

Abstract
Human immunodeficiency virus-1 (HIV-1) Tat is degraded in the host cell both by proteasomal and lysosomal pathways, but the specific molecules that engage with Tat from these pathways are not known. Because E3 ubiquitin ligases are the primary determinants of substrate specificity within the ubiquitin-dependent proteasomal degradation of proteins, we first sought to identify the E3 ligase associated with Tat degradation. Based on the intrinsic disordered nature of Tat protein, we focused our attention on host cell E3 ubiquitin ligase CHIP (C terminus of HSP70-binding protein). Co-transfection of Tat with a CHIP-expressing plasmid decreased the levels of Tat protein in a dose-dependent manner, without affecting the corresponding mRNA levels. Additionally, the rate of Tat protein degradation as measured by cycloheximide (CHX) chase assay was increased in the presence of CHIP. A CHIP mutant lacking the U-box domain, which is responsible for protein ubiquitination (CHIPΔU-box), was unable to degrade Tat protein. Furthermore, CHIP promoted ubiquitination of Tat by both WT as well as Lys-48–ubiquitin, which has only a single lysine residue at position 48. CHIP transfection in HIV-1 reporter TZM-bl cells resulted in decreased Tat-dependent HIV-1 long-terminal repeat (LTR) promoter transactivation as well as HIV-1 virion production. CHIP knockdown in HEK-293T cells using CRISPR-Cas9 led to higher virion production and enhanced Tat-mediated HIV-1 LTR promoter transactivation, along with stabilization of Tat protein. Together, these results suggest a novel role of host cell E3 ubiquitin ligase protein CHIP in regulating HIV-1 replication through ubiquitin-dependent degradation of its regulatory protein Tat.
Keywords: viral replication, viral protein, ubiquitylation (ubiquitination), protein degradation, proteasome
Introduction
The majority of cellular protein degradation occurs through the lysosomal and proteasomal pathways (1, 2). The proteasomal protein degradation is accomplished either by the attachment of ubiquitin to substrate protein followed by its degradation through the 26S proteasomal pathway or by the ubiquitin-independent 20S proteasomal pathway (3, 4). The ubiquitination of proteins occurs by sequential action of ubiquitin-activating enzyme E1, ubiquitin-conjugating enzyme E2, and ubiquitin ligase E3 using ubiquitin, substrate protein, and ATP. The ubiquitination process is finely regulated by E3 ubiquitin ligases that control its final step and are central to the recognition of the substrate proteins (5). Human genome contains >600 E3 ubiquitin ligase proteins, many of which are responsible for the ubiquitination of multiple substrate proteins (5, 6). In addition to protein degradation, ubiquitination of proteins also controls various other cellular processes like protein localization or protein–protein interaction. Different functions or fates of the ubiquitinated proteins are governed by the type of ubiquitin chain branches and by number of ubiquitin molecules attached (7, 8). Proteins ubiquitinated through lysine 48-linked ubiquitin chains are mainly targeted for degradation, whereas lysine 63–ubiquitinated proteins are targeted for altered localization or association (8). Proteins ubiquitinated through nonconventional ubiquitin chains, i.e. non-Lys-48 or non-Lys-63, are also reported to be degraded through the 26S proteasomal pathway (9, 10). Protein ubiquitination also play important roles in host–pathogen interactions, and the pathway is exploited by many viruses for their own survival and expansion. It is used in regulating viral replication, progeny virus generation, protection of viruses by the host immune system, and neutralization of host cell restriction factors (11, 12). HIV-1 Vif utilizes cellular ubiquitin ligase CULLIN5 to promote the ubiquitination and degradation of APOBEC3G, which causes hypermutation in the HIV-1 genome (13). Similarly, Vpr uses CULLIN4 for G2 cell cycle arrest for enhanced viral replication and virion production (14). Recently, we have shown that Vpr redirects the ubiquitin proteasome system by suppressing the whole-cell ubiquitination process and enhancing the ubiquitination of its substrates for optimal viral replication (15).
Replication and production of HIV-1 virions are primarily regulated by the regulatory protein Tat, which enhances viral replication by multiple orders by promoting the formation of full-length viral transcripts (16, 17). Tat protein is not a fully folded protein but is structurally disordered. The intrinsically disordered nature of Tat is important for its recruitment of host cell proteins for viral promoter transactivation and viral RNA synthesis (18). The presence of intrinsic disorder in Tat was demonstrated by multiple approaches, including CD and NMR spectroscopy. NMR studies have shown the lack of a fixed conformation and fast dynamics that provide the ability of Tat to interact with multiple proteins and nucleic acids (18, 19). Interaction of Tat with TAR RNA promotes folding of disordered Tat protein, and Tat interaction with TAR RNA maintains Tat in the folding competent state, which is important for binding of Tat with cellular factors for transactivation function (20). The level of Tat protein to control HIV-1 replication is extremely small, which is required for optimal replication and for causing pathogenicity (21).
In addition to viral replication, Tat also regulates other cellular and viral pathways to support pathogenicity of HIV-1. Tat plays a critical role in breaking the viral latency, and the secreted Tat protein induces the death of uninfected bystander cells (22, 23). Recent studies revealed multiple novel functions of Tat in addition to its role as HIV-1 LTR4 transcriptional activator. In the brains of HIV-1–infected patients, Tat causes neurotoxicity by promoting the aggregation of Aβ fibrils into rigid and mechanically-resistant thick fibers, which make pores in membranes; Tat also increases the adhesion capacity of these fibers to cell membranes thereby increasing the damage (24). Tat is also involved in gene translocation–mediated cancer formation in HIV-1–infected patients, as treatment to B lymphocytes with of Tat protein results in the elevation cellular RAG-1 gene expression, which causes DNA damage in the cells. DNA damage in the MYC gene locus results in the localization of MYC with immunoglobulin heavy chain gene IGH, which causes enhanced MYC expression and cellular transformation (25). Recent reports also show that Tat and RNA interaction in the cell regulates HIV-1 genome splicing at the major splice donor site (5′splice site) located in the untranslated leader of the HIV-1 transcript. Tat-mediated splicing results in optimal production of all viral RNAs and proteins (26). Nonprocessive transcription from HIV-1 LTR promoter produces short TAR RNAs, which act as precursors to miRNAs and are cleaved by DICER enzyme to yield miRNAs. Production of these miRNAs is stimulated by HIV-1 Tat, and hence it promotes miRNA formation from the HIV-1 genome without its cleavage from the viral genome (27). Tat protein is cleared from infected cells as it is degraded by multiple pathways. Recent studies show that HIV-1 Tat is degraded through the lysosomal pathway (28). Tat is also degraded through the ubiquitin-independent 20S proteasomal pathway due to its disordered nature (29, 30). Tat protein is reported to be ubiquitinated through the lysine 48–linked ubiquitin chain, which is then recognized by the 26S proteasome for its degradation (31). NRON, an lncRNA expressed in resting CD4+ T cells, promotes Tat degradation through the ubiquitin proteasome system (UPS). NRON directly links Tat to components of UPS (CUL4B and PSMD11) resulting in ubiquitination and subsequent proteasomal degradation (32). Similar to accessory proteins, Tat also hijacks cellular ubiquitination machinery. Tat binds to host cell E3 ubiquitin ligase UBE2O and hijacks it to promote ubiquitination of host cell P-TEFb kinase inhibitor HEXIM1. Ubiquitination of HEXIM1 leads to its cytoplasmic sequestration and the release of P-TEFb. The released P-TEFb is recruited to the HIV-1 promoter leading to transcriptional activation (33). Similarly, Tat recruits HDM2 (human MDM2), E3 ubiquitin ligase, to promote ubiquitination of interferon regulatory factor 1 (IRF-1) to inhibit host immune responses during HIV-1 infection (34).
The cellular U-box E3 ubiquitin ligase protein CHIP controls the stability of various cellular proteins by promoting their ubiquitination (35). CHIP is associated with molecular chaperones HSP90 and HSP70 through its TPR domain and is involved in the degradation of unfolded proteins through ubiquitination and proteasomal degradation (35, 36). CHIP promotes ubiquitination and degradation of prematurely terminated proteins, destabilized mutant proteins, as well as proteins involved in a diverse range of cellular functions (37, 38). Here, we report that HIV-1 Tat protein is degraded by CHIP and show that its U-box ubiquitin ligase domain is important for this process. CHIP promotes Tat degradation by its ubiquitination and subsequent proteasomal degradation. The Tat-mediated LTR promoter transactivation and virus production are also inhibited by CHIP. The knockdown of endogenous CHIP resulted in Tat protein stabilization with increased HIV-1 production. Thus, host cell E3 ubiquitin ligase CHIP controls HIV-1 replication by promoting ubiquitin-dependent proteasomal degradation of Tat.
