

Abstract
The urokinase receptor (uPAR) is a founding member of a small protein family with multiple Ly6/uPAR (LU) domains. The motif defining these LU domains contains five plesiotypic disulfide bonds stabilizing its prototypical three-fingered fold having three protruding loops. Notwithstanding the detailed knowledge on structure-function relationships in uPAR, one puzzling enigma remains unexplored. Why does the first LU domain in uPAR (DI) lack one of its consensus disulfide bonds, when the absence of this particular disulfide bond impairs the correct folding of other single LU domain-containing proteins? Here, using a variety of contemporary biophysical methods, we found that reintroducing the two missing half-cystines in uPAR DI caused the spontaneous formation of the corresponding consensus 7–8 LU domain disulfide bond. Importantly, constraints due to this cross-link impaired (i) the binding of uPAR to its primary ligand urokinase and (ii) the flexible interdomain assembly of the three LU domains in uPAR. We conclude that the evolutionary deletion of this particular disulfide bond in uPAR DI may have enabled the assembly of a high-affinity urokinase-binding cavity involving all three LU domains in uPAR. Of note, an analogous neofunctionalization occurred in snake venom α-neurotoxins upon loss of another pair of the plesiotypic LU domain half-cystines. In summary, elimination of the 7–8 consensus disulfide bond in the first LU domain of uPAR did have significant functional and structural consequences.
Keywords: receptor structure-function, surface plasmon resonance (SPR), protein evolution, urokinase receptor, disulfide, fibrinolysis, hydrogen exchange mass spectrometry, small-angle X-ray scattering (SAXS), plasminogen regulation, LU domain
Introduction
The urokinase-type plasminogen activator receptor (uPAR)2 is an extracellular membrane protein composed of three homologous Ly6/uPAR-type (LU) domains and a C-terminal glycosylphosphatidylinositol (GPI) membrane anchor (1). It serves to focalize plasminogen activation on cell surfaces via its high-affinity binding to the urokinase-type plasminogen activator (uPA) (2). In so doing, it facilitates extravascular fibrin surveillance reducing the adverse effects of chronic inflammation caused by unremitting fibrin deposition (3). Besides promoting pericellular proteolysis, the uPA·uPAR interaction also stimulates cell adhesion and migration via direct and indirect interactions with vitronectin and integrins (4–9). Elegant transgenic mouse models show that the interaction between uPA and uPAR promotes hepatic fibrin clearance (3) and improves neuronal recovery after either cerebral ischemia (10, 11) or spinal cord injury (12). Notwithstanding these beneficial effects, the uPA·uPAR interaction may also elicit detrimental pathological effects, particularly in relationship to chronic inflammation. In genetic mouse models, the interplay between uPA and uPAR augments the pathogenesis of collagen-induced arthritis (13, 14). In line with these causal correlations, high plasma levels of shed uPA/uPAR predict poor prognosis in several pathologic conditions with inflammatory lesions, e.g. bacterial infections (15, 16), kidney disease (17, 18), and invasive and metastatic solid cancers (19). The latter association spurred a considerable interest in developing uPAR-specific targeting strategies intended for use in cancer therapy (20–24). These initiatives are now being supplemented by the development of uPAR-targeting probes for noninvasive imaging of uPAR expression using either (i) positron emission tomography to guide patient staging (25–27) or (ii) near-IR fluorescence to guide precision cancer surgery by improving margin resection (28–30).
Crystal structures of uPAR solved in complex with its natural protein ligands (Fig. 1, C and D) (31–34), small molecule antagonists (21, 35, 36), or antibodies (31, 37) reveal that all three LU domains in uPAR combine to form a compact globular structure. This assembly creates (i) a large and hydrophobic uPA-binding cavity comprising elements from all three LU domains and (ii) a smaller peripheral binding site for the somatomedin B (SMB) domain of vitronectin at the interface between the first (DI) and second (DII) LU domain in uPAR. Although these binding sites are nonoverlapping they do, nevertheless, interact cooperatively. Prior uPA-binding thus increases uPAR's affinity for vitronectin (7) and in so doing it leads to increased cell adhesion and migration (5, 8, 9). We showed previously that the dynamic assembly of the LU domains in uPAR enables this allosteric regulation of ligand binding (5, 38). Combining our biophysical and functional data led us to conclude that uPA occupancy drives uPAR into a more closed and compact conformation and this increases the affinity for SMB (5). Locking uPAR permanently in this compact conformation, by introducing a nonnative disulfide bond between DI and DIII, by-passed the cooperativity of uPA binding and generated a constitutive high-affinity binding site for SMB (6, 39). Our data, furthermore, suggested that uPAR DI plays a dominating role in this flexibility (38). Given that this particular domain differs from the otherwise invariant LU-consensus motif, inasmuch as it lacks one of the 5 plesiotypic disulfide bonds (40), we speculate that the loss of this structural constraint could have been instrumental for evolving high-affinity uPA binding and gaining cooperativity in vitronectin binding. This proposition is not unprecedented as snake venom α-neurotoxins (representing secreted single LU-domain proteins) developed potent neurotoxicity toward synapsid targets and underwent neofunctionalization after deletion of a plesiotypic disulfide bond (41). In this study, we therefore introduced the lacking consensus disulfide bond in human uPAR DI and studied the consequences thereof on the structural flexibility and ligand binding properties of intact uPAR.
Figure 1.

Sequence alignment of LU domains in human uPAR and snake venom α-neurotoxins. A shows an alignment of primary sequences for the three LU domains in uPAR (Homo sapiens, Q03405) and the single LU domains in the snake venom toxins: demotoxin (Boiga dendrophilia, DQ366293), erabutoxin a (Laticuda semifasciata, P60775), and α-cobratoxin (Naja kauthia, P01391). Linker regions and extensions are omitted from the alignment (their presence are indicated by ·). Half-cystines are highlighted in yellow boxes along with their disulfide connectivity. Arrows indicate Thr51 and Val70 in uPAR DI. Residues located in the ligand-binding interface in crystal structures of ATF·uPAR (34) and α-cobratoxin·AChBP (71) complexes are highlighted in green, as are residues important for neurotoxicity of eraboutoxin a (72). The crystal structure of uPAR is shown in B as a cartoon representation (DI, cyan; DII, wheat; DIII, blue). C shows the ATF·uPAR complex with uPAR in a gray surface representation and ATF (containing GFD and a kringle domain) in cartoon representation. D shows the ATF·uPAR·SMB complex. E shows the LU domain in uPAR DI (residues 1–77) with β-strands in cyan and disulfide bonds as yellow sticks. A yellow hatched line between the Cα-atoms of Thr51 and Val70 illustrates one possible position of the lacking 7–8 disulfide bond. F shows the same structure tilted 90° to illustrate their structural constraint on the β-sheets and the proximity of the N-linked glycosylation site (Asn52) and the lacking 7–8 consensus disulfide bond. F shows the positons of the introduced disulfide bonds: Thr51–Val70 (*) and His47–Asn259 (**). Protein structures were created with PyMol (Schrödinger, LLC) using the PDB code 3BTI.
Results
Loss of a consensus disulfide bond in uPAR DI
Sequence alignments of the three homologous LU domains in human uPAR clearly show that the 5-disulfide bond signature, considered a plesiotypic trait of ancient three-fingered neurotoxins (41), is maintained in both uPAR DII and uPAR DIII (Fig. 1A). This primordial disulfide pattern was first identified in the basal-type α-neurotoxins (e.g. denmotoxin) that are present in venoms of nonfront-fanged snakes (Colubridae) that feed preferentially on nonmammalian prey (41, 42). In accordance with this feeding behavior, their α-neurotoxins have only weak affinity for mammalian nicotinic acetylcholine receptors (41, 42). In the more advanced elapid snakes, the α-neurotoxins gained high affinity for mammalian acetylcholine receptors via deletion of the 2–3 LU consensus disulfide bond in loop 1 (41, 42), as illustrated in Fig. 1A. Intriguingly, this sequence alignment and our experimental disulfide assignments (31, 32, 35, 40) reveal that uPAR DI unexpectedly lacks another of the invariant disulfide bonds that defines the ancient LU protein domain fold. In this case, the missing disulfide (denoted 7–8 in Fig. 1) is located at the base of loop 3 connecting β-strands E and F (Fig. 1, E and F) and it is absent from DIs of all known mammalian orthologues of uPAR (Fig. S1). The deletion of this particular disulfide bond in uPAR during evolution is remarkable given that single LU domain-containing proteins, such as GPIHBP1, CD59, and κ-bungarotoxin require this disulfide bond for their correct protein folding and function (43–45).
Reintroducing the missing disulfide bond in uPAR DI
To assign the most probable site(s) for reintroducing the primordial 7–8 consensus disulfide bond in uPAR DI, we examined primary sequence alignments of individual LU domains. This clearly pinpointed Thr51 and Val70 as the most promising candidate pair (Fig. 1A, Figs. S1 and S8). This notion was further substantiated by the pairwise Cβ–Cβ atom distances in seven different crystal structures available for uPAR in complex with various ligands (Table S5). The Cβ–Cβ distances of these Thr51–Val70 pairs were 6.4 ± 0.5 Å, which is slightly longer than the distances for the bona fide 7–8 consensus disulfide bonds present in DII (4.0 ± 0.2 Å) and DIII (3.9 ± 0.4 Å). Nonetheless, evaluations focused only on minimizing structural perturbations highlighted yet another possible candidate pair, as the Cβ–Cβ atoms for Lys50 and Val70 were only 5.0 ± 0.4 Å apart.
Based on these considerations, we chose to express both uPART51C-V70C and uPARK50C-V70C (residues 1–283) in Drosophila S2-cells and purify the secreted proteins. To confirm the oxidation status of the introduced cysteine residues (i.e. validating that they are indeed engaged in disulfide bond formation), we subjected uPAR to limited proteolysis with chymotrypsin under nondenaturing conditions. We optimized the conditions to hydrolyze predominately the Tyr87–Ser88 peptide bond in the linker region between DI and DII and to a lesser extent the Tyr57–Arg58 peptide bond located in loop 3 of DI. Mass spectrometry confirmed that all cysteine residues in these protein preparations were engaged in disulfide bonding (Table 1, Fig. S2).
Table 1.
