

Abstract
Apoptotic cells expose phosphatidylserine (PtdSer) on their surface, leading to efferocytosis, i.e. their engulfment by resident macrophages that express the PtdSer receptor T cell immunoglobulin mucin receptor 4 (TIM4) and TAM family receptor tyrosine kinase receptors (MERTK, AXL, and TYRO3). TAM family receptors stimulate cell proliferation, and the many aspects of the growth signaling pathway downstream of TAM family receptors have been elucidated previously. However, the signaling cascade for TAM receptor–mediated efferocytosis has been elusive. Here we observed that efferocytosis by mouse-resident peritoneal macrophages was blocked by inhibitors against the MERTK, mitogen-activated protein kinase/extracellular signal-regulated kinase kinase (MEK), AKT Ser/Thr kinase (AKT), focal adhesion kinase (FAK), or STAT6 pathway. Accordingly, apoptotic cells stimulated the phosphorylation of MERTK, ERK, AKT, FAK, and STAT6, but not of IκB or STAT5. A reconstituted efferocytosis system using MERTK- and TIM4-expressing NIH3T3-derived cells revealed that the juxtamembrane and C-terminal regions of MERTK have redundant roles in efferocytosis. The transformation of murine IL-3–dependent Ba/F3 cells (a pro-B cell line) with MERTK and TIM4 enabled them to proliferate in response to apoptotic cells in a PtdSer-dependent manner. This apoptotic cell–induced MERTK-mediated proliferation required both MERTK's juxtamembrane and C-terminal regions and was blocked by inhibitors of not only ERK, AKT, FAK, and STAT6 but also of NF-κB and STAT5 signaling. These results suggest that apoptotic cells stimulate distinct sets of signal transduction pathways via MERTK to induce either efferocytosis or proliferation.
Keywords: apoptosis, macrophage, signal transduction, phosphatidylserine, phosphorylation, cell death, cell signaling, cancer, efferocytosis, kinase
Introduction
Vast numbers of surplus or toxic cells are generated during animal development (1, 2). These cells undergo apoptosis, expose phosphatidylserine (PtdSer)2 on their surface as an “eat me” signal, and are cleared by phagocytes (3, 4). This process also occurs at the resolution phase of inflammation in adult tissues (5). In addition, large numbers of senescent cells, such as aged neutrophils and red blood cells, are cleared by macrophages in a PtdSer-dependent manner (3). The engulfment of apoptotic or senescent cells, called “efferocytosis (6),” is essential for preventing these cells from undergoing secondary necrosis, which can cause cells to release their contents, thereby activating the immune system (3, 5, 7, 8).
The PtdSer exposed on apoptotic and senescent cells is recognized by soluble PtdSer–binding proteins such as protein S (PROS), GAS6, and MFG-E8 (9–11) and by PtdSer receptors (for example, TIM1 and TIM4) expressed on phagocytes (12). PROS and GAS6 bind to PtdSer on apoptotic cells and to TAM family receptor kinases (TYRO3, AXL, and MERTK) on macrophages and act as a bridge between apoptotic cells and phagocytes (13, 14). Macrophages are the most prominent phagocytes that perform efferocytosis and express at least one of the TAM receptor kinases. We recently showed that a set of resident macrophages, such as resident peritoneal macrophages (rpMacs), Kupffer cells, and skin macrophages, express TIM4 and TAM kinase for efficient efferocytosis (15, 16). The extracellular region of TIM4 binds PtdSer with high affinity (12), and its cytoplasmic region is dispensable for efferocytosis (17). On the other hand, the affinity of PROS or GAS6 for PtdSer is weaker than that of TIM4 (16), but the TAM receptors' cytoplasmic region is indispensable for efferocytosis (18). These results support a two-step model for efferocytosis (19) in which TIM4 mediates tethering of the apoptotic cell to the macrophage, followed by TAM receptor–mediated internalization of the apoptotic cell.
TAM receptor kinases were originally identified as oncogenes expressed in various cancer cells, in particular myeloid leukemia cells (14, 20). PROS or GAS6 induces dimerization of TAM family receptors, which activates their kinase activity, followed by phosphorylation of tyrosine residues in their cytoplasmic region (21). Many signaling molecules, such as ERK, p38 MAPK, FAK, AKT, NF-κB, and STAT6, have been identified as downstream components of MERTK in MERTK-mediated growth promotion or chemoresistance of cancer cells (22–24). In contrast, the signaling cascade for TAM-mediated efferocytosis has been elusive.
In this report, we expressed MERTK and TIM4 in IL-3–dependent Ba/F3 cells and found that these cells survived in the absence of IL-3 in a PtdSer-dependent manner and that their growth was strongly enhanced by the presence of apoptotic cells. We then found that efferocytosis with resident peritoneal macrophages was inhibited by inhibitors against MEK, AKT, FAK, or STAT6 but not against NF-κB or STAT5 pathways. On the other hand, apoptotic cell–induced cell growth was efficiently blocked not only by inhibitors of MEK, AKT, FAK, or STAT6 but also by inhibitors against NF-κB or STAT5 pathways. Using NIH3T3-derived cell lines expressing TIM4 and MERTK mutants, we showed that MERTK's membrane-proximal and C-terminal tail regions were not required for efferocytosis, whereas apoptotic cell–stimulated growth signaling required the membrane-proximal and C-terminal tail regions of MERTK in addition to its kinase domain. These results indicate that apoptotic cells can stimulate cell growth via MERTK and that overlapping distinct signaling molecules are involved in MERTK-mediated efferocytosis versus MERTK-mediated growth promotion.
Results
Apoptotic cell–activated cell proliferation
We showed previously that apoptotic cells are engulfed by MERTK- and TIM4-expressing macrophages or NIH3T3 cells (16). Because MERTK is known to mediate growth signaling (20), we examined the effect of TIM4 on MERTK-mediated cell growth using mouse IL-3–dependent Ba/F3 cells. The Ba/F3 cells were transformed with MERTK or TIM4 alone or with both MERTK and TIM4 and cultured in RPMI 1640 medium containing 10% FCS and IL-3. The culture medium was then deprived of IL-3 overnight, and the transformants were kept in medium lacking IL-3. As shown in Fig. 1A, not only the parental and TIM4-expressing Ba/F3 cells but also the transformants expressing only MERTK died within 24 h, suggesting that the PROS in 10% serum (about 30 nm) (9) were not sufficient to support MERTK-mediated cell growth. On the other hand, about 60% of cells expressing both TIM4 and MERTK (TIM4/MERTK) survived for 24 h, and this percentage did not change after 72 h, suggesting that the same number of cells died and proliferated.
Figure 1.

PtdSer-dependent cell proliferation. A, PtdSer-dependent growth stimulation of TIM4- and MERTK-expressing Ba/F3 cells. Ba/F3 cells (2.5 × 105) expressing TIM4, MERTK, or both TIM4 and MERTK were cultured in 0.5 ml of RPMI 1640 medium containing 10% FBS. After incubation for the indicated periods, viable cells were counted after staining with trypan blue. Right panel, Ba/F3 cells expressing TIM4 and MERTK were cultured for 72 h in medium containing the indicated concentration of D89E, and the number of viable cells was expressed as the percentage of that in the absence of D89E. The experiments were carried out three times, and average values were plotted with S.D. (error bars). The values were statistically analyzed by Student's t test. **, p < 0.01; ***, p < 0.001. B, apoptotic cell–stimulated, PtdSer-dependent cell growth. Ba/F3 cells (2.5 × 105) expressing TIM4, MERTK, or both TIM4 and MERTK were cultured in 0.5 ml of medium for the indicated periods in the presence of 2.5 × 106 apoptotic thymocytes (left panel). Center and right panels, Ba/F3 cells (2.5 × 105) expressing both TIM4 and MERTK were cultured in 0.5 ml of medium for 72 h in the presence of the indicated concentration of apoptotic thymocytes (center panel) or in the presence of 6.25 × 105 apoptotic thymocytes and the indicated concentration of D89E. After incubation, the trypan blue–negative viable cells were counted. Right panel, the number of viable cells was expressed as the percentage of that in the absence of D89E. The experiments were carried in triplicate, and average values were plotted with S.D. (error bars). *, p < 0.01; **, p < 0.001; Student's t test. C, DNA synthesis of Ba/F3 cells expressing TIM4 and MERTK without IL-3. 2.5 × 105 Ba/F3 cells (a, d, and e) and Ba/F3 cells expressing MERTK and TIM4 (b and c) were cultured at 37 °C for 72 h without (a, b, and c) or with (d and e) IL-3 in the absence (a, b, d, and e) or presence (c) of 2.5 × 106 apoptotic thymocytes. The culture was supplemented with (a–d) or without (e) 10 μm BrdU and incubated further for 4 h. The incorporated BrdU was then detected with FITC-labeled anti-BrdU Ab.
