

Abstract
Eukaryotic elongation factor 2 kinase (eEF2K) negatively regulates the elongation stage of mRNA translation and is activated under different stress conditions to slow down protein synthesis. One effect of eEF2K is to alter the repertoire of expressed proteins, perhaps to aid survival of stressed cells. Here, we applied pulsed stable isotope labeling with amino acids in cell culture (SILAC) to study changes in the synthesis of specific proteins in human lung adenocarcinoma (A549) cells in which eEF2K had been depleted by an inducible shRNA. We discovered that levels of heat-shock protein 90 (HSP90) are increased in eEF2K-depleted human cells as well as in eEF2K-knockout (eEF2K−/−) mouse embryonic fibroblasts (MEFs). This rise in HSP90 coincided with an increase in the fraction of HSP90 mRNAs associated with translationally active polysomes, irrespective of unchanged total HSP90 levels. These results indicate that blocking eEF2K function can enhance expression of HSP90 chaperones. In eEF2K−/− mouse embryonic fibroblasts (MEFs), inhibition of HSP90 by its specific inhibitor AUY922 promoted the accumulation of ubiquitinated proteins. Notably, HSP90 inhibition promoted apoptosis of eEF2K−/− MEFs under proteostatic stress induced by the proteasome inhibitor MG132. Up-regulation of HSP90 likely protects cells from protein folding stress, arising, for example, from faster rates of polypeptide synthesis due to the lack of eEF2K. Our findings indicate that eEF2K and HSPs closely cooperate to maintain proper proteostasis and suggest that concomitant inhibition of HSP90 and eEF2K could be a strategy to decrease cancer cell survival.
Keywords: translation elongation factor, heat shock protein (HSP), proteasome, protein synthesis, apoptosis, cancer, elongation, eukaryotic elongation factor 2 kinase (eEF2K), molecular chaperone, mRNA translation
Introduction
Eukaryotic elongation factor 2 (eEF2)3 kinase (eEF2K) is an atypical protein kinase that phosphorylates eEF2 on Thr-56 and renders it unable to interact with ribosomes. By reducing the availability of active eEF2, eEF2K slows down the elongation stage of protein synthesis (1), which consumes the vast majority of the energy used (>99%) by protein synthesis. eEF2K is activated under diverse cell stresses such as hypoxia (2), low pH (3), nutrient deprivation (4), and glycolytic stress (5), which are hallmarks of tumor microenvironments, and where saving energy or nutrients such as amino acids would benefit tumor cell survival. eEF2K-knockout (eEF2K−/−) mice are healthy and viable (2), showing that eEF2K is not required for normal cell function under standard animal husbandry conditions. As such, it has been suggested that eEF2K inhibitors could hold therapeutic potential to treat some cancers while exerting minimal on-target side effects.
Notably, we recently reported that accelerated translation rates lead to enhanced levels of translation errors and that eEF2K serves to ensure accurate protein synthesis (6). In this study, using pulsed stable isotope labeling with amino acids in cell culture (pSILAC) combined with LC-MS/MS analysis, we discovered that knocking down or deleting eEF2K in cells increases the translational efficiencies of HSP90 mRNAs. This may help cells to cope with higher levels of erroneously translated and thus misfolded proteins. Strikingly, inhibition of HSP90 in eEF2K-null cells evoked apoptosis under conditions of proteolytic stress, suggesting that concomitant inhibition of HSP90 and eEF2K is detrimental to (cancer) cell survival.
Results
Knockdown or knockout of eEF2K increases HSP90 expression
To assess whether eEF2K affects the synthesis of specific proteins under metabolic stress conditions, we applied pSILAC (7) to label newly synthesized proteins metabolically and quantify their rates of (differential) accumulation by LC-MS/MS. A549 cells expressing an isopropyl β-d-1-thiogalactopyranoside (IPTG)-inducible short hairpin RNA (shRNA) against EEF2K were generated and treated with 2-deoxyglucose (2-DG), a nonmetabolizable glucose analog that induces metabolic stress by inhibiting glycolysis, to activate eEF2K (5). They were subsequently incubated in culture medium containing heavy lysine and arginine isotopologs for 2 h to label (“mass-tag”) newly synthesized proteins (Fig. S1A). Proteins were extracted and subjected to trypsin digestion after which the resulting peptides were analyzed by LC-MS/MS (Fig. S1B).
