were expressed more strongly in morula than at the 4-cell stage, indicating that they were among genes induced during EGA (Figures 4A, S4A, and S4B) and targeting TE subfamilies of similar ages (Figure 4B). Young KZFPs were also induced during the early phase of reprogramming of fibroblasts by OKSM expression (Figure S4C). Correspondingly, clusters containing these KZFP genes were enriched in KLF4 binding sites, many of which resided on TEs, and the forced expression of this TF in primed hESCs induced their histone acetylation and their transcription (Figures 4C and S4B). Of note, KLF4 overexpression ultimately led to H3K9me3 increase over hundreds of HERVH, HERVK, HERVL, and LINEs targeted by these KLF-activated KZFPs (as exemplified in Figure S4D).
Figure 4.

Evolutionary Recent KZFPs Tame TEENhancers during Human Early Embryogenesis
(A) Evolutionary young KZFPs are activated during EGA. Fold change expression of KZFP genes (classified by evolutionary age; Imbeault et al., 2017) between 4C and morula (data re-analyzed from Yan et al., 2013).
(B) Young KZFPs target contemporaneous TEs. Violin representation of evolutionary ages of KZFPs and their TE targets, determined by re-analyzing ChIP-exo data (Imbeault et al., 2017) and using the most significantly bound TE for each EGA-induced KZFP. TEs were classified by evolutionary age established through lineages comparison.
(C) KLF4 induces histone acetylation within evolutionary young KZFP clusters. (Top) Schematic representation of human chromosome 19 with KZFP gene clusters, individual units, and TEs distribution below are shown. (Bottom) Highlight of the H3K27ac profile of two regions in naive or primed hESCs with or without overexpression of KLF4 is shown. Underlying genetic entities are indicated by colored boxes.
(D) UpSet plot showing impact of ZNF611 overexpression in naive hESCs. Black dots crossed by black lines are the different combinations of intersections and are mutually exclusive of each other. Intersection combinations were plotted as bar chart representing the number of loci with increased H3K9me3 signal in naive hESCs overexpressing ZNF611 compared to GFP, split into subcategories according to whether they were known ZNF611 binding sites in 293T cells (ZNF611), SVAs (SVA), H3K27ac-depleted (H3K27ac ↓), expressed SVAs (exp SVA), and downregulated SVAs (exp SVA ↓). Heatmap illustrates H3K9me3 raw signal ±5 kb around all SVAs in naive hESCs overexpressing ZNF611 or GFP.
(E) Impact of activation domain fused to ZNF611 zinc fingers in primed hESCs. The VPR activation domain (comprising the corresponding regions of VP64, P65, and Rta) was fused to the zinc finger domain of ZNF611 and overexpressed in primed hESCs. Bars represent qRT-PCR expression levels (normalized to GAPDH). Error bars were established using SEM and p value using t test (∗∗∗ ≤ 0.001; ∗∗ ≤ 0.01; ∗ ≤ 0.05). Represented genes were selected on the following criteria: downregulated by ZNF611 overexpression in naive hESC; devoid of SVA inside of their gene body; and having a SVA enhancer within 3–30 kb of their TSS.
See also Figures S4 and S5.
Differential levels of H3K9me3 enrichment at given TE subgroups between naive and primed ESCs reflected the relative expression of their cognate KZFPs, as exemplified by the HERVH-recognizing ZNF90, ZNF257, ZNF534, and ZNF600 and by the SVA-targeting ZNF28 and ZNF611 (Figures S4B and S4E). Interestingly, the predictably low production of ZNF611 and ZNF28 proteins in naive ESCs stemmed from alternative splicing of their primary transcripts into internal SVA and Alu sequences, respectively, which precluded translation of their ZF-coding 3′ end (Figure S4F).
We used the SVA-targeting ZNF611, which can be traced back to the last common ancestor of old world monkeys and humans, as a paradigm to explore more thoroughly the impact of KZFPs on the activation of EGA- and naive hESC-specific genes by TEENhancers. We found that the forced expression of ZNF611 in naive hESCs resulted in a gain of H3K9me3 and a loss of H3K27ac over hundreds of SVAs (Figure 4D). This resulted in reduced expression not only of these TEs but also of several SVA-driven transpochimeric transcripts (fusions between TE- and gene-derived RNAs; Figure S5A) and more importantly of hundreds of SVA-close genes previously found to be repressed by the SVA-targeting CRISPRi system (Figures S5B and S5C). Most of these ZNF611-repressed genes did not exhibit significant changes in chromatin marks at their TSS (Figure S5D), indicating that the KZFP primarily acted by blocking their SVA-based enhancers. Conversely, expressing in primed hESCs a fusion protein between the VP64-p65-Rta (VPR) activator domain and the ZNF611 poly-ZF sequence induced the expression of several genes controlled by ZNF611-targeted enhancers in their naive counterparts (Figure 4E).
KZFP-Controlled TEENhancers Confer Species Specificity to Human Early Embryonic Transcription
The hundreds of genes downregulated in naive hESCs by both the SVA-targeting CRISPRi system and ZNF611 overexpression were genes induced during human EGA and more highly expressed in morula and naive ESCs than in their primed counterparts (Figure 5A), whereas the reverse trend was observed for genes anti-correlating SVA activation (Figure S5E). In addition, many genes downregulated by ZNF611 displayed relative RNA levels that were higher in human than in macaque morula, consistent with a model whereby they were under the influence of species-restricted TE-based enhancers active during human EGA. In contrast, these inter-species differences were absent in primed ESCs, where human TEENhancers were largely repressed (Figure 5B). Reciprocally, macaque-restricted HERVKs (LTR5RM/HERVK) were expressed during macaque EGA (Figure S5F), and genes close to these elements were relatively more expressed in macaque than in human EGA (Figure 5C). Together, these results demonstrate that TE-based regulatory sequences exert species-specific transcriptional influences detectable during the earliest phase of embryogenesis.
Figure 5.

KZFPs and TEENhancers Engineer Species-Specific Early Embryonic Regulatory Networks
(A) Relative expression of SVA-enhanced genes (common repressed by SVA-targeting CRISPRi and ZNF611 overexpression in naive hESCs) in morula versus 4C (EGA genes) and morula or naive hESCs or primordial germ cells versus primed hESCs (partly by re-analyzing data from Tang et al., 2015, Theunissen et al., 2016, and Yan et al., 2013). p values were established by two-sample t test.
(B) Comparative expression of same human SVA-enhanced genes in human versus macaque EGA (morula versus 4C) and primed ESCs (re-analyzing data from Fang et al., 2014, Theunissen et al., 2016, Wang et al., 2017, and Yan et al., 2013). Two-sample t tests were computed to generate p values.
(C) Relative expression of orthologous genes situated within 20 kb of macaque-restricted LTR5RM/ERVK or human-restricted LTR5Hs/HERVK during macaque versus human EGA. One-sample t tests were computed to generate p values (re-analyzing data from Wang et al., 2017 and Yan et al., 2013).
See also Figure S5.
