

Abstract
Many proteins, such as RNA-binding proteins, have complex folding landscapes. How cells maintain the solubility and folding state of such proteins, particularly under stress conditions, is largely unknown. Here, we argue that prion-like low-complexity regions (LCRs) are key regulators of protein solubility and folding. We discuss emerging evidence that prion-like LCRs are not, as commonly thought, autonomous aggregation modules that adopt amyloid-like conformations, but protein-specific sequences with chaperone-like functions. On the basis of recent findings, we propose that prion-like LCRs have evolved to regulate protein phase behavior and to protect proteins against proteotoxic damage.
Keywords: protein aggregation, protein misfolding, neurodegenerative disease, prion, chaperone, Prion-like protein, protein misfolding disease, protein phase separation, protein phase transition, RNA-binding protein
Introduction
All proteins are synthesized by the ribosome as linear chains of amino acids that fold into complex three-dimensional structures. These structures frequently have complex folding landscapes, and the native structures are often marginally stable. This marginal stability may be a result of the necessity of proteins to exert their functions (1). Protein functionality often requires the binding of ligands and switching between alternative conformations. At the same time, the conformational heterogeneity of the polypeptide chain increases the probability of protein misfolding and aggregation.
The cytoplasm of cells is a crowded environment that contains an extremely high concentration of proteins and other macromolecules (2). Proteins in such crowded environments show rich phase behavior. They can assemble into higher-order structures that adopt different physical states such as liquids, gels, and glasses, and these states often have functional roles as membraneless compartments or organelles (3–5). However, because many proteins are barely soluble in the highly crowded cytoplasm, the cytoplasm is always on the verge of transitioning into an aggregated state, especially under stress conditions.
How do cells ensure the integrity of proteins in an environment that is always on the verge of catastrophe? One solution is the group of molecular chaperones, proteins that help other proteins adopt and maintain the native state. Different classes of chaperones exist: sophisticated ATP-driven machines that assist protein folding, such as the GroE/Chaperonin, Hsp70, and Hsp90 chaperones, and ATP-independent holdases, such as small heat-shock proteins, that prevent aggregation of proteins by shielding them from aberrant interactions (6). The role of chaperones in regulating the solubility and folding of proteins is well established. But is this the only way in which cells can regulate the solubility and folding state of proteins?
In this review, we argue that regions of low sequence complexity (LCRs)2 play a key role in regulating the solubility and folding state of proteins. LCRs are intrinsically disordered and abundant in eukaryotic proteomes. The special class of prion-like LCRs has gained a lot of attention in recent years because these LCRs have been linked to various neurodegenerative diseases. The current view of prion-like LCRs is that they have evolved as autonomous protein aggregation modules that assemble into higher-order structures by adopting β-sheet–rich conformations. Here, we take an entirely different view and hypothesize that prion-like LCRs have evolved to counteract protein aggregation and promote protein solubility and folding. We propose that prion-like LCRs are protein-specific sequences with chaperone-like functions that regulate protein phase behavior and protect proteins from damage.
Discovery of prion-like LCRs: the case of yeast prions
Prion-like LCRs were first discovered in connection with prion proteins in budding yeast. The history of yeast prions begins with two genetic traits in budding yeast, called [URE3] (7, 8) and [PSI+] (9). These traits were inherited in a non-Mendelian fashion in mating experiments. Later studies showed that these unusual phenotypes are based on self-propagating aggregates of two proteins called Ure2 and Sup35 (10–13). Proteins that can form self-propagating aggregates are also known as prions.
Aggregation of the prion protein in humans causes a spectrum of devastating neurodegenerative diseases called prion diseases (14, 15). In yeast, prions are the basis of diverse heritable phenotypes, some of which may be advantageous (16–20). A unifying feature of prions in yeast and in humans is their ability to assemble into amyloid fibrils that are characterized by the presence of tightly packed cross–β-sheets that are arranged perpendicular to the fibril axis (21). The presence of amyloid is a common feature of many human disorders, including Alzheimer's and Parkinson's disease. Amyloid formation is a rare event that requires the formation of a nucleus (22). Once formed, this nucleus can convert other molecules of the same protein to the prion state. This self-templating ability allows prion aggregates to propagate between cells and tissues over long time scales and gives prions properties that are otherwise only known from nucleic acids, such as heritability, infectivity, and the formation of distinct strains.
Prions in yeast can confer adaptive advantages in some circumstances, but they can also be detrimental in others (16, 23, 24). For example, prion formation by the translation termination factor Sup35 inactivates the protein and mediates stop-codon readthrough (17, 18). This was proposed to be advantageous under some environmental conditions because it allows variation in gene expression. However, the adaptive significance of yeast prions has been questioned repeatedly. One early argument against the biological utility of yeast prions was the absence of prions in wild strains (25–28). Later work described heritable traits with hallmarks of prions in a large fraction of wild strains (29), but many of these prion phenotypes could not be reproduced (28). Thus, the adaptive value of prions remains a contentious issue.
Genetic and biochemical experiments showed that the prion behavior of the Sup35 and Ure2 prion proteins is restricted to specific domains. These so-called prion domains carry all the information for prion behavior and thus were genetically transferable to other proteins (30–33). Close inspection of prion domains revealed that they have a low sequence complexity and are enriched in polar amino acid residues such as glutamine, asparagine, serine, and tyrosine (34, 35). The low sequence complexity and the absence of hydrophobic amino acids suggested that prion domains are largely unfolded (36, 37). Importantly, scrambling prion domain sequences revealed that the amino acid composition and not the exact sequence determines its prion behavior (38, 39). Thus, a consensus view emerged that prion domains are modular, transferable, and randomizable sequences that can stochastically convert from a disordered state into a highly-ordered prion state.
The recognition that prion domains have distinctive amino acid compositions motivated the search for other prions in yeast. The first comprehensive screen was based on a Hidden Markov Model that was trained on the sequence of three known yeast prions (34). This algorithm identified ∼200 prion-like candidates in the yeast proteome. The candidates were tested for their prion properties by replacing the prion domain of Sup35 with candidate prion domains to determine whether they can mimic the [PSI+] phenotype. This led to the discovery of two dozen new prions. This screen was followed by a phenotypic screen that was agnostic to primary sequence (40). It yielded ∼80 protein-based traits with prion behavior. Importantly, many of these proteins were not enriched for glutamine and asparagine residues as the candidates identified in the previous screen. Despite this difference, the prion properties were mostly confined to LCRs. Together, these studies showed that yeast contains many LCRs with prion-like properties.
The picture that emerged over the years is that prion-like LCRs are independent functional units that encode self-templating aggregates. Consequently, scientists working with these proteins have taken a reductionist approach, focusing mostly on isolated LCRs or amyloidogenic peptide fragments. This view has shaped the thinking of researchers for many years and led to the pervasive idea that prion-like LCRs have evolved to function as autonomous protein aggregation modules. However, as we will argue in this review, this view may only apply to a minority of prion-like LCRs.