A total of 3,011 unique proteins were identified, of which the SILAC ratios of 973 proteins could reliably be quantified (see “Experimental procedures” and Table S1). We detected 78 protein groups that differed in their synthesis rates (p value <0.01) upon knockdown of eEF2K (Fig. S1B). Furthermore, synthesis of three members of the heat-shock protein molecular chaperone family (HSPA5, HSP90B1, and HSP90AA1) was up-regulated upon eEF2K knockdown (Fig. S1B). Of note, HSP90 proteins have been shown to interact with eEF2K, resulting in its stabilization (8). SDS-PAGE followed by Western blot analysis confirmed the higher HSP90 protein expression levels in IPTG-treated A549 cells and eEF2K−/− MEFs compared with their vehicle-treated or WT counterparts, by 1.5- and 2.0-fold, respectively. Also consistent with the pSILAC analysis, GAPDH levels were 1.6- and 2.3-fold lower in IPTG-treated A549 and eEF2K−/− MEFs, respectively (Fig. S1, C–E). In contrast, actin levels were unaltered (Fig. 1A). 2-DG treatment also led to a reduction in HSP90 protein levels in MEFs (Fig. 1, D and E) (6). It was shown previously (9) that TOP mRNAs (mRNAs that contain a 5′-terminal oligopyrimidine tract, many of which encode ribosomal proteins and translation factors) are recruited onto polysomes upon activation of eEF2K, thereby causing an increase in the relative synthesis of TOP mRNA–encoded proteins compared with others. This feature was clearly evident in our pSILAC data, in good agreement with the findings of Gismondi et al. (9), thereby validating our own findings.
Figure 1.

Knocking down or ablating eEF2K elevates HSP90 protein levels. A, lysates of A549 cells treated ±IPTG (to induce shRNA expression to knock down eEF2K) or eEF2K+/+ and eEF2K−/− MEFs were subjected to SDS-PAGE followed by WB analysis of the indicated proteins. B and C, quantification of HSP90 levels in A549 cells (B) or MEFs (C) from A. D, eEF2K+/+ and eEF2K−/− MEFs were cultured in low-glucose (5.5 mm) DMEM ± 10 mm 2-DG for 24 h before immunoblot analysis. E, quantification of HSP90 levels from D. P-eEF2, phosphorylated eEF2. *, 0.01 ≤ p < 0.05; **, 0.001 ≤ p < 0.01. Error bars represent S.D.
eEF2K knockout shifts HSP90 mRNAs into polysomal fractions
Higher HSP90 protein levels in eEF2K-null cells were not due to elevated levels of its mRNAs as, if anything, HSP90AA1 and HSP90B1 mRNA levels were slightly lower in eEF2K−/− MEFs (Fig. 2, A and B). To study further how eEF2K regulates HSP90 protein expression, we fractionated lysates from 2-DG–treated eEF2K+/+ and eEF2K−/− MEFs through sucrose gradient profiling (Fig. 2C). 2-DG treatment greatly reduced the proportion of ribosomes in translationally active polysomal fractions (Fig. S2). Interestingly, upon 2-DG–induced eEF2K activation (Fig. 1D), the amounts of HSP90AA1 and HSP90B1 mRNAs associated with active polysomes were higher in eEF2K−/− MEFs than in WT cells, and their association with monosomal or subpolysomal fractions was correspondingly lower (Fig. 2, C–E). In contrast, the distribution of a “control” mRNA, B2M (encoding β2-microglobulin), was essentially unchanged (Fig. 2F). These data indicate that eEF2K negatively regulates the translation of HSP90 mRNAs by impairing the association of their mRNAs with polysomes.
Figure 2.

eEF2K knockout in MEFs shifts HSP90AA1 and HSP90B1 mRNAs from polysomal to non/subpolysomal fractions. A and B, eEF2K+/+ and eEF2K−/− MEFs were treated as in Fig. 1E, and total cell mRNA was extracted and subjected to qPCR with primers for HSP90AA1 (A) and HSP90B1 (B). C–F, eEF2K+/+ and eEF2K−/− MEFs were treated as in Fig. 1E, and cell lysates were subjected to polysome analysis. A representative profile is shown in C, and positions of ribosomal/polysomal species are indicated. RT-qPCR was performed using specific primers for mouse HSP90AA1 (D), HSP90B1 (E), and B2M (F). *, 0.01 ≤ p < 0.05; **, 0.001 ≤ p < 0.01; ***, p < 0.001. Error bars represent S.D.
Concomitant inhibition of HSP90 and eEF2K induces protein ubiquitination and cell death
Higher levels of HSP90 may help cells to maintain adequate protein folding under conditions of accelerated polypeptide synthesis (translation elongation) caused by the absence of eEF2K. Thus, blocking HSP90 function may be particularly detrimental to protein homeostasis (proteostasis) in cells lacking eEF2K. To test this, we exposed eEF2K+/+ and eEF2K−/− MEFs to MG132, a proteasome inhibitor, in the presence or absence of the HSP90 inhibitor NVP-AUY922 (hereafter called AUY922) (10). MG132 evokes phosphorylation of eEF2 (Fig. 3A), indicating activation of eEF2K. Under conditions of impaired protein degradation, ubiquitinated proteins first accumulate in the fraction that is soluble in 1% (v/v) Triton and then aggregate and relocate into the insoluble fraction (11).
Figure 3.