Results
Tat protein is degraded through the ubiquitin proteasome pathway
To investigate the mechanism of HIV-1 Tat protein degradation, the Myc-Tat–expressing plasmid was transfected in HEK-293T cells. The cells were treated with CHX in the presence or absence of the proteasomal inhibitor MG132 or the lysosomal inhibitor ammonium chloride for different time periods and probed for Tat protein. CHX treatment resulted in rapid degradation of Tat. However, in the presence of MG132, the degradation is reduced, but treatment with ammonium chloride did not affect the rate of Tat protein degradation (Fig. 1, A and B). This stabilization of Tat by proteasomal inhibitor MG132 but not by ammonium chloride suggested that the degradation of Tat occurs predominantly through the proteasomal pathway.
Figure 1.

A, HEK-293T cells were transfected with Myc-Tat–expressing plasmid; 24 h post-transfection, the cells were either treated with CHX (100 μg/ml) alone or CHX with ammonium chloride or MG132 for the indicated time periods. Tat protein level was measured by immunoblotting. The blot shown is representative of three different independent experiments. B, Tat protein intensity was measured using ImageJ and plotted with reference to CHX treatment time.
CHIP enhances degradation of Tat by promoting its ubiquitination
CHIP is a U-box E3 ubiquitin ligase that catalyzes the ubiquitination reaction by transferring the ubiquitin molecule from E2 to the substrate (35, 36). The specificity of substrate is governed by their physical interaction with CHIP. The previous results of Tat stabilization by MG132 suggested that Tat might be ubiquitinated and degraded by the proteasomal pathway. Recent reports have also shown Lys-48 ubiquitination and degradation of Tat by the cellular E3 ubiquitin ligase (31, 32). Tat is an intrinsically unstructured protein with major structural disorder along its entire length (29). The intrinsically unstructured cellular proteins as well as proteins under the conditions of unfolding and misfolding are recognized by HSP70-associated U-box E3 ubiquitin ligase CHIP. These proteins are then ubiquitinated by CHIP and degraded through the 26S proteasome. We have hypothesized that CHIP, which is an abundantly expressed cellular E3 ubiquitin ligase, might be promoting degradation of Tat because HIV-1 Tat is completely unstructured. To test this hypothesis, HEK-293T cells were co-transfected with Myc-Tat- and CHIP- or CHIPΔU-expressing plasmids. The results showed that CHIP but not CHIPΔU resulted in the reduction of the levels of Tat protein (Fig. 2A and Fig. S4, A). Similar to CHIPΔU, expression of CHIPK30A, which does not bind with HSP70, was unable to reduce the levels of Tat protein (Fig. S1). To rule out the nonspecific effect of CHIP on the reduction of protein levels in transfected cells, HEK-293T cells were co-transfected with CHIP and Gag or Nef, and immunoblotting was carried out with their respective antibodies. Compared with the effect of CHIP on Tat, the levels of Gag and Nef proteins were largely unaffected by the expression of CHIP (Fig. 2, B and C and Fig. S4B). The p53 tumor suppressor protein is a known target of CHIP, and it promotes ubiquitination and degradation of p53 (40). To confirm the effect of CHIP on Tat, the endogenous p53 protein levels of CHIP-transfected cells were also probed as a positive control. As shown in Fig. 2D, the level of p53 was decreased in CHIP-transfected cells, which confirmed the functional activity of CHIP. To study the dose-dependent effect of CHIP on Tat protein, the indicated amounts of Tat were co-transfected with increasing doses of CHIP. The immunoblotting of Tat showed a dose-dependent decrease of Tat protein with CHIP (Fig. 2E). We then wanted to find out whether the decrease in the levels of Tat protein is at transcriptional or translational levels. Tat mRNA levels were unchanged in the same cells in response to the expression of CHIP (Fig. 2F). These results confirmed that CHIP specifically promoted the degradation of Tat protein, and it was not due to decreased transcription. Because Tat is reported to be degraded both through lysosomal and proteasomal pathways, hence to investigate the pathway involved with CHIP-induced Tat degradation, HEK-293T cells were transfected with Tat and CHIP followed by treatment with chloroquine or MG132. The result showed that CHIP reduces Tat in untreated and chloroquine-treated cells but not in MG132-treated cells, which suggested that the degradation of Tat by CHIP occurred through the proteasomal pathway (Fig. 2G). Red fluorescence protein–tagged HIV-1 Tat (RFP–Tat) was co-transfected with CHIP or CHIPΔU in HEK-293T cells, and the red fluorescence of the cells was measured, which showed a reduction by CHIP but not by CHIPΔU (Fig. S2). To investigate the effect of CHIP on the degradation kinetics of Tat, HEK-293T cells were co-transfected with Tat- and CHIP- or CHIPΔU-expressing plasmids, and the cells were treated with CHX for different time periods. The level of Tat protein was measured by immunoblotting. CHX treatment resulted in a time-dependent decrease of Tat, but the rate of degradation is significantly enhanced in the presence of CHIP (Fig. 2, H and I). CHIPΔU did not affect the rate of Tat protein degradation when compared with WT CHIP (Fig. 2, H and I).
Figure 2.

A, HEK-293T cells were co-transfected with Myc-Tat-, Myc-CHIP-, or Myc-CHIPΔU–expressing plasmids, and Tat protein was immunoblotted 36 h post-transfection. B, HIV-1 Gag was transfected with CHIP into HEK-293T cells, and 36 h after transfection, Gag was immunoblotted with anti-Gag antibody. C, HIV-1 Nef was co-transfected with CHIP or CHIPΔU in HEK-293T cells, and Nef was immunoblotted. D, CHIP was transfected into HEK-293T cells, and after 36 h, p53 protein was measured by immunoblotting. E, HEK-293T cells were co-transfected with Tat (05 μg) and increasing concentrations of CHIP (05, 1, and 2 μg). After 36 h, cells were harvested and divided into two parts. One part was lysed and immunoblotted for Tat protein. F, the other part of cells from E was used for total RNA isolation followed by cDNA synthesis, and HIV-1 Tat cDNA was amplified using Tat-specific primers by RT-PCR assay. G, HA-Tat was transfected with CHIP into HEK-293T cells, and after 24 h, cells were treated either with chloroquine or MG132 for 8 h, and Tat was immunoblotted. H, HEK-293T cells were co-transfected with Tat and CHIP or CHIPΔU, after 24 h, CHX treatment was done for the time periods as shown in figure, and Tat protein was immunoblotted. I, band intensity of Tat and GAPDH was measured and plotted with reference to CHX treatment times. The images shown here are representative of three different independent experiments.
To investigate whether HIV-1 Tat is ubiquitinated in the presence of MG132, HEK-293T cells were co-transfected with Tat- and ubiquitin-expressing plasmids followed by MG132 treatment. The ubiquitinated proteins were purified and separated on SDS-PAGE followed by immunoblotting for Tat. This experiment showed increased ubiquitination of Tat in the presence of MG132 (Fig. 3A). It was reported earlier that cellular E3 ubiquitin ligase MDM2 ubiquitinates Tat through lysine 63 linkages, which result in the enhancement of its transactivation potential (41). However, Lys-63–linked ubiquitination does not target the proteins for proteasomal degradation (5), and hence it does not accumulate after MG132 treatment. Thus, the accumulation of ubiquitinated Tat protein in response to MG132 treatment indicated Lys-48–linked ubiquitination of Tat. Because Lys-48–ubiquitinated proteins are degraded through proteasomes, blocking the proteasomal action should result in their accumulation. To assess whether CHIP promotes Tat ubiquitination, Tat was co-transfected with ubiquitin and CHIP in HEK-293T cells. MG132 treatment was carried out as described before, and ubiquitinated proteins were purified. Tat was immunoblotted, which showed that CHIP enhanced Tat protein ubiquitination (Fig. 3B). Furthermore, HEK-293T cells were transfected with Tat and ubiquitin in the presence of CHIP or CHIPΔU expression plasmids. The ubiquitinated Tat was probed by anti-HA antibody after Ni-NTA affinity purification. The result showed that ubiquitination of Tat in CHIP-transfected cells is significantly higher than observed with CHIPΔU-expressing cells (Fig. 3C). To investigate whether Tat protein ubiquitination by CHIP occurs by Lys-48–linked ubiquitin chains, HEK-293T cells were co-transfected with Tat, ubiquitin R48K, and CHIP. The transfected cells were treated with MG132, and ubiquitinated proteins were purified with Ni-NTA and immunoblotted for Tat protein. Expression of CHIP enhanced the ubiquitination of Tat in a dose-dependent manner by Lys-48–linked ubiquitin chains (Fig. 3D). CHIP is an E3 ubiquitin ligase that interacts with the substrate protein as well as E2 ubiquitin-conjugating enzyme leading to transfer of ubiquitin from E2 to the substrate. Thus, CHIP physically interacts with its substrate proteins. Our experiments have clearly shown that CHIP promotes degradation of Tat by its ubiquitination, and hence we hypothesize that it might be interacting with Tat. To investigate the interaction of CHIP with Tat, GST-pulldown assay was performed using Myc-Tat expressed in HEK-293T cells and bacterially expressed and purified GST–CHIP. GST was used as a negative control. GST pulldown assay was performed as described under “Experimental procedures,” and the eluted proteins were immunoblotted for Tat. Tat interacted specifically with GST–CHIP but not with GST confirming the interaction of Tat with CHIP (Fig. 3E). To investigate the role of chaperones in the interaction of Tat with CHIP, HA-Tat was co-transfected separately with Myc-CHIP, Myc-CHIP-K30A, and Myc-CHIPΔU in HEK-293T cells. Immunoprecipitation was carried out using anti-HA antibody followed by immunoblotting. Myc-CHIP and Myc-CHIPΔU but not Myc-CHIP K30A were detected in HA-Tat immunoprecipitated samples (Fig. 3F). Inability of interaction between Tat and CHIP K30A suggests that the association between Tat with CHIP depends on Hsp70. The mutation in chaperone-binding domain of CHIP resulted in the loss of interaction between CHIP and Tat.