Prion-like LCRs as aggregation-promoting modules
Out of the 100 prion candidates that were tested for prion properties, only a quarter formed prions (34). What is the function of those candidate LCRs that did not show prion behavior? LCRs without prion properties still formed assemblies upon overexpression, suggesting that the majority of LCRs are able to form assemblies, yet those are distinct and different from prion aggregates.
The prion algorithm was later applied to the human proteome. This effort revealed more than 200 human proteins that were compositionally similar to yeast prions (41–45). More than a quarter of these proteins are annotated as RNA-binding proteins. What is remarkable is that the aggregation of many of these prion-like RNA–binding proteins has been linked to age-related neurodegenerative diseases, including amyotrophic lateral sclerosis (ALS) and fronto-temporal dementia (FTD). These diseases involve the proteins TDP-43 (TAR DNA-binding protein), FUS (RNA-binding protein fused in sarcoma), TAF-15 (TATA-binding protein associated factor N2), EWSR1 (Ewing sarcoma breakpoint region 1), and hnRNPA1 (heterogeneous nuclear ribonucleoprotein A1) (46). Further studies showed that these proteins have an increased ability to aggregate. For example, TDP-43 normally localizes to the nucleus, but in ALS and FTD patients it mislocalizes to the cytoplasm and forms protein aggregates (47). The prion-like LCR of TDP-43 was subsequently shown to be aggregation-prone (48), and it frequently carries mutations in patients that increase the aggregation behavior of TDP-43 (46). Importantly, the aggregates formed by TDP-43 had properties of amyloid-like fibers, and they showed prion-like behavior in cell culture systems and in animals (49, 50).
Have the prion-like LCRs of these human proteins evolved to form prions? This seems unlikely given that the prion states of these proteins have so far only been associated with disease, suggesting that their LCRs must have other functional roles. Recent studies provided evidence that prion-like proteins may play a role in forming specialized compartments, referred to as membraneless compartments (3–5). Examples of such compartments are DNA damage repair sites and ribonucleoprotein (RNP) granules such as stress granules (SGs) that form in the cytoplasm upon stress. Evidence suggested that many prion-like proteins, including TDP-43 and FUS, localize to SGs, but how they contribute to the assembly of SGs remained mysterious for a while.
A first insight into the role of prion-like proteins in RNP granule formation was provided by a serendipitous discovery using the chemical b-isox (51–53). This chemical selectively precipitated components of RNP granules, including FUS and TDP-43. Further experiments showed that b-isox binds to prion-like LCRs and induces their conversion into a crystalline amyloid-like state. This led McKnight and co-workers (51–53) to hypothesize that prion-like LCRs form amyloid-like assemblies that drive RNP assembly. This conclusion was based on the finding that the LCR of FUS assembled into gel-like structures in vitro, and EM studies and X-ray diffraction patterns revealed that these gels contained amyloid-like fibrils.
One feature made these amyloid-like fibrils unusual: they were much more labile than previously reported amyloid structures (51, 52). Recent work has provided high-resolution structural insight into the assembled form of the isolated LCR of FUS (54–56). This revealed key structural elements called kinked β-sheets that assemble into extended protofilaments. Unlike the steric zippers of previously described prion fibrils, the kinked β-sheets interact only weakly through polar and aromatic side chains. This was proposed to allow reversibility and regulation of these structures through post-translational modifications such as phosphorylation (54, 56).
This new model for the role of prion-like LCRs deviates from the original yeast prion concept with respect to the lower stability of the structures. However, it still postulates that prion-like LCRs are the main determinants of specificity and are protein-independent aggregation modules that can be studied out of the context of the full-length protein.
LCRs as organizers of membrane-less organelles
The hypothesis that LCRs function as protein aggregation domains that polymerize through amyloid-like interactions is problematic for several reasons. One reason is that there is only little experimental evidence that LCRs polymerize under physiological conditions in living cells. Furthermore, most of the existing evidence is based on in vitro experiments that were performed under extreme conditions, such as high protein concentrations and nonphysiological buffer conditions (54, 56). This raises questions whether the gels and polymers observed in vitro are physiological structures or rather represent aberrant/pathological states. In fact, membrane-less compartments such as SGs are extremely dynamic in living human cells. Photobleaching experiments showed that some SG components like FUS and hnRNPA1 turn over on the order of seconds (57, 58). This was difficult to reconcile with the idea that these proteins polymerize to form relatively stable β-sheet–rich structures.
These discrepancies led researchers to look for other modes of assembly. Phase separation has emerged as an alternative to explain the formation of membraneless compartments. Phase separation occurs when a well-mixed solution of proteins demixes into two coexisting phases, one that is enriched for the protein and one that is depleted of it (3, 4). Indeed, experiments with FUS showed that this protein phase separates to form liquid droplets at physiological protein concentrations (Fig. 1A) (57). These droplets were highly dynamic and exhibited fusion and fission behavior and wetted surfaces. Importantly, NMR experiments demonstrated that the assembly of the FUS LCR into liquid droplets does not require the formation of cross–β-sheet structures (59). Instead, liquid droplet formation was proposed to be driven by weak and transient interactions among LCRs, involving, for example, dipole–dipole interactions between polar atoms and π–π interactions between aromatic residues (60). However, many structural biologists viewed these findings with skepticism. It seemed difficult to imagine that such interactions could provide sufficient specificity for the formation of RNP granules in the complex environment of the cell.
Figure 1.

A, schematic representation of a low-complexity region-containing RNA-binding protein (RNP). The LCR is depicted in magenta and the RBD in cyan. The RNP can self-assemble to form biomolecular condensates, such as liquid-like droplets. B, both the isolated LCR (left) and full-length RNP can self-assemble into liquid-like droplets, yet the LCR phase separates at much higher protein concentrations compared with the full-length protein. C, liquid droplets formed by the isolated LCR are predominantly stabilized by π–π interactions of aromatic residues. Alternatively, a kinked β-sheet structure is suggested to drive the assembly of liquid-like droplets of isolated LCR (middle). In contrast, liquid-like condensates of the full-length RNP are stabilized by the interaction between aromatic residues within the LCR and cation residues within the RBD. Note, the interaction of aromatic and cation stickers occurs in trans and may very well also occur in cis. Importantly, the full-length RNP phase separates at physiological concentrations.
The idea that LCRs are the key drivers of RNP granule formation was pervasive in 2015, when the first studies on reconstituted condensates were published. Indeed, early in vitro experiments to study protein phase behavior were often performed with isolated LCRs (59, 61–64). Although these LCRs phase-separated on their own, it required very high protein concentrations that were orders of magnitude higher than the physiological protein concentration (Fig. 1B). A recent study suggests that this focus on the LCRs may have been misleading (65). This study showed that condensation of FUS and related proteins involves heterotypic interactions among prion-like LCRs and other domains in the same or other proteins (Fig. 1B). Using extensive mutagenesis, the authors found that the interactions occur among tyrosine residues in the N-terminal prion-like LCR and arginine residues in the C-terminal RNA-binding domain. They proposed a sticker and spacer model in which the phase behavior of the FUS family of proteins is driven by specific interactions among associative motifs called stickers that are interspersed by spacers (Fig. 1C). These spacers mainly impart flexibility but make little contributions to the driving forces for phase separation. In the FUS family of proteins, the stickers are tyrosine and arginine residues, and the spacers are polar residues such as serine, glutamine, as well as glycine (Fig. 1C). This shows that phase separation of the FUS family of proteins depends on collective interactions among specific amino acid residues, many of which are located outside of prion-like LCRs.