The percentage of trypan blue–positive dead cells in the culture of TIM4/MERTK transformants was about 70%, and the presence of the D89E mutant of MFG-E8 that could mask PtdSer on apoptotic cells (11) dose-dependently inhibited cell survival (Fig. 1A). These results suggested that PtdSer on apoptotic cells generated by IL-3 deprivation supported the growth of neighboring cells. To examine this possibility, the Ba/F3 cell transformants were co-cultured with FASL-treated apoptotic thymocytes. As shown in Fig. 1B, apoptotic thymocytes stimulated the growth of Ba/F3 cells expressing both TIM4 and MERTK but not of cells expressing one or the other. This effect of apoptotic cells on the Ba/F3 transformants was dose-dependent and was reduced by masking PtdSer with D89E.
To further confirm that Ba/F3 cells expressing MERTK and TIM4 proliferated in response to apoptotic cells, they were cultured for 72 h in the absence of IL-3 and pulsed for 4 h with BrdU. As shown in Fig. 1C, more than 50% of Ba/F3 cells were labeled with BrdU in the presence of IL-3, whereas no BrdU-positive cells were observed in the absence of IL-3. On the other hand, about 0.8% of Ba/F3 cells expressing TIM4 and MERTK were labeled with BrdU in the absence of IL-3, and this percentage increased to 10.4% when the culture was supplemented with apoptotic thymocytes.
Effect of signal transducer inhibitors on efferocytosis and cell proliferation
Mouse rpMacs express both TIM4 and MERTK and efficiently engulf apoptotic cells in a TIM4- and MERTK-dependent manner (15, 16). To analyze the signaling molecules required for efferocytosis, rpMacs were treated with various inhibitors against signal transducers and then incubated with apoptotic thymocytes in the presence of the inhibitor. As shown in Fig. 2A, a low concentration of CH5451098, an inhibitor against MERTK and AXL (25), suppressed efferocytosis in a dose-dependent manner, confirming that MERTK is essential for efferocytosis by rpMacs (15, 16). Among the signaling components that are reported to be activated by TAM kinase (20, 22), inhibitors against MEK (PD98059) (26) and PI3K (LY294002) (27) for the AKT pathway and FAK (PF-00562271) (28) for the STAT6 pathway (AS1517499) (29) efficiently blocked efferocytosis. The concentrations of these inhibitors required to block efferocytosis were comparable with those reported to inhibit signal transduction in cells. In contrast, caffeic acid phenethyl ester and SH4–54, inhibitors against NF-κB (30) and STAT3/STAT5 (31), respectively, did not inhibit efferocytosis by rpMacs at concentrations that should inhibit cellular signal transduction.
Figure 2.

Effects of various inhibitors on MERTK-mediated efferocytosis and cell growth. A, effect of various inhibitors on efferocytosis by rpMacs. Resident peritoneal cells were pretreated with the indicated concentration of each inhibitor and subjected to an efferocytosis assay with pHrodo-labeled apoptotic thymocytes in the presence of the inhibitor. Efferocytosis (the percentage of pHrodo-positive cells) was determined by flow cytometry and is expressed relative to that observed without inhibitors. The experiment was done in triplicate, and the average values are plotted with S.D. (error bars). The values were statistically analyzed by Student's t test against the value without inhibitor. ##, p < 0.001. B, effect of various inhibitors on apoptotic cell–promoted cell proliferation. Apoptotic thymocytes were added to Ba/F3 cells expressing WT MERTK and TIM4, and the mixture was incubated in the presence of the indicated concentrations of the inhibitor. After incubation for 24 h, viable Ba/F3 cells were counted. The experiments were carried out three times, and the average values were plotted with S.D. (error bars). Student's t test against the value without inhibitor. The cell number is expressed relative to that observed in the presence of apoptotic cells without inhibitors. The cell number in the absence of inhibitors was 3–5 × 105 cells/ml. #, p < 0.01; ##, p < 0.001; Student's t test.
To examine which signal transducers were involved in apoptotic cell–stimulated MERTK-mediated cell growth, 2.5 × 105 Ba/F3 cell transformants expressing TIM4 and MERTK were cultured in the absence of IL-3 for 24 h with or without 2.5 × 106 apoptotic thymocytes in the presence of specific inhibitors for signal transducers. As shown in Fig. 2B, the number of TIM4/MERTK-Ba/F3 cells decreased to 60–70% in the absence of apoptotic thymocytes, whereas this number remained almost unchanged in their presence. In accordance with the requirement for MERTK's kinase activity, an inhibitor of MERTK's kinase activity blocked the apoptotic cell–stimulated cell growth in a dose-dependent manner. The inhibitors against MEK, AKT, FAK, and STAT6 pathways that blocked efferocytosis (Fig. 2A) also inhibited cell growth, although a higher concentration of the PI3K inhibitor was needed to inhibit cell growth than efferocytosis. Notably, inhibitors against NF-κB or STAT5 also efficiently blocked apoptotic cell–stimulated IL-3-independent growth of Ba/F3 cell transformants expressing TIM4 and MERTK.
Phosphorylation of signaling molecules activated by apoptotic cells
MERTK is autophosphorylated at tyrosine residues when it is activated (32). To examine whether apoptotic cells induced activation of MERTK, rpMacs from WT and MerTK−/− mice were incubated with or without a 10× excess of apoptotic thymocytes for 10 min. The cell lysates were then immunoprecipitated with an anti-mouse MERTK Ab, and the precipitates were analyzed by Western blotting. As shown in Fig. 3A, a band of about 200 kDa was detected with anti-MERTK in WT but not MerTK−/− (KO) rpMacs. Mouse MERTK is heavily glycosylated (15 N-glycosylation sites), which may explain why its apparent molecular weight was much greater than that calculated from its amino acid sequence (Mr = 110,155). Western blotting of the immunoprecipitates with an anti-phosphotyrosine mAb (clone 4G10) revealed a 200-kDa band in rpMacs that had been treated with apoptotic cells. These results indicated that apoptotic cells activated the tyrosine kinase activity of MERTK in rpMacs.
Figure 3.

Activation of signaling molecules induced by apoptotic cells. Adherent resident peritoneal cells (2.0 × 106 cells) from WT or MerTK−/− (KO) mice were incubated at 37 °C for 10 min with (+) or without (−) 1.0 × 107 apoptotic thymocytes, washed with PBS, and lysed in lysis buffer. A, MERTK was immunoprecipitated with anti-MERTK, dissolved in SDS sample buffer, and one-quarter of the aliquots were analyzed by Western blotting with an HRP anti-phosphotyrosine mAb (top panel) or a biotinylated anti-MERTK Ab followed by incubation with HRP–streptavidin (center panel). The membrane was stained with Coomassie Brilliant Blue (CBB, bottom panel). B, cell lysates from 1.5 × 106 cells were analyzed by Western blotting using antibodies against the indicated phosphorylated (top panel) or nonphosphorylated molecules (bottom panel), followed by HRP-conjugated anti-rabbit IgG. The phosphorylated amino acid residues recognized by the antibody are indicated in parentheses. Membranes were stained with CBB (bottom panel). #, nonspecific band. The western blots were performed several times, and the band intensity of the phosphorylated kinase was quantitated by densitometry. When addition of apoptotic thymocytes in WT macrophages caused an apparent change in band intensity, the -fold change is shown with S.D. *, p < 0.03; Student's t test.