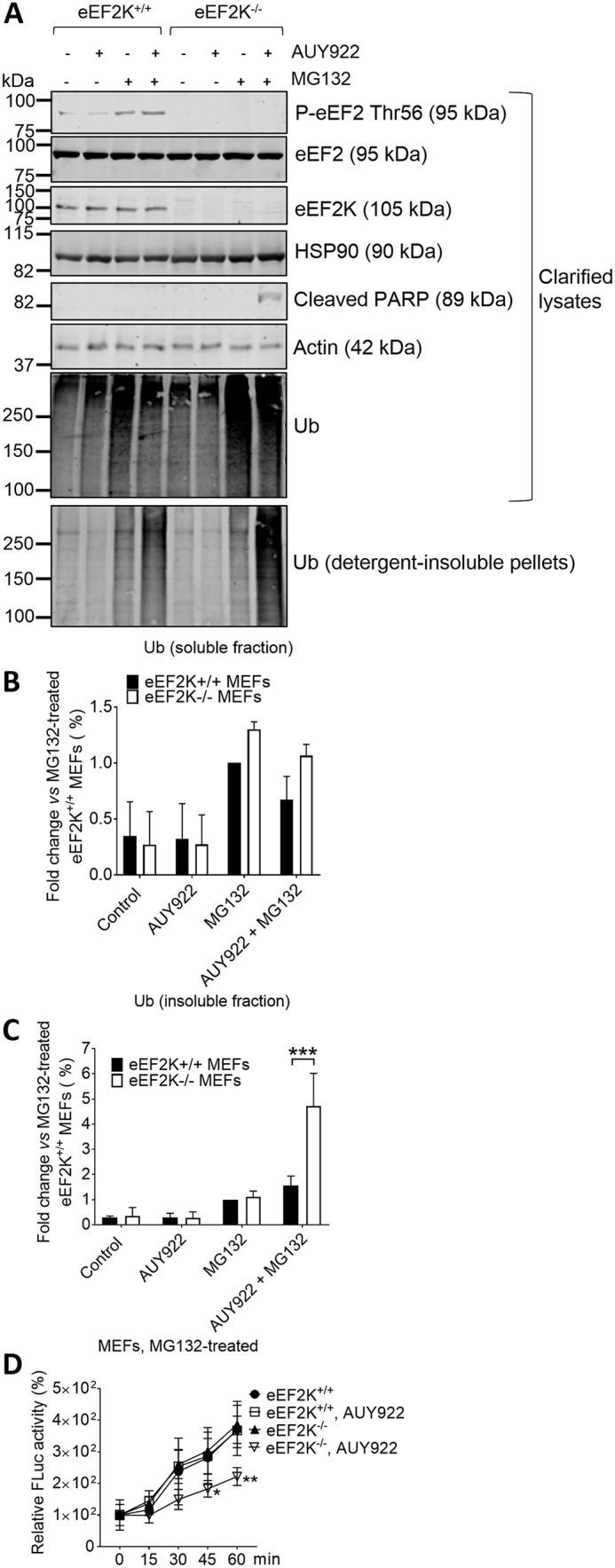
AUY922 increases levels of ubiquitinated proteins in 1% (v/v) Triton–insoluble pellets from eEF2K−/− MEFs. A–C, eEF2K+/+ and eEF2K−/− MEFs were cultured with vehicle or with 50 nm AUY922 and/or 3 μm MG132 for 24 h. Clarified cell lysates and detergent-insoluble pellets were subjected to SDS-PAGE followed by WB analysis of the indicated proteins (A). Levels of ubiquitin (Ub) in clarified lysates (B) and insoluble pellets (C) were quantified. D, MEFs were treated as in A, and cell lysates were incubated with previously heat-denatured Fluc at room temperature. Fluc refolding was assessed by measuring Fluc activity at the indicated time points. P-eEF2, phosphorylated eEF2. *, 0.01 ≤ p < 0.05; **, 0.001 ≤ p < 0.01; ***, p < 0.001. Error bars represent S.D.
As expected, after MG132 treatment, a large quantity of ubiquitinated proteins was found in clarified lysates, and a slight increase in ubiquitinated proteins in 1% (v/v) Triton X-100–insoluble pellets was also observed (Fig. 3, A–C). Inhibiting HSP90 activity by AUY922 increased levels of ubiquitinated proteins in the pellet fraction in eEF2K+/+ MEFs, but the effect was considerably enlarged in eEF2K−/− cells (Fig. 3, A–C), indicating a greater buildup of incorrectly folded proteins. Furthermore, this was accompanied with poly(ADP-ribose) polymerase cleavage (Fig. 3A), indicating apoptosis (12).
We observed a reduction in protein refolding capacity in eEF2K−/− MEF lysates from cells pretreated with MG132 and AUY922 (Fig. 3D), showing that HSP90 inhibition greatly impairs overall chaperone capacity in eEF2K-null cells. We also detected a large increase in early and late apoptotic AUY922-treated eEF2K−/− cells under proteotoxic stress induced by MG132 (Fig. 4), likely reflecting impaired proteostasis. Taken together, these data indicate that when elongation rates are enhanced, subsequent up-regulation of HSP90 chaperone expression plays an important role in maintaining proteostasis.