Figure 3.

A, Myc-Tat and His6-ubiquitin were transfected into HEK-293T cells, and after 24 h, the cells were treated with MG132 for 8 h. Ubiquitinated proteins were purified as described under “Experimental procedures,” and ubiquitination of Tat was detected with Myc antibody. The blot is representative of three different independent experiments. B, Myc-Tat was transfected along with His6-ubiquitin (6×His-Ub) in the absence or presence of CHIP; the cells were treated with MG132 for 8 h, and the ubiquitinated proteins were purified and immunoblotted with anti-Myc antibody. The blot is representative of three different independent experiments. C, HEK-293T cells were co-transfected with Tat and His6-ubiquitin, and CHIP or CHIPΔU MG132 treatment was done for 8 h followed by purification of ubiquitinated proteins using Ni-NTA, and Tat was detected by immunoblotting using anti-Myc antibody. D, HEK-293T cells were transfected with HA-Tat, His6-Myc-ubiquitin, R48K, and increasing doses of CHIP. After 24 h, MG132 was added for 8 h, and ubiquitinated proteins were purified using Ni-NTA. Tat ubiquitination was detected using anti-HA antibody. E, GST and GST–CHIP were expressed in E. coli DH5α, and Myc-Tat was expressed in HEK-293T. Pulldown assay was performed using cell lysates and GSH-agarose beads as described under “Experimental procedures” followed by immunoblotting with anti-Myc and anti-GST antibody. The blot is representative of three different independent experiments. F, HEK-293T cells were transfected with plasmids expressing HA-Tat either with Myc-CHIP, Myc-CHIP K30A, or Myc-CHIPΔU. Lysates were prepared, and rabbit-raised anti-HA antibody was added followed by pullout of antibody-binding proteins using protein-A–agarose beads. Immunoblotting of pulled out proteins was done using mouse-raised anti-Myc antibody for CHIP protein detection and rabbit anti-HA for Tat protein detection.
HIV-1 replication is inhibited by CHIP
Tat alone is responsible for activation of the HIV-1 promoter and regulates the replication of viral genome and expression of its proteins. Thus, an increase or decrease in the level of Tat must correlate with the extent of HIV-1 promoter activation and viral gene expression. To study the effect of CHIP on Tat-mediated HIV-1 LTR promoter transactivation, TZM-bl cells were transfected with Tat- and CHIP-expressing plasmids followed by measurement of luciferase activity. Expression of Tat in TZM-bl cells resulted in HIV-1 LTR promoter activation as expected. However, simultaneous expression of CHIP but not CHIPΔU led to the reduction of Tat-dependent HIV-1 LTR transactivation (Fig. 4A). The pNL4-3.Luc.R−E− expresses luciferase, which is an indirect measurement of HIV-1 replication or virus production. HEK-293T cells were co-transfected with pNL4-3.Luc.R−E− and CHIP or CHIPΔU followed by measurement of luciferase activity. We observed a significant decrease in luciferase activity in the presence of CHIP but not CHIPΔU confirming the role of CHIP in Tat protein degradation (Fig. 4B). The decrease of LTR luciferase activity by CHIP once again confirmed the destabilizing effect of CHIP on Tat protein, which supports our previous results. To assess the role of CHIP in virus production, HIV-1 proviral construct pNL4-3 was co-transfected with CHIP in HEK-293T followed by immunoblotting for Gag protein. The expression of CHIP resulted in the reduction of Gag protein levels (Fig. S3A). We also observed a dose-dependent reduction in the levels of Gag protein in CHIP-transfected cells (Fig. 4C). However, under similar conditions CHIPΔU did not decrease the levels of Gag protein (Fig. 4D). To investigate the effect of CHIP on HIV-1 production in CD4 T lymphocytes, pNL4-3 was co-transfected with CHIP or CHIPΔU in Jurkat cells. The transfected cells were incubated for 48 h followed by immunoblotting for Gag to measure HIV-1 production. CHIP-transfected cells showed reduction in Gag protein levels. However, there is no significant decrease in Gag level in CHIPΔU-transfected cells. The result shows that CHIP controls HIV-1 replication and virus production in CD4 T-lymphocytic Jurkat cell line (Fig. 4E). To study the effect of CHIP on HIV-1 production, we used APOBEC3G as a positive control. HEK293T cells were transfected with pNL4-3 and CHIP or APOBEC3G and immunoblotted for Gag to assess the effect of these proteins on virus production. We found that Gag was reduced both in APOBEC3G- and CHIP-transfected cells further confirming the negative role of CHIP on HIV-1 virion production (Fig. 4F). To further evaluate the role of CHIP on HIV-1 production, TZM-bl cells were transfected with CHIP followed by infection with VSV-G pseudotyped HIV-1. After 24 h, β-gal staining was carried out to measure HIV-1 virion production. The viral production was reduced by ∼60% in CHIP transfected TZM-bl cells (Fig. 4, G and H, and Fig. S3, B and C). HEK-293T cells were transfected with CHIP or CHIPΔU, and the cells were infected with VSV-G–pseudotyped HIV-1. The HIV-1 production was measured by immunoblotting the Gag protein. Gag protein was reduced significantly in CHIP-transfected cells (Fig. 4, I and J), whereas its level was unaffected in CHIPΔU-transfected cells (Fig. 4, K and L). Gag was also reduced in a dose-dependent manner in CHIP-transfected HEK-293T cells infected with VSV-G–pseudotyped HIV-1 (Fig. S3D). These results support the conclusion that CHIP reduces the Tat-mediated HIV-1 LTR transactivation by promoting Tat protein degradation leading to reduction of HIV-1 replication and virus production.
Figure 4.

A, TZM-bl cells were co-transfected with Tat along with CHIP- or CHIPΔU-expressing constructs, and 36 h post-transfection, cells were lysed, and luciferase activity was measured. As a transfection control, GFP was used, and the luciferase readings were normalized with GFP fluorescence and plotted. B, pNL4-3Luc.R−E− was transfected with CHIP- or CHIPΔU-expressing plasmids into TZM-bl cells, and luciferase activity was measured and plotted after normalization with GFP. C, pNL4-3 was co-transfected with increasing doses of CHIP in HEK-293T cells, and after 36 h, the Gag was immunoblotted. The blot is representative of three different independent experiments. D, increasing doses of CHIPΔU were co-transfected with 10 μg of pNL4-3. The Gag level was monitored by immunoblotting after 36 h of transfection. The blot is representative of three different independent experiments. E, Jurkat cells were transfected with pNL4-3 and CHIP or CHIPΔU, and the Gag protein levels were measured by immunoblotting after 48 h of transfection. F, HEK-293T cells were transfected with pNL4-3 and CHIP or A3G, and the Gag protein levels were measured by immunoblotting. The blot is representative of three different independent experiments. G, TZM-bl cells were transfected with CHIP, and after 24 h, the cells were infected with VSVG-pseudotyped HIV-1. Cells were further cultured for 24 h followed by staining for β-gal activity as described under “Experimental procedures.” The image is representative of three different independent experiments. H, β-gal staining intensity was measured by ImageJ and plotted. I, HEK-293T cells were transfected with CHIP, and after 24 h, the cells were infected with VSVG-pseudotyped HIV-1. Cells were cultured for 24 h, and immunoblotted for Gag. The blot is representative of three different independent experiments. J, Gag level was measured with ImageJ and plotted. K, HEK-293T cells were transfected with CHIPΔU, and after 24 h, the cells were infected with VSVG-pseudotyped HIV-1. Cells were further cultured for 24 h and immunoblotted for Gag. The blot is representative of three different independent experiments. L, Gag level was measured with ImageJ and plotted. The blot is representative of three different independent experiments.