The cell lysates from the apoptotic cell-treated rpMacs were then analyzed by Western blotting using antibodies against the phosphorylated signal transducers. Anti-phospho-ERK1/2 recognizes the phosphorylated Thr-202/Tyr-204 of MAPK (ERK1/2) generated by MAPK kinase (33). Activated AKT is detected by an antibody that recognizes phosphorylated Ser-473, which is phosphorylated by the mTOR-Rictor complex (34). FAK is a cytoplasmic tyrosine kinase and is activated by integrin clustering, leading to its auto-phosphorylation at Tyr-397 (35). STAT5 and STAT6 are transcription factors that are activated by various cytokines via Janus kinases, which phosphorylate Tyr-694 of STAT5 and Tyr-641 of STAT6 (36–38). Finally, IκB kinase (IKK) phosphorylates Ser-32 of IκB, leading to activation of the transcription factor NF-κB (39). As shown in Fig. 3B, ERK1/2 phosphorylated at Thr-202/Tyr-204, AKT phosphorylated at Ser-473, FAK phosphorylated at Tyr-397, and STAT6 phosphorylated at Tyr-641 were detected in rpMacs incubated with apoptotic cells but not in untreated cells. None of the phosphorylated signaling molecules were detected in MerTK−/− rpMacs, indicating that their apoptotic cell–induced phosphorylation was MERTK-dependent. In contrast, neither IκB phosphorylated at Ser-32 nor STAT5 phosphorylated at Tyr-694 was detected in apoptotic cell–treated rpMacs. These results indicated that apoptotic cells induced activation of ERK1/2, AKT, FAK, and STAT6 in rpMacs, which agreed with our observation that inhibitors against these signaling pathways suppressed efferocytosis (Fig. 2).
Different MERTK cytoplasmic domains for efferocytosis and cell growth
The cytoplasmic region of mouse MERTK consists of 386 amino acids, of which 271 form a tyrosine kinase domain (UniProt entry Q60805) (Fig. 4A). The lysine residue at position 614 of MERTK is involved in forming the cleft for ATP binding and is essential for the MERTK kinase activity (40). To confirm that MERTK's tyrosine kinase activity was essential for MERTK-mediated efferocytosis, lysine 614 was replaced with methionine (K614M) (Fig. 4A). The juxtamembrane region of tyrosine kinase receptors, including MERTK and AXL, is well-conserved and involved in clustering anionic lipids, such as for signal transduction (41). Mouse MERTK carries two tyrosine residues (Tyr-867 and Tyr-924) at its C-terminal tail region (Fig. 4A), and the phosphorylated Tyr-867 is reported to be involved in activation of NF-κB and PI3K (40). To examine the requirement of these regions for efferocytosis and cell growth, three mutants of MERTK lacking either the juxtamembrane (ΔN, amino acids 531–559), the C-terminal tail region (ΔC, amino acids 853–994), or both regions (ΔNΔC) were constructed.
Figure 4.

Effect of MERTK deletion mutations on efferocytosis and cell growth. A, Schematics of MERTK mutants. At the top, the structure of MERTK is shown schematically. Immunoglobulin-like domains (IG1 and IG2), fibronectin type III–like domains (FN1 and FN2), the transmembrane region (TM), and the tyrosine kinase domain are boxed, with the amino acid positions indicated at the borders. Amino acid positions 530 and 560 indicate the exon–intron junction used to construct the ΔN mutant. Three tyrosine residues at positions 544, 867, and 924, and a lysine residue at position 614 are indicated. In the K614M mutant, the lysine residue at 614 was mutated to methionine. In the ΔN and ΔC mutants, the amino acid region from 531 to 559 proximal to the transmembrane region and the amino acid region from 853 to 994 in the C terminus were deleted, respectively. In the ΔNΔC mutant, both the N-terminal juxtamembrane and the C-terminal regions were deleted. B, effect of MERTK mutations on efferocytosis. TKO cell transformants (6 × 104 cells) expressing WT or the indicated mutant MERTK together with TIM4 were incubated with 6 × 105 pHrodo-apoptotic thymocytes at 37 °C for 60 min and then analyzed by flow cytometry for pHrodo positivity. The experiments were carried out in triplicate, and the average percentage of pHrodo-positive cells was plotted as efferocytosis with S.D. (error bars). **, p < 0.01; Student's t test. C, effect of various mutations of MERTK on apoptotic cell–promoted cell growth. Ba/F3/TIM4 cell transformants (2.5 × 105 cells in 0.5 ml) expressing WT or the indicated mutant MERTK were incubated for 24, 48, or 72 h in the absence (left) or presence (right) of 2.5 × 106 apoptotic thymocytes, and viable Ba/F3 cells were counted. The experiments were performed in triplicate, and average values were plotted with S.D. (error bars). The values were statistically analyzed by Student's t test against that obtained with WT MERTK.
We reported previously that Axl−/−Tyro3−/−Gas6−/− NIH3T3 (TKO) cells expressing TIM4 and MERTK behave like rpMacs and efficiently engulf apoptotic cells (16). To examine the ability of MERTK mutants to engulf apoptotic cells, TKO cells expressing TIM4 were transformed with WT or mutant MERTK (Fig. S1) and then subjected to efferocytosis assays. As reported previously (16), TKO transformants expressing TIM4 and WT MERTK efficiently engulfed apoptotic cells (Fig. 4B). In contrast, the K614M mutant completely abolished the cells' ability to perform MERTK-mediated efferocytosis. On the other hand, although the ΔN and ΔC mutants supported efferocytosis as efficiently as WT MERTK, the efferocytosis activity of the ΔNΔC mutant carrying only the kinase domain in the cytoplasmic region (amino acids 582–852) (Fig. 4A) was significantly reduced (Fig. 4B), suggesting that either the N-terminal juxtamembrane region or the C-terminal tail region of MERTK was necessary for full efferocytosis activity.
To examine which of the regions of MERTK was responsible for apoptotic cell–stimulated cell proliferation, Ba/F3 cells expressing TIM4 were transformed with WT or mutant MERTK (Fig. S2). As shown in Fig. 4C, in addition to the kinase-dead mutants (K614M mutants), the deletion mutant of the N-terminal juxtamembrane region (ΔN) completely lost the ability to support IL-3–independent Ba/F3 cell growth. The ability of the C-terminal deletion mutant (ΔC) of MERTK to support cell growth was reduced to about 20% of the ability of WT MERTK. These results indicated that, unlike signal transduction for efferocytosis, not only tyrosine kinase activity but also signals from the juxtamembrane region and C-terminal tail region of MERTK were required for MERTK-supported cell growth.
Activation of signaling molecules via the kinase domain of MERTK
We next examined the effect of the MERTK mutations on its tyrosine kinase activity and on the activation of signaling molecules. As shown in Fig. 5A, the K614M mutant of MERTK was not autophosphorylated at tyrosine residues in TKO cells upon addition of apoptotic cells, confirming that the mutant lost the kinase activity. On the other hand, the ΔN or ΔC mutant was tyrosine-phosphorylated at a similar efficiency as WT MERTK by apoptotic cells, confirming that tyrosine kinase activity was not disrupted by the ΔN or ΔC mutation. Accordingly, addition of apoptotic cells stimulated phosphorylation of ERK1/2, AKT, and STAT6 in TKO cells expressing WT but not kinase-dead mutant MERTK (K614M) (Fig. 5B). The activated forms of the signaling molecules of ERK1/2 and AKT were observed in apoptotic cell–treated cells expressing the ΔN, ΔC, or ΔNΔC mutant. In contrast, STAT6 phosphorylated at Tyr-641 was observed in cells expressing the ΔN or ΔC mutant of MERTK but not in cells expressing the ΔNΔC mutant, suggesting that, in addition to the kinase activity of MERTK, a signal from the N-terminal juxtamembrane region or the C-terminal tail region of MERTK is required to activate STAT6. In contrast to ERK, AKT, and STAT6, FAK phosphorylated at Tyr-397 was observed in parental TKO cells without apoptotic cell stimulation (data not shown), suggesting that FAK might have been activated through a pathway other than MERTK, such as the integrin system, which was constitutively activated in TKO cells.
