

Abstract
Myoglobin is a monomeric heme protein expressed ubiquitously in skeletal and cardiac muscle and is traditionally considered to function as an oxygen reservoir for mitochondria during hypoxia. It is now well established that low concentrations of myoglobin are aberrantly expressed in a significant proportion of breast cancer tumors. Despite being expressed only at low levels in these tumors, myoglobin is associated with attenuated tumor growth and a better prognosis in breast cancer patients, but the mechanism of this myoglobin-mediated protection against further cancer growth remains unclear. Herein, we report a signaling pathway by which myoglobin regulates mitochondrial dynamics and thereby decreases cell proliferation. We demonstrate in vitro that expression of human myoglobin in MDA-MB-231, MDA-MB-468, and MCF7 breast cancer cells induces mitochondrial hyperfusion by up-regulating mitofusins 1 and 2, the predominant catalysts of mitochondrial fusion. This hyperfusion causes cell cycle arrest and subsequent inhibition of cell proliferation. Mechanistically, increased mitofusin expression was due to myoglobin-dependent free-radical production, leading to the oxidation and degradation of the E3 ubiquitin ligase parkin. We recapitulated this pathway in a murine model in which myoglobin-expressing xenografts exhibited decreased tumor volume with increased mitofusin, markers of cell cycle arrest, and decreased parkin expression. Furthermore, in human triple-negative breast tumor tissues, mitofusin and myoglobin levels were positively correlated. Collectively, these results elucidate a new function for myoglobin as a modulator of mitochondrial dynamics and reveal a novel pathway by which myoglobin decreases breast cancer cell proliferation and tumor growth by up-regulating mitofusin levels.
Keywords: myoglobin, breast cancer, reactive oxygen species (ROS), parkin, oxidative stress, cell cycle, heme protein, mitochondrial fusion, proliferation, tumor growth
Introduction
Myoglobin is a monomeric heme protein constitutively expressed in cardiac tissue and skeletal muscle at high micromolar concentrations (1) and to a lesser extent in smooth muscle cells (2). The traditional role of myoglobin is to store and deliver oxygen to mitochondria to facilitate cellular respiration in hypoxic conditions (3, 4). However, in recent years, novel functions of myoglobin, all catalyzed by its heme prosthetic group, have been uncovered. These include the regulation of tissue nitric oxide (NO) levels (5–7) and the production of cellular oxidants (8). Investigation of the physiological effect of these novel functions on mitochondrial function has largely been limited to elucidating myoglobin's role in regulating mitochondrial oxygen consumption and fatty acid metabolism (9–12), whereas the effects of myoglobin on mitochondrial structure and dynamics are unknown.
Mitochondria are dynamic organelles that continually reorganize within the cell to break and form intracellular networks through fission and fusion events. Although changes in these networks regulate energy production and are intimately linked to mitochondrial quality control, it is now established that mitochondrial dynamics can also modulate cell cycle progression and cell proliferation. For example, mitochondria fuse, dependent on the expression of the fusion proteins mitofusins 1 and 2 (MFN1 and -2),4 to form intracellular networks prior to G1–S phase. Conversely, these networks undergo fission to break these networks, dependent on the action of dynamin-related protein 1 (DRP1), to facilitate the G2–M transition (13–15). Highlighting the importance of changes in dynamics, mitochondrial hyperfusion inhibits the transition from G1 to S phase, mediating cell cycle arrest and attenuating cellular proliferation (16, 17). Although it is well established that mitochondrial dynamics regulate cell proliferation, the factors that control mitochondrial dynamics in rapidly proliferating cells remain unclear.
Accumulating studies now provide evidence that myoglobin is expressed at low concentrations in many types of highly aggressive human carcinomas, including breast, ovary, colon, lung, and head and neck cancers (18–22). In a study of 917 women with invasive breast cancer, ∼40% of tumors showed myoglobin expression (20). Notably, this expression was associated with a more favorable prognosis (increased overall survival in both estrogen receptor α–positive and –deficient tumors) (1, 20). Expression of myoglobin in these tumors is associated with alterations in mitochondrial β-oxidation; however, the role of myoglobin in modulating cell proliferation in these highly proliferative tumors has not been considered.