CHIP knockdown increases the stability of Tat as well as HIV-1 virion production
Human cells express a substantial amount of endogenous CHIP, and therefore to determine its effect on Tat protein stability, viral replication, and virion production, CHIP levels should be reduced sufficiently. To reduce endogenous CHIP, knockdown was performed using CRISPR-Cas9 as described under “Experimental procedures.” Two guide RNAs (sgRNA) targeting CHIP at 7–26 and 76–95 nucleotides at the N terminus were synthesized and cloned in pX458 plasmid (Fig. 5A). The sgRNA constructs were transfected in HEK-293T cells and selected by fluorescence-activated cell sorting (FACS) using green fluorescence because pX458 also expresses GFP. A scrambled control sgRNA targeting firefly luciferase was also designed and cloned in pX458 plasmid that was used as a control. The cells were cultured further, and the expression of CHIP was measured by immunoblotting that was found to be reduced by ∼95% in CHIP sgRNA-transfected cells but not in control sgRNA-transfected cells confirming the specificity of CHIP sgRNA 2 expression (Fig. 5B). The cell population was cultured and used for further experiments. The effect of endogenous CHIP on HIV-1 Tat protein was observed by transfecting Myc-Tat in both control and CHIP knockdown HEK-293T cells followed by immunoblotting for Tat protein. The levels of Tat protein were significantly increased in CHIP knockdown cells but not in control knockdown cells (Fig. 5C). Furthermore, to study the effect of CHIP on Tat protein degradation kinetics, Myc-Tat was expressed both in control and CHIP knockdown HEK-293T cells followed by CHX chase assay. The stability of Tat protein was increased in CHIP knockdown cells compared with control knockdown cells (Fig. 5, D and E). Furthermore, the functional activity of Tat was also measured in both WT and CHIP knockdown HEK-293T cells using pNL4-3.LucR−E−. Transfection of pNL4-3.LucR−E− in CHIP knockdown cells resulted in enhanced luciferase activity compared with control knockdown cells (Fig. 5F), showing the negative effect of CHIP on Tat. Effect of endogenous CHIP on virus production was measured by transfection of pNL4-3 in both control and CHIP knockdown HEK-293T cells followed by immunoblotting for Gag protein. The Gag protein levels were increased in CHIP knockdown HEK-293T cells confirming the negative regulation of CHIP on HIV-1 virus production (Fig. 5, G and H). Altogether, these results confirmed that CHIP promotes Tat protein degradation, which results in decreasing HIV-1 replication.
Figure 5.

A, diagram showing the position of sgRNA in CHIP cDNA. B, HEK-293T cells were transfected with CHIP sgRNA1- and sgRNA2-expressing pX458 plasmid; after 48 h, the GFP-expressing cells were subjected to FACS. Cells were lysed, and endogenous CHIP level was monitored by immunoblotting. C, Myc-Tat was transfected into WT or CHIP knockdown HEK-293T cells, and after 36 h, Tat protein was detected by immunoblotting. The blot is representative of three different independent experiments. D, both WT and CHIP knockdown HEK-293T cells were transfected with Myc-Tat, and after 24 h, CHX chase was performed, and Tat protein was immunoblotted. The blot is representative of three different independent experiments. E, Tat amount was measured by ImageJ and plotted with reference to CHX treatment time. F, pNL4-3Luc.R−E− was transfected into both WT and CHIP knockdown HEK-293T, and after 36 h the luciferase activity was measured. GFP was also transfected as a transfection control, and the graph was plotted after normalization of luciferase readings with GFP fluorescence. G, pNL4-3 was transfected both into WT and CHIP knockdown HEK-293T, and after 36 h of post-transfection, Gag was immunoblotted. The blot is representative of three different independent experiments. H, Gag protein level was measured with ImageJ and plotted.
Endogenous CHIP protein level is unaffected by HIV-1
Previous experiments have clearly shown the involvement of CHIP E3 ubiquitin ligase in the ubiquitination and degradation of HIV-1 Tat. Because of CHIP-mediated degradation of Tat, virus production was also reduced in the presence of CHIP but not CHIPΔU. To investigate the effect of HIV-1 infection on endogenous CHIP protein levels, HEK-293T cells were infected with VSVG-pseudotyped HIV-1, and the CHIP level was monitored by immunoblotting. The endogenous CHIP protein in HEK-293T remains more or less constant after the HIV-1 infection from 6 to 48 h post-infection. The Gag protein was also probed for confirming HIV-1 infection (Fig. 6A). To study the effect of HIV-1 infection on endogenous CHIP, its level was also measured after viral induction in latently infected cells. Latently infected CD4 T lymphocyte J1.1 and monocytic cell line U1 were treated with tumor necrosis α (TNFα) to induce viral production followed by immunoblotting for CHIP. Gag was also probed to monitor virus production in the cells. TNFα treatment resulted in an increased Gag protein level, which confirmed virus production. CHIP immunoblotting in these cells did not show any variation in the level of CHIP after virus induction (Fig. 6B). Both of these experiments show that the level of CHIP protein in the host cells is unaffected by virus production.
Figure 6.
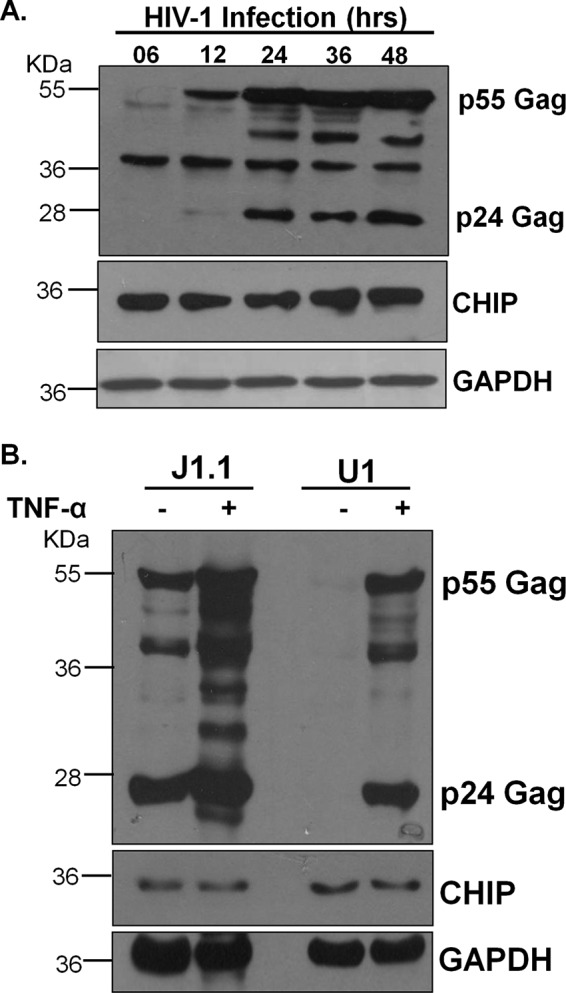
A, HEK-293T cells were infected with VSV-G–pseudotyped HIV-1, and the infected cells were harvested at the indicated time points. Cells were lysed and immunoblotted for CHIP, GAPDH, and HIV-1 Gag. The blot shown is representative of three different independent experiments. B, latently infected cells J1.1 and U1 were cultured in RPMI 1640 medium and stimulated by TNFα. After incubating 12 h in TNFα, the cells were transferred in fresh medium and further incubated for 24 h. Cell lysates were prepared, and proteins were separated on SDS-PAGE. CHIP, Gag, and GAPDH antigens were immunoblotted with respective antibodies.