Figure 5.
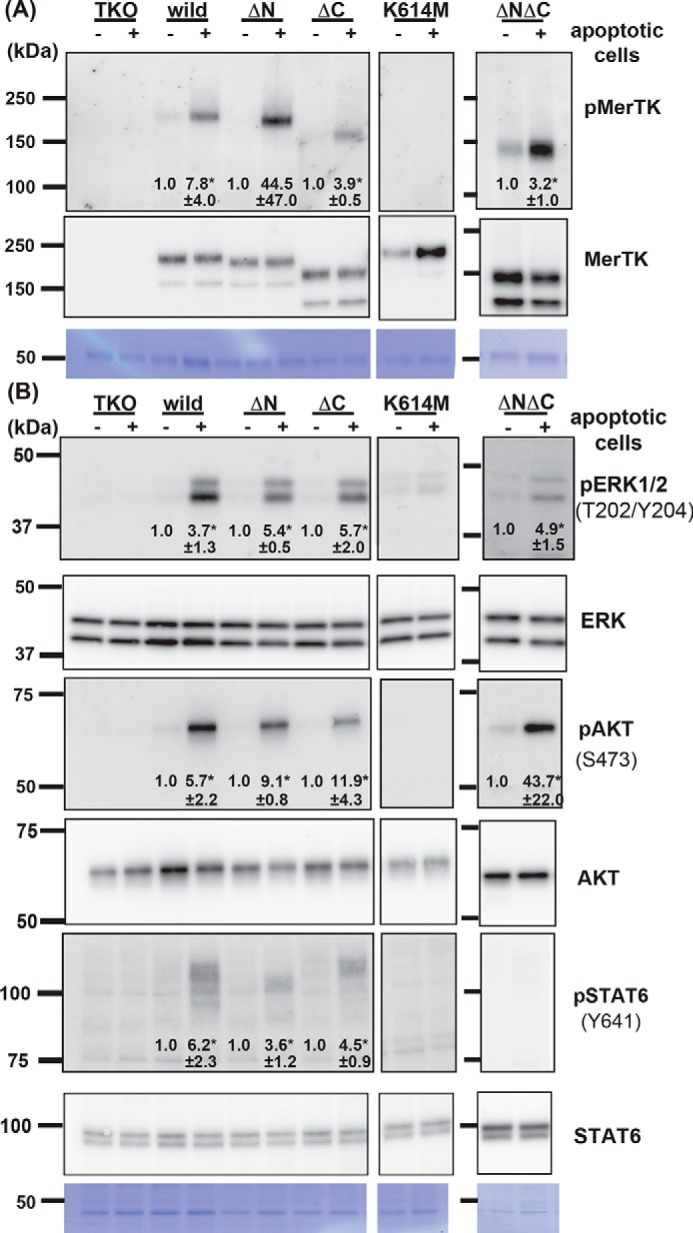
Effect of MERTK mutations on activation of MERTK and signaling molecules. TKO transformants (1.0 × 106 cells) expressing the WT or the indicated mutant MERTK were incubated with (+) or without (−) 1.0 × 107 apoptotic thymocytes at 37 °C for 10 min in DMEM containing 1 μg/ml protein S. After removing unengulfed thymocytes, the cells were lysed with 1.0 ml of lysis buffer containing 1% Triton X-100. A, MERTK in the cell lysates (1 ml) was immunoprecipitated with an anti-MERTK Ab and dissolved in 60 μl of SDS sample buffer, and 15-μl aliquots were subjected to Western blotting with an HRP-anti-phosphotyrosine mAb (top panel) or a biotinylated anti-MERTK Ab followed by streptavidin–HRP (bottom panel). Membranes were stained with CBB (bottom panel). B, cell lysates (15 μl) were analyzed by Western blotting using HRP antibodies against the indicated phosphorylated (top panel) or nonphosphorylated proteins (bottom panel). The phosphorylated amino acid residues recognized by the antibodies are shown in parentheses. Membranes were stained with CBB (bottom panel). Western blots were performed several times. The band intensity was quantified by densitometry and compared between those obtained with or without apoptotic cells. When the difference was apparent, the induction -fold is indicated with S.D. *, p < 0.05, Student's t test.
Discussion
Like other processes of programmed cell death, the removal of cell corpses has been genetically studied in Caenorhabditis elegans, in which dying cells are engulfed by neighboring cells (42). That study identified three partially redundant signaling pathways, CED-1/CED-6/CED7/CED-10, CED-2/CED-5/CED-12/CED-10, and ABL-1/API-1/CED-10 (43). Like mammalian cells, PtdSer is exposed on the surface of dying cells in C. elegans by the caspase (CED-3)–dependent phospholipid scramblase (CED-8), an ortholog of mammalian XKR8 (44, 45). CED-1 appears to indirectly bind PtdSer via a soluble bridging protein called TTR-52 (46). However, how CED-1 activates downstream signaling molecules has not been elucidated. A pathway consisting of CED12/ELMO and CED10/RAC has been reported to elicit efferocytosis in experiments using Chinese hamster ovary cell lines (47). In addition, brain-specific angiogenesis inhibitor 1 (BAI1), an adhesion G-protein–coupled receptor family receptor, has been proposed to activate the ELMO–RAC pathway for efferocytosis in this Chinese hamster ovary system. However, whether macrophages use this system for efferocytosis is not clear.
We recently reported that a set of resident mouse macrophages uses MERTK and TIM4 to elicit efficient efferocytosis (16). Roles of MERTK-activated phospholipase Cγ2 and FAK in efferocytosis have been reported previously using the J774 mouse macrophage cell line or HEK293T cells that were transiently transfected with MERTK cDNA (48–50). MERTK is known to activate many other signaling molecules, and we found here that at least the MEK/ERK, AKT, FAK, and STAT6 pathways were necessary for efficient efferocytosis. Efferocytosis takes place at lamellipodia of engulfing cells, where activated RAC1 is recruited to form a phagocytic cup with polymerized actin patches to encapsulate apoptotic cells (51). When the apoptotic cells are internalized, the phagocytic cup closes, accompanied by depolymerization of actin bundles. This elaborate process of efferocytosis has features in common with cell motility (52). Cell motility consists of multiple processes, including cell protrusion, cell retraction, adhesion, and vesicle exocytosis, and is regulated by multiple signaling molecules (53). For example, downstream of FAK, ERK localizes to lamellipodia and phosphorylates myosin light chain kinase to regulate turnover of focal adhesions (54). Meanwhile, ERK phosphorylates Paxillin, and phosphorylated Paxillin serves as a scaffold for FAK to activate PI3K for RAC activation (55). STATs are transcription factors and are activated by various cytokines via Janus kinases (56). However, recent studies indicate that some STAT members also act outside of the nucleus, such as at mitochondria and focal adhesions (56–58). Thus, it is likely that interplay occurs among these kinases (ERK, FAK, AKT, STAT6, and probably more) to regulate RAC1 to polymerize or depolymerize actin for efferocytosis. Using specific inhibitors for these kinases, it should be possible to dissect this process to study each step in more detail.
Here we found that apoptotic cells stimulated proliferation of cells expressing MERTK and TIM4. This growth-promoting activity was fully dependent on PtdSer. MERTK-mediated efferocytosis is known to produce transforming growth factor β to stimulate cell proliferation (59). Although this possibility cannot be ruled out, considering the oncogenic property of MERTK (14, 20), we prefer the hypothesis that apoptotic cells directly activate MERTK for cell proliferation. As shown for PtdSer-dependent efferocytosis (15, 16), it is likely that PtdSer-exposing apoptotic cells are recruited to MERTK-expressing cells by TIM4 for MERTK-mediated cell growth. The apoptotic cells are then cross-linked with MERTK on the responder cells via an interaction between PtdSer and PROS and activate MERTK to promote cell growth. Notably, TIM4 is expressed together with MERTK or AXL in various tumors, such as histiocytic sarcoma, histiocytic and dendritic cell neoplasms, lung cancer, and glioma (60–62), and has been proposed to contribute to tumorigenicity. It will be interesting to examine whether these primary tumor cells respond to apoptotic cells for their growth.