KZFP-Controlled TEENhancers Regulate Transcription in Developing and Adult Tissues
A recent study of human primordial germ cells (hPGCs) revealed the co-expression of KLF4 and a number of HERVK and SVA loci (Tang et al., 2015). Upon re-analyzing these data, we noted that this correlated with a higher expression of SVA-controlled genes, compared to levels recorded in primed ESCs (Figure 5A). Thus, gametogenesis seems also influenced by TEENhancers active during embryonic genome activation. We further noticed that numerous TEENhancer-controlled genes expressed during human EGA encode for products, the function of which is relevant later in development or in adult tissues, such as GPR176, a regulator of the circadian clock, the Parkinson disease-related kinase LRRK2, or the APOE lipoprotein important for liver and brain function. We thus asked whether TE-based regulatory sequences responsible for fostering EGA were also active at later stages. Upon scrutinizing SVA-based TEENhancers activated during EGA and in naive hESCs and repressed in epiblast and primed hESCs, we found that their sequences re-acquired H3K27ac activation marks in neurons differentiated from induced pluripotent stem cells, as well as in fetal and adult brain and liver (Figure 6A). Furthermore, we found that, although SVAs represent only one-thousandth of the human genome TE load, they constituted up to 17% of TE sequences detected as carrying active enhancer marks in fetal brain (Figure 6B). Finally, targeting these SVAs in induced pluripotent stem cell (iPSC)-derived neurons by CRISPRi led to downregulation of genes similarly repressed by this system in naive hESCs (Figure 6C). Thus, the exaptation of evolutionary recent TEs broadly disseminated in the human genome not only promotes EGA but also shapes transcriptional networks active later in development and in adult tissues.
Figure 6.

KZFP-Controlled Early Embryonic TEENhancers Could Act as Developmental and Tissue-Specific Regulators
(A) Histone methylation and acetylation profiles of TEENhancers in naive or primed hESCs and indicated tissues. Histone ChIP-seq raw signals (H3K9me3 and H3K27ac) at TEENhancers (SVA) obtained in naive, primed hESCs and IPS-induced neurons (i.neu.; this study) were superimposed to those described in brain and fetal liver (Yan et al., 2016); adult brain, including hypothalamus (hypo), pineal gland (pineal), and supramarginal gyrus (gyrus) (Vermunt et al., 2014); and adult liver (Trizzino et al., 2017).
(B) SVAs are overrepresented among TE-based enhancers in fetal brain. H3K27ac-bearing loci distribution between TSS (±500 bp) of coding genes and the non-TSS overlapping loci are shown: TEs (50% overlap from either TE or H3K27ac peak) or other regions. p value were computed for SVA with a permutation test (1,000 permutations; ∗∗∗ < 0.001).
(C) SVA-enhanced genes in naive hESCs are similarly controlled in neurons. Boxplot represents log2 fold change upon SVA-targeting CRISPRi expression in neurons obtained by iPSC differentiation, of downregulated genes in naive hESCs upon CRISPRi and ZNF611 overexpression (left), or other genes (right). p value was generated by two-sample t test.
Discussion
We found the chromatin of naive hESCs to be characterized by its high degree of accessibility and histone acetylation and further determined that this property stemmed largely from the activation of young TE loci also induced during embryonic genome activation. We further determined that members of the KLF family of transcription factors, notably KLF4 and KLF17, play a major role in this process, as a large fraction of naive-specific accessible chromatin domains harbor binding sites for these proteins, which can activate numerous HERVH and HERVK integrants, together with hundreds of the corresponding solo-LTRs (LTR7B/Y and LTR5Hs) and with SVAs, for the latter through their LTR5Hs-homologous SINE-R region. KLF4 was previously noted to stimulate LTR7/HERVH transcription during the forced re-programming of adult cells into iPSCs (Friedli et al., 2014, Ohnuki et al., 2014) and OCT4 to activate LTR7/HERVH and LTR5Hs/HERVK in early human embryos and hESCs (Grow et al., 2015, Lu et al., 2014). Here, we further defined that KLF4 and its functional homolog KLF17 are likely responsible for opening thousands of genomic loci during EGA, including many morula- and naive hESC-active TEs from the HERVH (LTR7B/Y), LTR5Hs/HERVK, and SVA subgroups. Most EGA and naive hESC-activated, TE-derived sequences contain binding sites for both KLF4 and OCT4, but we observed that the former is required for many of these targets to recruit the latter. KLFs are also involved in activating KZFPs that go on to repress TEs active during this period, as EGA and naive hESC-activated KZFP gene clusters harbor numerous KLF-responsive TEENhancers, the activity of which they ultimately repress. Reciprocally, activation of some human SVA inserts results in modifying the splicing pattern of the underlying genes, some of which code for their controlling KZFPs, constituting another feedback loop between KZFP repressors and their TE targets. A large majority of TE-derived sequences activated during human EGA and in naive hESCs behave as enhancers, even forming so-called super-enhancers. They rarely serve as promoters, in contrast with the mouse, where LTRs of ERVs, such as mouse endogenous retrovirus L (MERVL), drive a number of gene transcripts produced at the 2-cell stage, when EGA takes place in this species (De Iaco et al., 2017, Macfarlan et al., 2012). Some of the genes activated under the influence of these TEENhancers encode for activities protecting the nascent human embryo against invasion by both endogenous transposons and exogenous viruses. These include the HERVK-encoded Rec protein (Grow et al., 2015) and SAMHD1 (sterile alpha motif and histidine-aspartate domain-containing protein 1), which we found here to be controlled by a SVA-based enhancer in naive hESCs. As an inhibitor of a broad range of retroelements (Zhao et al., 2013), SAMHD1 likely is an important guardian of genome integrity during early embryogenesis.
Inheritable transposition events occur during early embryogenesis and in the germline, when chromatin is broadly opened and the genomic DNA widely accessible to the preintegration complexes of TEs. Accordingly, new TE integrants can insert and contribute to renewing the pool of TF binding sites over broad regions of the genome. The model commonly held so far was that, if these new TEs landed in places where they had a detrimental impact, the concerned individuals were rapidly eliminated by negative selection. Although this remains a generally valid model, our demonstration that KZFPs co-evolve with TEs to tame their transcriptional impact in early embryogenesis implies that a far greater proportion of TE-derived regulatory sequences can be co-opted, because their utility or toxicity for the host is no longer determined by their sole genomic location and immediate effect. In essence, rather than just limiting the spread of TEs, KZFPs increase their genomic tolerability, thus facilitating a genome-wide, TE-mediated turnover of regulatory sequences with pleiotropic functions.