Discussion
Tat is a master regulator of HIV-1 gene expression, and intracellular levels of Tat directly regulate the amount of virus produced in virus-infected cells (21). The intracellular Tat protein is degraded by multiple pathways, including ubiquitination and proteasomal degradation (28–31, 32). The stabilization of Tat by the treatment of proteasomal inhibitor MG132 confirmed the role of proteasome in Tat degradation. Earlier studies have shown that Tat is ubiquitinated through Lys-48–linked ubiquitin chains for proteasomal degradation (31), and we also confirmed the same in our experiments (Fig. 7). Recently, it was also shown that HIV-1 Tat is ubiquitinated through Lys-48–linked ubiquitin chains with the help of long-noncoding RNA NRON, which promotes interaction of Tat with cellular E3 ubiquitin ligase. NRON-mediated decrease of the cellular Tat level promotes viral latency in the resting T cells (32). Tat is also ubiquitinated through Lys-63–linked ubiquitin chains for enhancing its transcriptional activity (41). Lys-63–linked Tat ubiquitination is also carried out by cellular E3 ubiquitin ligase, MDM2, and Tat translocates MDM2 into the nucleus to protect it from proteasomal degradation (42). Tat also uses MDM2 for suppressing immune activation by promoting IRF-1 degradation (34). Tat is an intrinsically unstructured protein (18–20) because it is rapidly degraded by the 20S proteasome (29, 30). The stability of several intrinsically unfolded proteins is also reported to be regulated by CHIP-dependent ubiquitination and proteasomal degradation (38). The CHIP-mediated down-regulation of Tat and the reduction of Tat t½ in CHX chase assay clearly suggest the important role of CHIP in promoting ubiquitin-dependent proteasomal degradation of Tat. GST-pulldown assay showed the specific association of Tat with CHIP, which suggested that CHIP might be binding and promoting Tat ubiquitination. The stability of cellular protein is central to the regulation of cellular processes, and their degradation involves various E3 ubiquitin ligases. The p53 tumor suppressor protein is known to be ubiquitinated by multiple ubiquitin E3 ligases under different cellular conditions. We hypothesized that HIV-1 Tat stability might be similarly regulated by various degradation pathways. We had earlier shown that the degradation of Tat is also regulated by HIV-1 Rev through indirect means by the host cell protein NQO1 (22). Rev down-regulates the cellular level of NQO1, which results in Tat destabilization as NQO1 protects Tat from 20S proteasomal degradation (29, 30). The down-regulation of Tat by CHIP but not Nef and Gag in co-transfection assay systems suggested the specificity of CHIP toward Tat degradation. Other HIV-1 proteins are also degraded by cellular E3 ubiquitin ligases. MDM2 has been earlier shown to promote ubiquitination and degradation of Vif (43).
Figure 7.

Model showing the ubiquitination of HIV-1 Tat by CHIP. The multiply-spliced HIV-1 mRNA is translated resulting in expression of Tat leading to transcriptional activation of HIV-1 genome. Tat protein is degraded through the lysosomal and 20S proteasomal pathway. Tat is also ubiquitinated by host cell E3 ligase CHIP leading to its Lys-48–linked ubiquitination. Ubiquitinated Tat is recognized by 26S proteasome and degraded resulting in the inhibition of HIV-1 replication and virion production.
Regulation of Tat stability by autophagic (lysosomal), 20S proteasomal, and CHIP-mediated ubiquitination and 26S proteasomal degradation does not rule out the possibility of involvement of other cellular E3 ligases in HIV-1 Tat degradation. Fine regulation of protein levels in the cell also involves their deubiquitination by the cellular deubiquitinase enzymes. We recently showed that HIV-1 Tat is deubiquitinated by the cellular deubiquitinase enzyme USP7 by removing Lys-48–linked ubiquitin chains from ubiquitinated Tat and provides stability to Tat protein in virus-infected cells. Tat deubiquitination by USP7 led to Tat protein stability resulting in enhanced viral replication. Hence, in virus-infected cells, Tat is regulated by both the cellular E3 ubiquitin ligases and deubiquitinases for its optimal function in viral replication. In the CHIP-expressing cells, reduction of Tat-dependent HIV-1 LTR promoter activation measured by luciferase activity as well as reduction of Gag in provirus-transfected or virus-infected cells by CHIP clearly established the role of CHIP in Tat degradation. Thus, CHIP functions as an inhibitory factor for HIV-1. The decreased rate of Tat degradation and the enhancement of HIV-1 production after endogenous CHIP knockdown using the CRISPR-Cas9 method further confirmed the inhibitory role of CHIP for HIV-1. Thus, CHIP is protecting host cells from HIV-1 replication by facilitating Tat degradation. Negative effect of CHIP on Tat and HIV-1 suggests that endogenous CHIP is critically important in Tat destabilization and HIV-1 control in the virus-infected cells. Thus, in this study we have clearly established that CHIP mediates Tat degradation and subsequently inhibits HIV-1 production. Therefore, CHIP is an attractive target for inhibiting HIV-1 replication and pathogenesis. In summary, it can be argued that intracellular stability of HIV-1 Tat is a complex process involving multiple mechanisms.
Experimental procedures
Cell culture and transfection
HEK-293T (human embryonic kidney-293T cells) and TZM-bl (human microglial cells) were maintained in Dulbecco's modified Eagle's medium (Himedia Laboratories, India) with heat-inactivated 10% fetal bovine serum (Biological Industries, Israel) and 100 units of penicillin, 0.1 mg of streptomycin, and 0.25 μg of amphotericin B per ml at 37 °C in the presence of 5% CO2 in a humidified incubator. TZM-bl cells contain β-gal, and the luciferase gene downstream of HIV-1 LTR promoter was obtained from AIDS Research Reagent Program, National Institutes of Health (44). Jurkat, human CD4 T lymphocytic cell line, J1.1, latently infected Jurkat, and the U1 latently-infected human monocytic cell line were maintained in RPMI 1640 medium (Himedia Laboratories, India) with heat-inactivated 10% fetal bovine serum (Biological Industries, Israel) and 100 units of penicillin, 0.1 mg of streptomycin, and 0.25 μg of amphotericin B per ml at 37 °C in the presence of 5% CO2 in a humidified incubator. Transfections were performed using Lipofectamine 2000 (Invitrogen) or polyethyleneimine (PEI, Mr 25,000; Polysciences Inc.) linear reagents using the manufacturer's protocol. For virus production from latently infected cells, J1.1 and U1 cells were induced with 20 ng/ml tumor necrosis factor-α for 12 h.
Plasmids
Myc-tagged HIV-1 Tat expression plasmid pCMV-Myc-Tat was prepared by cloning pNL4-3–derived Tat gene in pCMV-Myc plasmid (Clontech) as described before (29). pcDNA3.1-HA-Tat was obtained from Addgene (45). His6-Myc-CHIP, Myc-CHIP K30A, and Myc-CHIPΔU were obtained from Cam Patterson (38). His6-Ubiquitin was kindly provided by Dmitri Xirodimas, Dundee University, Scotland, UK (46). HA-ubiquitin R48K, which contains lysine residue only at position 48 and all of the other lysines mutated, was kindly provided by Shigeru Yanagi (47) and was subcloned in pCMV-Myc plasmid to yield His6-Myc ubiquitin R48K. The ubiquitin mutant with all seven lysines substituted with alanine, HA-ubiquitin KO, was provided by Ted Dawson, Johns Hopkins University (48). Codon-optimized HIV-1 Gag (Gag-Opt) (49), HIV-1 proviral construct pNL4-3, and luciferase reporter HIV-1 proviral construct pNL4-3.Luc.R−E− (50) were obtained through the AIDS Reference and Reagent Program, National Institutes of Health, Bethesda. CRISPR-Cas9 expression vector pX458 was kindly provided by Feng Zhang, Massachusetts Institute of Technology (51). GST–CHIP was cloned in GST-expressing pGEX-2T plasmid.
Immunoblot analysis
For immunoblotting purposes, the cells were harvested and resuspended in RIPA lysis buffer (1% Nonidet P-40, 20 mm Tris-Cl, pH 7.5, 150 mm NaCl, 1 mm Na2EDTA, 1 mm EGTA, 1% sodium deoxycholate, 1 mm Na3VO4), followed by centrifugation. The supernatant was collected, and the protein concentration was estimated using BCA protein assay kit (Pierce, Thermo Fisher Scientific). The lysates were separated on SDS-PAGE and transferred onto nitrocellulose membrane, followed by blocking of membranes with 5% nonfat dry milk (Himedia Laboratories, India) in 1× PBS (PBS: 137 mm NaCl, 2.7 mm KCl, 10 mm Na2HPO4, 1.8 mm KH2PO4) and washed three times with 1× PBS containing 0.1% Tween 20 (PBST). Primary antibody was added to the membranes and incubated overnight, followed by washing with PBST and further incubation in the horseradish peroxide (HRP)–conjugated secondary antibody. The blots were developed using enhanced chemiluminescent reagent.