Verification of disulfide bond status by MS
uPAR DI (residues 1–87) was excised from intact uPAR1–283 by limited chymotrypsin digestion and the molecular masses were determined by LC-ESI-MS and maximum entropy (MaxEnt1) deconvolution of the charge state distributions of the proteins (settings, Gaussian FWHM: 1.0 and resolution: 0.25 Da/channel). The calculated masses are from the primary sequences including a paucimannosidic glycan (Man3GlcNAc2Fuc; 1,038.5 Da) tethered to Asn52 in S2-cells produced uPAR (42).
| uPAR1–87 | Measured mass | Calculated mass | ΔMass |
|---|---|---|---|
| wt | 10,792.00 | 10,792.04 | −0.04 |
| K50C–V70C | 10,769.05 | 10,769.01 | 0.04 |
| T51C–V70C | 10,796.15 | 10,796.08 | 0.07 |
| 10,814.35 | 10,796.08 | 18.27a | |
| C6S–C12S | 10,762.20 | 10,761.93 | 0.27 |
a This mass difference represents uPAR1–87 with a hydrolyzed peptide bond (Tyr57–Arg58), but the fragments (1–57 and 58–87) remain covalently attached via the introduced disulfide bond between Cys51 and Cys70. The corresponding mass spectra are displayed in Fig. S2.
Introducing the 7–8 disulfide bond desensitizes DI to enzymatic deglycosylation and limited proteolysis in intact uPAR
We suspected that the inherent flexibility of DI as well as its assembly with DII and DIII were likely to be perturbed by the constraints introduced by the additional disulfide bond. Several independent lines of experimental evidence support this assumption.
Introducing the 7–8 disulfide bond rendered the glycan attached to DI (Asn52) resistant toward PNGase F-mediated release, when incubating intact uPAR with the deglycosidase under nondenaturing conditions. As shown in Fig. 2A, PNGase F readily removed the glycan attached to DI from full-length uPARwt, but was unable to do so in uPART51C-V70C. Likewise, uPARK50C-V70C was a very poor substrate for PNGase F. Notably, a similar resilience toward PNGase F was acquired by uPARwt when it was driven into its closed conformation either by uPA binding (46) or by insertion of a remote interdomain disulfide bond (uPARH47C-N259C, Fig. 1G) (6). The accessibility of the glycan on Asn52 to PNGase F was thus markedly compromised by the structural constraints originating from the introduction of the 7–8 disulfide bond between β-strands IE and IF (Fig. 1F) and this recapitulated the properties observed for uPAR in complex with uPA. All glycans attached to DII or DIII proved resistant to PNGase F in all tested uPAR variants.
Figure 2.
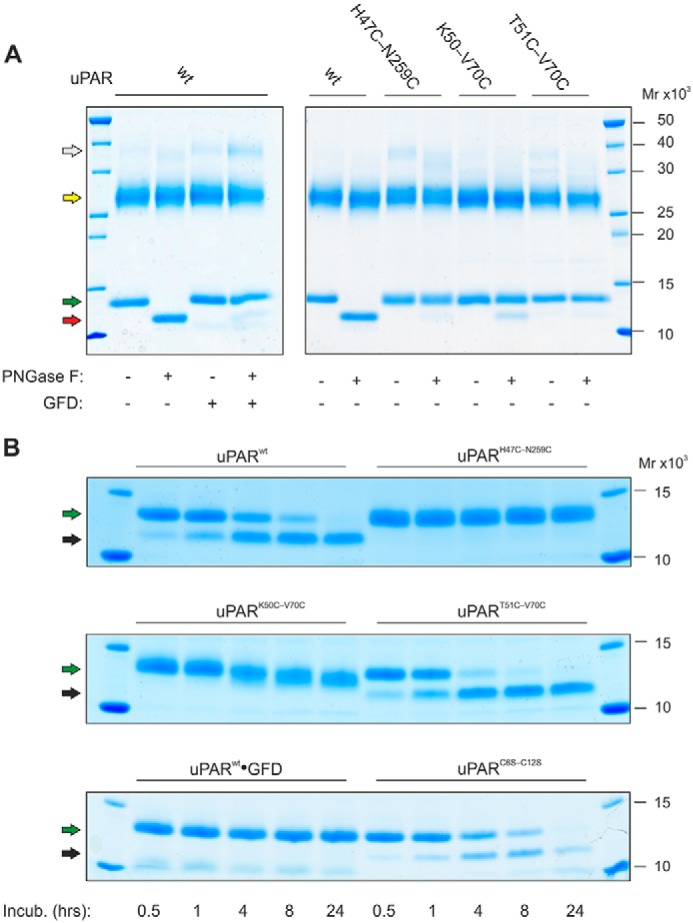
The presence of a 7–8 disulfide bond alters the sensitivity of uPAR to deglycosylation and limited proteolysis. A shows the enzymatic removal of the glycan tethered to Asn52 in intact uPAR under nondenaturing conditions by PNGase F. To illustrate the selective deglycosylation of DI, subsequent incubation with chymotrypsin liberated DI from DIIDIII before analysis by SDS-PAGE of the reduced and alkylated samples. Second to sixth lanes show that in uPARwt the glycan is readily removed, except when uPAR is bound to GFD. Seventh to 12th lanes show that the glycan in uPARH47C-N259C, uPARK50C-V70C, and uPART51-V70C is refractive to PNGase F. B shows the sensitivity of various uPAR variants to cleavage by chymotrypsin under nondenaturing conditions (E:S of 1:750 (w/w)). Colored arrows in A and B highlight the different uPAR fragments: white, intact uPAR; yellow, DIIDIII; green, DI residues 1–87; red, DI residues 1–87 without any glycan; black, DI residues 1–57. Noncropped SDS-PAGE gels are shown in Fig. S3.
Probing the various uPAR mutants by limited proteolysis with chymotrypsin revealed that the cleavage efficiency of the exposed linker region between DI and DII (Tyr87–Ser88) was largely unaffected by the constraints from the additional disulfide bonds or by occupancy with GFD (Fig. 2B). In contrast, the sensitivity toward a secondary cleavage site at Tyr57–Arg58 within loop 3 proved markedly different between the various uPAR mutants. Although uPART51C-V70C appeared slightly more prone to this cleavage compared with uPARwt, uPARH47C-N259C was refractory. We suspect that this resistance reflects that DI remains fully integrated with DIIDIII in uPARH47C-N259C despite having a cleaved linker region between DI and DII (6). Maintaining the compact globular three-domain assembly of intact uPAR would thus shield Tyr57–Arg58 from proteolysis. Aligned with that proposition, we observed that GFD occupancy also prevents the cleavage at this position (Fig. 2B). An unexpected fuzziness in the electrophoretic mobility of the 1–57 fragment from uPARK50C-V70C complicated the kinetic evaluation of the second chymotrypsin cleavage event in this mutant. The origin of this aberrant mobility remains unclear. Mass spectrometry of uPARK50C-V70C digested for the 24 h showed only the expected mass of DI with one internal peptide cleavage (+18 Da). Furthermore, this mass collapsed completely into the expected mass for the 1–57 fragment upon reduction (data not shown).
One plausible mechanism explaining these differences in the sensitivity of Tyr57–Arg58 to chymotrypsin cleavage is that they report on the half-life of the assembled three LU domains after the initial Tyr87–Ser88 cleavage. In one extreme case, DI would remain covalently tethered to DIIDIII thus sterically shielding Tyr57–Arg58 (e.g. in uPARH47C-N259C). In another case, little or no DI would remain attached to DIIDIII thus allowing significant cleavage at this position (e.g. in uPART51C-V70C). To test this possibility, we performed size exclusion chromatography of samples freshly treated with chymotrypsin (Fig. 3). The elution profiles from the size exclusion column revealed: (i) that uPAR DI released by limited proteolysis from uPARwt remains partly associated to DIIDIII (Fig. 3A); (ii) that prior occupancy with GFD greatly enhances this association (Fig. 3C); and (iii) that introduction of the 7–8 consensus disulfide bond completely eliminates the noncovalent association of DI and DIIDIII (Fig. 3, B and D). Combined, these data provide further evidence supporting the notion that the constraints from the 7–8 disulfide bond lower the propensity for the globular assembly of DI-DIIDIII, which to some extent relies on a prominent contribution from βIE to the DI-DII interface (Fig. 1, B and E).
Figure 3.

Size exclusion chromatography reveals that DI remains partly attached to DIIDIII in chymotrypsin-cleaved uPARwt but not in the presence of the 7–8 disulfide bond. A–F show the elution profiles of various uPAR mutants subjected to limited chymotrypsin cleavage from a SuperdexTM 75 HR10/300 size exclusion chromatography (injected 50 μl of 1 mg of uPAR/ml). The insets show silver-stained SDS-polyacrylamide gels of the relevant fractions analyzed after reduction and alkylation (the light gray line at the bottom of the chromatograms identifies the analyzed fractions). Asterisks identify peak fractions and the yellow arrows show DI associated to DIIDIII; white arrows show detached DI. A, uPARwt (note, 20–30% of DI co-elutes with DIIDIII). B, uPART51C-V70C (no co-elution). C, uPARwt in the presence of a 4-fold molar excess of GFD (note, >90% of DI co-elutes with DIIDIII and the peak eluted earlier indicative of the formation of a trimolecular DI·GFD·DIIDIII complex). D, uPARK50C-V70C (no co-elution). E, uPARH47C-N259C (note, 100% DI co-elutes with DIIDIII due to the covalent tether between DI and DIII). F, uPARC6S-C12S (note, 5–10% of DI co-elutes with DIIDIII).