Many groups have studied the signal transduction pathway for MERTK-mediated cell growth and reported that not only the kinase domain but also the juxtamembrane and C-terminal tail regions are necessary to mediate the signal (20). The tyrosine residues in these regions are autophosphorylated, and phosphorylated tyrosines serve as binding sites for signaling molecules. In fact, we found that deleting either the juxtamembrane or the C-terminal tail region of MERTK severely reduced its growth-promoting activity. This was in sharp contrast to the effect of these mutations on efferocytosis. In addition to the signaling molecules required for efferocytosis, signals from the NF-κB and STAT6 pathways were required for MERTK-mediated cell growth, suggesting that the juxtamembrane region and C-terminal tail regions are responsible for these signaling pathways. It is also possible that signaling through the MEK, AKT, FAK, and STAT6 pathways has different roles in growth promotion and efferocytosis. Because MERTK is strongly expressed in various tumor cells, inhibitors against MERTK are considered to be anti-tumor agents (63–65). However, these inhibitors would also block MERTK-mediated efferocytosis, which could lead to autoimmune disease (3, 66). Our results indicating that MERTK-mediated cell growth requires additional signaling pathways may be useful for developing safer and more useful reagents for cancer therapy.
Experimental procedures
Mice, cell lines, recombinant proteins, antibodies, and reagents
C57BL/6J mice were purchased from Japan SLC and CLEA Japan. MerTK−/− mice were from The Jackson Laboratory. All animal experiments were approved by the Animal Care and Use Committee of the Research Institute for Microbial Diseases (Osaka University, Osaka, Japan).
Mouse IL-3–dependent Ba/F3 cells were maintained in RPMI 1640 medium containing 10% FBS and 45 units/ml IL-3 as described previously (67). TKO NIH3T3 cells were described previously (16). TKO NIH3T3 cells and Plat-E cells (68) were maintained in DMEM containing 10% FBS.
Recombinant leucine-zippered human Fas ligand (FASL) was produced in COS7 cells and purified as described previously (69). In brief, COS7 cells were transfected with a FASL expression plasmid by electroporation and cultured in DMEM containing 1% FBS for 48 h. The supernatant was subjected to (NH4)2SO4 precipitation at 60% saturation and dialyzed against PBS. The hamster anti-TIM4 Ab (clone Kat5–18) was described previously (12). Other antibodies and reagents were as follows: biotinylated goat anti-mouse MERTK (R&D Systems); rabbit anti-ERK1/2 and anti-phospho-ERK1/2 (Thr-202/Tyr-204), anti-p38 and anti-phospho-p38 (Thr-180/Tyr-182), anti-AKT and anti-phospho-AKT (Ser-473), anti-FAK and anti-phospho-FAK (Tyr-397), anti-IκB and phospho-IκB (Ser-32), anti-STAT5 and anti-phospho-STAT5 (Tyr-694), and anti-STAT6 and anti-phospho-STAT6 (Tyr-641) (Cell Signaling Technology); HRP-conjugated mouse anti-phosphotyrosine (4G10, Merck Millipore); HRP-mouse anti-FLAG (Sigma-Aldrich); and pHrodo Red succinimidyl ester (pHrodo, Life Technologies). The following chemicals were used to inhibit signaling pathways: PD98059 (MEK), PF-00562271 (FAK), caffeic acid phenethyl ester (NF-κB), and SH4–54 (STAT5, Selleck Chemicals), LY294002 (PI3K, Cell Signaling Technology); AS1517499 (STAT6, Sigma-Aldrich), and CH5451098 (MERTK and AXL, Chugai Pharmaceutical Co. Ltd.).
Transformation
Lentiviral expression vectors (CSII-EF, pCAG-HIVgp, pENV-IRES-puro, and pRSV-Rev) were from H. Miyoshi (Riken Resource Center). Mouse MERTK cDNA (NM_008587.1) was described previously (15). Deletion mutants of MERTK, ΔN (Δ531–559), ΔC (Δ853–894), and ΔNΔC (Δ531–559 and Δ853–894), were constructed using In-Fusion HD Cloning Kits (Takara Clontech). The kinase-dead K614M mutant (40) was prepared by recombinant PCR (70). MERTK and its mutants were FLAG-tagged at the C terminus and inserted into the CSII-EF vector. pMxs-puro-TIM4 and pNEF-BOS-EX-TIM4 were described previously (12, 15).
The Ba/F3 transformants expressing MERTK and TIM4 (15) and TKO transformants expressing MERTK and TIM4 (16) were described previously. MERTK mutants were expressed in Ba/F3-TIM4 or TKO-TIM4 cells using a lentiviral vector system. Briefly, HEK293T cells were co-transfected using FuGENE 6 (Promega) with the CSII-EF vector carrying cDNA for MERTK or its mutants (pCAG-HIVgp, pENV-IRES-puro, and pRSV-Rev). After culturing for 48 h, viruses in the supernatant were used to infect TKO cells. For infection of Ba/F3 cells, viruses in the supernatant were concentrated by centrifugation at 6000 × g for 16 h at 4 °C and used for spin infection. Transformants were stained with anti-MERTK and, when necessary, sorted using a FACSAria II (BD Biosciences).
Efferocytosis assay
Efferocytosis was assayed as described previously (15, 16) with rpMacs or TKO cells as phagocytes and with apoptotic thymocytes as prey. In brief, thymocytes from 4-week-old mice were treated at 37 °C for 1.5 h with 100 units/ml FASL and washed with PBS. The cells were stained with 0.1 μg/ml pHrodo for 30 min at room temperature and washed with DMEM containing 10% FBS. For efferocytosis with rpMacs, peritoneal cells were isolated from 8- to 10-week old mice and seeded at 5 × 105 cells on a 12-well plate. After incubation for 2 h at 37 °C in DMEM containing 10% FBS, the cells were washed with PBS to remove nonadherent cells and subjected to the efferocytosis assay with 2.5 × 106 pHrodo-labeled apoptotic thymocytes. To examine the effect of inhibitors, rpMacs were preincubated with the inhibitor for 30 min, and then the efferocytosis assay was performed at 37 °C for 60 min in the presence of the inhibitor. For the efferocytosis assay with TKO cells, 6 × 104 TKO cells were seeded in a 24-well plate and cultured for 24 h at 37 °C. pHrodo-labeled apoptotic thymocytes (6 × 105) were added to TKO cells, and the mixture was incubated at 37 °C for 60 min in DMEM containing 10% FBS. After incubation, the TKO cells were washed with PBS, detached with trypsin, stained with 0.5 μm SYTOX Blue (Life Technologies) in 20 mm 2-(cyclohexylamino)ethanesulfonic acid buffer (pH 9.0) containing 150 mm NaCl and analyzed by flow cytometry with a FACSCanto II (BD Biosciences).
Immunoprecipitation and Western blotting
TKO transformants (1 × 106 cells) in a 6-cm plate were incubated at 37 °C for 6 h in DMEM containing 10% FBS and starved overnight in serum-free DMEM. The cells were then incubated with 1 × 107 apoptotic thymocytes in 2 ml of DMEM containing 1 μg/ml PROS for 10 min at 37 °C, washed with cold PBS, and lysed in 1 ml of lysis buffer (25 mm Tris-HCl (pH 7.4), 150 mm NaCl, 1 mm EGTA, 1% Triton X-100, 5% glycerol, a mixture of protease inhibitors (Complete Mini EDTA-free, Roche), and a mixture of phosphatase inhibitors (PhosSTOP, Roche)). After centrifugation at 15,000 rpm for 10 min at 4 °C, the supernatants were mixed with protein G Dynabeads (Life Technologies, 10 μl/sample) to which the goat anti-MERTK Ab had been conjugated by incubation for 3 h at 4 °C in TBS-T (25 mm Tris-HCl buffer (pH 7.5), 137 mm NaCl, 2.7 mm KCl, and 0.1% Tween 20). The mixture was rotated for 3 h at 4 °C, washed with lysis buffer, suspended in 30 μl of sample buffer (63 mm Tris-HCl (pH 6.8), 10% glycerol, 2% SDS, 0.1% bromphenol blue, and 2% β-mercaptoethanol) and incubated at 95 °C for 5 min. After removing the beads, the eluates were subjected to Western blotting.