Antibodies
Anti-c-Myc mAb (Clontech; 1:1000), anti-HA tag polyclonal antibody (Clontech; 1:1000), anti-GAPDH antibody (Cell Signaling Technology; 1:10,000), anti-His6 mAb (Sigma; 1:1000), anti-p53 polyclonal antibody (SCBT; 1:2000), anti-GST (SCBT; 1:2000), anti-p24 mAb (AIDS Reagent Program, National Institutes of Health; 1:3000), anti-rabbit IgG conjugated to HRP (Jackson ImmunoResearch; 1:10,000), and anti-mouse IgG conjugated to HRP (Jackson ImmunoResearch; 1:10,000) were used for the Western blotting experiments.
CHX chase assay
The degradation kinetics of Tat protein in the presence of CHIP overexpression or knockdown effect was studied by CHX chase assay as described before (52). Plasmids expressing Tat and CHIP were transfected into HEK-293T cells, and 24 h post-transfection the cells were treated with CHX (100 μg/ml; Sigma) for the indicated time periods and harvested. The lysate was prepared and resolved on 12% SDS-PAGE followed by immunoblotting as described above.
Luciferase reporter assay
To study the effect of CHIP on the replication of the HIV-1, luciferase expressing HIV-1 proviral construct pNL4-3.Luc.R−E− was co-transfected with CHIP, and 36 h post-transfection, the cells were harvested and lysed in Passive Lysis Buffer (Promega). The supernatant was collected after centrifugation of the lysate, and the luciferase activity was measured in the luminometer (TECAN) using the luciferase reporter assay kit (Promega). For normalization, GFP-expressing plasmid pEGFP-N1 was co-transfected in each sample, and the luciferase reading was divided by GFP values in each sample.
RT-PCR
The mRNA level of a gene is a measure of its expression in the cell and is quantified by RT-PCR assay. To carry out RT-PCR, Tat- and CHIP-expressing plasmids were transfected into HEK-293T cells for 36 h, and subsequently, the total RNA was isolated by TRIzol extraction as described by the manufacturer (Invitrogen). Reverse transcription was performed using the RNA obtained after TRIzol treatment to obtain cDNA using the cDNA synthesis kit (Promega). RNA (1 μg) was mixed with random primers followed by incubation at 70 °C for 15 min. The mixture was kept at 4 °C, and the reverse transcription mix containing 1× reaction buffer (MgCl2, dNTPs, RNasin, and reverse transcriptase) was added. The reaction mixture was incubated at 25 °C for 5 min and 42 °C for 1 h. The cDNA was obtained and used as a template for the PCR amplification of the Tat gene using primers of Tat. The Tat forward primer 5′-ATGGAGCCAGTAGATCCTAGACTAGAG-3′ and the Tat reverse primer 5′-CGTCGCTGTCTCCGCTTCTTCCT-3′ were used.
We have standardized the semi-quantitative PCR and found that 15 cycles is in the log range of amplification, where the amount of template is directly proportional to the band intensities of PCR products. Thus, the PCR was carried out for 15 cycles, and we compared the amount of Tat in the presence and absence of CHIP. GAPDH was used as a control and amplified using the following primers: GAPDH forward primer, 5′-ACCACCATGGAGAAGGCTGG-3′, and GAPDH reverse primer, 5′-CTCAGTGTAGCCCAGGATGC-3′.
In vivo ubiquitination assay
To study the effect of CHIP on the ubiquitination of Tat, in vivo ubiquitination assay was performed as described previously by us (52). HEK-293T cells were co-transfected with plasmids encoding CHIP, Tat, and His6-ubiquitin, as described in the figures, and 24 h after transfection cells were treated with MG132 for 8 h. The cells were harvested and lysed in the denaturing lysis buffer containing guanidine hydrochloride and centrifuged. The supernatant was used to purify the His6-ubiquitin–tagged proteins using Ni-NTA affinity chromatography. The purified proteins were separated on SDS-PAGE and immunoblotted for Tat protein.
GST-pulldown assay
Plasmid expressing GST and GST–CHIP were transformed in Escherichia coli DH5α. Gene expression was induced by 0.4 μm isopropyl 1-thio-β-d-galactopyranoside treatment for 4 h. Expression of the proteins was verified by analyzing the cell lysate on SDS-PAGE and immunoblotted using anti-GST antibody. Myc-Tat–expressing plasmid was also transfected simultaneously in HEK-293T cells for 36 h, and cell lysate was prepared. For pulldown assay, lysate containing Myc-Tat protein was mixed with GST or GST–CHIP and incubated overnight with rotation at 4 °C followed by addition of GSH-agarose beads for 1 h. The beads were washed, and the proteins were eluted by boiling the beads in SDS loading buffer. The proteins were resolved on 12% SDS-PAGE and immunoblotted using anti-Myc and anti-GST antibody.
Cloning of sgRNA in CRISPR-Cas9 vector
The knockdown of endogenous CHIP was performed using CRISPR-Cas9 plasmid containing CHIP sgRNA (51). The sgRNA was designed using the sgRNA designer tool provided by the Broad Institute, Massachusetts Institute of Technology. Two pairs of DNA oligonucleotides corresponding to the N-terminal region of the CHIP cDNA sequence were synthesized. Similarly scrambled control oligonucleotides from firefly (Photinus pyralis) luciferase gene were also synthesized. The sequences of sgRNAs are provided in Table S1. For cloning of sgRNA, each pair of oligonucleotides was mixed in equal proportions in 10× phosphonucleotide kinase (PNK) buffer, 10 μm ATP, PNK enzyme and incubated at 37 °C for 40 min. This mixture was further incubated followed by addition of 0.1 m NaCl and followed by further incubation at 65 °C for 20 min. For annealing, the nucleotide mixture was boiled at 100 °C for 5 min and left to cool at room temperature. The phosphorylated and annealed oligonucleotides were diluted and ligated to the BbsI-digested pSpCas9 (BB)-2A-GFP (pX458) plasmid (51). The positive clones were confirmed by sequencing. Plasmid DNA was then transfected into HEK-293T cells and subjected to FACS for GFP.
VSV-G pseudotyped pNL4-3–derived HIV-1 preparation
To prepare the vesicular stomatitis virus envelope glycoprotein (VSV-G)-pseudotyped HIV-1, 18 μg of pNL4-3 was co-transfected with 2 μg of VSV-G–expressing plasmid in a 100-mm cell culture dish of HEK-293T cells using Lipofectamine 2000 (Invitrogen). Cell culture medium (supernatant) harboring the virus particles was collected 48 h post-transfection. The supernatant was filtered through a 0.45-μm pore-size filter and used for infecting the cells. Infectivity of the virus was determined by β-gal staining of HIV-1 reporter cell line TZM-bl. The viral stock was stored at −80 °C.
Virus production from latently infected cells
The latently infected J1.1 and U1 cells were maintained in RPMI 1640 medium and were treated with 20 ng/ml TNFα (39, 53). After 12 h, the medium was replaced with fresh complete RPMI 1640 medium, and the cells were further incubated for 24 h. Subsequently, the lysate was used to measure p24 level by immunoblotting as an indicator of virus production. The CHIP was also immunoblotted using anti-CHIP antibody to measure the endogenous level in the virus-producing cells.
Co-immunoprecipitation assay
Protein–protein interaction was studied by co-immunoprecipitation. Plasmids encoding the HA-Tat and the Myc-CHIP variants were transfected into HEK-293T cells, and after 24 h the cells were harvested and lysed in CelLytic M (cell lysis reagent from Sigma). Antibody was added to cell lysate and incubated at 4 °C overnight with rotation followed by addition of 0.1% BSA-saturated protein A–agarose beads and further incubated for 2–3 h. Subsequently, the beads were pelleted and washed with IP buffer (Sigma) four times. The 2× SDS-PAGE loading buffer was added to the beads and boiled for 5 min. Protein was resolved on 12% SDS-PAGE and immunoblotted.
Author contributions
A. A. and A. C. B. conceptualization; A. A. validation; A. A. investigation; A. A. and S. R. F. methodology; A. A. writing-original draft; A. A., S. R. F., and A. C. B. writing-review and editing; A. C. B. resources; A. C. B. supervision; A. C. B. funding acquisition.
Supplementary Material
Acknowledgments
Many of reagents used in this study, including TZM-bl, J1.1, and U1 cell lines and plasmids were obtained from AIDS Reference and Reagent Program, National Institutes of Health, Bethesda, MD. We are indebted to Malcolm Martin (National Institutes of Health) for pNL4-3, Nathaniel Landau (The Rockefeller University) for pNL4-3Luc.R−E−, Lung-Ji Chang (University of Florida) for pHEF-VSVG, and Beatrice H. Hahn (University of Pennsylvania) for Gag-opt plasmid. We are thankful to Dimitris Xirodimas (University of Dundee, Dundee, Scotland, United Kingdom) for His6-ubiquitin expression plasmid and Shigeru Yanagi (Tokyo University of Pharmacy and Life Sciences, Tokyo, Japan) for HA-ubiquitin Lys-48. We are thankful to Cam Patterson (Weill-Cornell Medical Center, New York) for His6-Myc-CHIP, Myc-CHIP K30A, Myc-CHIPΔU box, and GST–CHIP and Feng Zhang (Broad Institute, Massachusetts Institute of Technology, Boston, MA) for CRISPR-Cas9 (pX458) plasmid.