The presence of a collective molecular grammar that is encoded by intrinsically disordered domains suggests that specificity can arise in the absence of distinct structures. However, whether the protein-intrinsic molecular grammar generates enough specificity for the formation of RNP granules in cells is unclear. Indeed, recent findings suggest that domains other than LCRs play important roles in condensate formation. For example, binding of RNA to prion-like RNA-binding proteins has major effects on protein solubility and phase behavior in living cells (66, 67). Enzymatic removal of RNA from the nucleus led to rapid phase separation of FUS and related RNA-binding proteins, suggesting that nuclear RNA keeps these proteins in an unassembled state. Moreover, condensate formation by FUS was shown to depend on high local concentrations of specific RNAs such as Neat1 (66). Thus, it is conceivable that phase separation in cells is primarily driven by RNA scaffolds that recruit FUS and other family members, increasing their local protein concentration so that these proteins can phase separate.
FUS phase separation is also very sensitive to post-translational modifications. For example, methylation of arginine residues within the FUS RBD inhibits phase separation and reduces FUS partitioning into SGs (68). Moreover, the association of FUS with its nuclear import factor karyopherin-β2 suppresses FUS phase separation (68–70). Importantly, the import factor binding sites in FUS overlap with the residues involved in phase separation, thus providing an explanation for its inhibitory effect on FUS phase separation (70). Strikingly, association of FUS with its import factor is not only sufficient to suppress phase separation, but it is even able to dissolve hydrogels formed by FUS and related proteins (69). This strengthens the notion that interactions among the PLD and the RBD of FUS compete with a diverse set of interactions and post-translational modifications that regulate phase separation.
Does this mean that cross–β-interactions are not required for FUS condensate formation? Several studies reported that the liquid droplet state of FUS and other prion-like proteins is unstable and converts with time into a gel-like or solid-like fibrillar state (57, 58). Importantly, the transition into more solid states was accelerated by disease-associated mutations. This suggests that gels and fibrils of FUS reflect pathological states. In agreement with this idea, mutations that were predicted to disrupt the cross–β-structures of the FUS LCR (54) inhibited gel and fibril formation, but did not affect the formation of liquid droplets by phase separation (65). This suggests that FUS liquid droplet formation is not critically dependent on cross–β-sheets and indicates that these cross–β-structures rather stabilize droplets or may even be involved in a pathological transition. Therefore, it seems possible that cross–β-sheet interactions sometimes play a role in compartment formation, but the physiological relevance of cross–β-sheet interactions, particularly for FUS, remains unclear.
In summary, these findings suggest that LCRs are key modules that regulate the solubility and the phase behavior of prion-like proteins. In the FUS family of proteins, LCRs function as intrinsically disordered regions that support collective interactions among adhesive amino acid motifs. Importantly, the LCRs do not have to self-interact to promote condensation, as it is still widely assumed, but function by interacting with other parts of these proteins. This suggests that LCRs are not independent modules that drive protein assembly but protein-specific modifier sequences. These modifier LCRs synergize with other protein regions and their sequences likely coevolve with these regions to regulate protein solubility and phase behavior.
PLDs as regulators of protein solubility and phase behavior
Key insights into the idea that prion-like LCRs are modifiers of protein phase behavior has come from studies with budding yeast. In contrast to human cells, yeast cells live in very unstable and stressful environments. This leads to sudden changes in internal conditions, such as pH, osmotic conditions, ionic strength, or temperature. Such fluctuations have a strong impact on the material properties of the cytoplasm, sometimes causing a phase transition of the cytoplasm that can induce a complete stiffening of the cell (71).
The mechanism underlying this phase transition of the cytoplasm is best understood for starved yeast cells. When yeast cells are deprived of energy, they can no longer remove protons from the cytosol through ATP-driven pumps, which leads to a rapid drop of the cytosolic pH from neutral to acidic values (71, 72). The pH change decreases the solubility of proteins, thus causing macromolecular assembly and the formation of intracellular structures such as SGs (71, 73). However, in contrast to SGs in mammalian cells, yeast SGs have solid-like properties (73). The stiffening of the cytoplasm is thought to result from the large number of proteins that adopt a solid-like state in stressed yeast cells. Importantly, many of the proteins that assemble into higher-order structures upon stress are prion-like proteins.
One of the prion-like proteins that assembles into higher-order structures in stressed yeast is poly(A)-binding protein (Pab1), a known component of SGs. A recent study showed that Pab1 phase separates and forms gels in vitro upon exposure to physiological stress conditions such as temperature increase and low pH (74). Interestingly, phase separation of Pab1 was driven by determinants in the RNA-binding RNA recognition motifs (RRMs) and not by Pab1's LCR. Importantly, mutations in this proline-rich LCR profoundly changed the biophysical properties of Pab1 phase separation, but it did not prevent it. Together, this provides strong evidence that Pab1 phase separates upon stress and that phase separation is modulated but not caused by its LCR.
Another recent study focused on the prion-like SG protein poly(U)-binding protein Pub1 (75). Similar to Pab1, physiological stresses such as changes in temperature and pH caused the formation of Pub1 condensates. Removal of the proline-rich LCRs in Pub1 rendered the protein more insoluble and increased its propensity to form condensates. The presented data are consistent with a model in which the RRMs drive Pub1 self-assembly, whereas the prion-like LCRs of Pub1 function as modifiers of phase separation. This suggests that in Pub1, the LCRs have an important solubilizing and regulatory function, but they do not drive condensate formation.
Together, these findings point toward a general principle where cells harness the criticality of phase separation to detect sudden fluctuations in physical or chemical parameters. This suggests that Pub1 and Pab1 have been shaped by evolution to detect specific changes in the environment. Condensate formation by Pab1 and Pub1 has been proposed to be adaptive, presumably by releasing previously repressed RNAs that encode stress-protective factors (74–76). The ability of Pub1 and Pab1 to sense environmental changes requires that the solubility of these proteins is tuned to the conditions that exist in growing cells. At the same time, the protein solubility has to be close to the critical threshold for phase separation, so that the proteins can condense when the conditions change. This suggests that there is a strong evolutionary pressure to adjust the solubility of Pub1 and Pab1 to a level that is favorable for both growth and rapid stress sensing.