Flexibility in the globular three-domain assembly of uPAR by small angle X-ray scattering
To gain further insights into possible differences in the interdomain flexibility between the various disulfide-constrained uPAR conformers, we performed small angle X-ray scattering (SAXS) analyses. We analyzed both uPAR and uPAR·ATF complexes to compare the domain flexibility before and after ligand-induced compaction of the receptor. To maximize sample monodispersity, we fractionated uPAR and uPAR·ATF complexes by size exclusion chromatography before collecting batch scattering data by SAXS. From the normalized scattering data, we first derived the intra-particle distance distribution function, p(r) providing model-independent information on the shape parameters (Fig. 4). Comparison of the different unoccupied uPAR mutants revealed that only uPARH47C-N259C exhibits a symmetrical, bell-shaped p(r) function indicative of a compact and globular structure with a radius of gyration (Rg) of 22.4 ± 0.1 Å and a maximal particle dimension (Dmax) of 70 ± 5 Å (Table S2). In contrast, reintroducing the missing 7–8 consensus disulfide in uPAR DI did not lead to large scale changes in the overall shape parameters, the p(r) functions for uPARK50C-V70C and uPART51C-V70C are almost superimposable onto that of uPARwt (Fig. 4B). Upon ATF binding, all uPAR disulfide conformers compacted into similar sized particles with a Dmax of 90 Å (Fig. 4D, Tables S1–S4). A small shift to higher distances in the p(r) function of the uPARwt·ATF complex is observed relative to ATF complexes with the uPAR disulfide mutants, but whether this reflects a significant structural difference is unclear. Nonetheless, transforming the scattering data into dimensionless Kratky plots provides a clear ranking of the unoccupied uPAR disulfide conformers into three groups according to their degree of flexibility. This transformation of the SAXS data are particularly useful to obtain a semi-quantitative analysis of the propensity of a given protein to adopt a globular fold (represented by a bell-shaped curve with peak at 1.104 for qRg = √3) or be intrinsically disordered (represented by a hyperbolic curve with a plateau around qRg values of 1.5–2.0). From the Kratky plots presented in Fig. 5A, uPARwt exhibits the greatest flexibility and uPARH47C-N259C is stable and globular. Both uPAR variants with an intact 7–8 disulfide bond show an intermediate flexibility (Fig. 5A). In the presence of ATF, this difference in flexibility largely disappears, as illustrated by the Kratky plots in Fig. 5B. SAXS-driven ensemble modeling of the scattering data by the ensemble optimization method (EOM) yielded a similar conclusion regarding the flexibility of unoccupied uPAR (Fig. 5, C and D). In this analysis, a homogeneous ensemble of relatively compact structures provides an excellent fit to the scattering data for uPARH47C-N259C with low flexibility metrics (Rflex of 47%, relative to the threshold of randomness: 85%, determined from the random pool) calculated from the probability distributions (47). In contrast, a heterogeneous ensemble comprising both extended and compact conformations with a relatively high Rflex of 81% is required to fit the scattering data for uPARwt. Intermediate ensemble compositions with Rflex values of 74 and 73% provide good fits to uPART51C-V70C and uPARK50C-V70C, respectively. These properties are also evident from the variations in shape observed in the refined ab initio surface envelopes reconstructed from the experimental SAXS data (Fig. S4). Extended models are generated for the highly flexible variants (uPARwt, uPART51C-V70C, and uPARK50C-V70C), in contrast to the compact models generated for the less flexible uPARH47C-N259C and all uPAR·ATF complexes.
Figure 4.

Comparison of molecular shape parameters of the uPAR disulfide variants by SAXS. A and B show the SAXS data and real-space distance-distribution functions of the uPAR disulfide variants. C and D show the SAXS data and real-space distribution functions of the uPAR variants in complex with ATF. Shown are experimental data (circles) along with fits of representative ab initio models, reconstructed using DAMMIF (dotted lines), the corresponding shape models are shown in Fig. S4. The lower panels in A and C show the error-weighted residual differences between the model fits and the experimental data.
Figure 5.

Assessing the flexibility of the various uPAR disulfide conformers. Dimensionless Kratky plots of the normalized scattering data for unoccupied uPAR (A) and the corresponding complexes with ATF (B) reveal the relative flexibility of uPAR with different disulfide constraints. C and D show that ensemble representations provide a good description of the relative flexibility of the unoccupied uPAR variants. Fits of optimized ensembles (dotted lines) determined by EOM to the experimental data (circles) are shown (C). The size (Rg) distributions of optimized ensembles (solid lines) relative to a pool of random conformations (shaded area) highlight the decreased flexibility of uPARH47C-N259C (panel D). Increases in the populations of the more compact conformations and changes in the width of the size distributions, relative to that of uPARwt, are observed for each uPAR variant. The lower panel in C shows the error-weighted residual differences between the ensemble fits and the experimental data.
Dynamics of uPAR DI
Previously we showed with hydrogen-deuterium exchange MS (HDX-MS) that uPAR DI undergoes a pronounced change in flexibility during the compaction of intact uPAR that occurs on uPA binding (38). In particular, peptides spanning the third loop of DI (i.e. βIE and βIF) experience significant reductions in their deuterium uptake when uPAR is driven into its compact state by uPA binding (38). Due to the covalent tethering of βIE and βIF by the 7–8 consensus disulfide bond in LU domains, we suspected that it could stabilize the β-sheet between strands E and F in uPAR DI and this could in part be reconciled with the reduced flexibility observed in the Kratky plot of the SAXS data (Fig. 5A). To probe this proposition further, we performed a continuous deuterium labeling of the different uPAR disulfide variants in the presence or absence of saturating levels of GFD and determined the deuterium uptake values by MS after pepsin digestion with special emphasis on peptide(57–66). The deuterium uptake plots for this peptide reveals a considerable variability in the flexibility of βIE and βIF in the different disulfide variants (Fig. 6A). Interestingly, the ranking of the deuterium uptake recapitulates to some extent the flexibility assigned by Kratky plots of the scattering data, i.e. uPART51C-V70C ≥ uPARwt > uPARK50C-V70C > uPARH47C-N259C. Yet one notable difference was apparent. The deuterium uptake for residues 57–66 differed significantly between the two uPAR variants with introduced 7–8 disulfide bonds, uPART51C-V70C exhibiting the far greater uptake thus resembling uPARwt (Fig. 6A). All isotope envelopes are unimodal at the shortest exchange time (10 s) in the presence and absence of GFD (Fig. S6), demonstrating that the amount of misfolded protein is negligible (48). This in turn signifies that the faster exchange kinetics in uPART51C-V70C as compared with uPARK50C-V70C reflects increased dynamics rather than irreversible protein misfolding (Fig. S6).
Figure 6.

Domain flexibility of loop 3 in DI of the various uPAR disulfide variants tested by HDX-MS. A shows the deuterium uptake plots for the peptic peptide(57–66) from unoccupied uPAR at 25 °C and pH 7.4. Shown are data from uPARwt (blue), uPARH47C-N259C (red), uPARK50C-V70C (green), and uPART51C-V70C (orange). For comparison the data for uPARwt·GFD complexes are shown (light gray). This peptide display the larges differences observed between the different uPAR variants. B shows the corresponding deuterium uptake plots for the GFD-bound uPAR. In this graph, the uptake of unoccupied uPARwt is shown (light gray). The hatched black line represents the peptide derived from the fully deuterated protein. C highlights the position of the peptic peptide(57–66) in intact uPAR (blue) where it represents the β-hairpin formed by β-strands βIE and βIF (using PDB code 3BT1). Uptake values are shown with standard deviations. Deuterium uptake plots for other peptic peptides are shown in Fig. S5.
These differences in exchange rates of the 57–66 fragment among the tested uPAR disulfide conformers is almost erased in the corresponding uPAR·GFD complexes (Fig. 6B). The compaction of uPAR by ligand binding thus reduces the macromolecular interdomain flexibility as well as the intradomain flexibility in DI, as monitored by SAXS and HDX-MS, respectively. Deuterium uptake plots for other regions in uPAR are shown in Fig. S5, but none of those show as prominent effects as peptide(57–66) (reporting on βIE and βIF).
Binding kinetics of uPA to the uPAR disulfide variants
To determine the kinetic rate constants for the uPA·uPAR interactions by surface plasmon resonance, we developed a capturing system, which enabled a homogenous presentation of uPAR via an antibody-mediated noncovalent tether onto the biosensor chip. To accomplish this, we chose ATN-615 as capturing mAb for uPAR because its functional epitope is located distant to the uPA-binding cavity (Fig. S7) and it forms a very stable complex with uPAR (koff being 6 × 10−5 s−1, Fig. 7A). As shown in Fig. 7 and Table 2, this experimental setup provided kinetic data of high-end quality and it revealed a tight interaction between uPARwt and ATF1–143 (KD is 20 pm with a kon of 1 × 107 m−1 s−1 and a koff of 2 × 10−4 s−1). As shown in Table 2, all the tested uPAR mutants had comparable association rate constants (kon), but they differed significantly in dissociation rate constants (koff). Notably, reintroduction of the 7–8 consensus disulfide bond at the position defined by sequence predictions led to a >40-fold decrease in the stability of the corresponding uPART51C-V70C·ATF complex as reflected by the greater koff value (Fig. 7, B versus E). Importantly, mutating the positions chosen for the 7–8 disulfide bonds individually to alanine did not recapitulate this gross impairment in stability (i.e. T51A and V70A). In contrast, reintroducing the disulfide bond at the position judged to cause minimal structural perturbation led to a moderate 3.5-fold increase in the koff for the corresponding uPARK50C-V70C·ATF complex (Fig. 7D).
Figure 7.

Kinetics of uPAR·ATF interactions as assessed by surface plasmon resonance. A shows the principle in our capture protocol for assessing the rate constants of an oriented uPAR·ATF interaction by three rounds of single cycle kinetics. Initially, amine-coupled rabbit anti-mouse IgG (RAM) captures the high-affinity anti-uPAR mAb ATN-615 (31), which subsequently captures 50 nm uPAR yielding a binding stoichiometry of approximately two uPAR molecules bound per ATN-615. Finally, injections of five serial 2-fold dilutions of ATF without intermitting regeneration yields the binding curves. The inset shows a cartoon representation of the experimental setup. The double blank referenced sensorgrams are shown for uPARwt (B), uPARH47C-N259C (C), uPARK50C-V70C (D), uPART51C-V70C (E), and uPARC6S-C12S (F). Different colors of the sensorgrams represents different ATF concentrations used for the single cycle setup: 0.03–0.5 nm (blue), 0.06–1.0 nm (red), 0.12–2.0 nm (green), and 0.25–4.0 nm (purple). The thin black line superimposed on each curve represents the experimental fit to a simple bimolecular interaction and the corresponding residuals are in the bottom of the panels.
Table 2.