This work was supported by Department of Biotechnology and Indian Council of Medical Research of Government of India Grants BT/PR8322/Med/-14/1245/2006, BT/PR1800/AGR/36/676/2011, and HIV/50/142/9/2011-ECD-II. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S4 and Table S1.
- LTR
- long terminal repeat
- CHIP
- C terminus of Hsp70-binding protein Hsp70
- CHX
- cycloheximide
- Ni-NTA
- nickel-nitrilotriacetic acid
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- TNFα
- tumor necrosis factor-α
- UPS
- ubiquitin proteasome system
- HRP
- horseradish peroxide
- PNK
- phosphonucleotide kinase
- VSV-G
- vesicular stomatitis virus envelope glycoprotein.
References
- 1. Dunn W. A., Jr. (1994) Autophagy and related mechanisms of lysosome-mediated protein degradation. Trends Cell Biol. 4, 139–143 [DOI] [PubMed] [Google Scholar]
- 2. Voges D., Zwickl P., and Baumeister W. (1999) The 26S proteasome: a molecular machine designed for controlled proteolysis. Annu. Rev. Biochem. 68, 1015–1068 10.1146/annurev.biochem.68.1.1015 [DOI] [PubMed] [Google Scholar]
- 3. Asher G., Reuven N., and Shaul Y. (2006) 20S proteasomes and protein degradation “by default.” Bioessays 28, 844–849 10.1002/bies.20447 [DOI] [PubMed] [Google Scholar]
- 4. Gsponer J., Futschik M. E., Teichmann S. A., and Babu M. M. (2008) Tight regulation of unstructured proteins: from transcript synthesis to protein degradation. Science 322, 1365–1368 10.1126/science.1163581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Glickman M. H., and Ciechanover A. (2002) The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol. Rev. 82, 373–428 10.1152/physrev.00027.2001 [DOI] [PubMed] [Google Scholar]
- 6. Li W., Bengtson M. H., Ulbrich A., Matsuda A., Reddy V. A., Orth A., Chanda S. K., Batalov S., and Joazeiro C. A. (2008) Genome-wide and functional annotation of human E3 ubiquitin ligases identifies MULAN, a mitochondrial E3 that regulates the organelle's dynamics and signaling. PLoS ONE 3, e1487 10.1371/journal.pone.0001487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Herrmann J., Lerman L. O., and Lerman A. (2007) Ubiquitin and ubiquitin-like proteins in protein regulation. Circ. Res. 100, 1276–1291 10.1161/01.RES.0000264500.11888.f0 [DOI] [PubMed] [Google Scholar]
- 8. Welchman R. L., Gordon C., and Mayer R. J. (2005) Ubiquitin and ubiquitin-like proteins as multifunctional signals. Nat. Rev. Mol. Cell Biol. 6, 599–609 10.1038/nrm1700 [DOI] [PubMed] [Google Scholar]
- 9. Xu P., Duong D. M., Seyfried N. T., Cheng D., Xie Y., Robert J., Rush J., Hochstrasser M., Finley D., and Peng J. (2009) Quantitative proteomics reveals the function of unconventional ubiquitin chains in proteasomal degradation. Cell 137, 133–145 10.1016/j.cell.2009.01.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Boban M., Ljungdahl P. O., and Foisner R. (2015) Atypical ubiquitylation in yeast targets lysine-less Asi2 for proteasomal degradation. J. Biol. Chem. 290, 2489–2495 10.1074/jbc.M114.600593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gao G., and Luo H. (2006) The ubiquitin-proteasome pathway in viral infections. Can. J. Physiol. Pharmacol. 84, 5–14 10.1139/y05-144 [DOI] [PubMed] [Google Scholar]
- 12. Randow F., and Lehner P. J. (2009) Viral avoidance and exploitation of the ubiquitin system. Nat. Cell Biol. 11, 527–534 10.1038/ncb0509-527 [DOI] [PubMed] [Google Scholar]
- 13. Sheehy A. M., Gaddis N. C., Choi J. D., and Malim M. H. (2002) Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 418, 646–650 10.1038/nature00939 [DOI] [PubMed] [Google Scholar]
- 14. Belzile J. P., Duisit G., Rougeau N., Mercier J., Finzi A., and Cohen E. A. (2007) HIV-1 Vpr-mediated G2 arrest involves the DDB1-CUL4AVPRBP E3 ubiquitin ligase. PLoS Pathog. 3, e85 10.1371/journal.ppat.0030085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Arora S., Verma S., and Banerjea A. C. (2014) HIV-1 Vpr redirects host ubiquitination pathway. J. Virol. 88, 9141–9152 10.1128/JVI.00619-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ott M., Geyer M., and Zhou Q. (2011) The control of HIV transcription: keeping RNA polymerase II on track. Cell Host Microbe 10, 426–435 10.1016/j.chom.2011.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Muniz L., Egloff S., Ughy B., Jády B. E., and Kiss T. (2010) Controlling cellular P-TEFb activity by the HIV-1 transcriptional transactivator Tat. PLoS Pathog. 6, e1001152 10.1371/journal.ppat.1001152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shojania S., and O'Neil J. D. (2010) Intrinsic disorder and function of the HIV-1 Tat protein. Protein Pept. Lett. 17, 999–1011 10.2174/092986610791498993 [DOI] [PubMed] [Google Scholar]
- 19. Shojania S., and O'Neil J. D. (2006) HIV-1 Tat is a natively unfolded protein: the solution conformation and dynamics of reduced HIV-1 Tat-(1–72) by NMR spectroscopy. J. Biol. Chem. 281, 8347–8356 10.1074/jbc.M510748200 [DOI] [PubMed] [Google Scholar]
- 20. Kim J. M., Choi H. S., and Seong B. L. (2017) The folding competence of HIV-1 Tat mediated by interaction with TAR RNA. RNA Biol. 14, 926–937 10.1080/15476286.2017.1311455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Verhoef K., Koper M., and Berkhout B. (1997) Determination of the minimal amount of Tat activity required for human immunodeficiency virus type 1 replication. Virology 237, 228–236 10.1006/viro.1997.8786 [DOI] [PubMed] [Google Scholar]
- 22. Karn J. (2011) The molecular biology of HIV latency: breaking and restoring the Tat-dependent transcriptional circuit. Curr. Opin. HIV AIDS 6, 4–11 10.1097/COH.0b013e328340ffbb [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Minami R., Yamamoto M., Takahama S., Miyamura T., Watanabe H., and Suematsu E. (2006) RCAS1 induced by HIV-Tat is involved in the apoptosis of HIV-1 infected and uninfected CD4+ T cells. Cell. Immunol. 243, 41–47 10.1016/j.cellimm.2006.11.003 [DOI] [PubMed] [Google Scholar]
- 24. Hategan A., Bianchet M. A., Steiner J., Karnaukhova E., Masliah E., Fields A., Lee M. H., Dickens A. M., Haughey N., Dimitriadis E. K., and Nath A. (2017) HIV Tat protein and amyloid-β peptide form multifibrillar structures that cause neurotoxicity. Nat. Struct. Mol. Biol. 24, 379–386 10.1038/nsmb.3379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Germini D., Tsfasman T., Klibi M., El-Amine R., Pichugin A., Iarovaia O. V., Bilhou-Nabera C., Subra F., Bou Saada Y., Sukhanova A., Boutboul D., Raphaël M., Wiels J., Razin S. V., Bury-Moné S., et al. (2017) HIV Tat induces a prolonged MYC relocalization next to IGH in circulating B-cells. Leukemia 31, 2515–2522 10.1038/leu.2017.106 [DOI] [PubMed] [Google Scholar]
- 26. Mueller N., Pasternak A. O., Klaver B., Cornelissen M., Berkhout B., and Das A. T. (2018) The HIV-1 Tat protein enhances splicing at the major splice donor site. J. Virol. 92, e01855–17 10.1128/JVI.01855-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Harwig A., Jongejan A., van Kampen A. H., Berkhout B., and Das A. T. (2016) Tat-dependent production of an HIV-1 TAR-encoded miRNA-like small RNA. Nucleic Acids Res. 44, 4340–4353 10.1093/nar/gkw167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sagnier S., Daussy C. F., Borel S., Robert-Hebmann V., Faure M., Blanchet F. P., Beaumelle B., Biard-Piechaczyk M., and Espert L. (2015) Autophagy restricts HIV-1 infection by selectively degrading Tat in CD4+ T lymphocytes. J. Virol. 89, 615–625 10.1128/JVI.02174-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lata S., Ali A., Sood V., Raja R., and Banerjea A. C. (2015) HIV-1 Rev downregulates Tat expression and viral replication via modulation of NAD(P)H:quinine oxidoreductase 1 (NQO1). Nat. Commun. 6, 7244 10.1038/ncomms8244 [DOI] [PubMed] [Google Scholar]