For Western blotting, the samples were separated by 7.5% or 10% SDS-PAGE and transferred to a PVDF membrane (Merck Millipore). After incubation for 1 h at room temperature in blocking buffer consisting of TBS-T and 5% skim milk or 5% BSA (Probumin, Merck Millipore), the membranes were incubated overnight at 4 °C with primary antibody in blocking buffer. The membranes were then incubated for 1 h at room temperature with the secondary antibody, and signals were detected with Immobilon Western Chemiluminescent HRP substrate (Merck Millipore).
Proliferation assay
Ba/F3 cells (5 × 106 cells) were incubated at 37 °C overnight in 10 ml of RPMI 1640 medium containing 10% FBS and 45 units/ml mouse IL-3, washed with RPMI 1640 medium containing 10% FBS, and cultured overnight in RPMI 1640 medium containing 10% FBS. The cells were then cultured at 2.5 × 105 cells/ml in 24-well plates with 2.5 × 106 apoptotic thymocytes in RPMI 1640 medium containing 10% FBS and counted with a hemocytometer after staining with trypan blue. DNA synthesis was assayed by incorporation of BrdU. In brief, Ba/F3 cells were cultured for 72 h in the absence of IL-3, pulsed for 4 h with 10 μm BrdU (Sigma-Aldrich), and fixed at room temperature for 20 min with 1% paraformaldehyde. After permeabilization with 0.3% saponin, the cells were treated at 37 °C for 1 h with 300 μg/ml DNase I and stained with anti-BrdU (Abcam), followed by incubation with Alexa Fluor 488–anti-rat IgG (Molecular Probes).
Statistical analysis
All data were expressed as the mean with S.D. Differences between groups were examined for statistical significance using Student's t test.
Author contributions
C. N. data curation; C. N. and S. N. funding acquisition; C. N. investigation; C. N. writing-original draft; Y. Y. methodology; K. S. and S. N. supervision; K. S. and S. N. writing-review and editing.
Supplementary Material
Acknowledgments
We thank Chugai Pharmaceutical Co., Ltd. for providing CH5451098 and M. Fujii for secretarial assistance.
This work was supported in part by Grant-in-Aid for Research Activity Start-Up from Japan Society for the Promotion of Science (JSPS) 16H06943 (to C. N.) and Grants-in-Aid for Scientific Research (S) from JSPS 15H05785 and Core Research for Evolutional Science and Technology from the Japan Science and Technology Agency JPMJCR14M4 (to S. N.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1 and S2.
- PtdSer
- phosphatidylserine
- PROS
- protein S
- rpMac
- resident peritoneal macrophage
- ERK
- extracellular signal–regulated kinase
- MAPK
- mitogen-activated protein kinase
- FAK
- focal adhesion kinase
- FASL
- Fas ligand
- Ab
- antibody
- TKO
- triple knockout
- cDNA
- complementary DNA
- MEK
- mitogen-activated protein kinase/extracellular signal–regulated kinase kinase
- IKK
- IκB kinase.
References
- 1. Fuchs Y., and Steller H. (2011) Programmed cell death in animal development and disease. Cell 147, 742–758 10.1016/j.cell.2011.10.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Vaux D. L., and Korsmeyer S. J. (1999) Cell death in development. Cell 96, 245–254 10.1016/S0092-8674(00)80564-4 [DOI] [PubMed] [Google Scholar]
- 3. Nagata S. (2018) Apoptosis and the clearance of apoptotic cells. Annu. Rev. Immunol. 36, 489–517 10.1146/annurev-immunol-042617-053010 [DOI] [PubMed] [Google Scholar]
- 4. Birge R. B., Boeltz S., Kumar S., Carlson J., Wanderley J., Calianese D., Barcinski M., Brekken R. A., Huang X., Hutchins J. T., Freimark B., Empig C., Mercer J., Schroit A. J., Schett G., and Herrmann M. (2016) Phosphatidylserine is a global immunosuppressive signal in efferocytosis, infectious disease, and cancer. Cell Death Differ. 23, 962–978 10.1038/cdd.2016.11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Arandjelovic S., and Ravichandran K. S. (2015) Phagocytosis of apoptotic cells in homeostasis. Nat. Immunol. 16, 907–917 10.1038/ni.3253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. deCathelineau A. M., and Henson P. M. (2003) The final step in programmed cell death: phagocytes carry apoptotic cells to the grave. Essays Biochem. 39, 105–117 10.1042/bse0390105 [DOI] [PubMed] [Google Scholar]
- 7. Muñoz L. E., Lauber K., Schiller M., Manfredi A. A., and Herrmann M. (2010) The role of defective clearance of apoptotic cells in systemic autoimmunity. Nat. Rev. Rheumatol. 6, 280–289 10.1038/nrrheum.2010.46 [DOI] [PubMed] [Google Scholar]
- 8. Kawano M., and Nagata S. (2018) Lupus-like autoimmune disease caused by a lack of Xkr8, a caspase-dependent phospholipid scramblase. Proc. Natl. Acad. Sci. U.S.A. 115, 2132–2137 10.1073/pnas.1720732115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hafizi S., and Dahlbäck B. (2006) Gas6 and protein S: vitamin K-dependent ligands for the Axl receptor tyrosine kinase subfamily. FEBS J. 273, 5231–5244 10.1111/j.1742-4658.2006.05529.x [DOI] [PubMed] [Google Scholar]
- 10. Nakano T., Ishimoto Y., Kishino J., Umeda M., Inoue K., Nagata K., Ohashi K., Mizuno K., and Arita H. (1997) Cell adhesion to phosphatidylserine mediated by a product of growth arrest-specific gene 6. J. Biol. Chem. 272, 29411–29414 10.1074/jbc.272.47.29411 [DOI] [PubMed] [Google Scholar]
- 11. Hanayama R., Tanaka M., Miwa K., Shinohara A., Iwamatsu A., and Nagata S. (2002) Identification of a factor that links apoptotic cells to phagocytes. Nature 417, 182–187 10.1038/417182a [DOI] [PubMed] [Google Scholar]
- 12. Miyanishi M., Tada K., Koike M., Uchiyama Y., Kitamura T., and Nagata S. (2007) Identification of TIM4 as a phosphatidylserine receptor. Nature 450, 435–439 10.1038/nature06307 [DOI] [PubMed] [Google Scholar]
- 13. Lemke G. (2017) Phosphatidylserine is the signal for TAM receptors and their ligands. Trends Biochem. Sci. 42, 738–748 10.1016/j.tibs.2017.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rothlin C. V., Carrera-Silva E. A., Bosurgi L., and Ghosh S. (2015) TAM receptor signaling in immune homeostasis. Annu. Rev. Immunol. 33, 355–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nishi C., Toda S., Segawa K., and Nagata S. (2014) TIM4- and MerTK-mediated engulfment of apoptotic cells by mouse resident peritoneal macrophages. Mol. Cell. Biol. 34, 1512–1520 10.1128/MCB.01394-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yanagihashi Y., Segawa K., Maeda R., Nabeshima Y-I., and Nagata S. (2017) Mouse macrophages show different requirements for phosphatidylserine receptor TIM4 in efferocytosis. Proc. Natl. Acad. Sci. U.S.A. 114, 8800–8805 10.1073/pnas.1705365114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Park D., Hochreiter-Hufford A., and Ravichandran K. (2009) The phosphatidylserine receptor TIM-4 does not mediate direct signaling. Curr. Biol. 19, 346–351 10.1016/j.cub.2009.01.042 [DOI] [PubMed] [Google Scholar]
- 18. Scott R. S., McMahon E. J., Pop S. M., Reap E. A., Caricchio R., Cohen P. L., Earp H. S., and Matsushima G. K. (2001) Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature 411, 207–211 10.1038/35075603 [DOI] [PubMed] [Google Scholar]