A corollary of the contribution of the KZFP-TE system to the dissemination of regulatory sequences is the high degree of species specificity that it confers to transcriptional networks. For instance, the overall similarity of human and rhesus macaque EGAs contrasts with the striking divergence of their cis-acting TE and trans-acting KZFP regulators. The same breadth of genome activation is observed in both cases, but species-specific differences are seen in the relative expression of some genes, which coincide with the genomic location of equally species-restricted TE enhancers recognized by non-orthologous sets of KZFPs. That such a critical developmental step is so differentially controlled in two primates whose ancestors diverged less than 30 million years ago illustrates the formidable evolutionary dynamism of the TE/KZFP system. Owing to the global plasticity of EGA, where probably only a subset of genes needs to be expressed with a critical degree of precision, these species-specific differences do not translate into distinguishable phenotypes at this developmental stage. However, because many of the TE enhancers activated during EGA go on to govern the expression of genes important later in development or for the physiology of adult tissues, it is likely that the TE/KZFP regulatory system significantly contributes not only to the mechanistic but also to the phenotypic speciation of all higher vertebrates, including humans.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-H3K9me3 - Rabbit Polyclonal | Diagenode | Diagenode Cat# pAb-056-050; RRID:AB_2616051 |
| anti-H3K27ac - Rabbit Polyclonal | Abcam | Abcam Cat# ab4729; RRID:AB_2118291 |
| anti-KLF4 - Goat Polyclonal Goat | R&D Systems | RRID:AB_2130224 |
| anti-OCT4 - Rabbit polyclonal | Abcam | Abcam Cat# ab19857; RRID:AB_445175) |
| Bacterial and Virus Strains | ||
| CRISPRi - pLV hU6-sgRNA hUbC-dCas9-KRAB-T2a-Puro | Addgene | #71236; RRID:Addgene_71236 |
| lenti sgRNA(MS2)_zeo backbone | Addgene | #62205; RRID:Addgene_61427 |
| CRISPRa - EF1a-NLS-dCas9(N863)-VP64-2A-Blast-WPRE | Addgene | #61425; RRID:Addgene_61425 |
| CRISPRa - EF1a-MS2-p65-HSF1-2A-Hygro-WPRE | Addgene | #61426; RRID:Addgene_61426 |
| FpG5 - Enhancer reporter vector | Addgene | #69443; RRID:Addgene_69443 |
| pAIB-GFP-IRES-BSD | De Iaco et al., 2017 | N/A |
| pAIB-KLF4-IRES-BSD | This paper | N/A |
| pAIB-KLF17-IRES-BSD | This paper | N/A |
| pRLL-GFP-IRES-BSD | This paper | N/A |
| pRLL-ZNF611-IRES-BSD | This paper | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| N2 | Thermo Fisher Scientific | #17502048 |
| B27 | Thermo Fisher Scientific | #17504044 |
| hLIF | Peprotech | #300-05 |
| Activin A | Peprotech | #120-1 |
| WH-4-023 | SelleckChem | #S7565 |
| PD0325901 | Stemgent | #04-0006 |
| CHIR99021 | Stemgent | #04-0004 |
| SB590885 | R&D system | # 2650 |
| Doxycycline | Sigma-Aldrich | #D9891 |
| IM-12 | Enzo life Sciences | #BML-WN102-0005 |
| Y-27632 | Abcam | # ab120129 |
| Deposited Data | ||
| ATAC-seq - naive/primed hESC | This paper | GEO: GSE117395 |
| ATAC-seq -naive hESC ± CRISPRi against SVA/LTR5Hs | This paper | GEO: GSE117395 |
| ChIP-seq - KLF4 in naive hESC | This paper | GEO: GSE117395 |
| ChIP-seq - H3K27ac in naive/primed hESC | This paper | GEO: GSE117395 |
| ChIP-seq - KLF4 in HAP1 + OKS | This paper | GEO: GSE117395 |
| ChIP-seq - H3K27ac in induced neurons | This paper | GEO: GSE117395 |
| ChIP-seq - H3K9me3/H3K27ac in primed hESC + GFP, KLF4 or KLF17 | This paper | GEO: GSE117395 |
| ChIP-seq - H3K9me3/H3K27ac in naive hESC ± CRISPRi against SVA/LTR5Hs | This paper | GEO: GSE117395 |
| ChIP-seq - H3K9me3/H3K27ac in naive hESC + GFP or ZNF611 | This paper | GEO: GSE117395 |
| RNA-seq - primed hESC + GFP, KLF4 or KLF17 | This paper | GEO: GSE117395 |
| RNA-seq - naive hESC ± CRISPRi against SVA/LTR5Hs | This paper | GEO: GSE117395 |
| RNA-seq - induced neurons ± CRISPRi against SVA/LTR5Hs | This paper | GEO: GSE117395 |
| RNA-seq - naive hESC ± CRISPRi against LTR7YB | This paper | GEO: GSE117395 |
| RNA-seq - naive hESC + GFP or ZNF611 | This paper | GEO: GSE117395 |
| Experimental Models: Cell Lines | ||
| H1 - Human Embryonic Stem Cells | Male - Human Embryo - From Krause lab | N/A |
| WIBR3 - Human Embryonic Stem Cells | Female - Human Embryo - From Jaenisch lab | N/A |
| HEK293T | Female - Embryonic Kidney | N/A |
| HAP1 | Male - derived from KBM-7 (chronic myeloid leukemia) - From Horizon discovery | N/A |
| Primary Dermal Fibroblast Normal; Human, Neonatal (HDFn) | Male - Neonatal - From ATCC | PCS-201-010 |
| Oligonucleotides | ||
| See Table S3. | This paper | N/A |
| Software and Algorithms | ||
| FlowJo - FACS analysis | FlowJo, LLC | v8.8.7 |
| Bowtie2 - Mapping DNA-sequencing | Langmead and Salzberg, 2012 | v2.2 |
| MarkDuplicates - Remove PCR duplicates | Picard tools | v1.1 |
| Seqminer - Data visualiztation | Ye et al., 2014 | v1.4 |
| IGV - Data visualiztation | Robinson et al., 2011 | v2.3 |
| Samtools - Processing post-mapping | Li et al., 2009 | v1.7 |
| Homer - Enrichment analysis | Heinz et al., 2010 | v3 |
| Intervene - Intersection analysis | Khan and Mathelier, 2017 | v0.6 |
| MACS1.4 & MACS2 - Peak calling | Zhang et al., 2008 | N/A |
| hg19 & RheMac8 - Genome Assembly | N/A | |
| TopHat - Mapping RNA-sequencing | Kim et al., 2013 | 2.0.11 |
| HTSeq-count - RNA-seq reads counting | Anders et al., 2015 | 0.6.1 |
| multiBamCov - Bedtools | Quinlan and Hall, 2010 | v2.27.1 |
| limma - Bioconductor | Gentleman et al., 2004 | Bioconductor version 3.7 |
| UCSC liftOver tool | Karolchik et al., 2012 | N/A |
| Shuffle - Bedtools | Quinlan and Hall, 2010 | v2.27.1 |
| getfasta tool - Bedtools | Quinlan and Hall, 2010 | v2.27.1 |
| MAFFT | Katoh et al., 2002 | 7.310 |
| HISAT2 - Mapping RNA-sequencing | Kim et al., 2015 | 2.1.0 |
| ETE toolkit | Huerta-Cepas et al., 2016 | v3 |
| Other | ||
| DNase-seq - pre-implantation embryo | Gao et al., 2018 | GSA: CRA000297 |
| DNase-seq - Roadmap tissues | Roadmap consortium | GEO: GSE18927 |
| ChIA-PET - SMC1 in naive/primed hESC | Ji et al., 2016 | GEO: GSE69643 |
| ChIP-seq - OCT4 in naive/primed hESC | Ji et al., 2016 | GEO: GSE69646 |
| ChIP-seq - OCT4/KLF4 + OKSM in Human dermal fibroblast | Ohnuki et al., 2014 | GEO: GSE56569 |
| ChIP-seq - H3K27ac in fetal brain/liver | Yan et al., 2016 | GEO: GSE63634 |
| ChIP-seq - H3K27ac in adult liver | Trizzino et al., 2017 | SRA: SRP091949 |
| ChIP-seq - H3K27ac in adult brain | Vermunt et al., 2014 | GEO: GSE40465 |
| RNA-seq - Human Primordial Germ Cells | Tang et al., 2015 | SRA: SRP057098 |
| RNA-seq - Human dermal fibroblast reprogrammation by OKSM | Ohnuki et al., 2014 | GEO: GSE56569 |
| RNA-seq - single-cell Human embryo | Yan et al., 2013 | GEO: GSE36552 |
| RNA-seq - single-cell Rhesus Macaque embryo | Wang et al., 2017 | GEO: GSE86938 |
| RNA-seq - primed Rhesus Macaque and Human ESC | Fang et al., 2014 | GEO: GSE61420 |
Contact for Reagent And Resource Sharing
Further information and requests for resources and reagents should be directed to the Lead Contact, Didier Trono (didier.trono@epfl.ch).