Figure 4.

AUY922 induces apoptosis in eEF2K−/− MEFs under proteolytic stress. eEF2K+/+ and eEF2K−/− MEFs were treated as in Fig. 3A. A, cell viability and morphology were assessed by microscopic analysis. Scale bars, 500 μm. B, cells were dispersed, stained with annexin V (FITC-A) and PI (PI-A), and then analyzed by flow cytometry. C, quantification of B. ***, p < 0.001. Error bars represent S.D.
Discussion
Proteostasis involves tightly-regulated and balanced processes that are essential to cell and organ health span. The processes that modulate proteostasis include the synthesis, maturation, and degradation of proteins. It follows that mRNA translation requires effective mechanisms to ensure the correct folding and assembly of newly synthesized polypeptides into functional proteins; dysregulation of this crucial process in cells contributes to development of protein misfolding disorders such as Alzheimer's disease, Huntington's disease, and amyotrophic lateral sclerosis (ALS) among others (13).
Conn and Qian (14) and we (6) recently reported that mTOR complex 1 (mTORC1) (6, 14), ribosomal protein S6 kinases (14), and eEF2K (6) function to optimize quality control during protein synthesis. For example, faster translation elongation resulting from eEF2K inhibition can lead to the production of mistranslated proteins (6), and conversely, inhibition of mTORC1 by rapamycin slows down translation elongation, resulting in more accurate translation (14). Faster elongation will increase the load of newly synthesized polypeptides that need to be properly folded. Misfolded proteins are degraded, but it is not clear how mTORC1 controls protein degradation (15, 16); neither we (6) nor Conn and Qian (14) saw any detectable changes in chaperone capacity or proteasome activity upon inhibition of mTORC1 or eEF2K. However, because translation quantity is inversely related with quality, it is reasonable to speculate that alterations in translation speed and hence accuracy may also impact other proteostasis regulatory mechanisms to ensure proper protein production.
Previously, we showed that levels of certain proteins involved in cell migration are decreased upon eEF2K knockdown in A549 cells (17). To measure the rates of de novo synthesis at the proteome-wide level, we applied pSILAC (which enables the identification and quantification of newly synthesized proteins). Consistent with results from our study of steady-state protein levels (17), we only observed changes in the synthesis of a small proportion of proteins (78 of the 973 quantifiable proteins) when comparing control and eEF2K knockdown cells (Fig. S1). Among them, levels of HSP90 chaperones were increased in A549 cells upon eEF2K knockdown and in eEF2K−/− MEFs (Fig. 1). This observation, however, was not associated with increases in HSP90 mRNA levels but instead resulted from increased translational efficiency of HSP-encoding mRNAs (HSP90AA1 and HSP90B1) (Fig. 2) overall, ensuring proper protein folding under accelerated rates of translation elongation. The mechanism by which depletion of eEF2K, and thus faster elongation, increases translation of HSP90 mRNAs is not clear. One possibility is that their coding sequences contain rare codons for which levels of the cognate tRNAs are low; this appears not to be the case. Another is that it relates to features of their 5′-untranslated regions (UTRs), regions that often control translation initiation. Notably, the HSP90 5′-UTR is known to possess extensive secondary structure (18) and may therefore make the translation of their mRNAs more susceptible to modulation during elongation, although the mechanisms that link control of elongation to modulation of initiation remain unclear (see Ref. 19).
Conversely, suppressing HSP90 activity using AUY922 led to the accumulation of ubiquitinated proteins (Fig. 3) in eEF2K−/− cells and to apoptosis (Fig. 4), implying that eEF2K and HSP90 jointly act to maintain proper proteostasis. It has previously been reported that HSP90 physically interacts with eEF2K, and disrupting the HSP90–eEF2K interaction by geldanamycin reduced eEF2K protein levels in glioblastoma cells (8), suggestive of the requirement of HSP90 for the proper folding and stability of eEF2K. Thus, eEF2K and chaperones such as HSP90 form a mutually complementary system to ensure proper protein folding and protection of cells against buildup of misfolded polypeptides. Furthermore, given the growing interest in HSP90 and eEF2K inhibitors as anticancer agents, a combination therapy involving both types of inhibitors may offer a promising and effective strategy for therapy of some types of cancer.
Experimental procedures
Chemicals and reagents
All reagents were from Sigma-Aldrich unless specified. Bradford assay reagent was from Bio-Rad. IPTG was from Promega (Alexandria, New South Wales, Australia). AUY922 was kindly provided by Professor Lisa Butler (South Australian Health and Medical Research Institute, Adelaide, South Australia, Australia) (20).