Herein we test the hypothesis that myoglobin modulates mitochondrial dynamics to inhibit breast cancer cell proliferation, which is associated with attenuation of tumor growth. We demonstrate that myoglobin expression causes mitochondrial hyperfusion, leading to cell cycle arrest and inhibition of breast cancer cell proliferation. Mechanistically, we demonstrate that this mitochondrial fusion is due to myoglobin-dependent oxidation and degradation of the E3 ubiquitin ligase parkin, leading to up-regulation of mitofusins 1 and 2, catalysts of mitochondrial fusion. We show that this pathway is recapitulated in a murine model in which myoglobin expression attenuates tumor growth. Additionally, analysis of human triple-negative breast cancer (TNBC) tissues confirms our findings that myoglobin expression is associated with increased MFN1 expression. We therefore reveal a novel role for myoglobin in regulating mitochondrial structure and cell proliferation. The implications of this pathway for cancer cell biology and development of treatment are discussed along with the potential role of this signaling mechanism in human muscle physiology.
Results
Myoglobin expression significantly decreases breast cancer cell proliferation in vitro
In the first series of experiments, we sought to determine whether myoglobin expression altered proliferation of breast cancer cells. MDA-MB-231 cells, which do not endogenously express myoglobin, were stably transfected to express either GFP-tagged full-length human myoglobin (Mb) or GFP-tagged myoglobin apoprotein (which has identical globin protein structure but lacks the heme center (apo-Mb). Western blot analysis and densitometry confirmed that WT MDA-MB-231 cells showed no myoglobin expression, whereas the transfected cells expressed myoglobin (Fig. 1A). Calculation of myoglobin concentration from spectrophotometric absorbance measurements demonstrated that that the myoglobin-expressing MDA-MB-231 cells expressed ∼0.8 μm myoglobin (data not shown).
Figure 1.

Myoglobin expression significantly reduces proliferation and migration of breast cancer cells in vitro. A, representative Western blot and densitometry quantification of myoglobin expression in WT MDA-MB-231 cells, MDA-MB-231 cells transfected to express GFP-tagged apo-Mb, and MDA-MB-231 cells transfected to express GFP-tagged human Mb. Apomyoglobin-expressing MDA-MB-231 cells serve as a heme-null control. n = 3 per group. ****, p < 0.0001. B, cellular proliferation normalized to WT MDA-MB-231 cells 48 h after [3H]thymidine incorporation for WT MDA-MB-231 cells, apomyoglobin-expressing MDA-MB-231 cells, and myoglobin-expressing MDA-MB-231 cells. n = 6 per group. **, p < 0.005; ****, p < 0.0001. C, representative Western blot and proliferation rate of WT MDA-MB-231 and myoglobin-expressing MDA-MB-231 cells transfected with scrambled (SCR) or myoglobin (Mb −/−)-targeted siRNA. Data are normalized to WT MDA-MB-231 scrambled siRNA cell proliferation. n = 9 per group. ***, p < 0.001; ****, p < 0.0001. D, representative Western blot demonstrating myoglobin expression and proliferation rate of WT MDA-MB-468 cells transfected with scrambled (SCR) or myoglobin (Mb −/−)-targeted siRNA. Data are normalized to WT MDA-MB-231 scrambled siRNA cell proliferation. n = 12 per group. ****, p < 0.0001. Error bars represent S.E.
Using this cell model, we tested whether myoglobin expression affects cellular proliferation. Measurement of cellular [3H]thymidine incorporation at 48 h demonstrated that the proliferation rate of myoglobin-expressing MDA-MB-231 cells was significantly decreased (40 ± 7%) compared with WT MDA-MB-231 cells (Fig. 1B). Importantly, cells stably expressing apomyoglobin proliferated at the same rate as WT MDA-MB-231 cells, demonstrating that the myoglobin-dependent decrease in proliferation requires the protein's heme moiety (Fig. 1B). To further confirm that myoglobin expression was responsible for decreased cell proliferation, cells were transfected with either scrambled or myoglobin-targeted siRNA, and proliferation was measured. As expected, silencing of myoglobin had no effect on the proliferation rate of WT MDA-MB-231 cells but restored the proliferation rate of the myoglobin-expressing MDA-MB-231 cells to the level of the WT MDA-MB-231 cells (Fig. 1C).
To test whether myoglobin-dependent inhibition of proliferation was specific to MDA-MB-231 cells, a second cell type, MCF7, was stably transfected with human myoglobin. Similar to MDA-MB-231 cells, WT MCF7 cells did not express myoglobin, but these levels were increased with stable transfection (Fig. S1A). Also similar to MDA-MB-231 cells, measurement of proliferation after 72 h by crystal violet staining demonstrated that the proliferation rate of myoglobin-expressing MCF7 cells was significantly decreased (26 ± 2.5%) compared with WT MC7 cells (Fig. S1B).