Experimental Model and Subject Details
Human ESC usage has been approved by the Swiss Federal Office of Public Health, the Canton of Vaud Ethics Committee (Autorization Number R-FP-S-2-0009-0000) and registered in the European Human Pluripotent Stem Cell Registry (hPSCreg). Conventional (primed) human ESC lines were maintained in mTSER for H1 (Male) and IPS on Matrigel, for WIBR3 (Female) on irradiated inactivated mouse embryonic fibroblast (MEF) feeders in human ESC medium (hESM) and passaged with collagenase and dispase, followed by sequential sedimentation steps in hESM to remove single cells while naive ES cells, primed H1 and IPS were passaged by Accutase in single cells. hES media composition: DMEM/F12 supplemented with 15% fetal bovine serum, 5% KnockOut Serum Replacement, 2 mM L-glutamine, 1% nonessential amino acids, 1% penicillin-streptomycin, 0.1 mM β-mercaptoethanol and 4 ng/ml FGF2. Naive media composition: 500 mL of medium was generated by including: 240 mL DMEM/F12, 240 mL Neurobasal, 5 mL N2 supplement, 10 mL B27 supplement, 2 mM L-glutamine, 1% nonessential amino acids, 0.1 mM β-mercaptoethanol, 1% penicillin-streptomycin, 50 μg/ml BSA. In addition for 4i/LA: PD0325901 (1 μM), SB590885 (0.5 μM), WH4-023 (1 μM), Activin A (10 ng/mL), 20 ng/ml hLIF, Y-27632 (10 μM) and IM-12 (0-1 μM). In addition for KN/2i media: PD0325901 (1 μM), CHIR99021 (1 μM), 20 ng/ml hLIF and Doxycycline (2 μg/ml). For conversion of primed human ESC lines (WIBR3), we seeded 2-3e105 trypsinized single cells on an MEF feeder layer in hESM supplemented with ROCK inhibitor Y-27632 (10 μM). Two days later, medium was switched to 4i/LA (+/− IM12)-containing naive hESM (Theunissen et al., 2016). WIBR3dPE cells (OCT4 GFP knock-in depleted for its primed specific Proximal Enhancer (dPE) were converted in naive with DOX-inducible KLF2 and NANOG transgenes and maintained in 2i/L/DOX (Theunissen et al., 2014). Primed conversion was performed under physiological oxygen conditions (5% O2, 3% CO2) and then passaged in classical cell culture incubator at 37°C with 5% CO2. Primary Dermal Fibroblast Normal; Human, Neonatal (HDFn, ATCC ® PCS-201-010) were cultivated following manufacturer’s protocol. HAP1 and HEK293T were cultivated in DMEM supplemented with 10% fetal bovin serum, Penicillin/Streptomycin, Glutamine.
Method Details
Transcription factor overexpression experiments
GFP, KLF4, KLF17 coding ORF were cloned with C-ter HA tag into pAIB blasticydin resistant lentiviral vector (backbone from (De Iaco et al., 2017)) and for GFP and ZNF611 coding ORF were cloned with C-ter HA tag into a homemade derived blasticydin resistant form of pRRL-pGK lentiviral vector. Primed H1 were transduced with GFP, KLF4 or KLF17-containing lentiviral vectors and split after 48h then selected using blasticydin for the 3 following days. Naive WIBR3dPE hESC cells in KN/2iL media were transduced with GFP or ZNF611-containing lentiviral vectors, split after 96h, then selected for a couple of passages with blasticydin on irradiated Mouse Embryonic Blasticidin-resistant (MMMbz).
CRISPRi experiments
sgRNA designed was perform taking Dfam consensus of LTR7BY and LTR5Hs/SVA common sequence. Specificity was predicted with CRISPOR software (Haeussler et al., 2016). Naive hES WIBR3dPE cells in KN/2i media were transduced with dCAS9-KRAB lentiviral vector. Naive cells were selected using 0.5 μg/mL of Puromycin on DR4 irradiated MEF cells, amplify and then harvest after 3-4 passages. IPS were selected using 0.5 μg/mL of Puromycin then differentiated into induced neurons as describe in (Busskamp et al., 2014). Human Fibroblast were selected using 1 μg/mL of Puromycin then somatic reprogramming experiment was performed using CytoTune-iPS 2.0 Sendai Reprogramming Kit on human fibroblast (ATCC ® PCS-201-010) following manufacturer’s protocol.
Enhancer reporter experiment
We used a lentiviral vector containing a minimal promoter followed of GFP cDNA (FpG5, Addgene #69443) containing the LTR5Hs/SVA common fragment amplified from a SVA (chr19:20248081-20249469, hg19). Then a single mutation was generated using Agilent Technologies QuikChange II XL (Cat#200522-5). H1 hESC were transduced first by CRISPRa activators then by either of these enhancer-containing vectors followed by FACS to select cells with basal level of GFP. Primed H1 were transduced without or with sgRNA (targeting upstream of the KLF-motif), KLF4 or KLF17-containing lentiviral vectors and analyzed using FACS after 5 days.