Kinetics of uPAR·ATF interactions and KD of SMB binding
Analyses with single cycle protocols provided association (kon) and dissociation (koff) rate constants for the interactions between ATF in solution and different uPAR mutants captured on mAb ATN-615. This setup uses 3–4 rounds of single cycle injections each including five serial 2-fold dilutions of ATF1–135, which in combination covers the concentration range of 0.03–2 nm. Fitting with non-linear regression to a simple bimolecular interaction model yielded the kinetic rate constants and the stoichiometry was calculated as the molar ratio between captured ligand and calculated Rmax for the analyte. Fig. 7 shows the corresponding sensorgrams. Equilibrium binding of SMB to uPAR and uPAR·ATF complexes were measured by SPR after capture to the mAb R24 as described in Fig. 8. Standard deviations (shown as ±) refer to parameters derived directly from the fitting procedures.
| uPAR | uPAR·ATF |
uPAR·SMB |
uPAR·ATF·SMB |
|||
|---|---|---|---|---|---|---|
| kon | koff | KD | Stoichiometry | KD | KD | |
| 106m−1s−1 | 10−4 s−1 | nm | μm | |||
| Wt | 11 ± 0.009 | 2.0 ± 0.002 | 0.019 | 0.94 | 3.3 ± 0.1 | 1.8 ± 0.04 |
| T51C–V70C | 10 ± 0.021 | 84 ± 0.19 | 0.800 | 0.88 | 4.8 ± 0.2 | 0.8 ± 0.03 |
| K50C–V70C | 15 ± 0.011 | 7.2 ± 0.005 | 0.050 | 0.67 | 5.9 ± 0.6 | 1.5 ± 0.04 |
| H47C–N259C | 12 ± 0.008 | 1.8 ± 0.001 | 0.015 | 0.94 | 2.0 ± 0.1 | 1.8 ± 0.09 |
| C6S–C12S | 8.4 ± 0.015 | 2.5 ± 0.004 | 0.030 | 0.75 | 3.6 ± 0.1 | 1.9 ± 0.05 |
| H47A | 7.6 ± 0.053 | 2.0 ± 0.001 | 0.027 | 0.72 | 3.3 ± 0.1 | 1.7 ± 0.04 |
| K50A | 13 ± 0.011 | 4.1 ± 0.003 | 0.031 | 0.67 | 3.5 ± 0.1 | 1.4 ± 0.04 |
| T51A | 7.7 ± 0.005 | 6.0 ± 0.004 | 0.078 | 0.79 | 3.9 ± 0.1 | 2.0 ± 0.05 |
| V70A | 4.3 ± 0.002 | 2.2 ± 0.001 | 0.050 | 0.55 | 5.3 ± 0.8 | 2.7 ± 0.34 |
| R91D | 18 ± 0.002 | 2.2 ± 0.002 | 0.013 | 0.88 | No binding | No binding |
| D140A | 11 ± 0.009 | 17 ± 0.015 | 0.170 | 0.67 | 2.8 ± 0.1 | 1.4 ± 0.03 |
| N259A | 8.1 ± 0.006 | 1.3 ± 0.001 | 0.016 | 0.79 | 2.9 ± 0.1 | 1.7 ± 0.05 |
SMB binding to the various disulfide variants of uPAR
To assess the low-affinity binding between the SMB domain of vitronectin and uPAR by surface plasmon resonance (SPR) in solution, we used a slightly different format. In this setup, 2-fold serial dilutions of SMB reached equilibrium binding with uPAR or uPAR·ATF complexes in solution before they were captured on the sensor surface by the anti-uPAR mAb R24 (Fig. 8A). The virtue of this system is that it uses relatively high concentrations of uPAR and ATF (100 and 150 nm), which makes certain that ATF saturates uPAR despite some of the analyzed disulfide mutants having impaired uPA binding, e.g. uPART51C-V70C. In accordance with previous studies (5–7), uPARwt bound SMB with a KD of 3 μm, whereas no binding of SMB to uPARR91D could be detected (Fig. 7B).
Figure 8.

Equilibrium binding of SMB to uPAR and uPAR·ATF complexes by surface plasmon resonance. A shows the principle in our capture protocol for assessing the equilibrium dissociation constant (KD) between uPAR or uPAR·ATF complexes and SMB in solution. In brief, the amine-coupled anti-uPAR mAb R24 captures 100 nm uPAR or 100 nm uPAR·ATF complexes in the presence of 2-fold serial dilutions of SMB (0.3–10 μm; replicates at 1.25 and 2.5 μm) in a multicycle setup. The inset shows the section of the sensorgrams where the dose-dependent binding of SMB is recorded. The SMB-binding isotherms for uPAR and uPAR·ATF complexes are shown for uPARwt and uPARR91D (B), uPARH47C-N259C (C), uPARK50C-V70C (D), uPART51C-V70C (E), and uPARC6S-C12S (F). Solid lines represent SMB-binding isotherms for uPAR and dotted lines those for the corresponding uPAR·ATF complexes.
As shown in Table 2 and Fig. 7, the introduction of the 7–8 consensus disulfide bond in uPAR DI led to a minor decline in the affinity of SMB for intact uPAR. With one exception, all uPAR mutants exhibited an increased affinity for SMB upon saturation with ATF. As reported previously (6), the affinity of uPARH47C-N259C for SMB did not change upon ATF binding as it already presents the higher affinity without bound ATF (Fig. 7C). The present SPR platform is thus capable of detecting the allosteric effect of ATF saturation on the SMB binding to uPAR in solution, albeit the magnitude of this effect is slightly less pronounced compared with the previous solid-phase based detection systems (6, 7). Nonetheless, this analysis clearly showed that the allosteric impact of ATF on SMB binding remain intact in both uPART51C-V70C and uPARK50C-V70C showing that reintroduction of the 7–8 consensus disulfide bond in uPAR DI does not uncouple the beneficial effect of ATF on SMB binding. In fact, binding of ATF managed to compensate for the lower inherent affinity of uPART51C-V70C and uPARK50C-V70C for SMB and restored an affinity comparable with that of uPARwt·ATF (Fig. 7, B versus D and E).
Impact of deleting the 2–3 LU disulfide bond in uPAR DI
Finally, we explored the impact of deleting the 2–3 disulfide bond in uPAR DI, the event that presumably led to neofunctionalization of this LU domain in snake venom α-neurotoxins (41). In all our functional tests, the uPARC6S-C12S mutant behaved essentially as uPARwt despite having only three of the five plesiotypic LU domain disulfides in DI. We found that (i) DI associate with DIIDIII after chymotrypsin cleavage of the linker region in uPARC6S-C12S (Fig. 3F); (ii) the affinity of uPARC6S-C12S for uPA and SMB is comparable with those of uPARwt (Figs. 7F and 8F); and (iii) ATF-binding stimulates SMB binding to the same extent as uPARwt (Fig. 8F). These data indicate that deletion of the 2–3 disulfide bond in uPAR DI has no deleterious effect on the known ligand interactions with uPAR.
Discussion
Loss and gain of disulfide-bonded cysteine residues (half-cystines) occurs very infrequently during evolution. One possible reason for this low frequency is that missense mutations of consensus half-cystines rarely survive selection and become permanently integrated in the genomes due to the deleterious effects of the reactive free thiol group in the partner half-cystine (49). Interestingly, one study found that in 99% of the cases, where missense mutations of half-cystines were maintained during evolution, both pairs of half-cystines were in fact replaced in concert (50). Circumstantial evidence from human genetics on LU domains align well with this proposition. One example illustrating this relationship is the pathological outcome of natural missense mutations in GPIHBP1. This protein has a single LU domain and it plays an essential role in intravascular triglyceride hydrolysis by shuttling the lipoprotein lipase to the capillary lumen (51). Individuals with a dysfunctional GPIHBP1 develop severe chylomicronemia. Notably, the majority of the missense mutations in human GPIHBP1 causing disease involves the replacement of single half-cystines (43) or introduction of an unpaired single cysteine (52). A similar pattern emerges for disease-causing mutations in the secreted single LU-domain protein SLURP1, where the dysfunctional protein is associated with a human skin disorder called mal de Meleda (53).
Notwithstanding the need for a concerted replacement of both half-cystines to eliminate a given disulfide bond, such rare events have in fact occurred in the evolution and diversification of LU domain proteins. The evolution of snake venom α-neurotoxins clearly emphasizes this relationship, where the deletion of the 2–3 disulfide bond allegedly coincided with neofunctionalization and development of high potency toward synapsid neuronal acetylcholine receptors (41). One possible mechanism underlying this association is that the deletion of this particular disulfide bond would relax the LU domain scaffold, which allowed subsequent exploitation of new binding interfaces.
In the present study, we found that elimination of the 2–3 disulfide bond in the first LU domain of uPAR did not have notable functional consequences. Another report showed that elimination of this disulfide bond in the single LU domain protein CD59 also had no impact on its complement regulatory function (44). Based on these studies, it would thus appear that the plesiotypic 2–3 disulfide bond is not essential for the overall structural integrity of LU domain proteins, which would explain why its deletion could cause functional diversification in α-neurotoxins.
Paradoxically, the one plesiotypic LU domain disulfide bond that actually is lost in uPAR is the 7–8 disulfide in DI. In contrast to the 2–3 disulfide, this disulfide bond appears to be indispensable for the correct folding and function of single LU domain proteins in general (45, 51, 53, 54). Furthermore, the deletion of this particular disulfide bond occurs in very few proteins and exclusively in the first LU domain of proteins with 2 or more LU domains e.g. uPAR/PLAUR (31, 35, 40), C4.4A/LYPD3 (55, 56), and Haldisin/LYPD5 (57). Bearing in mind that flexibility between the individual LU domains in uPAR plays an important role for ligand binding (5, 6, 38) it becomes highly pertinent to ask the question: did deletion of this plesiotypic 7–8 disulfide in uPAR DI facilitate the evolution of a high-affinity ligand-binding cavity for uPA by enabling a dynamic assembly of its three LU domains?
To address this question, we first needed to identify the putative positions of the two deleted half-cystines. Our comprehensive alignments of more than 50 annotated orthologous uPAR sequences from the class of Mammalia revealed an invariant distance (5 amino acids) between the 6th and 7th cysteines in the second and third LU domains of all orthologues (not shown, but Fig. 1A, and Figs. S1 and S8 provide representative alignments). Moreover, the 5th and 7th positions after the 6th consensus cysteine were invariantly a lysine and a glycosylated asparagine in uPAR DI (equivalent to Lys50 and Asn52 in human uPAR DI). In contrast, the 6th position, the assumed position of the missing 7th consensus cysteine, varied between species (Fig. S1). The 8th and 9th consensus cysteines were always neighboring residues in those uPAR domains, where all 5 plesiotypic disulfide bonds were preserved. Importantly, uPAR-like orthologues annotated from the class of Reptilia including lizards, snakes, turtles, and crocodilians (Fig. S8) faithfully replicate these properties. It is therefore beyond any reasonable doubt that the correct position, from an evolutionary point view, for the introduction of the missing 7–8 disulfide bond in human uPAR DI is indeed Thr51 and Val70.