Cell culture, treatment, and lysis
A549 (human lung carcinoma) cells expressing inducible shRNA against eEF2K were generously provided by Janssen Pharmaceutica NV (Beerse, Belgium) (3). eEF2K-knockout MEFs from eEF2K+/+ and eEF2K−/− mice (2) were prepared from embryos at embryonic day 13.5. The experimental procedures were approved by the South Australian Health and Medical Research Institute Animal Ethics Committee (ethics approval number SAM220) and carried out in accordance with the Australian code for the care and use of animals for scientific purposes (2013). Cells were cultured in high-glucose (25 mm) Dulbecco's modified Eagle's medium (DMEM; Life Technologies) supplemented with 10% (v/v) fetal bovine serum, 100 units/ml penicillin, and 0.1 mg/ml streptomycin and maintained at 37 °C in humidified air with 5% (v/v) CO2. For pSILAC labeling, cells were cultured in low-glucose (5.5 mm) DMEM (supplemented with dialyzed fetal bovine serum) containing 0.71 mm l-[13C6]Arg and 0.41 mm l-[13C6,15N2]Lys (Fig. S1A). To induce eEF2K knockdown, 5 days before sampling 1 μm IPTG was added to A549 cells to induce expression of the shRNA against eEF2K. Control cells were treated with the corresponding vehicle. Cells were collected by trypsinization, washed twice with PBS (Thermo Fisher Scientific, Bremen, Germany), and lysed in ice-cold lysis buffer (1% (v/v) Triton X-100, 20 mm Tris-HCl (pH 7.5), 150 mm NaCl, 1 mm EDTA, 1 mm EGTA, 2.5 mm NaH2P2O7, 1 mm β-glycerophosphate, 1 mm dithiothreitol (DTT), 1 mm Na3VO4, and protease inhibitor mixture (1×)). Lysates were centrifuged at 16,000 × g at 4 °C for 10 min, and total protein concentrations in the supernatants were quantified by Bradford assay. Where indicated, insoluble pellets were dissolved in 1× Laemmli sample buffer (62.5 μm Tris-HCl (pH 6.8), 1% (w/v) SDS, 10% (v/v) glycerol, 10 μm DTT, and 5 μg/ml bromphenol blue).
pSILAC labeling and LC-MS/MS analysis
As described in Fig. S1A, A549 cells were cultured in growth medium with or without IPTG (to induce shRNA eEF2K expression) for 5 days, medium was replaced by Arg/Lys-free DMEM for 1 h, and cells were then incubated with 10 mm 2-DG for 1 h before the addition of 0.71 mm l-[13C6]Arg and 0.41 mm l-[13C6,15N2]Lys and further incubation for 2 h. Cell (5 × 106) pellets were resuspended in PBS with equal volumes of 8 m guanidine chloride HCl lysis buffer, lysed by three freeze–thaw rounds in liquid N2, and processed for LC-MS/MS analysis as described previously (21). All samples were introduced into an LC-MS/MS system through an Ultimate 3000 RSLC nano LC (Thermo Fisher Scientific) in line–connected to a Q Exactive HF mass spectrometer (Thermo Fisher Scientific) (22).
Proteome data analysis
Raw data files were searched with MaxQuant (23) using the Andromeda search engine (version 1.5.4.1) (24), and MS/MS spectra were searched against the Swiss-Prot database (taxonomy, Homo sapiens; 20,198 entries; August 2016 version). Potential contaminants present in the contaminants.fasta file that comes with MaxQuant were automatically added. The precursor mass tolerance was set to 20 ppm for the first search (used for nonlinear mass recalibration) and to 4.5 ppm for the main search. Trypsin/P (i.e. cleavages between lysine/arginine-proline residues were allowed) was selected as enzyme setting, and up to two missed cleavages were allowed. Methionine oxidation was searched as a fixed modification, whereas N-terminal protein acetylation was set as a variable modification. The false discovery rate for peptide and protein identification, estimated by using the reversed search sequences, was set to 1%, and the minimum peptide length was set to 7. The minimum score threshold for both modified and unmodified peptides was set to 40. The match between runs function with a match time window of 0.7 min and an alignment time window of 20 min was enabled, and proteins were quantified by the MaxLFQ algorithm (25) integrated in the MaxQuant software. For SILAC ratio quantification, the requantification feature was enabled, multiplicity was set at 2, and the relative quantification of the peptides against their SILAC-labeled counterparts (carrying +6Arg and/or +8Lys) was performed by MaxQuant. Only unique peptides were used for quantification, and we required proteins to be quantified with at least two ratio counts. For basic data analysis, normalization, statistics, and annotation enrichment analysis, the freely available open-source bioinformatics platform Perseus (version 1.5.3.2) (26) was used. Data analysis was done using the proteinGroups.txt file from MaxQuant. Potential contaminants as well as reversed sequences and proteins only identified by site (thus only by a peptide carrying a modified residue) were removed from the data set. The replicate samples were grouped, and the normalized heavy over light protein ratios (i.e. in the case of normalized protein ratios, the median of ratio subpopulations was shifted to 1) were log2-transformed. A multiple-sample analysis of variance test was applied with the S0 parameter set to 0.1 and a p value threshold of 0.01 to assign proteins with significantly different pSILAC protein ratios between groups, thereby reflecting differential protein synthesis rates. Only proteins with minimally two valid quantification values (i.e. SILAC ratios) in at least one group were considered, and missing values were imputed from a normal distribution around the detection limit (with 0.3 spread and 1.8 downshift). Proteins with differential pSILAC ratios were selected for subsequent Z-score normalization and k-means clustering.