ChIP-seq
Cells were cross-linked for 10 minutes at room temperature by the addition of one-tenth of the volume of 11% formaldehyde solution to the PBS followed by quenching with glycine. Cells were washed twice with PBS, then the supernatant was aspirated and the cell pellet was conserved in −80°C. Pellets were lysed, resuspended in 1mL of LB1 on ice for 10 min (50 mM HEPES-KOH pH 7.4, 140 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 10% Glycerol, 0.5% NP40, 0.25% Tx100, protease inhibitors), then after centrifugation resuspend in LB2 on ice for 10 min (10 mM Tris pH 8.0, 200 mM NaCl, 1 mM EDTA, 0.5 mM EGTA and protease inhibitors). After centrifugation, resuspend in LB3 (10 mM Tris pH 8.0, 200 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 0.1% NaDOC, 0.1% SDS and protease inhibitors) for histone marks and SDS shearing buffer (10 mM Tris pH8, EDTA 1mM, SDS 0.15% and protease inhibitors) for transcription factor and sonicated (Covaris settings: 5% duty, 200 cycle, 140 PIP, 20 min), yielding genomic DNA fragments with a bulk size of 100-300bp. Coating of the beads with the specific antibody and carried out during the day at 4°C, then chromatin was added overnight at 4°C for histone marks while antibody for transcription factor is incubated with chromatin first with 1% Triton and 150mM NaCl. Subsequently, washes were performed with 2x Low Salt Wash Buffer (10 mM Tris pH 8, 1 mM EDTA, 150mM NaCl, 0.15% SDS), 1x High Salt Wash Buffer (10 mM Tris pH 8, 1 mM EDTA, 500 mM NaCl, 0.15% SDS), 1x LiCl buffer (10 mM Tris pH 8, 1 mM EDTA, 0.5 mM EGTA, 250 mM LiCl, 1% NP40, 1% NaDOC) and 1 with TE buffer. Final DNA was purified with QIAGEN Elute Column. Up to 10 nanograms of ChIPed DNA or input DNA (Input) were prepared for sequencing. Library was quality checked by DNA high sensitivity chip (Agilent). Quality controlled samples were then quantified by picogreen (Qubit® 2.0 Fluorometer, Invitrogen). Cluster amplification and following sequencing steps strictly followed the Illumina standard protocol. Sequenced reads were de-multiplexed to attribute each read to a DNA sample and then aligned to reference human genome hg19 with bowtie2 (with parameters–end-to-end). PCR duplicates removal (MarkDuplicates using picard tools and parameters: VALIDATION_STRINGENCY = LENIENT REMOVE_DUPLICATES = true), samples were downsampled (DownsampleSam using picard tools) to the lowest dataset count. Heatmaps and profile averages were calculated using Seqminer 1.4 (Ye et al., 2014) over 5kb windows around the peak/repeat center from BAM files. Screenshots were made with bigwig from BAM files, then BAM files where filtered MAPQ > 10 except for KLF4/OCT4 ChIP-seq to remove multimapped reads for any counting and peak calling produce by MACS1.4 (–nomodel–shiftsize 75). Differential analysis between conditions has been performed with VOOM as described in the RNA-seq section using unique reads (filter for MAPQ > 10), counted on the union of all peaks of a same experiment. Samples were normalized for sequencing depth using the counts on the union peaks as library size and using the TMM method as it is implemented in the limma package of Bioconductor (Gentleman et al., 2004). Enrichment analysis over TE subfamilies was performed with HOMER software (Heinz et al., 2010). Intersection of multiple bed files were performed using Intervene (Khan and Mathelier, 2017).
Chromatin accessibility
ATAC-seq was performed as in (Buenrostro et al., 2013) on primed WIRB3 and WIBR3dPE; naive WIBR3 and WIBR3dPE in 4iLA and KN/2iL media respectively; and in WIBR3dPE in KN/2iL media upon dCAS9-KRAB overexpression containing or not a guide RNA targeting SVA/LTR5Hs. Library were made using Nextera DNA Library Prep Kit (Illumina #FC-121-1030). ATAC-seq and DNase-seq reads were mapped to the human (hg19) genome using bowtie2. Mitochondrial reads were removed. Then accessible sites were called using MACS2, only peaks with a score higher than 5 (–log10 p value) were kept. Then differential analysis between conditions was done using unique reads (filter for MAPQ > 10), counted on the union of all peaks of a same experiment.
qRT-PCR/RNA-sequencing
Total RNA from cell lines was isolated with a High Pure RNA Isolation Kit (Roche). cDNA was prepared with SuperScript II reverse transcriptase (Invitrogen). Sequencing library were performed with SMARTer Stranded Total RNA-seq, Pico input (ref 635006) or Illumina Truseq Stranded mRNA LT.
RNA-seq analysis
Reads were mapped to the human (hg19) or macaque (RheMac8) genome using TopHat. Gene counts were generated using HTSeq-count. For repetitive sequences, an in-house curated version of the Repbase database was used (fragmented LTR and internal segments belonging to a single integrant were merged). TEs counts were generated using the multiBamCov tool from the bedtools software. Only uniquely mapped reads were used for counting on genes and repetitive sequences integrants. TEs overlapping exons or that did not have at least one sample with 20 reads were discarded from the analysis. Normalization for sequencing depth has been done for both, genes and TEs, using the counts on genes as library size using the TMM method as it is implemented in the limma package of Bioconductor (Gentleman et al., 2004). Differential gene expression analysis was performed using Voom (Law et al., 2014) as it has been implemented in the limma package of Bioconductor. A moderated t test (as implemented in the limma package of R) was used to test significance. P values were corrected for multiple testing using the Benjamini-Hochberg’s method. For counting on TE subfamilies, we counted the reads on the repetitive sequences without filtering out for multi-mapped and added-up per subfamily. Interspecies RNA-seq normalization was performed as in (Brawand et al., 2011). In short, we calculated standard RPKM expression values (that were then log2-transformed) for the orthologous genes as defined by the ensembl database. We then normalized these expression values by a scaling procedure. Specifically, among the genes with expression values in the interquartile range, we identified the 100 genes that have the most-conserved ranks among samples and assessed their median expression levels in each sample. We then derived scaling factors that adjust these medians to a common value. Finally, these factors were used to scale expression values of all genes in the samples.
Synteny analysis
Synteny analysis. Batch coordinate conversion between human (hg38) and 47 different species was obtained through UCSC liftOver tool (option -minMatch = 0.5), which relies on whole-genome alignments with BLASTZ. The age of sequences was assumed to be the divergence time between human and the farthest species showing it with at least 50% homology. Peaks synteny were compared to synteny of 100 random set of peaks (obtained through Bedtools suite Shuffle tool with –chrom and –noOverlapping options) for statistical comparison. For TEs, matched sequences were considered syntenic only if a TE with similar Repbase subfamily annotation than in Human was present in the foreign species at the syntenic genomic location.
Multiple sequence alignment plot
Fasta sequences from LTR5Hs and SVA_D TE families were extracted from the hg19 genome assembly using bedtools getfasta tool (Quinlan and Hall, 2010). SVAD (> 200bp) and LTR5Hs (> 100bp) sequences were aligned using MAFFT (Katoh et al., 2002). Regions in the alignment consisting of more than 95% of gaps were trimmed out. For each selected integrand, the KLF4 ChIP-seq signal was extracted from the bigwig coverage file and scaled to the interval [0,1] before being plotted on top of the alignment alongside the average ChIP-seq signal.
Chimeric transcript analysis
RNA-seq were aligned on the hg19 genome using HISAT2 (Kim et al., 2015) with parameters:–rna-strandness RF–seed 42. Then, transcripts spanning between genes and TE were extracted from the transcriptome data. The so-called transpo chimeras were then split into two groups: the one starting on TEs and the one containing TEs. Finally, the chimeras in the groups were counted and added up per family.
KZFP phylogeny and conservation
KZFP ages were retrieved from (Imbeault et al., 2017) by clustering with a threshold similarity score of 60% between any two zinc-finger arrays. Age was established by the most evolutionary distant KZFP present in the same cluster. KZFP phylogeny: Fasta sequences were downloaded from the UniProt website using the following search criteria: annotation:(type”:positional domain” krab) family”:zinc finger” AND organism”:Homo sapiens (Human) [9606].” Several KZFP sequences from the cluster 9 were manually added to this list, as its KRAB domain is not annotated in UniProt. All KZFP sequences were aligned using MAFFT with parameters –reorder –auto. The phylogenic tree was build using the ETE toolkit (Huerta-Cepas et al., 2016) using the command: ete3 build with parameters–no-seq-rename -w none-trimal05-none-fasttree_default. The tree was then parsed, colored and annotated using the ete3 python module.