The corresponding uPART51C-V70C mutant expressed well in S2-cells and all cysteines were engaged in the expected disulfide bonding in the purified protein. Nonetheless, our biochemical and biophysical analysis on the purified uPART51C-V70C revealed that the disulfide constrained DI behaves very different to that of uPARwt. Among other features, we observed with size exclusion chromatography that the introduction of the 7–8 disulfide abrogated the interdomain interaction between DI and DIIDIII in uPART51C-V70C (Fig. 3). Importantly, we show that the penalty for introducing the missing disulfide in uPAR DI is a >40-fold reduction in its affinity for uPA, which would provide uPART51C-V70C with a KD that is at least 40-fold above the plasma concentration of pro-uPA in humans (58). It would therefore appear that the evolutionary deletion of the plesiotypic 7–8 disulfide bond in uPAR DI was essential for creating a high-affinity binding site for uPA via a flexible assembly of all three LU domains in uPAR. This proposition is well aligned with studies on the co-evolution of uPA and uPAR (32, 59). Based on crystal structures of human and murine uPAR·ATF complexes and extensive mutagenesis (3, 31, 32, 60), the functional hotspot residues in the β-hairpin of GFD for uPAR binding is well characterized in these species. The molecular basis for the species selectivity in the uPAR·uPA interaction between primates and nonprimate mammals is represented by the concerted replacement of the Asn22 → Tyr23 and Trp30 → Arg31 dyad in human and mouse uPA (32) (Fig. S9). Moreover, the majority of the important residues in the uPAR-binding motif of GFD from mammals are also conserved within the class of Reptilia (Fig. S9A) and this class is also the earliest class where a bona fide uPAR orthologue with three LU domains can be traced (59). In this phylogenetic class, uPAR DI has already lost its plesiotypic 7–8 disulfide (Fig. S8). In the class of Sarcopterygii, a uPAR-like protein with three LU domains has recently been identified in the African lungfish Protopterus (59). This protein is unique in the sense that it retains the full signature of an ancient LU domain with all five plesiotypic disulfide bonds present in all three domains (Fig. S8). Examination of the β-hairpin of GFD in the corresponding uPA orthologue reveals that the binding motif known from Mammalia and Reptilia has not yet evolved (Fig. S9A). It is therefore possible that this interaction is not operational in species belonging to lobe-finned fishes, but further functional studies on purified proteins is required before a definitive conclusion can be made.
Evolution of the low-affinity interaction between uPAR and the SMB domain of vitronectin follows a slightly different path. Although the hotspot residues in SMB for the vitronectin interaction with human uPAR is highly conserved all the way to Sarcopterygii (Fig. S10A), the hotspot residue in uPAR (i.e. Arg91 in the linker region between DI and DII) is only conserved within Mammalia (Fig. S10B). It is therefore likely that the co-evolution, which shaped the uPAR-binding site in SMB, originally was driven by the interaction between vitronectin and the cognate uPA-inhibitor plasminogen activator inhibitor type 1, in which the hotspot residue for SMB-binding (i.e. Arg101) is conserved in lobe-finned fishes, bony fishes, and cartilaginous fishes (61). Later in evolution, the class of Mammalia presumably developed the uPAR·vitronectin interaction through convergent evolution.
Experimental procedures
Chemicals and purified proteins
Recombinant human uPAR (residues 1–283) and a panel of mutants thereof were expressed and secreted by Drosophila melanogaster S2 cells and purified from the culture supernatants by affinity chromatography (62). The SMB domain of human vitronectin (residues 1–48) with a C-terminal His6 purification tag was expressed by Pichia pastoris and purified and characterized as described (63). Human pro-uPA1–411 (as the catalytic inactive S356A mutant) and its receptor binding N-terminal fragment ATF1–143 were produced by D. melanogaster S2 cells and purified by affinity chromatography (60). The growth factor-like domain of uPA, GFD (residues 1–48), was a kind gift from Dr. S. Rosenberg. Mouse monoclonal anti-human uPAR antibodies were produced either locally (R24) (5) or was a kind gift from Dr. A. Mazar (ATN-615) (31). Recombinant peptide-N4-(acetyl-β-glucosaminyl)-asparagine amidase (PNGase F; E.C. 3.5.1.52) was from Roche Applied Science (25 units/μg) and α-chrymotrypsin was from Worthington.
Generation of uPAR1–87 by limited proteolysis with chymotrypsin
The N-terminal LU domain of human uPAR (DI, residues 1–87) was excised by limited proteolysis with chymotrypsin using conditions that preferentially led to hydrolysis of the Tyr87–Ser88 peptide bond in uPAR. In brief, 45 μg of intact uPAR was incubated with 9 ng of chymotrypsin (E:S of 1:5000) in 54 μl of PBS, pH 7.4, for 120 min at 25 °C and the released uPAR DI1–87 was isolated by size exclusion chromatography with a SuperdexTM 75 HR10/300 columnTM (GE Healthcare) operated at 0.5 ml/min in PBS (40). Time course experiments were conducted at higher chymotrypsin ratios (E:S of 1:750) for up to 24 h, which led to an additional cleavage at Tyr57–Arg58 in uPARwt.
Deglycosylation of intact uPAR under native conditions
Purified uPAR variants (10 μg in 10 μl of PBS) were incubated for 1 h at 25 °C with 1 unit of PNGase F. To discriminate between effects on DI and DIIDIII, the PNGase F-treated samples were further incubated for 1 h with 2 ng of chymotrypsin (1:5000 (w/w)) to cleave the Tyr87–Ser88 peptide bond in the linker region. Heating at 95 °C in SDS-PAGE sample buffer containing 20 mm DTT stopped the reaction. After cooling and addition of 50 mm iodoacetamide, samples were analyzed by SDS-PAGE and visualized by Coomassie G250 staining.
Surface plasmon resonance
We determined the binding kinetics for the uPA·uPAR interactions with SPR measurements on a Biacore T200TM system (GE Healthcare). To accomplish this, we immobilized a polyclonal rabbit anti-mouse immunoglobulin antibody (GE Healthcare) as the first capture layer (30 μg/ml in 10 mm sodium acetate, pH 5.0) on a CM5 sensor chip using N-hydroxysuccinimide and N-ethyl-N-(3-(diethylamino)propyl)-carbodiimide. This yielded a surface density of 1100 resonance units (RU), which corresponds to 7.3 fmol of mAb/mm2 (assuming one RU ∼ 1 pg/mm2). Injection of 1 m ethanolamine inactivated excess NHS-esters. To prepare the second capture layer, we injected 50 nm of a high-affinity mouse monoclonal anti-uPAR mAb (ATN-615) for 350 s at 20 μl/min in the active flow cell only, which led to ∼0.8 fmol of mAb/mm2 (120 RU). The last capture step involved a 300-s injection of 50 nm uPAR at 20 μl/min in both flow cells, which led to a capture level of 1.6–2.0 uPAR molecules bound per ATN-615 in the active flow cell. Kinetic rate constants for the various uPA·uPAR interactions were determined with single cycle protocols by which five serial 2-fold dilutions of the interaction partner (ATF) were injected for 200 s without intervening regeneration and followed by a longer dissociation phase (1,000–3,000 s dependent on the dissociation rate constant koff). These real-time interactions were measured at 50 μl/min in 10 mm HEPES, 150 mm NaCl, 3 mm EDTA, and 0.05% (v/v) surfactant P-20 at pH 7.4 at 20 °C. Three consecutive injections of 10 mm glycine/HCl, pH 1.7, at the end of each single cycle regenerated the chip.
Fitting of the double blank referenced data by nonlinear regression to a bimolecular interaction model, assuming pseudo-first order reaction conditions, yielded the association (kon) and dissociation (koff) rate constants, the KD (koff/kon), as well as the binding capacity (Rmax). We used the evaluation software supplied with the instrument for global fitting (BiacoreT200 EvaluationTM 3.0).
Our experimental protocol for measuring equilibrium binding between SMB and uPAR or uPAR·ATF complexes relies entirely on steady state conditions in solution between 100 nm uPAR ± 150 nm ATF and serial 2-fold dilutions of SMB (0.3–10 μm). Binding of SMB was monitored by the mass increase of uPAR or uPAR·ATF complexes during capture by the anti-uPAR mAb R24 as a function of the added SMB concentration (37). We used amine chemistry to immobilize R24 directly on a CM5 chip at a density of 651 RU (4.3 fmol of mAb/mm2). We designed this atypical binding protocol to minimize the effects of the vastly different inherent stabilities of the bimolecular uPARwt·ATF and uPART51C-V70C·ATF complexes (see Table 2). Using concentrations of uPAR and ATF that were at least 100-fold above the highest measured KD we made sure that uPAR was saturated with ATF during the steady state binding to SMB in solution.
Hydrogen-deuterium exchange
Purified uPAR was diluted in PBS to a final concentration of 30 μm in the absence or presence of 2-fold molar excess of GFD1–48, and subsequent incubation for 15 min at 25 °C secured complete complex formation. To initiate isotopic exchange, samples were diluted 10-fold in PBS exchange D2O buffer (10 mm Na2HPO4, 150 mm NaCl in D2O, pHread 7.2), which resulted in 90% D2O (v/v) in the final labeling solutions. Aliquots of 60 pmol of uPAR were withdrawn after 10, 100, and 1000 s of incubation at 25 °C. We used acidification and lowered the temperature to efficiently prevent further exchange by adding 1 volume of ice-cold quench buffer (0.1 M Na2HPO4, 0.8 m tris-(2-carboxyethyl)phosphine, 2 m urea in H2O, pH 2.5) to the withdrawn sample and kept it on ice for 3 min to allow reduction of disulfide bonds. Subsequently, the quenched samples were snap frozen in liquid nitrogen and stored at −80 °C until analysis. All samples were labeled in triplicates except uPART51C-V70C, which was measured in duplicates. To prepare an undeuterated control, the samples were prepared as described above except we replaced the PBS exchange D2O buffer with the corresponding protiated solvent. We prepared a full-deuterium control by incubating the sample in the exchange buffer for 72 h at 37 °C before quenching.