SDS-PAGE and Western blot (WB) analysis
Western blot analysis were performed as described previously (7). Primary antibodies used were: HSP90 (Sigma-Aldrich, catalog number 04-594), β-actin (Sigma-Aldrich, catalog number A5316), eEF2 (New England Biolabs, Hitchin, Herts, UK, catalog number 2332), ubiquitin (New England Biolabs, catalog number 3936), poly(ADP-ribose) polymerase (New England Biolabs, catalog number 9542), and phosphorylated eEF2 Thr-56 and eEF2K (custom-made by Eurogentec, Seraing, Belgium). Fluorescently tagged secondary antibodies were from Thermo Fisher Scientific. Blots were scanned using a LI-COR Odyssey imaging system (Millenium Science, Mulgrave, Victoria, Australia).
Real-time quantitative RT-PCR (qPCR) amplification analysis
Total RNA was extracted using TRIzol (Life Technologies). cDNA was produced using the ImProm-II reverse transcription system (Promega) with oligo(dT)15. For qPCR analysis of sucrose gradient fractions, 1.2-kb kanamycin RNA provided by the reverse transcription kit was used as an internal control. qPCR was performed using the following primers (5′–3′): mouse HSP90AA1: forward, CTGACGGACCCCAGTAAACT; reverse, CCTGCAAAGCCTCCATGAAG; HSP90B1: forward, AGTCGGGAAGCAACAGAGAA; reverse, TCTCCATGTTGCCAGACCAT; B2M, forward, CTGCTACGTAACACAGTTCCACCC; reverse, CATGATGCTTGATCACATGTCTCG. Samples were analyzed with SYBR Green dye mix (Life Technologies) on an ABI Step One Plus qPCR instrument (Applied Biosystems, Cheshire, UK). For total RNA analysis, B2M was used as the normalization control. The comparative threshold cycle (CT) method was applied to quantify mRNAs present in each sample.
Polysome analysis
Polysome analysis was performed as described previously (7). For RNA extraction, 1% (w/v) SDS and 0.15 mg/ml proteinase K were added to each fractions, 1:3 (v/v) phenol:chloroform, pH 4.5, was then added to the samples to extract RNA, and RNA was precipitated from the aqueous phase by the addition of 70% (v/v) isopropanol. RNA pellets were washed once with 80% (v/v) ethanol before dissolving in RNase/DNase–free water for further analysis.
In vitro refolding assay
Recombinant firefly luciferase (Fluc) (Promega) was diluted in lysis buffer at 50 μg/ml and then denatured at 42 °C at 1000 rpm for 15 min. Denatured Fluc was added to cell lysates for a final concentration of 16.5 μg/ml. Refolding was conducted at room temperature at 1,000 rpm for the indicated periods of time. Fluc activity was monitored with a luciferase reporter assay system (Promega) on a Glomax Discover multimode microplate reader (Promega) following the manufacturer's instructions. Fluc activity in lysis buffer alone was used as a control to exclude spontaneous refolding of denatured Fluc.
Annexin V/propidium iodide (PI) staining
Following removal of culture media, the cells were washed once with PBS and then incubated in 1× trypsin/EDTA (0.5%) for 1 min at 37 °C. Growth medium was added, and the cells were gently dispersed by pipetting and centrifuged at 200 × g for 5 min at room temperature. The medium was removed, and annexin V/PI staining was performed using the Annexin-V-FLUOS Staining kit (Sigma-Aldrich) according to the manufacturer's instructions. The intensity of fluorescence signals was recorded using a FACSCantoTM II flow cytometer (BD Biosciences), and data were analyzed using FlowJo software version 10.2 (BD Biosciences).
Statistical analysis
Statistical analyses were performed using one-way or two-way analysis of variance with an unpaired Student's t test with the means of three independent experiments unless otherwise specified. Data are presented as means ± S.D. GraphPad Prism software was used to calculate p values: *, 0.01 ≤ p < 0.05; **, 0.001 ≤ p < 0.01; ***, p < 0.001.
Author contributions
J. X., P. V. D., and C. G. P. conceptualization; J. X. and P. V. D. data curation; J. X., P. V. D., and D. F. formal analysis; J. X., P. V. D., and D. F. investigation; J. X. methodology; J. X. and C. G. P. writing-original draft; P. V. D. and C. G. P. funding acquisition; P. V. D., D. F., and C. G. P. writing-review and editing; D. F. validation; C. G. P. supervision; C. G. P. project administration.