Quantification and Statistical Analysis
The details of the statistical tests have been explained in each figure and in the above “Method Details” part.
We performed two-sided t test for group comparisons (F1e, FS1b, FS1g, F2b-e, FS2b-d, F3b-c, F3e, FS3b-e, FS3g-i, FS3k, F4a, F4e, FS4c-e, F5a-c, FS5b-f and F6c) and wilcoxon test were normality could not be assumed in F1b. Permutation tests were used in F1a, FS1a, F2c, FS3g and F6b. Hypergenometric tests were computed (FS1h, FS3g-i and FS5c) to compare proportions. Standard Error of the Mean (SEM) has been used for error bars (F2d, FS2b, F3e and F4e). The Benjamini and Hochberg method was used to adjust for multiple testing (F1b, FS1b, F2b-c, F2e, FS2c, FS3d, FS4d-e, FS5b and FS5d). Pearson correlation was computed in FS2c.
Differential ATAC-seq enrichment was analyzed using ATAC-seq from WIBR3dPE in KN/2iL and WIBR3 in 4iLA (naive hESC) or hESM media (primed hESC). Differential enrichment of H3K9me3 and ATAC-seq upon CRISPRi against LTR5Hs/SVA in WIBR3dPE (KN/2iL media) naive hESC were analyzed on duplicate and triplicate experiments respectively. Differential expression RNA-seq analysis upon CRISPRi against LTR5Hs/SVA and LTR7YB in WIBR3dPE (KN/2iL media) were analyzed with two different sgRNA each in quadruplicate and duplicate respectively. Differential expression RNA-seq analysis upon GFP, KLF4, KLF17 overexpression in H1 primed hESC cells or GFP and ZNF611 overexpression in WIBR3dPE (KN/2iL media) naive hESC were performed in duplicates and triplicates for GFP and KLF4 in H1 primed cells. Differential enrichment of H3K9me3 and H3K27ac upon GFP, KLF4, KLF17 overexpression in H1 primed hESC cells or GFP and ZNF611 overexpression in WIBR3dPE (KN/2iL media) naive hESC were performed in duplicates. Other experiments as GFP signal quantification of F2d (n = 6), ChIP-qPCR of FS2b (n = 3), RT-qPCR analysis of F4e (n = 9) were performed in H1 primed hESC, while RT-qPCR analysis of F3e (n = 3) were performed in fibroblast cells.
Data and Software Availability
The accession number for the RNA-seq, ChIP-seq and ATAC-seq reported in this paper is GEO: GSE117395.
Acknowledgments
We thank A. Necsulea, C. Raclot, M. Friedli, P.-Y. Helleboid, and A. De Iaco for technical and scientific advice; T. Pontis for the graphical abstract; A. Coluccio and C.C. Bolt for critical reading of the manuscript; and the EPFL Flow Cytometry and Genomics core facilities and the University of Lausanne Genomic Technologies Facility for help with cell sorting and sequencing. This study was supported by grants from the Swiss National Science Foundation and the European Research Council (KRABnKAP, no. 268721; Transpos-X, no. 694658) to D.T.; by fellowships from the EPFL/Marie Skłodowska-Curie Fund, the Association pour la Recherche sur le Cancer (ARC), and the Fondation Bettencourt to J.P.; and by NIH grants R37HD045022, R01-NS088538, and R01-MH to R.J.
Author Contributions
J.P. and D.T. conceived the study and designed experiments; J.P. performed most wet experiments with the technical help of S.O.; and T.W.T. and P.T. contributed to hESC- and iPSC-to-neurons-related studies, respectively. J.P., E.P., J.D., and A.C. completed the bioinformatics analyses, and J.P. and D.T. wrote the manuscript, with review and corrections by all authors.
Declaration of Interests
The authors declare no competing interests.
Published: April 18, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.stem.2019.03.012.
Supplemental Information
References
- Anders S., Pyl P.T., Huber W. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brawand D., Soumillon M., Necsulea A., Julien P., Csárdi G., Harrigan P., Weier M., Liechti A., Aximu-Petri A., Kircher M. The evolution of gene expression levels in mammalian organs. Nature. 2011;478:343–348. doi: 10.1038/nature10532. [DOI] [PubMed] [Google Scholar]
- Britten R.J., Davidson E.H. Repetitive and non-repetitive DNA sequences and a speculation on the origins of evolutionary novelty. Q. Rev. Biol. 1971;46:111–138. doi: 10.1086/406830. [DOI] [PubMed] [Google Scholar]
- Buenrostro J.D., Giresi P.G., Zaba L.C., Chang H.Y., Greenleaf W.J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods. 2013;10:1213–1218. doi: 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busskamp V., Lewis N.E., Guye P., Ng A.H.M., Shipman S.L., Byrne S.M., Sanjana N.E., Murn J., Li Y., Li S. Rapid neurogenesis through transcriptional activation in human stem cells. Mol. Syst. Biol. 2014;10:760. doi: 10.15252/msb.20145508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro-Diaz N., Ecco G., Coluccio A., Kapopoulou A., Yazdanpanah B., Friedli M., Duc J., Jang S.M., Turelli P., Trono D. Evolutionally dynamic L1 regulation in embryonic stem cells. Genes Dev. 2014;28:1397–1409. doi: 10.1101/gad.241661.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalopin D., Naville M., Plard F., Galiana D., Volff J.-N. Comparative analysis of transposable elements highlights mobilome diversity and evolution in vertebrates. Genome Biol. Evol. 2015;7:567–580. doi: 10.1093/gbe/evv005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuong E.B., Elde N.C., Feschotte C. Regulatory activities of transposable elements: from conflicts to benefits. Nat. Rev. Genet. 2017;18:71–86. doi: 10.1038/nrg.2016.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collier A.J., Panula S.P., Schell J.P., Chovanec P., Plaza Reyes A., Petropoulos S., Corcoran A.E., Walker R., Douagi I., Lanner F., Rugg-Gunn P.J. Comprehensive cell surface protein profiling identifies specific markers of human naive and primed pluripotent states. Cell Stem Cell. 2017;20:874–890.e7. doi: 10.1016/j.stem.2017.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordaux R., Batzer M.A. The impact of retrotransposons on human genome evolution. Nat. Rev. Genet. 2009;10:691–703. doi: 10.1038/nrg2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Iaco A., Planet E., Coluccio A., Verp S., Duc J., Trono D. DUX-family transcription factors regulate zygotic genome activation in placental mammals. Nat. Genet. 2017;49:941–945. doi: 10.1038/ng.3858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ecco G., Imbeault M., Trono D. KRAB zinc finger proteins. Development. 2017;144:2719–2729. doi: 10.1242/dev.132605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang R., Liu K., Zhao Y., Li H., Zhu D., Du Y., Xiang C., Li X., Liu H., Miao Z. Generation of naive induced pluripotent stem cells from rhesus monkey fibroblasts. Cell Stem Cell. 2014;15:488–497. doi: 10.1016/j.stem.2014.09.004. [DOI] [PubMed] [Google Scholar]