Determining deuterium uptake by UPLC-ESI-MS
Quenched and reduced uPAR samples (60 pmol) were thawed and immediately injected into a cooled (0 °C) nanoACQUITY UPLC reversed-phased chromatographic system equipped with HDX technology (Waters, Milford, MA), using an ice-cold syringe to minimize back-exchange. The sample entered a 100-μl injection loop prior to the on-line digestion of the protein, which was carried out by a 2.0 × 20-mm column (IDEX Upchurch Analytical Guard Column, Oak Harbor, WA) packed with agarose-immobilized pepsin (Thermo Scientific Pierce, Rockford, IL), located in a compartment with a temperature of 20 °C. The on-line digestion occurred at a flow rate of 300 μl/min in 0.23% (v/v) formic acid in H2O and the generated peptic peptides were trapped on a short guard column and desalted for 3 min (ACQUITY UPLC BEH C18 VanGuard Pre-Column, 1.7 μm, 2.1 × 5 mm, Waters). Subsequently, the peptides were separated at a flow rate of 40 μl/min on a 1.0 × 50-mm analytical column (ACQUITY UPLC BEH C18, 1.7 μm, 1.0 × 50 mm, Waters) with a 12-min linear gradient from 5–50% (v/v) acetonitrile containing 0.23% (v/v) formic acid. In some experiments, we used a longer analytical column (100 mm).
Eluted peptides were introduced into a quadrupole TOF mass spectrometer (Synapt G2, Waters Corp.) by electrospray ionization. Acquired MS spectra was lock-mass corrected against leucine enkephalin and calibrated against sodium iodide.
Extensive cleaning reduced carryover between runs to less than 5%. In brief, we washed the injection loop with 200 μl of 50% (v/v) MeOH in H2O containing 0.23% (v/v) formic acid, followed by 200 μl of 0.23% (v/v) formic acid followed by a blank gradient.
The peptic peptides from uPAR were identified by collision-induced dissociation with data independent (MSE) acquisition mode. Protein Lynx Global Server 3.0 (PLGS) software (Waters) searched and identified the peptic peptides from uPAR. Data processing with DynamX 3.0 (Waters) determined the deuterium content in each peptide.
Small-angle X-ray scattering
Synchrotron radiation X-ray scattering data were collected on the EMBL P12 beamline of the storage ring PETRA III (DESY, Hamburg) and EMBL X33 beamline of the storage ring DORIS (DESY, Hamburg) (Tables S1–S4), using PILATUS 2M and 1M pixel detectors (DECTRIS, Switzerland), respectively. Batch experiments of uPAR and uPAR·ATF complexes (purified by size exclusion chromatography) were measured in 20 mm PBS, 5% glycerol, pH 7.4, ± 50 mm NaSO4, while flowing through a temperature controlled capillary (P12: 1.2-mm inner diameter, X33: 1.7-mm inner diameter) at 10 °C. Twenty frames of 0.05-s exposure time (P12) or four frames of 30-s exposure time (X33) were collected. The sample-to-detector distance was 2.7 m (P12) and 3.1 m (X33), covering a range of momentum transfer 0.002 Å−1 ≤ s ≥ 0.5 Å−1 and 0.008 Å−1 ≤ s ≥ 0.6 Å−1 (s = 4πsinθ/λ, where 2θ is the scattering angle, and λ = 1.24 Å is the X-ray wavelength), respectively. Based on comparison of successive frames, no detectable radiation damage was observed. Data from the detector were normalized to the transmitted beam intensity, averaged, placed on absolute scale relative to water, and the scattering of buffer solutions subtracted. All data manipulations were performed using PRIMUSqt and the ATSAS software package (64). The forward scattering I(0) and radius of gyration, Rg were determined from Guinier analysis (65) assuming that at very small angles (s ≤ 1.3/Rg) the intensity is represented as I(s) = I(0)exp(−(sRg)2/3)). These parameters were also estimated from the full scattering curves using the indirect Fourier transform method implemented in the program GNOM (66), along with the distance distribution function p(r) and the maximum particle dimensions Dmax. Molecular masses of solutes were estimated from I(0) by computation of partial specific volume and the contrast between the glycosylated protein sequence and the chemical components of the solution using the MULCH server (http://smb-research.smb.usyd.edu.au/NCVWeb/)3 (67). Computation of theoretical scattering intensities was performed using the program CRYSOL (68).
Ab initio shape determination
Low resolution shapes were reconstructed from SAXS data using the programs DAMMIF (69), which represents the macromolecule as a densely packed interconnected configuration of beads or chain-like ensemble of dummy residues, respectively, that best fits the experimental data Iexp(s) by minimizing the discrepancy,
| (Eq. 1) |
where N is the number of experimental points, c is a scaling factor, and Icalc(sj) and σ(sj) are the calculated intensity and the experimental error at the momentum transfer sj, respectively. Multiple modeling runs were conducted to verify the stability of the solution, and to establish the most typical 3D reconstructions according to a spatial discrepancy measure using DAMAVER (70).
Ensemble modeling
Crystal structures of uPAR and uPAR·ATF complex (PDB IDs 2FD6, 3U74, and 3U73) were used as template for flexible modeling of the uPAR domain structures in solution. Glycosylation was introduced into the models using the GLYCOSYLATION routine of ATSAS (64). The program EOM (47) was used to generate a pool of 10,000 uPAR conformations with flexible linkers defined between the three LU domains. A generic algorithm was then employed to select subsets of conformers from the pool that best fit the experimental scattering data for each construct. The flexibility metric, Rflex, was determined for each ensemble as described previously (47), where Rflex = 100% indicates maximum flexibility/uncertainty. SAXS data has been deposited at the SASBDB (www.sasbdb.org)3 with accession codes: SASDAT4, SASDAX4, SASDAU4, SASDF82, SASDF92, SASDFA2, SASDFB2, and SASDFC2.
Author contributions
J. M. L., H. D. M., K. Z. L.-E., T. J. D. J., and M. P. data curation; J. M. L., H. D. M., K. Z. L.-E., and M. P. formal analysis; J. M. L., H. D. M., and K. Z. L.-E. investigation; J. M. L., H. D. M., and K. Z. L.-E. visualization; J. M. L., H. D. M., and K. Z. L.-E. methodology; J. M. L. and M. P. writing-original draft; J. M. L., H. D. M., K. Z. L.-E., T. J. D. J., and M. P. writing-review and editing; T. J. D. J. and M. P. supervision; M. P. conceptualization; M. P. project administration.
Supplementary Material
Acknowledgments
We are grateful to Gry Ellis Rasmussen for excellent technical assistance, Kristian K. Kristensen for running the batch SAXS samples, Simon Mysling for running the early HDX-MS analyses, and Henrik Gårdsvoll for preparing the expression vectors for the uPAR mutants. SAXS data were collected at the EMBL P12 beamline of the storage ring PETRA III and the EMBL X33 beamline of the storage ring DORIS (DESY, Hamburg, Germany).
The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S10 and Tables S2–S5.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- uPAR
- urokinase-type plasminogen activator receptor
- ATF
- amino-terminal fragment of uPA
- LU
- Ly6/uPAR domain
- HDX-MS
- hydrogen–deuterium exchange mass spectrometry
- RU
- resonance units
- SPR
- surface plasmon resonance
- uPA
- urokinase-type plasminogen activator
- uPAR
- uPA receptor
- GPI
- glycosylphosphatidylinositol
- SMB
- somatomedin B
- EOM
- ensemble optimization method
- GFD
- growth factor-like domain
- PNGase F
- peptide-N4-(acetyl-β-glucosaminyl)-asparagine amidase
- PDB
- Protein Data Bank.
References
- 1. Ploug M., Rønne E., Behrendt N., Jensen A. L., Blasi F., and Danø K. (1991) Cellular receptor for urokinase plasminogen activator: carboxyl-terminal processing and membrane anchoring by glycosylphosphatidylinositol. J. Biol. Chem. 266, 1926–1933 [PubMed] [Google Scholar]
- 2. Kjaergaard M., Hansen L. V., Jacobsen B., Gardsvoll H., and Ploug M. (2008) Structure and ligand interactions of the urokinase receptor (uPAR). Front. Biosci. 13, 5441–5461 [DOI] [PubMed] [Google Scholar]
- 3. Connolly B. M., Choi E. Y., Gårdsvoll H., Bey A. L., Currie B. M., Chavakis T., Liu S., Molinolo A., Ploug M., Leppla S. H., and Bugge T. H. (2010) Selective abrogation of the uPA-uPAR interaction in vivo reveals a novel role in suppression of fibrin-associated inflammation. Blood 116, 1593–1603 10.1182/blood-2010-03-276642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. De Lorenzi V., Sarra Ferraris G. M., Madsen J. B., Lupia M., Andreasen P. A., and Sidenius N. (2016) Urokinase links plasminogen activation and cell adhesion by cleavage of the RGD motif in vitronectin. EMBO Rep. 17, 982–998 10.15252/embr.201541681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gårdsvoll H., Jacobsen B., Kriegbaum M. C., Behrendt N., Engelholm L., Østergaard S., and Ploug M. (2011) Conformational regulation of urokinase receptor function: impact of receptor occupancy and epitope-mapped monoclonal antibodies on lamellipodia induction. J. Biol. Chem. 286, 33544–33556 10.1074/jbc.M111.220087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gårdsvoll H., Kjaergaard M., Jacobsen B., Kriegbaum M. C., Huang M., and Ploug M. (2011) Mimicry of the regulatory role of urokinase in lamellipodia formation by introduction of a non-native interdomain disulfide bond in its receptor. J. Biol. Chem. 286, 43515–43526 10.1074/jbc.M111.300020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gårdsvoll H., and Ploug M. (2007) Mapping of the vitronectin-binding site on the urokinase receptor: involvement of a coherent receptor interface consisting of residues from both domain I and the flanking interdomain linker region. J. Biol. Chem. 282, 13561–13572 10.1074/jbc.M610184200 [DOI] [PubMed] [Google Scholar]