Prion-like LCRs were previously described as autonomous domains that contain all information for prion behavior and promote amyloid-like aggregation. Given the new findings discussed above, can we generalize that LCRs function as modulators of protein solubility? To address this question, we revisited the functional role of the prion domain of the canonical prion protein Sup35. We found that Sup35 indeed forms condensates by pH-induced phase separation as a response to sudden stress in vitro and in vivo (77). In agreement with an important role of the prion domain for the overall protein solubility, the isolated catalytic guanosine triphosphatase (GTPase) domain of Sup35 already exhibited reduced solubility under nonstress conditions and aggregated in the absence of the prion domain upon stress. Consequently, cells lacking the prion domain exhibited impaired translational activity and a growth defect when recovering from stress. These data demonstrate that the prion domain rescues the essential GTPase domain of Sup35 from irreversible aggregation: 1) by increasing the overall solubility of the essential but aggregation-prone GTPase domain, and 2) by extending its phase behavior from irreversible aggregation toward reversible condensation. Thus, the prion domain ensures that the translation termination factor remains functional during harsh environmental conditions. Importantly, the ability to form condensates is shared among distantly related budding yeast and fission yeast, suggesting that condensate formation and not prion formation is the conserved and ancestral function of the prion domain of Sup35. In agreement with this, previous studies have shown that prion properties appear only sporadically in distantly related species, whereas the prion-like LCRs are conserved (78–81).
The picture that emerges is that the folded domains of LCR-containing proteins exhibit a strong tendency to self-associate under various conditions. The necessity of these folded domains to interact with a natural ligand, such as RNA, nucleotides, and other binding partners, may impose evolutionary constraints that make these domains highly aggregation-prone. It appears that LCRs act as regulatory elements that inhibit aberrant behavior of these domains, allowing these proteins to maintain protein structure and function in normal conditions and upon stress (Fig. 2, A and B). In agreement with this notion, it had previously been recognized that the aggregation of huntingtin exon 1 is quantitatively modulated by its flanking regions (82). Like the LCRs of Pab1 and Pub1, the C-terminal flanking region of exon 1 is proline-rich and increases the solubility of huntingtin, thus reducing the overall driving force for aggregation (82). In fact, cells frequently seem to use LCRs to harness the criticality of protein phase separation and mount adaptive stress responses. In summary, these findings suggest that prion and prion-like LCRs are modifiers of protein phase transitions that adjust the solubility of proteins and protect proteins from misfolding, thus allowing cells to respond to sudden changes in physico-chemical conditions.
Figure 2.
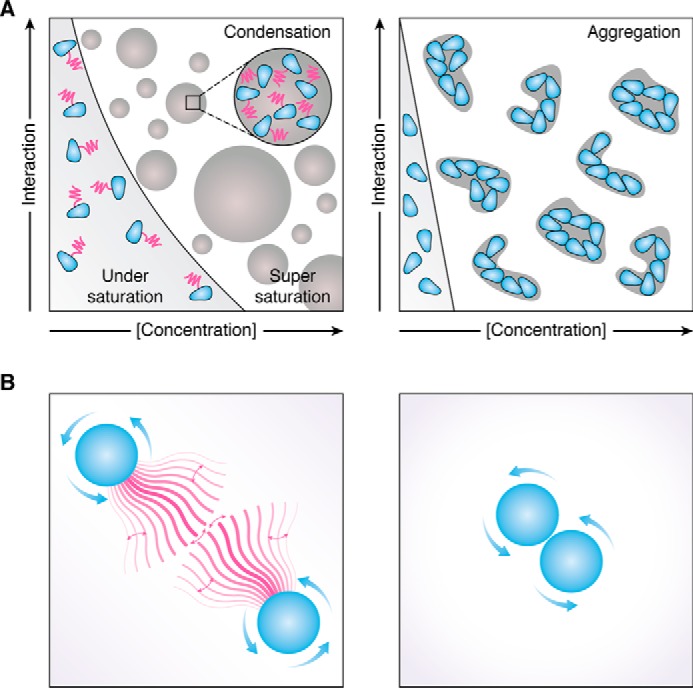
A, schematic representation of the phase behavior of a protein (blue) with LCR (magenta) (left) and the same protein without LCR (right). In the presence of the LCR the protein remains soluble over a wide range of concentrations and other parameters regulating its self-interaction (e.g. ionic strength, pH, temperature, and post-translational modifications). Upon crossing the saturation concentration, the LCR inhibits the strong self-assembly and allows the assemblies to adopt a reversible liquid-like or gel-like state. In the absence of the LCR (right), the soluble concentration range is substantially smaller, and the protein forms irreversible aggregates upon crossing the saturation concentration. B, LCR (magenta) could act as an entropic brush that extends the protein (left) and thus effectively increases the solubility and reversibility of assemblies. Collapse and extension of the brush further modulate the interaction. In the absence of the LCR (right), the proteins can approach each other much more closely.
Autonomous aggregation modules or modifiers of phase behavior?
The findings discussed above force us to develop a new view of prion-like LCRs. Most prion-like LCRs do not function as autonomous modules that drive assembly reactions. Rather, they function as modifier sequences that regulate the solubility of phase-separating proteins and modulate the material properties of condensates.
In agreement with this idea, prion-like LCRs are frequently associated with proteins that are supersaturated (83, 84). Supersaturation refers to a state of a protein solution in which the solution is kinetically trapped in a soluble state, despite the protein concentration being higher than its thermodynamic solubility. Upon breaking supersaturation, protein aggregates form, and the concentration of the protein decreases until it reaches thermodynamic solubility. Protein aggregation is caused by the folded non-LCR part of the proteins, which often have complex and only marginally stable folds. This marginal stability may arise from a need to interact with ligands (1, 85). For example, Pub1 and Pab1 carry multiple RRMs, which must associate with RNAs; Sup35 has a C-terminal GTPase domain that requires GTP-binding for stability. We speculate that prion-like LCRs have been added to these proteins to allow a deeper level of supersaturation despite their aggregation-prone nature. As a result, these proteins can now be soluble at higher levels than normally tolerated (Fig. 2, A and B). However, these proteins are still very sensitive to changing conditions and will thus become insoluble when the physical-chemical environment changes suddenly.
Does this mean that prion domains have not been selected for their prion properties? Considering the example of Sup35 and other related prions, there indeed seems to be a very high kinetic barrier to amyloid and prion formation. Induction of the prion state usually requires overproduction of the protein at levels that are orders of magnitude higher than normal. This is because protein overproduction increases the likelihood that these proteins will adopt a self-templating fold. Although some studies showed that prions are also detectable under physiological conditions, the large majority of these prion states appears only at very low frequencies. Thus, it seems that prion proteins have evolved a high kinetic barrier to prevent the formation of self-sustaining amyloids (22, 86, 87). By contrast, the barrier for the formation of condensates such as liquids, gels, or glasses is very low. This barrier is readily broken by post-translational modifications or changes in salt, pH, or temperature, and it can be harnessed by cells to promote adaptive responses.
Why are RNA- and DNA-binding proteins so frequently associated with prion-like LCRs? We speculate that solubility may be a special problem for this group of proteins because of their complex structure and abundance. For example, FUS has a long C-terminal region that contains several different RNA-binding domains. In addition, this region contains many RGG repeats resulting in a high amount of positively charged amino acids, a feature that drastically reduces protein solubility (88). The tyrosine-rich LCR may have been added to FUS to increase its solubility and allow controlled condensate formation by phase separation. The fact that prion-like RNA-binding proteins such as FUS are highly supersaturated may also explain why they cause diseases. Because of their supersaturated state, the appearance of self-templating aggregates must be prevented at all costs. However, once a self-templating aggregate has formed, these proteins enter into a state of catastrophic self-sustaining aggregation. Only under these conditions do the prion-like LCRs turn into autonomous aggregation domains that replicate highly-ordered amyloid folds.