Supplementary Material
Acknowledgment
We thank Dr. Randall H. Grose from the Australian Cancer Research Foundation (ACRF) Innovative Cancer Imaging Facility at South Australian Health and Medical Research Institute for technical support.
This work was supported by Prof. Kris Gevaert, VIB Center for Medical Biotechnology. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1 and S2 and Table S1.
- eEF2
- eukaryotic elongation factor 2
- eEF2K
- eukaryotic elongation factor 2 kinase
- pSILAC
- pulsed stable isotope labeling with amino acids in cell culture
- HSP90
- heat-shock protein 90
- MEF
- mouse embryonic fibroblast
- IPTG
- isopropyl β-d-1-thiogalactopyranoside
- 2-DG
- 2-deoxyglucose
- mTORC1
- mTOR complex 1
- WB
- Western blot
- qPCR
- real-time quantitative RT-PCR
- Fluc
- firefly luciferase
- PI
- propidium iodide.
References
- 1. Carlberg U., Nilsson A., and Nygård O. (1990) Functional properties of phosphorylated elongation factor 2. Eur. J. Biochem. 191, 639–645 10.1111/j.1432-1033.1990.tb19169.x [DOI] [PubMed] [Google Scholar]
- 2. Moore C. E., Mikolajek H., Regufe da Mota S., Wang X., Kenney J. W., Werner J. M., and Proud C. G. (2015) Elongation factor 2 kinase is regulated by proline hydroxylation and protects cells during hypoxia. Mol. Cell. Biol. 35, 1788–1804 10.1128/MCB.01457-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Xie J., Mikolajek H., Pigott C. R., Hooper K. J., Mellows T., Moore C. E., Mohammed H., Werner J. M., Thomas G. J., and Proud C. G. (2015) Molecular mechanism for the control of eukaryotic elongation factor 2 kinase by pH: role in cancer cell survival. Mol. Cell. Biol. 35, 1805–1824 10.1128/MCB.00012-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Leprivier G., Remke M., Rotblat B., Dubuc A., Mateo A. R., Kool M., Agnihotri S., El-Naggar A., Yu B., Somasekharan S. P., Faubert B., Bridon G., Tognon C. E., Mathers J., Thomas R., et al. (2013) The eEF2 kinase confers resistance to nutrient deprivation by blocking translation elongation. Cell 153, 1064–1079 10.1016/j.cell.2013.04.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wang X., Xie J., da Mota S. R., Moore C. E., and Proud C. G. (2015) Regulated stability of eukaryotic elongation factor 2 kinase requires intrinsic but not ongoing activity. Biochem. J. 467, 321–331 10.1042/BJ20150089 [DOI] [PubMed] [Google Scholar]
- 6. Xie J., de Souza Alves V., von der Haar T., O'Keefe L., Lenchine R. V., Jensen K. B., Liu R., Coldwell M. J., Wang X., and Proud C. G. (2019) Regulation of the elongation phase of protein synthesis enhances translation accuracy and modulates lifespan. Curr. Biol. 29, 737–749.e5 10.1016/j.cub.2019.01.029 [DOI] [PubMed] [Google Scholar]
- 7. Huo Y., Iadevaia V., Yao Z., Kelly I., Cosulich S., Guichard S., Foster L. J., and Proud C. G. (2012) Stable isotope-labelling analysis of the impact of inhibition of the mammalian target of rapamycin on protein synthesis. Biochem. J. 444, 141–151 10.1042/BJ20112107 [DOI] [PubMed] [Google Scholar]
- 8. Yang J., Yang J. M., Iannone M., Shih W. J., Lin Y., and Hait W. N. (2001) Disruption of the EF-2 kinase/Hsp90 protein complex: a possible mechanism to inhibit glioblastoma by geldanamycin. Cancer Res. 61, 4010–4016 [PubMed] [Google Scholar]
- 9. Gismondi A., Caldarola S., Lisi G., Juli G., Chellini L., Iadevaia V., Proud C. G., and Loreni F. (2014) Ribosomal stress activates eEF2K-eEF2 pathway causing translation elongation inhibition and recruitment of terminal oligopyrimidine (TOP) mRNAs on polysomes. Nucleic Acids Res. 42, 12668–12680 10.1093/nar/gku996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Eccles S. A., Massey A., Raynaud F. I., Sharp S. Y., Box G., Valenti M., Patterson L., de Haven Brandon A., Gowan S., Boxall F., Aherne W., Rowlands M., Hayes A., Martins V., Urban F., et al. (2008) NVP-AUY922: a novel heat shock protein 90 inhibitor active against xenograft tumor growth, angiogenesis, and metastasis. Cancer Res. 68, 2850–2860 10.1158/0008-5472.CAN-07-5256 [DOI] [PubMed] [Google Scholar]