- 8. Ferraris G. M., Schulte C., Buttiglione V., De Lorenzi V., Piontini A., Galluzzi M., Podestà A., Madsen C. D., and Sidenius N. (2014) The interaction between uPAR and vitronectin triggers ligand-independent adhesion signalling by integrins. EMBO J. 33, 2458–2472 10.15252/embj.201387611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Madsen C. D., Ferraris G. M., Andolfo A., Cunningham O., and Sidenius N. (2007) uPAR-induced cell adhesion and migration: vitronectin provides the key. J. Cell Biol. 177, 927–939 10.1083/jcb.200612058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Diaz A., Merino P., Manrique L. G., Ospina J. P., Cheng L., Wu F., Jeanneret V., and Yepes M. (2017) A cross talk between neuronal urokinase-type plasminogen activator (uPA) and astrocytic uPA receptor (uPAR) promotes astrocytic activation and synaptic recovery in the ischemic brain. J. Neurosci. 37, 10310–10322 10.1523/JNEUROSCI.1630-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Merino P., Diaz A., Jeanneret V., Wu F., Torre E., Cheng L., and Yepes M. (2017) Urokinase-type plasminogen activator (uPA) binding to the uPA receptor (uPAR) promotes axonal regeneration in the central nervous system. J. Biol. Chem. 292, 2741–2753 10.1074/jbc.M116.761650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Seeds N., Mikesell S., Vest R., Bugge T., Schaller K., and Minor K. (2011) Plasminogen activator promotes recovery following spinal cord injury. Cell Mol. Neurobiol. 31, 961–967 10.1007/s10571-011-9701-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Thornton S., Raghu H., Cruz C., Frederick M. D., Palumbo J. S., Mullins E. S., Almholt K., Usher P. A., and Flick M. J. (2017) Urokinase plasminogen activator and receptor promote collagen-induced arthritis through expression in hematopoietic cells. Blood Adv. 1, 545–556 10.1182/bloodadvances.2016004002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Almholt K., Hebsgaard J. B., Nansen A., Andersson C., Pass J., Rono B., Thygesen P., Pelzer H., Loftager M., Lund I. K., Hoyer-Hansen G., Frisch T., Jensen C. H., Otte K. S., et al. (2018) Antibody-mediated neutralization of uPA proteolytic function reduces disease progression in mouse arthritis models. J. Immunol. 200, 957–965 10.4049/jimmunol.1701317 [DOI] [PubMed] [Google Scholar]
- 15. Wiersinga W. J., Kager L. M., Hovius J. W., van der Windt G. J., de Vos A. F., Meijers J. C., Roelofs J. J., Dondorp A., Levi M., Day N. P., Peacock S. J., and van der Poll T. (2010) Urokinase receptor is necessary for bacterial defense against pneumonia-derived septic melioidosis by facilitating phagocytosis. J. Immunol. 184, 3079–3086 10.4049/jimmunol.0901008 [DOI] [PubMed] [Google Scholar]
- 16. Gyetko M. R., Aizenberg D., and Mayo-Bond L. (2004) Urokinase-deficient and urokinase receptor-deficient mice have impaired neutrophil antimicrobial activation in vitro. J. Leukoc. Biol. 76, 648–656 10.1189/jlb.0104023 [DOI] [PubMed] [Google Scholar]
- 17. Wada T., and Nangaku M. (2015) A circulating permeability factor in focal segmental glomerulosclerosis: the hunt continues. Clin. Kidney J. 8, 708–715 10.1093/ckj/sfv090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wei C., Möller C. C., Altintas M. M., Li J., Schwarz K., Zacchigna S., Xie L., Henger A., Schmid H., Rastaldi M. P., Cowan P., Kretzler M., Parrilla R., Bendayan M., Gupta V., et al. (2008) Modification of kidney barrier function by the urokinase receptor. Nat. Med. 14, 55–63 10.1038/nm1696 [DOI] [PubMed] [Google Scholar]
- 19. Lund I. K., Illemann M., Thurison T., Christensen I. J., and Høyer-Hansen G. (2011) uPAR as anti-cancer target: evaluation of biomarker potential, histological localization, and antibody-based therapy. Curr. Drug Targets 12, 1744–1760 10.2174/138945011797635902 [DOI] [PubMed] [Google Scholar]
- 20. Xu D., Bum-Erdene K., Si Y., Zhou D., Ghozayel M. K., and Meroueh S. O. (2017) Mimicking intermolecular interactions of tight protein-protein complexes for small-molecule antagonists. ChemMedChem. 12, 1794–1809 10.1002/cmdc.201700572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rullo A. F., Fitzgerald K. J., Muthusamy V., Liu M., Yuan C., Huang M., Kim M., Cho A. E., and Spiegel D. A. (2016) Re-engineering the immune response to metastatic cancer: antibody-recruiting small molecules targeting the urokinase receptor. Angew. Chem. Int. Ed. Engl. 55, 3642–3646 10.1002/anie.201510866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jing Y., Chavez V., Ban Y., Acquavella N., El-Ashry D., Pronin A., Chen X., and Merchan J. R. (2017) Molecular effects of stromal-selective targeting by uPAR-retargeted oncolytic virus in breast cancer. Mol. Cancer Res. 15, 1410–1420 10.1158/1541-7786.MCR-17-0016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pilbeam K., Wang H., Taras E., Bergerson R. J., Ettestad B., DeFor T., Borgatti A., Vallera D. A., and Verneris M. R. (2018) Targeting pediatric sarcoma with a bispecific ligand immunotoxin targeting urokinase and epidermal growth factor receptors. Oncotarget 9, 11938–11947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liu S., Ma Q., Fattah R., Bugge T. H., and Leppla S. H. (2017) Anti-tumor activity of anthrax toxin variants that form a functional translocation pore by intermolecular complementation. Oncotarget 8, 65123–65131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ploug M. (2013) Structure-driven design of radionuclide tracers for non-invasive imaging of uPAR and targeted radiotherapy: the tale of a synthetic peptide antagonist. Theranostics 3, 467–476 10.7150/thno.3791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Persson M., Skovgaard D., Brandt-Larsen M., Christensen C., Madsen J., Nielsen C. H., Thurison T., Klausen T. L., Holm S., Loft A., Berthelsen A. K., Ploug M., Pappot H., Brasso K., Kroman N., Højgaard L., and Kjaer A. (2015) First-in-human uPAR PET: imaging of cancer aggressiveness. Theranostics 5, 1303–1316 10.7150/thno.12956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Persson M., Hosseini M., Madsen J., Jørgensen T. J., Jensen K. J., Kjaer A., and Ploug M. (2013) Improved PET imaging of uPAR expression using new 64Cu-labeled cross-bridged peptide ligands: comparative in vitro and in vivo studies. Theranostics 3, 618–632 10.7150/thno.6810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Christensen A., Juhl K., Persson M., Charabi B. W., Mortensen J., Kiss K., Lelkaitis G., Rubek N., von Buchwald C., and Kjaer A. (2017) uPAR-targeted optical near-infrared (NIR) fluorescence imaging and PET for image-guided surgery in head and neck cancer: proof-of-concept in orthotopic xenograft model. Oncotarget 8, 15407–15419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Boonstra M. C., Van Driel P. B. A. A., Keereweer S., Prevoo H. A. J. M., Stammes M. A., Baart V. M., Löwik C., Mazar A. P., van de Velde C. J. H., Vahrmeijer A. L., and Sier C. F. M. (2017) Preclinical uPAR-targeted multimodal imaging of locoregional oral cancer. Oral Oncol. 66, 1–8 10.1016/j.oraloncology.2016.12.026 [DOI] [PubMed] [Google Scholar]
- 30. Kurbegovic S., Juhl K., Chen H., Qu C., Ding B., Leth J. M., Drzewiecki K. T., Kjaer A., and Cheng Z. (2018) Molecular targeted NIR-II probe for image-guided brain tumor surgery. Bioconjug. Chem. 29, 3833–3840 10.1021/acs.bioconjchem.8b00669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Huai Q., Mazar A. P., Kuo A., Parry G. C., Shaw D. E., Callahan J., Li Y., Yuan C., Bian C., Chen L., Furie B., Furie B. C., Cines D. B., and Huang M. (2006) Structure of human urokinase plasminogen activator in complex with its receptor. Science 311, 656–659 10.1126/science.1121143 [DOI] [PubMed] [Google Scholar]
- 32. Lin L., Gårdsvoll H., Huai Q., Huang M., and Ploug M. (2010) Structure-based engineering of species selectivity in the interaction between urokinase and its receptor: implication for preclinical cancer therapy. J. Biol. Chem. 285, 10982–10992 10.1074/jbc.M109.093492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Huai Q., Zhou A., Lin L., Mazar A. P., Parry G. C., Callahan J., Shaw D. E., Furie B., Furie B. C., and Huang M. (2008) Crystal structures of two human vitronectin, urokinase and urokinase receptor complexes. Nat. Struct. Mol. Biol. 15, 422–423 10.1038/nsmb.1404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Barinka C., Parry G., Callahan J., Shaw D. E., Kuo A., Bdeir K., Cines D. B., Mazar A., and Lubkowski J. (2006) Structural basis of interaction between urokinase-type plasminogen activator and its receptor. J. Mol. Biol. 363, 482–495 10.1016/j.jmb.2006.08.063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Llinas P., Le Du M. H., Gårdsvoll H., Danø K., Ploug M., Gilquin B., Stura E. A., and Ménez A. (2005) Crystal structure of the human urokinase plasminogen activator receptor bound to an antagonist peptide. EMBO J. 24, 1655–1663 10.1038/sj.emboj.7600635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liu D., Xu D., Liu M., Knabe W. E., Yuan C., Zhou D., Huang M., and Meroueh S. O. (2017) Small molecules engage hot spots through cooperative binding to inhibit a tight protein-protein interaction. Biochemistry 56, 1768–1784 10.1021/acs.biochem.6b01039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhao B., Gandhi S., Yuan C., Luo Z., Li R., Gårdsvoll H., de Lorenzi V., Sidenius N., Huang M., and Ploug M. (2015) Stabilizing a flexible interdomain hinge region harboring the SMB binding site drives uPAR into its closed conformation. J. Mol. Biol. 427, 1389–1403 10.1016/j.jmb.2015.01.022 [DOI] [PubMed] [Google Scholar]
- 38. Mertens H. D., Kjaergaard M., Mysling S., Gårdsvoll H., Jørgensen T. J., Svergun D. I., and Ploug M. (2012) A flexible multidomain structure drives the function of the urokinase-type plasminogen activator receptor (uPAR). J. Biol. Chem. 287, 34304–34315 10.1074/jbc.M112.398404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Xu X., Gårdsvoll H., Yuan C., Lin L., Ploug M., and Huang M. (2012) Crystal structure of the urokinase receptor in a ligand-free form. J. Mol. Biol. 416, 629–641 10.1016/j.jmb.2011.12.058 [DOI] [PubMed] [Google Scholar]