- Friedli M., Turelli P., Kapopoulou A., Rauwel B., Castro-Díaz N., Rowe H.M., Ecco G., Unzu C., Planet E., Lombardo A. Loss of transcriptional control over endogenous retroelements during reprogramming to pluripotency. Genome Res. 2014;24:1251–1259. doi: 10.1101/gr.172809.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuentes D.R., Swigut T., Wysocka J. Systematic perturbation of retroviral LTRs reveals widespread long-range effects on human gene regulation. eLife. 2018;7:e35989. doi: 10.7554/eLife.35989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao L., Wu K., Liu Z., Yao X., Yuan S., Tao W., Yi L., Yu G., Hou Z., Fan D. Chromatin accessibility landscape in human early embryos and its association with evolution. Cell. 2018;173:248–259.e15. doi: 10.1016/j.cell.2018.02.028. [DOI] [PubMed] [Google Scholar]
- Gentleman R.C., Carey V.J., Bates D.M., Bolstad B., Dettling M., Dudoit S., Ellis B., Gautier L., Ge Y., Gentry J. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Göke J., Lu X., Chan Y.-S., Ng H.-H., Ly L.-H., Sachs F., Szczerbinska I. Dynamic transcription of distinct classes of endogenous retroviral elements marks specific populations of early human embryonic cells. Cell Stem Cell. 2015;16:135–141. doi: 10.1016/j.stem.2015.01.005. [DOI] [PubMed] [Google Scholar]
- Grow E.J., Flynn R.A., Chavez S.L., Bayless N.L., Wossidlo M., Wesche D.J., Martin L., Ware C.B., Blish C.A., Chang H.Y. Intrinsic retroviral reactivation in human preimplantation embryos and pluripotent cells. Nature. 2015;522:221–225. doi: 10.1038/nature14308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H., Zhu P., Yan L., Li R., Hu B., Lian Y., Yan J., Ren X., Lin S., Li J. The DNA methylation landscape of human early embryos. Nature. 2014;511:606–610. doi: 10.1038/nature13544. [DOI] [PubMed] [Google Scholar]
- Guo G., von Meyenn F., Rostovskaya M., Clarke J., Dietmann S., Baker D., Sahakyan A., Myers S., Bertone P., Reik W. Epigenetic resetting of human pluripotency. Development. 2017;144:2748–2763. doi: 10.1242/dev.146811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeussler M., Schönig K., Eckert H., Eschstruth A., Mianné J., Renaud J.-B., Schneider-Maunoury S., Shkumatava A., Teboul L., Kent J. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biology. 2016;17:148. doi: 10.1186/s13059-016-1012-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huerta-Cepas J., Serra F., Bork P. ETE 3: reconstruction, analysis, and visualization of phylogenomic data. Mol. Biol. Evol. 2016;33:1635–1638. doi: 10.1093/molbev/msw046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntley S., Baggott D.M., Hamilton A.T., Tran-Gyamfi M., Yang S., Kim J., Gordon L., Branscomb E., Stubbs L. A comprehensive catalog of human KRAB-associated zinc finger genes: insights into the evolutionary history of a large family of transcriptional repressors. Genome Res. 2006;16:669–677. doi: 10.1101/gr.4842106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imbeault M., Helleboid P.-Y., Trono D. KRAB zinc-finger proteins contribute to the evolution of gene regulatory networks. Nature. 2017;543:550–554. doi: 10.1038/nature21683. [DOI] [PubMed] [Google Scholar]
- Jacobs F.M.J., Greenberg D., Nguyen N., Haeussler M., Ewing A.D., Katzman S., Paten B., Salama S.R., Haussler D. An evolutionary arms race between KRAB zinc-finger genes ZNF91/93 and SVA/L1 retrotransposons. Nature. 2014;516:242–245. doi: 10.1038/nature13760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji X., Dadon D.B., Powell B.E., Fan Z.P., Borges-Rivera D., Shachar S., Weintraub A.S., Hnisz D., Pegoraro G., Lee T.I. 3D chromosome regulatory landscape of human pluripotent cells. Cell Stem Cell. 2016;18:262–275. doi: 10.1016/j.stem.2015.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karolchik D., Hinrichs A.S., Kent W.J. The UCSC Genome Browser. Curr. Protoc. Bioinformatics. 2012;Chapter 1 doi: 10.1002/0471250953.bi0104s40. Unit 1.4. [DOI] [PubMed] [Google Scholar]
- Katoh K., Misawa K., Kuma K., Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30:3059–3066. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan A., Mathelier A. Intervene: a tool for intersection and visualization of multiple gene or genomic region sets. BMC Bioinformatics. 2017;18:287. doi: 10.1186/s12859-017-1708-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D., Langmead B., Salzberg S.L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods. 2015;12:357–360. doi: 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D., Pertea G., Trapnell C., Pimentel H., Kelley R., Salzberg S.L. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law C.W., Chen Y., Shi W., Smyth G.K. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biology. 2014;15:R29. doi: 10.1186/gb-2014-15-2-r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homre N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H., Chang L.-H., Sun Y., Lu X., Stubbs L. Deep vertebrate roots for mammalian zinc finger transcription factor subfamilies. Genome Biol. Evol. 2014;6:510–525. doi: 10.1093/gbe/evu030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X., Sachs F., Ramsay L., Jacques P.-É., Göke J., Bourque G., Ng H.-H. The retrovirus HERVH is a long noncoding RNA required for human embryonic stem cell identity. Nat. Struct. Mol. Biol. 2014;21:423–425. doi: 10.1038/nsmb.2799. [DOI] [PubMed] [Google Scholar]
- Macfarlan T.S., Gifford W.D., Driscoll S., Lettieri K., Rowe H.M., Bonanomi D., Firth A., Singer O., Trono D., Pfaff S.L. Embryonic stem cell potency fluctuates with endogenous retrovirus activity. Nature. 2012;487:57–63. doi: 10.1038/nature11244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui T., Leung D., Miyashita H., Maksakova I.A., Miyachi H., Kimura H., Tachibana M., Lorincz M.C., Shinkai Y. Proviral silencing in embryonic stem cells requires the histone methyltransferase ESET. Nature. 2010;464:927–931. doi: 10.1038/nature08858. [DOI] [PubMed] [Google Scholar]
- McClintock B. Intranuclear systems controlling gene action and mutation. Brookhaven Symp. Biol. 1956:58–74. [PubMed] [Google Scholar]