Thus, we conclude that in most prion-like proteins, the formation of self-templating aggregates is prevented by the presence of a high kinetic barrier. However, this does not exclude that evolution sometimes lowers this kinetic barrier to allow for regulated formation of self-sustaining aggregates. In fact, this seems to be true for some prion-like proteins, such as Xvelo and Rim4 (89, 90). Future studies will have to determine which LCRs have evolved to function as true prion domains and which have evolved to modify the solubility and folding of proteins. We predict that LCRs with chaperone-like functions are highly abundant in nature and are frequently found in organisms that live in unstable environments. How these organisms use their LCRs to explore new and unstable environments will be a fascinating research direction in the future.
Acknowledgments
We thank Christiane Iserman, Edgar Boczek, Jie Wang, Jordina Guillén-Boixet, and Shovamayee Maharana for critical comments on the manuscript.
This work was supported by the Max Planck Society, European Research Council Grants 725836 and 643417, Bundesministerium für Bildung und Forschung Grants 01ED1601A and 031A359A, and the Joint Programme Neurodegenerative Disease (CureALS). This article is part of the thematic series, Phase separation of RNA-binding proteins in physiology and disease. The authors declare that they have no conflicts of interest with the contents of this article.
- LCR
- low-complexity region
- RNP
- ribonucleoprotein
- SG
- stress granule
- FUS
- fused in sarcoma
- RRM
- RNA recognition motif
- RBD
- RNA-binding domain
- ALS
- amyotrophic lateral sclerosis
- FTD
- fronto-temporal dementia.
References
- 1. Foit L., Morgan G. J., Kern M. J., Steimer L. R., von Hacht A. A., Titchmarsh J., Warriner S. L., Radford S. E., and Bardwell J. C. (2009) Optimizing protein stability in vivo. Mol. Cell 36, 861–871 10.1016/j.molcel.2009.11.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ellis R. J. (2001) Macromolecular crowding: an important but neglected aspect of the intracellular environment. Curr. Opin. Struct. Biol. 11, 114–119 10.1016/S0959-440X(00)00172-X [DOI] [PubMed] [Google Scholar]
- 3. Banani S. F., Lee H. O., Hyman A. A., and Rosen M. K. (2017) Biomolecular condensates: organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 18, 285–298 10.1038/nrm.2017.7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shin Y., and Brangwynne C. P. (2017) Liquid phase condensation in cell physiology and disease. Science 357, eaaf4382 10.1126/science.aaf4382 [DOI] [PubMed] [Google Scholar]
- 5. Alberti S. (2017) The wisdom of crowds: regulating cell function through condensed states of living matter. J. Cell Sci. 130, 2789–2796 10.1242/jcs.200295 [DOI] [PubMed] [Google Scholar]
- 6. Balchin D., Hayer-Hartl M., and Hartl F. U. (2016) In vivo aspects of protein folding and quality control. Science 353, aac4354 10.1126/science.aac4354 [DOI] [PubMed] [Google Scholar]
- 7. Aigle M., and Lacroute F. (1975) Genetical aspects of [URE3], a non-mitochondrial, cytoplasmically inherited mutation in yeast. Mol. Gen. Genet. 136, 327–335 10.1007/BF00341717 [DOI] [PubMed] [Google Scholar]
- 8. Lacroute F. (1971) Non-mendelian mutation allowing ureidosuccinic acid uptake in yeast. J. Bacteriol. 106, 519–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cox B. S. (1965) Ψ, a cytoplasmic suppressor of super-suppressor in yeast. Heredity 20, 505 10.1038/hdy.1965.65 [DOI] [Google Scholar]
- 10. Wickner R. B. (1994) [URE3] as an altered URE2 protein: evidence for a prion analog in Saccharomyces cerevisiae. Science 264, 566–569 10.1126/science.7909170 [DOI] [PubMed] [Google Scholar]
- 11. Patino M. M., Liu J. J., Glover J. R., and Lindquist S. (1996) Support for the prion hypothesis for inheritance of a phenotypic trait in yeast. Science 273, 622–626 10.1126/science.273.5275.622 [DOI] [PubMed] [Google Scholar]
- 12. Ter-Avanesyan M. D., Dagkesamanskaya A. R., Kushnirov V. V., and Smirnov V. N. (1994) The SUP35 omnipotent suppressor gene is involved in the maintenance of the non-Mendelian determinant [psi+] in the yeast Saccharomyces cerevisiae. Genetics 137, 671–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chernoff Y. O., Lindquist S. L., Ono B., Inge-Vechtomov S. G., and Liebman S. W. (1995) Role of the chaperone protein Hsp104 in propagation of the yeast prion-like factor (psipositive). Science 268, 880–884 10.1126/science.7754373 [DOI] [PubMed] [Google Scholar]
- 14. Aguzzi A., and Lakkaraju A. K. K. (2016) Cell biology of prions and prionoids: a status report. Trends Cell Biol. 26, 40–51 10.1016/j.tcb.2015.08.007 [DOI] [PubMed] [Google Scholar]
- 15. Prusiner S. B. (1982) Novel proteinaceous infectious particles cause scrapie. Science 216, 136–144 10.1126/science.6801762 [DOI] [PubMed] [Google Scholar]
- 16. Newby G. A., and Lindquist S. (2013) Blessings in disguise: biological benefits of prion-like mechanisms. Trends Cell Biol. 23, 251–259 10.1016/j.tcb.2013.01.007 [DOI] [PubMed] [Google Scholar]
- 17. True H. L., and Lindquist S. L. (2000) A yeast prion provides a mechanism for genetic variation and phenotypic diversity. Nature 407, 477–483 10.1038/35035005 [DOI] [PubMed] [Google Scholar]
- 18. True H. L., Berlin I., and Lindquist S. L. (2004) Epigenetic regulation of translation reveals hidden genetic variation to produce complex traits. Nature 431, 184–187 10.1038/nature02885 [DOI] [PubMed] [Google Scholar]
- 19. Tuite M. F., and Serio T. R. (2010) The prion hypothesis: from biological anomaly to basic regulatory mechanism. Nat. Rev. Mol. Cell Biol. 11, 823–833 10.1038/nrm3007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jarosz D. F., Brown J. C. S., Walker G. A., Datta M. S., Ung W. L., Lancaster A. K., Rotem A., Chang A., Newby G. A., Weitz D. A., Bisson L. F., and Lindquist S. (2014) Cross-kingdom chemical communication drives a heritable, mutually beneficial prion-based transformation of metabolism. Cell 158, 1083–1093 10.1016/j.cell.2014.07.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Eisenberg D., and Jucker M. (2012) The amyloid state of proteins in human diseases. Cell 148, 1188–1203 10.1016/j.cell.2012.02.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Serio T. R., Cashikar A. G., Kowal A. S., Sawicki G. J., Moslehi J. J., Serpell L., Arnsdorf M. F., and Lindquist S. L. (2000) Nucleated conformational conversion and the replication of conformational information by a prion determinant. Science 289, 1317–1321 10.1126/science.289.5483.1317 [DOI] [PubMed] [Google Scholar]