- 40. Ploug M., Kjalke M., Rønne E., Weidle U., Høyer-Hansen G., and Danø K. (1993) Localization of the disulfide bonds in the NH2-terminal domain of the cellular receptor for human urokinase-type plasminogen activator: a domain structure belonging to a novel superfamily of glycolipid-anchored membrane proteins. J. Biol. Chem. 268, 17539–17546 [PubMed] [Google Scholar]
- 41. Sunagar K., Jackson T. N., Undheim E. A., Ali S. A., Antunes A., and Fry B. G. (2013) Three-fingered RAVERs: rapid accumulation of variations in exposed residues of snake venom toxins. Toxins (Basel) 5, 2172–2208 10.3390/toxins5112172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pawlak J., Mackessy S. P., Fry B. G., Bhatia M., Mourier G., Fruchart-Gaillard C., Servent D., Ménez R., Stura E., Ménez A., and Kini R. M. (2006) Denmotoxin, a three-finger toxin from the colubrid snake Boiga dendrophila (mangrove catsnake) with bird-specific activity. J. Biol. Chem. 281, 29030–29041 10.1074/jbc.M605850200 [DOI] [PubMed] [Google Scholar]
- 43. Beigneux A. P., Fong L. G., Bensadoun A., Davies B. S., Oberer M., Gårdsvoll H., Ploug M., and Young S. G. (2015) GPIHBP1 missense mutations often cause multimerization of GPIHBP1 and thereby prevent lipoprotein lipase binding. Circ. Res. 116, 624–632 10.1161/CIRCRESAHA.116.305085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Petranka J., Zhao J., Norris J., Tweedy N. B., Ware R. E., Sims P. J., and Rosse W. F. (1996) Structure-function relationships of the complement regulatory protein, CD59. Blood Cells Mol. Dis. 22, 281–296 10.1006/bcmd.1996.0111 [DOI] [PubMed] [Google Scholar]
- 45. Grant G. A., Luetje C. W., Summers R., and Xu X. L. (1998) Differential roles for disulfide bonds in the structural integrity and biological activity of κ-bungarotoxin, a neuronal nicotinic acetylcholine receptor antagonist. Biochemistry 37, 12166–12171 10.1021/bi981227y [DOI] [PubMed] [Google Scholar]
- 46. Ploug M., Rahbek-Nielsen H., Nielsen P. F., Roepstorff P., and Dano K. (1998) Glycosylation profile of a recombinant urokinase-type plasminogen activator receptor expressed in Chinese hamster ovary cells. J. Biol. Chem. 273, 13933–13943 10.1074/jbc.273.22.13933 [DOI] [PubMed] [Google Scholar]
- 47. Tria G., Mertens H. D., Kachala M., and Svergun D. I. (2015) Advanced ensemble modelling of flexible macromolecules using X-ray solution scattering. IUCrJ. 2, 207–217 10.1107/S205225251500202X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Trelle M. B., Pedersen S., Osterlund E. C., Madsen J. B., Kristensen S. R., and Jorgensen T. J. (2017) An asymmetric runaway domain swap antithrombin dimer as a key intermediate for polymerization revealed by hydrogen/deuterium-exchange mass spectrometry. Anal. Chem. 89, 616–624 [DOI] [PubMed] [Google Scholar]
- 49. Wong J. W., Ho S. Y., and Hogg P. J. (2011) Disulfide bond acquisition through eukaryotic protein evolution. Mol. Biol. Evol. 28, 327–334 10.1093/molbev/msq194 [DOI] [PubMed] [Google Scholar]
- 50. Rubinstein R., and Fiser A. (2008) Predicting disulfide bond connectivity in proteins by correlated mutations analysis. Bioinformatics 24, 498–504 10.1093/bioinformatics/btm637 [DOI] [PubMed] [Google Scholar]
- 51. Fong L. G., Young S. G., Beigneux A. P., Bensadoun A., Oberer M., Jiang H., and Ploug M. (2016) GPIHBP1 and plasma triglyceride metabolism. Trends Endocrinol. Metab. 27, 455–469 10.1016/j.tem.2016.04.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Plengpanich W., Young S. G., Khovidhunkit W., Bensadoun A., Karnman H., Ploug M., Gårdsvoll H., Leung C. S., Adeyo O., Larsson M., Muanpetch S., Charoen S., Fong L. G., Niramitmahapanya S., and Beigneux A. P. (2014) Multimerization of glycosylphosphatidylinositol-anchored high density lipoprotein-binding protein 1 (GPIHBP1) and familial chylomicronemia from a serine-to-cysteine substitution in GPIHBP1 Ly6 domain. J. Biol. Chem. 289, 19491–19499 10.1074/jbc.M114.558528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Adeyo O., Oberer M., Ploug M., Fong L. G., Young S. G., and Beigneux A. P. (2015) Heterogeneity in the properties of mutant secreted lymphocyte antigen 6/urokinase receptor-related protein 1 (SLURP1) in Mal de Meleda. Br. J. Dermatol. 173, 1066–1069 10.1111/bjd.13868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mevorach D. (2015) Paroxysmal nocturnal hemoglobinuria (PNH) and primary p.Cys89Tyr mutation in CD59: differences and similarities. Mol. Immunol. 67, 51–55 10.1016/j.molimm.2015.03.005 [DOI] [PubMed] [Google Scholar]
- 55. Hansen L. V., Gårdsvoll H., Nielsen B. S., Lund L. R., Danø K., Jensen O. N., and Ploug M. (2004) Structural analysis and tissue localization of human C4.4A: a protein homologue of the urokinase receptor. Biochem. J. 380, 845–857 10.1042/bj20031478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kriegbaum M. C., Jacobsen B., Füchtbauer A., Hansen G. H., Christensen I. J., Rundsten C. F., Persson M., Engelholm L. H., Madsen A. N., Di Meo I., Lund I. K., Holst B., Kjaer A., Lærum O. D., Füchtbauer E. M., and Ploug M. (2016) C4.4A gene ablation is compatible with normal epidermal development and causes modest overt phenotypes. Sci. Rep. 6, 25833 10.1038/srep25833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gårdsvoll H., Kriegbaum M. C., Hertz E. P., Alpízar-Alpízar W., and Ploug M. (2013) The urokinase receptor homolog haldisin is a novel differentiation marker of stratum granulosum in squamous epithelia. J. Histochem. Cytochem. 61, 802–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Huber K., Kirchheimer J., and Binder B. R. (1984) Characterization of a specific anti-human urokinase antibody: development of a sensitive competition radioimmunoassay for urokinase antigen. J. Lab. Clin. Med. 103, 684–694 [PubMed] [Google Scholar]
- 59. Chana-Muñoz A., Jendroszek A., Sønnichsen M., Wang T., Ploug M., Jensen J. K., Andreasen P. A., Bendixen C., and Panitz F. (2019) Origin and diversification of the plasminogen activation system among chordates. BMC Evol. Biol. 19, 27 10.1186/s12862-019-1353-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Gårdsvoll H., Gilquin B., Le Du M. H., Ménez A., Jørgensen T. J., and Ploug M. (2006) Characterization of the functional epitope on the urokinase receptor: complete alanine scanning mutagenesis supplemented by chemical cross-linking. J. Biol. Chem. 281, 19260–19272 10.1074/jbc.M513583200 [DOI] [PubMed] [Google Scholar]
- 61. Jendroszek A., Sønnichsen M. S., Muñoz A. C., Leyman K., Christensen A., Petersen S. V., Wang T., Bendixen C., Panitz F., Andreasen P. A., and Jensen J. K. (2017) Latency transition of plasminogen activator inhibitor type 1 is evolutionarily conserved. Thromb. Haemost. 117, 1688–1699 10.1160/TH17-02-0102 [DOI] [PubMed] [Google Scholar]
- 62. Gårdsvoll H., Werner F., Søndergaard L., Danø K., and Ploug M. (2004) Characterization of low-glycosylated forms of soluble human urokinase receptor expressed in Drosophila Schneider 2 cells after deletion of glycosylation-sites. Protein Expr. Purif 34, 284–295 10.1016/j.pep.2003.12.002 [DOI] [PubMed] [Google Scholar]
- 63. Kjaergaard M., Gårdsvoll H., Hirschberg D., Nielbo S., Mayasundari A., Peterson C. B., Jansson A., Jørgensen T. J., Poulsen F. M., and Ploug M. (2007) Solution structure of recombinant somatomedin B domain from vitronectin produced in Pichia pastoris. Protein Sci. 16, 1934–1945 10.1110/ps.072949607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Petoukhov M. V., Franke D., Shkumatov A. V., Tria G., Kikhney A. G., Gajda M., Gorba C., Mertens H. D., Konarev P. V., and Svergun D. I. (2012) New developments in the ATSAS program package for small-angle scattering data analysis. J. Appl. Crystallogr 45, 342–350 10.1107/S0021889812007662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Guinier A. (1939) La diffraction des rayons X aux très petits angles: application à l'étude de phénomènes ultramicroscopiques. Ann. Phys. 11, 161–237 10.1051/anphys/193911120161 [DOI] [Google Scholar]
- 66. Svergun D. I. (1992) Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 25, 495–503 10.1107/S0021889892001663 [DOI] [Google Scholar]
- 67. Whitten A. E., Cai S., and Trewhella J. (2008) MULCh: modules for the analysis of small-angle neutron contrast variation data from biomolecular assemblies. J. Appl. Crystallogr. 41, 222–226 10.1107/S0021889807055136 [DOI] [Google Scholar]
- 68. Svergun D. I., Barberato C., and Koch M. H. J. (1995) CRYSOL: a program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Crystallogr. 28, 768–773 10.1107/S0021889895007047 [DOI] [Google Scholar]
- 69. Franke D., and Svergun D. I. (2009) DAMMIF, a program for rapid ab initio shape determination in small-angle scattering. J. Appl. Crystallogr. 42, 342–346 10.1107/S0021889809000338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Volkov V. V., and Svergun D. I. (2003) Uniqueness of ab-initio shape determination in small-angle scattering. J. Appl. Crystallogr. 36, 860–864 10.1107/S0021889803000268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Bourne Y., Talley T. T., Hansen S. B., Taylor P., and Marchot P. (2005) Crystal structure of a Cbtx-AChBP complex reveals essential interactions between snake α-neurotoxins and nicotinic receptors. EMBO J. 24, 1512–1522 10.1038/sj.emboj.7600620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Ducancel F., Mérienne K., Fromen-Romano C., Trémeau O., Pillet L., Drevet P., Zinn-Justin S., Boulain J. C., and Ménez A. (1996) Mimicry between receptors and antibodies. Identification of snake toxin determinants recognized by the acetylcholine receptor and an acetylcholine receptor-mimicking monoclonal antibody. J. Biol. Chem. 271, 31345–31353 10.1074/jbc.271.49.31345 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.