- 11. Mimnaugh E. G., Xu W., Vos M., Yuan X., Isaacs J. S., Bisht K. S., Gius D., and Neckers L. (2004) Simultaneous inhibition of hsp 90 and the proteasome promotes protein ubiquitination, causes endoplasmic reticulum-derived cytosolic vacuolization, and enhances antitumor activity. Mol. Cancer Ther. 3, 551–566 10.4161/cbt.3.6.846 [DOI] [PubMed] [Google Scholar]
- 12. Gobeil S., Boucher C. C., Nadeau D., and Poirier G. G. (2001) Characterization of the necrotic cleavage of poly(ADP-ribose) polymerase (PARP-1): implication of lysosomal proteases. Cell Death Differ. 8, 588–594 10.1038/sj.cdd.4400851 [DOI] [PubMed] [Google Scholar]
- 13. Scheckel C., and Aguzzi A. (2018) Prions, prionoids and protein misfolding disorders. Nat. Rev. Genet. 19, 405–418 10.1038/s41576-018-0011-4 [DOI] [PubMed] [Google Scholar]
- 14. Conn C. S., and Qian S. B. (2013) Nutrient signaling in protein homeostasis: an increase in quantity at the expense of quality. Sci. Signal. 6, ra24 10.1126/scisignal.2003520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhao J., Zhai B., Gygi S. P., and Goldberg A. L. (2015) mTOR inhibition activates overall protein degradation by the ubiquitin proteasome system as well as by autophagy. Proc. Natl. Acad. Sci. U.S.A. 112, 15790–15797 10.1073/pnas.1521919112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhang Y., Nicholatos J., Dreier J. R., Ricoult S. J., Widenmaier S. B., Hotamisligil G. S., Kwiatkowski D. J., and Manning B. D. (2014) Coordinated regulation of protein synthesis and degradation by mTORC1. Nature 513, 440–443 10.1038/nature13492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Xie J., Shen K., Lenchine R. V., Gethings L. A., Trim P. J., Snel M. F., Zhou Y., Kenney J. W., Kamei M., Kochetkova M., Wang X., and Proud C. G. (2018) Eukaryotic elongation factor 2 kinase upregulates the expression of proteins implicated in cell migration and cancer cell metastasis. Int. J. Cancer 142, 1865–1877 10.1002/ijc.31210 [DOI] [PubMed] [Google Scholar]
- 18. Ahmed R., and Duncan R. F. (2004) Translational regulation of Hsp90 mRNA. AUG-proximal 5′-untranslated region elements essential for preferential heat shock translation. J. Biol. Chem. 279, 49919–49930 10.1074/jbc.M404681200 [DOI] [PubMed] [Google Scholar]
- 19. Proud C. G. (2018) Phosphorylation and signal transduction pathways in translational control. Cold Spring Harb. Perspect. Biol. a033050 10.1101/cshperspect.a033050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Centenera M. M., Gillis J. L., Hanson A. R., Jindal S., Taylor R. A., Risbridger G. P., Sutherland P. D., Scher H. I., Raj G. V., Knudsen K. E., Yeadon T., Australian Prostate Cancer BioResource, Tilley W. D., and Butler L. M. (2012) Evidence for efficacy of new Hsp90 inhibitors revealed by ex vivo culture of human prostate tumors. Clin. Cancer Res. 18, 3562–3570 10.1158/1078-0432.CCR-12-0782 [DOI] [PubMed] [Google Scholar]
- 21. Jonckheere V., Fijałkowska D., and Van Damme P. (2018) Omics assisted N-terminal proteoform and protein expression profiling on methionine aminopeptidase 1 (MetAP1) deletion. Mol. Cell. Proteomics 17, 694–708 10.1074/mcp.RA117.000360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Varland S., Aksnes H., Kryuchkov F., Impens F., Van Haver D., Jonckheere V., Ziegler M., Gevaert K., Van Damme P., and Arnesen T. (2018) N-terminal acetylation levels are maintained during acetyl-CoA deficiency in Saccharomyces cerevisiae. Mol. Cell. Proteomics 17, 2309–2323 10.1074/mcp.RA118.000982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cox J., and Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 10.1038/nbt.1511 [DOI] [PubMed] [Google Scholar]
- 24. Cox J., Neuhauser N., Michalski A., Scheltema R. A., Olsen J. V., and Mann M. (2011) Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 10, 1794–1805 10.1021/pr101065j [DOI] [PubMed] [Google Scholar]
- 25. Cox J., Hein M. Y., Luber C. A., Paron I., Nagaraj N., and Mann M. (2014) Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteomics 13, 2513–2526 10.1074/mcp.M113.031591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tyanova S., Temu T., Sinitcyn P., Carlson A., Hein M. Y., Geiger T., Mann M., and Cox J. (2016) The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 13, 731–740 10.1038/nmeth.3901 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.