- Ohnuki M., Tanabe K., Sutou K., Teramoto I., Sawamura Y., Narita M., Nakamura M., Tokunaga Y., Nakamura M., Watanabe A. Dynamic regulation of human endogenous retroviruses mediates factor-induced reprogramming and differentiation potential. Proc. Natl. Acad. Sci. USA. 2014;111:12426–12431. doi: 10.1073/pnas.1413299111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastor W.A., Liu W., Chen D., Ho J., Kim R., Hunt T.J., Lukianchikov A., Liu X., Polo J.M., Jacobsen S.E., Clark A.T. TFAP2C regulates transcription in human naive pluripotency by opening enhancers. Nat. Cell Biol. 2018;20:553–564. doi: 10.1038/s41556-018-0089-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson J.T., Thorvaldsdóttir H., Winckler W., Guttman M., Lander E.S., Getz G., Mesirov J.P. Integrative genomics viewer. Nature Biotechnology. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe H.M., Jakobsson J., Mesnard D., Rougemont J., Reynard S., Aktas T., Maillard P.V., Layard-Liesching H., Verp S., Marquis J. KAP1 controls endogenous retroviruses in embryonic stem cells. Nature. 2010;463:237–240. doi: 10.1038/nature08674. [DOI] [PubMed] [Google Scholar]
- Smith Z.D., Chan M.M., Humm K.C., Karnik R., Mekhoubad S., Regev A., Eggan K., Meissner A. DNA methylation dynamics of the human preimplantation embryo. Nature. 2014;511:611–615. doi: 10.1038/nature13581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suntsova M., Gogvadze E.V., Salozhin S., Gaifullin N., Eroshkin F., Dmitriev S.E., Martynova N., Kulikov K., Malakhova G., Tukhbatova G. Human-specific endogenous retroviral insert serves as an enhancer for the schizophrenia-linked gene PRODH. Proc. Natl. Acad. Sci. USA. 2013;110:19472–19477. doi: 10.1073/pnas.1318172110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takashima Y., Guo G., Loos R., Nichols J., Ficz G., Krueger F., Oxley D., Santos F., Clarke J., Mansfield W. Resetting transcription factor control circuitry toward ground-state pluripotency in human. Cell. 2014;158:1254–1269. doi: 10.1016/j.cell.2014.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang W.W.C., Dietmann S., Irie N., Leitch H.G., Floros V.I., Bradshaw C.R., Hackett J.A., Chinnery P.F., Surani M.A. A unique gene regulatory network resets the human germline epigenome for development. Cell. 2015;161:1453–1467. doi: 10.1016/j.cell.2015.04.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakore P.I., D’Ippolito A.M., Song L., Safi A., Shivakumar N.K., Kabadi A.M., Reddy T.E., Crawford G.E., Gersbach C.A. Highly specific epigenome editing by CRISPR-Cas9 repressors for silencing of distal regulatory elements. Nat. Methods. 2015;12:1143–1149. doi: 10.1038/nmeth.3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theunissen T.W., Powell B.E., Wang H., Mitalipova M., Faddah D.A., Reddy J., Fan Z.P., Maetzel D., Ganz K., Shi L. Systematic identification of culture conditions for induction and maintenance of naive human pluripotency. Cell Stem Cell. 2014;15:471–487. doi: 10.1016/j.stem.2014.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theunissen T.W., Friedli M., He Y., Planet E., O’Neil R.C., Markoulaki S., Pontis J., Wang H., Iouranova A., Imbeault M. Molecular criteria for defining the naive human pluripotent state. Cell Stem Cell. 2016;19:502–515. doi: 10.1016/j.stem.2016.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trizzino M., Park Y., Holsbach-Beltrame M., Aracena K., Mika K., Caliskan M., Perry G.H., Lynch V.J., Brown C.D. Transposable elements are the primary source of novelty in primate gene regulation. Genome Res. 2017;27:1623–1633. doi: 10.1101/gr.218149.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turelli P., Castro-Diaz N., Marzetta F., Kapopoulou A., Raclot C., Duc J., Tieng V., Quenneville S., Trono D. Interplay of TRIM28 and DNA methylation in controlling human endogenous retroelements. Genome Res. 2014;24:1260–1270. doi: 10.1101/gr.172833.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermunt M.W., Reinink P., Korving J., de Bruijn E., Creyghton P.M., Basak O., Geeven G., Toonen P.W., Lansu N., Meunier C. Large-scale identification of coregulated enhancer networks in the adult human brain. Cell Rep. 2014;9:767–779. doi: 10.1016/j.celrep.2014.09.023. [DOI] [PubMed] [Google Scholar]
- Wang X., Liu D., He D., Suo S., Xia X., He X., Han J.J., Zheng P. Transcriptome analyses of rhesus monkey preimplantation embryos reveal a reduced capacity for DNA double-strand break repair in primate oocytes and early embryos. Genome Res. 2017;27:567–579. doi: 10.1101/gr.198044.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf D., Goff S.P. Embryonic stem cells use ZFP809 to silence retroviral DNAs. Nature. 2009;458:1201–1204. doi: 10.1038/nature07844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf G., Yang P., Füchtbauer A.C., Füchtbauer E.-M., Silva A.M., Park C., Wu W., Nielsen A.L., Pedersen F.S., Macfarlan T.S. The KRAB zinc finger protein ZFP809 is required to initiate epigenetic silencing of endogenous retroviruses. Genes Dev. 2015;29:538–554. doi: 10.1101/gad.252767.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamane M., Ohtsuka S., Matsuura K., Nakamura A., Niwa H. Overlapping functions of Krüppel-like factor family members: targeting multiple transcription factors to maintain the naïve pluripotency of mouse embryonic stem cells. Development. 2018;145:dev162404. doi: 10.1242/dev.162404. [DOI] [PubMed] [Google Scholar]
- Yan L., Yang M., Guo H., Yang L., Wu J., Li R., Liu P., Lian Y., Zheng X., Yan J. Single-cell RNA-seq profiling of human preimplantation embryos and embryonic stem cells. Nat. Struct. Mol. Biol. 2013;20:1131–1139. doi: 10.1038/nsmb.2660. [DOI] [PubMed] [Google Scholar]
- Yan L., Guo H., Hu B., Li R., Yong J., Zhao Y., Zhi X., Fan X., Guo F., Wang X. Epigenomic landscape of human fetal brain, heart, and liver. J. Biol. Chem. 2016;291:4386–4398. doi: 10.1074/jbc.M115.672931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang P., Wang Y., Macfarlan T.S. The role of KRAB-ZFPs in transposable element repression and mammalian evolution. Trends Genet. 2017;33:871–881. doi: 10.1016/j.tig.2017.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye T., Ravens S., Krebs A.R., Tora L. Interpreting and visualizing ChIP-seq data with the seqMINER software. Methods Mol. Biol. 2014;1150:141–152. doi: 10.1007/978-1-4939-0512-6_8. [DOI] [PubMed] [Google Scholar]
- Zhao K., Du J., Han X., Goodier J.L., Li P., Zhou X., Wei W., Evans S.L., Li L., Zhang W. Modulation of LINE-1 and Alu/SVA retrotransposition by Aicardi-Goutières syndrome-related SAMHD1. Cell Rep. 2013;4:1108–1115. doi: 10.1016/j.celrep.2013.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W., Liu X.S. Model-based analysis of ChIP-seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou S., Liu Y., Ma Y., Zhang X., Li Y., Wen J. C9ORF135 encodes a membrane protein whose expression is related to pluripotency in human embryonic stem cells. Sci. Rep. 2017;7:45311. doi: 10.1038/srep45311. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the RNA-seq, ChIP-seq and ATAC-seq reported in this paper is GEO: GSE117395.