- 23. Shorter J., and Lindquist S. (2005) Prions as adaptive conduits of memory and inheritance. Nat. Rev. Genet. 6, 435–450 10.1038/nrg1616 [DOI] [PubMed] [Google Scholar]
- 24. Harvey Z. H., Chen Y., and Jarosz D. F. (2018) Protein-based inheritance: epigenetics beyond the chromosome. Mol. Cell 69, 195–202 10.1016/j.molcel.2017.10.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wickner R. B., Edskes H. K., Bateman D., Kelly A. C., and Gorkovskiy A. (2011) The yeast prions [PSI+] and [URE3] are molecular degenerative diseases. Prion 5, 258–262 10.4161/pri.17748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. McGlinchey R. P., Kryndushkin D., and Wickner R. B. (2011) Suicidal [PSI+] is a lethal yeast prion. Proc. Natl. Acad. Sci. U.S.A. 108, 5337–5341 10.1073/pnas.1102762108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nakayashiki T., Kurtzman C. P., Edskes H. K., and Wickner R. B. (2005) Yeast prions [URE3] and [PSI+] are diseases. Proc. Natl. Acad. Sci. U.S.A. 102, 10575–10580 10.1073/pnas.0504882102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wickner R. B., Shewmaker F. P., Bateman D. A., Edskes H. K., Gorkovskiy A., Dayani Y., and Bezsonov E. E. (2015) Yeast prions: structure, biology, and prion-handling systems. Microbiol. Mol. Biol. Rev. 79, 1–17 10.1128/MMBR.00041-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Halfmann R., Jarosz D. F., Jones S. K., Chang A., Lancaster A. K., and Lindquist S. (2012) Prions are a common mechanism for phenotypic inheritance in wild yeasts. Nature 482, 363–368 10.1038/nature10875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li L., and Lindquist S. (2000) Creating a protein-based element of inheritance. Science 287, 661–664 10.1126/science.287.5453.661 [DOI] [PubMed] [Google Scholar]
- 31. Santoso A., Chien P., Osherovich L. Z., and Weissman J. S. (2000) Molecular basis of a yeast prion species barrier. Cell 100, 277–288 10.1016/S0092-8674(00)81565-2 [DOI] [PubMed] [Google Scholar]
- 32. Edskes H. K., Gray V. T., and Wickner R. B. (1999) The [URE3] prion is an aggregated form of Ure2p that can be cured by overexpression of Ure2p fragments. Proc. Natl. Acad. Sci. U.S.A. 96, 1498–1503 10.1073/pnas.96.4.1498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Osherovich L. Z., and Weissman J. S. (2001) Multiple Gln/Asn-rich prion domains confer susceptibility to induction of the yeast [PSI+] prion. Cell 106, 183–194 10.1016/S0092-8674(01)00440-8 [DOI] [PubMed] [Google Scholar]
- 34. Alberti S., Halfmann R., King O., Kapila A., and Lindquist S. (2009) A systematic survey identifies prions and illuminates sequence features of prionogenic proteins. Cell 137, 146–158 10.1016/j.cell.2009.02.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Michelitsch M. D., and Weissman J. S. (2000) A census of glutamine/asparagine-rich regions: implications for their conserved function and the prediction of novel prions. Proc. Natl. Acad. Sci. U.S.A. 97, 11910–11915 10.1073/pnas.97.22.11910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mukhopadhyay S., Krishnan R., Lemke E. A., Lindquist S., and Deniz A. A. (2007) A natively unfolded yeast prion monomer adopts an ensemble of collapsed and rapidly fluctuating structures. Proc. Natl. Acad. Sci. U.S.A. 104, 2649–2654 10.1073/pnas.0611503104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Baxa U., Ross P. D., Wickner R. B., and Steven A. C. (2004) The N-terminal prion domain of Ure2p converts from an unfolded to a thermally resistant conformation upon filament formation. J. Mol. Biol. 339, 259–264 10.1016/j.jmb.2004.03.033 [DOI] [PubMed] [Google Scholar]
- 38. Ross E. D., Edskes H. K., Terry M. J., and Wickner R. B. (2005) Primary sequence independence for prion formation. Proc. Natl. Acad. Sci. U.S.A. 102, 12825–12830 10.1073/pnas.0506136102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ross E. D., Baxa U., and Wickner R. B. (2004) Scrambled prion domains form prions and amyloid. Mol. Cell. Biol. 24, 7206–7213 10.1128/MCB.24.16.7206-7213.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chakrabortee S., Byers J. S., Jones S., Garcia D. M., Bhullar B., Chang A., She R., Lee L., Fremin B., Lindquist S., and Jarosz D. F. (2016) Intrinsically disordered proteins drive emergence and inheritance of biological traits. Cell 167, 369–381.e12 10.1016/j.cell.2016.09.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lancaster A. K., Nutter-Upham A., Lindquist S., and King O. D. (2014) PLAAC: a web and command-line application to identify proteins with prion-like amino acid composition. Bioinformatics 30, 2501–2502 10.1093/bioinformatics/btu310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Couthouis J., Hart M. P., Shorter J., DeJesus-Hernandez M., Erion R., Oristano R., Liu A. X., Ramos D., Jethava N., Hosangadi D., Epstein J., Chiang A., Diaz Z., Nakaya T., Ibrahim F., et al. (2011) A yeast functional screen predicts new candidate ALS disease genes. Proc. Natl. Acad. Sci. U.S.A. 108, 20881–20890 10.1073/pnas.1109434108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Malinovska L., Kroschwald S., and Alberti S. (2013) Protein disorder, prion propensities, and self-organizing macromolecular collectives. Biochim. Biophys. Acta 1834, 918–931 10.1016/j.bbapap.2013.01.003 [DOI] [PubMed] [Google Scholar]
- 44. Kim H. J., Kim N. C., Wang Y.-D., Scarborough E. A., Moore J., Diaz Z., MacLea K. S., Freibaum B., Li S., Molliex A., Kanagaraj A. P., Carter R., Boylan K. B., Wojtas A. M., Rademakers R., et al. (2013) Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 495, 467–473 10.1038/nature11922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. King O. D., Gitler A. D., and Shorter J. (2012) The tip of the iceberg: RNA-binding proteins with prion-like domains in neurodegenerative disease. Brain Res. 1462, 61–80 10.1016/j.brainres.2012.01.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Taylor J. P., Brown R. H. Jr., and Cleveland D. W. (2016) Decoding ALS: from genes to mechanism. Nature 539, 197–206 10.1038/nature20413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Neumann M., Sampathu D. M., Kwong L. K., Truax A. C., Micsenyi M. C., Chou T. T., Bruce J., Schuck T., Grossman M., Clark C. M., McCluskey L. F., Miller B. L., Masliah E., Mackenzie I. R., Feldman H., et al. (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133 10.1126/science.1134108 [DOI] [PubMed] [Google Scholar]