- 19. Hoffmann P. R., deCathelineau A. M., Ogden C. A., Leverrier Y., Bratton D. L., Daleke D. L., Ridley A. J., Fadok V. A., and Henson P. M. (2001) Phosphatidylserine (PS) induces PS receptor-mediated macropinocytosis and promotes clearance of apoptotic cells. J. Cell Biol. 155, 649–659 10.1083/jcb.200108080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Graham D. K., DeRyckere D., Davies K. D., and Earp H. S. (2014) The TAM family: phosphatidylserine-sensing receptor tyrosine kinases gone awry in cancer. Nat. Rev. Cancer 14, 769–785 10.1038/nrc3847 [DOI] [PubMed] [Google Scholar]
- 21. Lemke G. (2013) Biology of the TAM receptors. CSH Perspect. Biol. 5, a009076 10.1101/cshperspect.a009076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cummings C. T., Deryckere D., Earp H. S., and Graham D. K. (2013) Molecular pathways: MERTK signaling in cancer. Clin. Cancer Res. 19, 5275–5280 10.1158/1078-0432.CCR-12-1451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kasikara C., Kumar S., Kimani S., Tsou W.-I., Geng K., Davra V., Sriram G., Devoe C., Nguyen K. N., Antes A., Krantz A., Rymarczyk G., Wilczynski A., Empig C., Freimark B., et al. (2017) Phosphatidylserine sensing by TAM receptors regulates AKT-dependent chemoresistance and PD-L1 expression. Mol. Cancer Res. 15, 753–764 10.1158/1541-7786.MCR-16-0350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schoumacher M., and Burbridge M. (2017) Key roles of AXL and MER receptor tyrosine kinases in resistance to multiple anticancer therapies. Curr. Oncol. Rep. 19, 19 10.1007/s11912-017-0579-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pettazzoni P., Viale A., Shah P., Carugo A., Ying H., Wang H., Genovese G., Seth S., Minelli R., Green T., Huang-Hobbs E., Corti D., Sanchez N., Nezi L., Marchesini M., et al. (2015) Genetic events that limit the efficacy of MEK and RTK inhibitor therapies in a mouse model of KRAS-driven pancreatic cancer. Cancer Res. 75, 1091–1101 10.1158/0008-5472.CAN-14-1854,10.1158/1538-7445.AM2015-1091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Alessi D. R., Cuenda A., Cohen P., Dudley D. T., and Saltiel A. R. (1995) PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J. Biol. Chem. 270, 27489–27494 10.1074/jbc.270.46.27489 [DOI] [PubMed] [Google Scholar]
- 27. Vlahos C. J., Matter W. F., Hui K. Y., and Brown R. F. (1994) A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002). J. Biol. Chem. 269, 5241–5248 [PubMed] [Google Scholar]
- 28. Roberts W. G., Ung E., Whalen P., Cooper B., Hulford C., Autry C., Richter D., Emerson E., Lin J., Kath J., Coleman K., Yao L., Martinez-Alsina L., Lorenzen M., Berliner M., et al. (2008) Antitumor activity and pharmacology of a selective focal adhesion kinase inhibitor, PF-562,271. Cancer Res. 68, 1935–1944 10.1158/0008-5472.CAN-07-5155 [DOI] [PubMed] [Google Scholar]
- 29. Nagashima S., Yokota M., Nakai E., Kuromitsu S., Ohga K., Takeuchi M., Tsukamoto S., and Ohta M. (2007) Synthesis and evaluation of 2-{[2-(4-hydroxyphenyl)-ethyl]amino}pyrimidine-5-carboxamide derivatives as novel STAT6 inhibitors. Bioorg. Med. Chem. 15, 1044–1055 10.1016/j.bmc.2006.10.015 [DOI] [PubMed] [Google Scholar]
- 30. Natarajan K., Singh S., Burke T. R. Jr., Grunberger D., and Aggarwal B. B. (1996) Caffeic acid phenethyl ester is a potent and specific inhibitor of activation of nuclear transcription factor NF-κB. Proc. Natl. Acad. Sci. U.S.A. 93, 9090–9095 10.1073/pnas.93.17.9090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Haftchenary S., Luchman H. A., Jouk A. O., Veloso A. J., Page B. D., Cheng X. R., Dawson S. S., Grinshtein N., Shahani V. M., Kerman K., Kaplan D. R., Griffin C., Aman A. M., Al-Awar, Weiss S., and Gunning P. T. (2013) Potent targeting of the STAT3 protein in brain cancer stem cells: a promising route for treating glioblastoma. ACS Med. Chem. Lett. 4, 1102–1107 10.1021/ml4003138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ling L., Templeton D., and Kung H. J. (1996) Identification of the major autophosphorylation sites of Nyk/Mer, an NCAM-related receptor tyrosine kinase. J. Biol. Chem. 271, 18355–18362 10.1074/jbc.271.31.18355 [DOI] [PubMed] [Google Scholar]
- 33. Nishimoto S., and Nishida E. (2006) MAPK signalling: ERK5 versus ERK1/2. EMBO Rep. 7, 782–786 10.1038/sj.embor.7400755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hresko R. C., and Mueckler M. (2005) mTOR.RICTOR is the Ser473 kinase for Akt/protein kinase B in 3T3-L1 adipocytes. J. Biol. Chem. 280, 40406–40416 10.1074/jbc.M508361200 [DOI] [PubMed] [Google Scholar]
- 35. Sulzmaier F. J., Jean C., and Schlaepfer D. D. (2014) FAK in cancer: mechanistic findings and clinical applications. Nat. Rev. Cancer 14, 598–610 10.1038/nrc3792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gouilleux F., Wakao H., Mundt M., and Groner B. (1994) Prolactin induces phosphorylation of Tyr694 of Stat5 (MGF), a prerequisite for DNA binding and induction of transcription. EMBO J. 13, 4361–4369 10.1002/j.1460-2075.1994.tb06756.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Reich N. C. (2013) STATs get their move on. JAK-STAT 2, e27080–27010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mikita T., Campbell D., Wu P., Williamson K., and Schindler U. (1996) Requirements for interleukin-4-induced gene expression and functional characterization of Stat6. Mol. Cell. Biol. 16, 5811–5820 10.1128/MCB.16.10.5811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hinz M., and Scheidereit C. (2014) The IκB kinase complex in NF-κB regulation and beyond. EMBO Rep. 15, 46–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Georgescu M. M., Kirsch K. H., Shishido T., Zong C., and Hanafusa H. (1999) Biological effects of c-Mer receptor tyrosine kinase in hematopoietic cells depend on the Grb2 binding site in the receptor and activation of NF-κB. Mol. Cell. Biol. 19, 1171–1181 10.1128/MCB.19.2.1171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hedger G., Sansom M. S., and Koldsø H. (2015) The juxtamembrane regions of human receptor tyrosine kinases exhibit conserved interaction sites with anionic lipids. Sci. Rep. 5, 9198 10.1038/srep09198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Reddien P. W., and Horvitz H. R. (2004) The engulfment process of programmed cell death in Caenorhabditis elegans. Annu. Rev. Cell Dev. Biol. 20, 193–221 10.1146/annurev.cellbio.20.022003.114619 [DOI] [PubMed] [Google Scholar]
- 43. Pinto S. M., and Hengartner M. O. (2012) Cleaning up the mess: cell corpse clearance in Caenorhabditis elegans. Curr. Opin. Cell Biol. 24, 881–888 10.1016/j.ceb.2012.11.002 [DOI] [PubMed] [Google Scholar]
- 44. Suzuki J., Denning D. P., Imanishi E., Horvitz H. R., and Nagata S. (2013) Xk-related protein 8 and CED-8 promote phosphatidylserine exposure in apoptotic cells. Science 341, 403–406 10.1126/science.1236758 [DOI] [PubMed] [Google Scholar]