- 30. Ali A., and Banerjea A. C. (2016) Curcumin inhibits HIV-1 by promoting Tat protein degradation. Sci. Rep. 6, 27539 10.1038/srep27539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang L., Qin J., Li Y., Wang J., He Q., Zhou J., Liu M., and Li D. (2014) Modulation of the stability and activities of HIV-1 Tat by its ubiquitination and carboxyl-terminal region. Cell. Biosci. 4, 61 10.1186/2045-3701-4-61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li J., Chen C., Ma X., Geng G., Liu B., Zhang Y., Zhang S., Zhong F., Liu C., Yin Y., Cai W., and Zhang H. (2016) Long noncoding RNA NRON contributes to HIV-1 latency by specifically inducing tat protein degradation. Nat. Commun. 7, 11730 10.1038/ncomms11730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Faust T. B., Li Y., Bacon C. W., Jang G. M., Weiss A., Jayaraman B., Newton B. W., Krogan N. J., D'Orso I., and Frankel A. D. (2018) The HIV-1 Tat protein recruits a ubiquitin ligase to reorganize the 7SK snRNP for transcriptional activation. Elife 7, e31879 10.7554/eLife.31879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Remoli A. L., Marsili G., Perrotti E., Acchioni C., Sgarbanti M., Borsetti A., Hiscott J., and Battistini A. (2016) HIV-1 Tat recruits HDM2 E3 ligase to target IRF-1 for ubiquitination and proteasomal degradation. MBio 7, e01528–16 10.1128/mBio.01528-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ballinger C. A., Connell P., Wu Y., Hu Z., Thompson L. J., Yin L. Y., and Patterson C. (1999) Identification of CHIP, a novel tetratricopeptide repeat-containing protein that interacts with heat shock proteins and negatively regulates chaperone functions. Mol. Cell. Biol. 19, 4535–4545 10.1128/MCB.19.6.4535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jiang J., Ballinger C. A., Wu Y., Dai Q., Cyr D. M., Höhfeld J., and Patterson C. (2001) CHIP is a U-box-dependent E3 ubiquitin ligase: identification of Hsc70 as a target for ubiquitylation. J. Biol. Chem. 276, 42938–42944 10.1074/jbc.M101968200 [DOI] [PubMed] [Google Scholar]
- 37. McDonough H., and Patterson C. (2003) CHIP: a link between the chaperone and proteasome systems. Cell Stress Chaperones 8, 303–308 10.1379/1466-1268(2003)008andlt;0303:CALBTCandgt;2.0.CO;2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Murata S., Minami Y., Minami M., Chiba T., and Tanaka K. (2001) CHIP is a chaperone-dependent E3 ligase that ubiquitylates unfolded protein. EMBO Rep. 2, 1133–1138 10.1093/embo-reports/kve246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Folks T. M., Justement J., Kinter A., Dinarello C. A., and Fauci A. S. (1987) Cytokine-induced expression of HIV-1 in a chronically infected promonocyte cell line. Science 238, 800–802 10.1126/science.3313729 [DOI] [PubMed] [Google Scholar]
- 40. Esser C., Scheffner M., and Höhfeld J. (2005) The chaperone-associated ubiquitin ligase CHIP is able to target p53 for proteasomal degradation. J. Biol. Chem. 280, 27443–27448 10.1074/jbc.M501574200 [DOI] [PubMed] [Google Scholar]
- 41. Brès V., Kiernan R. E., Linares L. K., Chable-Bessia C., Plechakova O., Tréand C., Emiliani S., Peloponese J. M., Jeang K. T., Coux O., Scheffner M., and Benkirane M. (2003) A non-proteolytic role for ubiquitin in Tat-mediated transactivation of the HIV-1 promoter. Nat. Cell Biol. 5, 754–761 10.1038/ncb1023 [DOI] [PubMed] [Google Scholar]
- 42. Raja R., Ronsard L., Lata S., Trivedi S., and Banerjea A. C. (2017) HIV-1 Tat potently stabilises Mdm2 and enhances viral replication. Biochem. J. 474, 2449–2464 10.1042/BCJ20160825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Izumi T., Takaori-Kondo A., Shirakawa K., Higashitsuji H., Itoh K., Io K., Matsui M., Iwai K., Kondoh H., Sato T., Tomonaga M., Ikeda S., Akari H., Koyanagi Y., Fujita J., and Uchiyama T. (2009) MDM2 is a novel E3 ligase for HIV-1 Vif. Retrovirology 6, 1 10.1186/1742-4690-6-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Platt E. J., Wehrly K., Kuhmann S. E., Chesebro B., and Kabat D. (1998) Effects of CCR5 and CD4 cell surface concentrations on infections by macrophagetropic isolates of human immunodeficiency virus type 1. J. Virol. 72, 2855–2864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cujec T. P., Okamoto H., Fujinaga K., Meyer J., Chamberlin H., Morgan D. O., and Peterlin B. M. (1997) The HIV transactivator TAT binds to the CDK-activating kinase and activates the phosphorylation of the carboxy-terminal domain of RNA polymerase II. Genes Dev. 11, 2645–2657 10.1101/gad.11.20.2645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Xirodimas D. P., Stephen C. W., and Lane D. P. (2001) Cocompartmentalization of p53 and Mdm-2 is a major determinant for Mdm-2-mediated degradation of p53. Exp. Cell Res. 270, 66–77 10.1006/excr.2001.5314 [DOI] [PubMed] [Google Scholar]
- 47. Sugiura A., Nagashima S., Tokuyama T., Amo T., Matsuki Y., Ishido S., Kudo Y., McBride H. M., Fukuda T., Matsushita N., Inatome R., and Yanagi S. (2013) MITOL regulates endoplasmic reticulum–mitochondria contacts via mitofusin2. Mol. Cell 51, 20–34 10.1016/j.molcel.2013.04.023 [DOI] [PubMed] [Google Scholar]
- 48. Lim K. L., Chew K. C., Tan J. M., Wang C., Chung K. K., Zhang Y., Tanaka Y., Smith W., Engelender S., Ross C. A., Dawson V. L., and Dawson T. M. (2005) Parkin mediates nonclassical, proteasomal-independent ubiquitination of synphilin-1: implications for Lewy body formation. J. Neurosci. 25, 2002–2009 10.1523/JNEUROSCI.4474-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gao F., Li Y., Decker J. M., Peyerl F. W., Bibollet-Ruche F., Rodenburg C. M., Chen Y., Shaw D. R., Allen S., Musonda R., Shaw G. M., Zajac A. J., Letvin N., and Hahn B. H. (2003) Codon usage optimization of HIV type 1 subtype C gag, pol, env, and nef genes: in vitro expression and immune responses in DNA-vaccinated mice. AIDS Res. Hum. Retroviruses 19, 817–823 10.1089/088922203769232610 [DOI] [PubMed] [Google Scholar]
- 50. He J., Choe S., Walker R., Di Marzio P., Morgan D. O., and Landau N. R. (1995) Human immunodeficiency virus type 1 viral protein R (Vpr) arrests cells in the G2 phase of the cell cycle by inhibiting p34cdc2 activity. J. Virol. 69, 6705–6711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ran F. A., Hsu P. D., Wright J., Agarwala V., Scott D. A., and Zhang F. (2013) Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281–2308 10.1038/nprot.2013.143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Verma S., Ali A., Arora S., and Banerjea A. C. (2011) Inhibition of β-TrcP–dependent ubiquitination of p53 by HIV-1 Vpu promotes p53–mediated apoptosis in human T cells. Blood 117, 6600–6607 10.1182/blood-2011-01-333427 [DOI] [PubMed] [Google Scholar]
- 53. Perez V. L., Rowe T., Justement J. S., Butera S. T., June C. H., and Folks T. M. (1991) An HIV-1-infected T cell clone defective in IL-2 production and Ca2+ mobilization after CD3 stimulation. J. Immunol. 147, 3145–3148 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.