- 48. Johnson B. S., Snead D., Lee J. J., McCaffery J. M., Shorter J., and Gitler A. D. (2009) TDP-43 is intrinsically aggregation-prone, and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity. J. Biol. Chem. 284, 20329–20339 10.1074/jbc.M109.010264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nonaka T., Masuda-Suzukake M., Arai T., Hasegawa Y., Akatsu H., Obi T., Yoshida M., Murayama S., Mann D. M., Akiyama H., and Hasegawa M. (2013) Prion-like properties of pathological TDP-43 aggregates from diseased brains. Cell Rep. 4, 124–134 10.1016/j.celrep.2013.06.007 [DOI] [PubMed] [Google Scholar]
- 50. Feiler M. S., Strobel B., Freischmidt A., Helferich A. M., Kappel J., Brewer B. M., Li D., Thal D. R., Walther P., Ludolph A. C., Danzer K. M., and Weishaupt J. H. (2015) TDP-43 is intercellularly transmitted across axon terminals. J. Cell Biol. 211, 897–911 10.1083/jcb.201504057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kato M., and McKnight S. L. (2018) A solid-state conceptualization of information transfer from gene to message to protein. Annu. Rev. Biochem. 87, 351–390 [DOI] [PubMed] [Google Scholar]
- 52. Kato M., Han T. W., Xie S., Shi K., Du X., Wu L. C., Mirzaei H., Goldsmith E. J., Longgood J., Pei J., Grishin N. V., Frantz D. E., Schneider J. W., Chen S., Li L., et al. (2012) Cell-free formation of RNA granules: low complexity sequence domains form dynamic fibers within hydrogels. Cell 149, 753–767 10.1016/j.cell.2012.04.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Han T. W., Kato M., Xie S., Wu L. C., Mirzaei H., Pei J., Chen M., Xie Y., Allen J., Xiao G., and McKnight S. L. (2012) Cell-free formation of RNA granules: bound RNAs identify features and components of cellular assemblies. Cell 149, 768–779 10.1016/j.cell.2012.04.016 [DOI] [PubMed] [Google Scholar]
- 54. Murray D. T., Kato M., Lin Y., Thurber K. R., Hung I., McKnight S. L., and Tycko R. (2017) Structure of FUS protein fibrils and its relevance to self-assembly and phase separation of low-complexity domains. Cell 171, 615–627.e16 10.1016/j.cell.2017.08.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hughes M. P., Sawaya M. R., Boyer D. R., Goldschmidt L., Rodriguez J. A., Cascio D., Chong L., Gonen T., and Eisenberg D. S. (2018) Atomic structures of low-complexity protein segments reveal kinked β sheets that assemble networks. Science 359, 698–701 10.1126/science.aan6398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Luo F., Gui X., Zhou H., Gu J., Li Y., Liu X., Zhao M., Li D., Li X., and Liu C. (2018) Atomic structures of FUS LC domain segments reveal bases for reversible amyloid fibril formation. Nat. Struct. Mol. Biol. 25, 341–346 10.1038/s41594-018-0050-8 [DOI] [PubMed] [Google Scholar]
- 57. Patel A., Lee H. O., Jawerth L., Maharana S., Jahnel M., Hein M. Y., Stoynov S., Mahamid J., Saha S., Franzmann T. M., Pozniakovski A., Poser I., Maghelli N., Royer L. A., Weigert M., et al. (2015) A liquid-to-solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell 162, 1066–1077 10.1016/j.cell.2015.07.047 [DOI] [PubMed] [Google Scholar]
- 58. Molliex A., Temirov J., Lee J., Coughlin M., Kanagaraj A. P., Kim H. J., Mittag T., and Taylor J. P. (2015) Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 163, 123–133 10.1016/j.cell.2015.09.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Burke K. A., Janke A. M., Rhine C. L., and Fawzi N. L. (2015) Residue-by-residue view of in vitro FUS granules that bind the C-terminal domain of RNA polymerase II. Mol. Cell 60, 231–241 10.1016/j.molcel.2015.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Brangwynne C. P., Tompa P., and Pappu R. V. (2015) Polymer physics of intracellular phase transitions. Nat. Phys. 11, 899–904 10.1038/nphys3532 [DOI] [Google Scholar]
- 61. Ryan V. H., Dignon G. L., Zerze G. H., Chabata C. V., Silva R., Conicella A. E., Amaya J., Burke K. A., Mittal J., and Fawzi N. L. (2018) Mechanistic view of hnRNPA2 low-complexity domain structure, interactions, and phase separation altered by mutation and arginine methylation. Mol. Cell 69, 465–479.e7 10.1016/j.molcel.2017.12.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Protter D. S. W., Rao B. S., Van Treeck B., Lin Y., Mizoue L., Rosen M. K., and Parker R. (2018) Intrinsically disordered regions can contribute promiscuous interactions to RNP granule assembly. Cell Rep. 22, 1401–1412 10.1016/j.celrep.2018.01.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lin Y., Protter D. S., Rosen M. K., and Parker R. (2015) Formation and maturation of phase-separated liquid droplets by RNA-binding proteins. Mol. Cell 60, 208–219 10.1016/j.molcel.2015.08.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lin Y., Currie S. L., and Rosen M. K. (2017) Intrinsically disordered sequences enable modulation of protein phase separation through distributed tyrosine motifs. J. Biol. Chem. 292, 19110–19120 10.1074/jbc.M117.800466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wang J., Choi J. M., Holehouse A. S., Lee H. O., Zhang X., Jahnel M., Maharana S., Lemaitre R., Pozniakovsky A., Drechsel D., Poser I., Pappu R. V., Alberti S., and Hyman A. A. (2018) A molecular grammar underlying the driving forces for phase separation of prion-like RNA binding proteins. Cell 174, 688–699.e16 10.1016/j.cell.2018.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Maharana S., Wang J., Papadopoulos D. K., Richter D., Pozniakovsky A., Poser I., Bickle M., Rizk S., Guillén-Boixet J., Franzmann T. M., Jahnel M., Marrone L., Chang Y.-T., Sterneckert J., Tomancak P., et al. (2018) RNA buffers the phase separation behavior of prion-like RNA binding proteins. Science 360, 918–921 10.1126/science.aar7366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Langdon E. M., Qiu Y., Ghanbari Niaki A., McLaughlin G. A., Weidmann C. A., Gerbich T. M., Smith J. A., Crutchley J. M., Termini C. M., Weeks K. M., Myong S., and Gladfelter A. S. (2018) mRNA structure determines specificity of a polyQ-driven phase separation. Science 360, 922–927 10.1126/science.aar7432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Hofweber M., Hutten S., Bourgeois B., Spreitzer E., Niedner-Boblenz A., Schifferer M., Ruepp M.-D., Simons M., Niessing D., Madl T., and Dormann D. (2018) Phase separation of FUS is suppressed by its nuclear import receptor and arginine methylation. Cell 173, 706–719.e13 10.1016/j.cell.2018.03.004 [DOI] [PubMed] [Google Scholar]