- 45. Chen Y.-Z., Mapes J., Lee E.-S., Skeen-Gaar R. R., and Xue D. (2013) Caspase-mediated activation of Caenorhabditis elegans CED-8 promotes apoptosis and phosphatidylserine externalization. Nat. Commun. 4, 2726 10.1038/ncomms3726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kang Y., Zhao D., Liang H., Liu B., Zhang Y., Liu Q., Wang X., and Liu Y. (2012) Structural study of TTR-52 reveals the mechanism by which a bridging molecule mediates apoptotic cell engulfment. Genes Dev. 26, 1339–1350 10.1101/gad.187815.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gumienny T. L., Brugnera E., Tosello-Trampont A. C., Kinchen J. M., Haney L. B., Nishiwaki K., Walk S. F., Nemergut M. E., Macara I. G., Francis R., Schedl T., Qin Y., Van Aelst L., Hengartner M. O., and Ravichandran K. S. (2001) CED-12/ELMO, a novel member of the CrkII/Dock180/Rac pathway, is required for phagocytosis and cell migration. Cell 107, 27–41 10.1016/S0092-8674(01)00520-7 [DOI] [PubMed] [Google Scholar]
- 48. Todt J. C., Hu B., and Curtis J. L. (2004) The receptor tyrosine kinase MerTK activates phospholipase C2 during recognition of apoptotic thymocytes by murine macrophages. J. Leukocyte Biol. 75, 705–713 10.1189/jlb.0903439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tibrewal N., Wu Y., D'mello V., Akakura V. R., George T. C., Varnum B., and Birge R. B. (2008) Autophosphorylation docking site Tyr-867 in Mer receptor tyrosine kinase allows for dissociation of multiple signaling pathways for phagocytosis of apoptotic cells and down-modulation of lipopolysaccharide-inducible NF-κB transcriptional activation. J. Biol. Chem. 283, 3618–3627 10.1074/jbc.M706906200 [DOI] [PubMed] [Google Scholar]
- 50. Wu Y., Singh S., Georgescu M.-M., and Birge R. B. (2005) A role for Mer tyrosine kinase in alphavbeta5 integrin-mediated phagocytosis of apoptotic cells. J. Cell Sci. 118, 539–553 10.1242/jcs.01632 [DOI] [PubMed] [Google Scholar]
- 51. Nakaya M., Kitano M., Matsuda M., and Nagata S. (2008) Spatiotemporal activation of Rac1 for engulfment of apoptotic cells. Proc. Natl. Acad. Sci. U.S.A. 105, 9198–9203 10.1073/pnas.0803677105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Elliott M. R., and Ravichandran K. S. (2016) The dynamics of apoptotic cell clearance. Dev. Cell 38, 147–160 10.1016/j.devcel.2016.06.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tanimura S., and Takeda K. (2017) ERK signalling as a regulator of cell motility. J. Biochem. 162, 145–154 10.1093/jb/mvx048 [DOI] [PubMed] [Google Scholar]
- 54. Webb D. J., Donais K., Whitmore L. A., Thomas S. M., Turner C. E., Parsons J. T., and Horwitz A. F. (2004) FAK-Src signalling through paxillin, ERK and MLCK regulates adhesion disassembly. Nat. Cell Biol. 6, 154–161 10.1038/ncb1094 [DOI] [PubMed] [Google Scholar]
- 55. Ishibe S., Joly D., Liu Z.-X., and Cantley L. G. (2004) Paxillin serves as an ERK-regulated scaffold for coordinating FAK and Rac activation in epithelial morphogenesis. Mol. Cell 16, 257–267 10.1016/j.molcel.2004.10.006 [DOI] [PubMed] [Google Scholar]
- 56. Stark G. R., and Darnell J. E. Jr. (2012) The JAK-STAT pathway at twenty. Immunity 36, 503–514 10.1016/j.immuni.2012.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sehgal P. B. (2013) Non-genomic STAT5-dependent effects at the endoplasmic reticulum and Golgi apparatus and STAT6-GFP in mitochondria. JAK-STAT 2, e24860–24810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Silver D. L., Naora H., Liu J., Cheng W., and Montell D. J. (2004) Activated signal transducer and activator of transcription (STAT) 3: localization in focal adhesions and function in ovarian cancer cell motility. Cancer Res. 64, 3550–3558 10.1158/0008-5472.CAN-03-3959 [DOI] [PubMed] [Google Scholar]
- 59. Stanford J. C., Young C., Hicks D., Owens P., Williams A., Vaught D. B., Morrison M. M., Lim J., Williams M., Brantley-Sieders D. M., Balko J. M., Tonetti D., Earp H. S. 3rd, and Cook R. S. (2014) Efferocytosis produces a prometastatic landscape during postpartum mammary gland involution. J. Clin. Invest. 124, 4737–4752 10.1172/JCI76375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Dorfman D. M., Hornick J. L., Shahsafaei A., and Freeman G. J. (2010) The phosphatidylserine receptors, T cell immunoglobulin mucin proteins 3 and 4, are markers of histiocytic sarcoma and other histiocytic and dendritic cell neoplasms. Hum. Pathol. 41, 1486–1494 10.1016/j.humpath.2010.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zhang Q., Wang H., Wu X., Liu B., Liu W., Wang R., Liang X., Ma C., and Gao L. (2015) TIM-4 promotes the growth of non-small-cell lung cancer in a RGD motif-dependent manner. Br. J. Cancer 113, 1484–1492 10.1038/bjc.2015.323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Xu L., Xiao H., Xu M., Zhou C., Yi L., and Liang H. (2011) Glioma-derived T cell immunoglobulin- and mucin domain-containing molecule-4 (TIM4) contributes to tumor tolerance. J. Biol. Chem. 286, 36694–36699 10.1074/jbc.M111.292540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Schlegel J., Sambade M. J., Sather S., Moschos S. J., Tan A.-C., Winges A., DeRyckere D., Carson C. C., Trembath D. G., Tentler J. J., Eckhardt S. G., Kuan P.-F., Hamilton R. L., Duncan L. M., Miller C. R., et al. (2013) MERTK receptor tyrosine kinase is a therapeutic target in melanoma. J. Clin. Invest. 123, 2257–2267 10.1172/JCI67816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. McIver A. L., Zhang W., Liu Q., Jiang X., Stashko M. A., Nichols J., Miley M. J., Norris-Drouin J., Machius M., DeRyckere D., Wood E., Graham D. K., Earp H. S., Kireev D., Frye S. V., and Wang X. (2017) Discovery of macrocyclic pyrimidines as MerTK-specific inhibitors. Chem. Med. Chem. 12, 207–213 10.1002/cmdc.201600589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Cummings C. T., Zhang W., Davies K. D., Kirkpatrick G. D., Zhang D., DeRyckere D., Wang X., Frye S. V., Earp H. S., and Graham D. K. (2015) Small molecule inhibition of MERTK is efficacious in non-small cell lung cancer models independent of driver oncogene status. Mol. Cancer Ther. 14, 2014–2022 10.1158/1535-7163.MCT-15-0116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Cohen P. L., Caricchio R., Abraham V., Camenisch T. D., Jennette J. C., Roubey R. A., Earp H. S., Matsushima G., and Reap E. A. (2002) Delayed apoptotic cell clearance and lupus-like autoimmunity in mice lacking the c-mer membrane tyrosine kinase. J. Exp. Med. 196, 135–140 10.1084/jem.20012094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Toda S., Hanayama R., and Nagata S. (2012) Two-step engulfment of apoptotic cells. Mol. Cell. Biol. 32, 118–125 10.1128/MCB.05993-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Morita S., Kojima T., and Kitamura T. (2000) Plat-E: an efficient and stable system for transient packaging of retroviruses. Gene Ther. 7, 1063–1066 10.1038/sj.gt.3301206 [DOI] [PubMed] [Google Scholar]
- 69. Shiraishi T., Suzuyama K., Okamoto H., Mineta T., Tabuchi K., Nakayama K., Shimizu Y., Tohma J., Ogihara T., Naba H., Mochizuki H., and Nagata S. (2004) Increased cytotoxicity of soluble Fas ligand by fusing isoleucine zipper motif. Biochem. Biophys. Res. Commun. 322, 197–202 10.1016/j.bbrc.2004.07.098 [DOI] [PubMed] [Google Scholar]
- 70. Higuchi R. (1990) Recombinant PCR. in PCR Protocols: A Guide to Methods and Applications (Michael A. I., David H. G., John J. S., and Thomas J. W., eds) pp. 177–188, Academic Press, San Diego, CA [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.