- 69. Guo L., Kim H. J., Wang H., Monaghan J., Freyermuth F., Sung J. C., O'Donovan K., Fare C. M., Diaz Z., Singh N., Zhang Z. C., Coughlin M., Sweeny E. A., DeSantis M. E., Jackrel M. E., et al. (2018) Nuclear-import receptors reverse aberrant phase transitions of RNA-binding proteins with prion-like domains. Cell 173, 677–692.e20 10.1016/j.cell.2018.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Yoshizawa T., Ali R., Jiou J., Fung H. Y. J., Burke K. A., Kim S. J., Lin Y., Peeples W. B., Saltzberg D., Soniat M., Baumhardt J. M., Oldenbourg R., Sali A., Fawzi N. L., Rosen M. K., and Chook Y. M. (2018) Nuclear import receptor inhibits phase separation of FUS through binding to multiple sites. Cell 173, 693–705.e22 10.1016/j.cell.2018.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Munder M. C., Midtvedt D., Franzmann T., Nüske E., Otto O., Herbig M., Ulbricht E., Müller P., Taubenberger A., Maharana S., Malinovska L., Richter D., Guck J., Zaburdaev V., and Alberti S. (2016) A pH-driven transition of the cytoplasm from a fluid- to a solid-like state promotes entry into dormancy. Elife 5, e09347 10.7554/eLife.09347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Dechant R., Binda M., Lee S. S., Pelet S., Winderickx J., and Peter M. (2010) Cytosolic pH is a second messenger for glucose and regulates the PKA pathway through V-ATPase. EMBO J. 29, 2515–2526 10.1038/emboj.2010.138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kroschwald S., Maharana S., Mateju D., Malinovska L., Nüske E., Poser I., Richter D., and Alberti S. (2015) Promiscuous interactions and protein disaggregases determine the material state of stress-inducible RNP granules. Elife 4, e06807 10.7554/eLife.06807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Riback J. A., Katanski C. D., Kear-Scott J. L., Pilipenko E. V., Rojek A. E., Sosnick T. R., and Drummond D. A. (2017) Stress-triggered phase separation is an adaptive, evolutionarily tuned response. Cell 168, 1028–1040.e19 10.1016/j.cell.2017.02.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Kroschwald S., Munder M. C., Maharana S., Franzmann T. M., Richter D., Ruer M., Hyman A. A., and Alberti S. (2018) Different material states of Pub1 condensates define distinct modes of stress adaptation and recovery. Cell Rep. 23, 3327–3339 10.1016/j.celrep.2018.05.041 [DOI] [PubMed] [Google Scholar]
- 76. Rabouille C., and Alberti S. (2017) Cell adaptation upon stress: the emerging role of membrane-less compartments. Curr. Opin. Cell Biol. 47, 34–42 10.1016/j.ceb.2017.02.006 [DOI] [PubMed] [Google Scholar]
- 77. Franzmann T. M., Jahnel M., Pozniakovsky A., Mahamid J., Holehouse A. S., Nüske E., Richter D., Baumeister W., Grill S. W., Pappu R. V., Hyman A. A., and Alberti S. (2018) Phase separation of a yeast prion protein promotes cellular fitness. Science 359, eaa05654 10.1126/science.aao5654 [DOI] [PubMed] [Google Scholar]
- 78. Edskes H. K., Khamar H. J., Winchester C.-L., Greenler A. J., Zhou A., McGlinchey R. P., Gorkovskiy A., and Wickner R. B. (2014) Sporadic distribution of prion-forming ability of Sup35p from yeasts and fungi. Genetics 198, 605–616 10.1534/genetics.114.166538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Wickner R. B., Edskes H. K., Shewmaker F., Kryndushkin D., and Nemecek J. (2009) Prion variants, species barriers, generation and propagation. J. Biol. 8, 47 10.1186/jbiol148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Edskes H. K., Engel A., McCann L. M., Brachmann A., Tsai H.-F., and Wickner R. B. (2011) Prion-forming ability of Ure2 of yeasts is not evolutionarily conserved. Genetics 188, 81–90 10.1534/genetics.111.127217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Safadi R. A., Talarek N., Jacques N., and Aigle M. (2011) Yeast prions: could they be exaptations? The URE2/[URE3] system in Kluyveromyces lactis. FEMS Yeast Res. 11, 151–153 10.1111/j.1567-1364.2010.00700.x [DOI] [PubMed] [Google Scholar]
- 82. Crick S. L., Ruff K. M., Garai K., Frieden C., and Pappu R. V. (2013) Unmasking the roles of N- and C-terminal flanking sequences from exon 1 of huntingtin as modulators of polyglutamine aggregation. Proc. Natl. Acad. Sci. U.S.A. 110, 20075–20080 10.1073/pnas.1320626110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Hayden E., Cone A., and Ju S. (2017) Supersaturated proteins in ALS. Proc. Natl. Acad. Sci. U.S.A. 114, 5065–5066 10.1073/pnas.1704885114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Ciryam P., Kundra R., Morimoto R. I., Dobson C. M., and Vendruscolo M. (2015) Supersaturation is a major driving force for protein aggregation in neurodegenerative diseases. Trends Pharmacol. Sci. 36, 72–77 10.1016/j.tips.2014.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Baldwin A. J., Knowles T. P., Tartaglia G. G., Fitzpatrick A. W., Devlin G. L., Shammas S. L., Waudby C. A., Mossuto M. F., Meehan S., Gras S. L., Christodoulou J., Anthony-Cahill S. J., Barker P. D., Vendruscolo M., and Dobson C. M. (2011) Metastability of native proteins and the phenomenon of amyloid formation. J. Am. Chem. Soc. 133, 14160–14163 10.1021/ja2017703 [DOI] [PubMed] [Google Scholar]
- 86. Glover J. R., Kowal A. S., Schirmer E. C., Patino M. M., Liu J. J., and Lindquist S. (1997) Self-seeded fibers formed by Sup35, the protein determinant of [PSI+], a heritable prion-like factor of S. cerevisiae. Cell 89, 811–819 10.1016/S0092-8674(00)80264-0 [DOI] [PubMed] [Google Scholar]
- 87. Khan T., Kandola T. S., Wu J., Venkatesan S., Ketter E., Lange J. J., Rodríguez Gama A., Box A., Unruh J. R., Cook M., Halfmann R. (2018) Quantifying nucleation in vivo reveals the physical basis of prion-like phase behavior. Mol. Cell. 71, 155–168.e7 10.1016/j.molcel.2018.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Kramer R. M., Shende V. R., Motl N., Pace C. N., and Scholtz J. M. (2012) Toward a molecular understanding of protein solubility: increased negative surface charge correlates with increased solubility. Biophys. J. 102, 1907–1915 10.1016/j.bpj.2012.01.060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Berchowitz L. E., Kabachinski G., Walker M. R., Carlile T. M., Gilbert W. V., Schwartz T. U., and Amon A. (2015) Regulated formation of an amyloid-like translational repressor governs gametogenesis. Cell 163, 406–418 10.1016/j.cell.2015.08.060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Boke E., Ruer M., Wühr M., Coughlin M., Lemaitre R., Gygi S. P., Alberti S., Drechsel D., Hyman A. A., and Mitchison T. J. (2016) Amyloid-like self-assembly of a cellular compartment. Cell 166, 637–650 10.1016/j.cell.2016.06.051 [DOI] [PMC free article] [PubMed] [Google Scholar]