To confirm that myoglobin-dependent inhibition of proliferation was not an artifact of overexpressing myoglobin in the MDA-MB-231 and MCF7 cell lines, we next measured the proliferation rate in a breast cancer cell line that endogenously expresses myoglobin, MDA-MB-468. Genetic silencing of myoglobin increased the proliferation rate of the MDA-MB-468 cells compared with WT cells (Fig. 1D), confirming that myoglobin expression regulates cellular proliferation rate.
Myoglobin expression causes cell cycle arrest at G1 to S phase transition
Because cell cycle progression determines cell proliferation rate, we compared the changes in the cell cycle between WT and myoglobin-expressing cells. Flow cytometric analysis of cell cycle stages showed that a significantly higher percentage of myoglobin-expressing MDA-MB-231 cells (53.43 ± 31.87%) were in the G1 phase compared with WT MDA-MB-231 cells (45.20 ± 1.83%; Fig. 2, A and B). Two critical regulators of the G1 to S phase transition are cyclin E and the cyclin-dependent kinase inhibitor, p21. Cyclin E expression is required for cells to enter S phase, whereas p21 expression inhibits S phase entry by binding to and inhibiting cyclin E/Cdk2 activity. Analysis showed that myoglobin-expressing MDA-MB-231 cells have significantly higher p21 expression and approximately a 2-fold lower cyclin E expression than WT MDA-MB-231 cells (Fig. 2, C and D). Notably, this increase in p21 and decrease in cyclin E were recapitulated in myoglobin-expressing MCF7 cells as well (Fig. S1, C and D). Taken together, these data are consistent with cell cycle arrest present at the G1 to S phase transition in myoglobin-expressing breast cancer cells.
Figure 2.

Myoglobin expression causes cell cycle arrest at the G1 to S phase transition. A and B, representative flow cytometry of WT MDA-MB-231 cells (left) and myoglobin-expressing MDA-MB-231 cells (right) (A) and quantification to measure the percentage of cells in the three major phases of the cell cycle (G1, S, and G2) in WT MDA-MB-231 cells and myoglobin-expressing MDA-MB-231 cells (B). n = 6 per group. #, p < 0.05; **, p < 0.001. C, Western blot analysis and densitometry quantification of the expression of the G1 to S phase protein p21 in WT MDA-MB-231 cells and myoglobin-expressing MDA-MB-231 cells. n = 4. #, p < 0.05. D, Western blot analysis and densitometry quantification of the expression of the G1 to S phase protein cyclin E in WT MDA-MB-231 cells and myoglobin-expressing MDA-MB-231 cells. n = 3. #, p < 0.05. Error bars represent S.E.
Myoglobin expression causes mitochondrial fusion
It is established that mitochondrial dynamics regulate progression through the cell cycle. For example, mitochondrial fusion occurs at the G1 to S phase transition and is required for S phase entry. However, physiologically, these fusion events are short-lived, and sustained continuous fusion (hyperfusion) at this stage of the cell cycle results in cell cycle arrest (16). Thus, we sought to determine whether the G1/S phase arrest in myoglobin-expressing cells was concomitant with changes in mitochondrial dynamics. Immunofluorescence using TOM20 to visualize the outer mitochondrial membrane revealed interconnected networks of elongated mitochondria in myoglobin-expressing MDA-MB-231 cells in contrast to smaller, punctate mitochondria in WT cells (Fig. 3A). This increased mitochondrial fusion present in the myoglobin-expressing cells was confirmed by EM (Fig. 3B). Quantification of the ratio of mitochondrial area to perimeter showed a significant increase in mitochondrial interconnectivity in myoglobin-expressing cells compared with WT cells, indicative of increased mitochondrial fusion in the myoglobin-expressing cells (Fig. 3C).
Figure 3.

Myoglobin expression causes mitochondrial fusion resulting in reduced cell proliferation. A, immunofluorescence imaging of the mitochondrial marker TOM20 that stains the outer mitochondrial membrane in WT MDA-MB-231 cells (left) and myoglobin-expressing MDA-MB-231 cells (right). B, EM imaging of WT MDA-MB-231 cells (left) and myoglobin-expressing MDA-MB-231 cells (right). Red arrows point to fused mitochondria. C, quantification of the EM images of mitochondrial interconnectivity was calculated as the area to perimeter ratio. n = 37 measurements in the WT MDA-MB-231 group and 28 measurements in the myoglobin-expressing MDA-MB-231 group. ****, p < 0.0001. D, representative Western blots of mitochondrial fusion (MFN1 and MFN2) and mitochondrial fission (DRP1) protein expressions in WT MDA-MB-231 cells, apomyoglobin-expressing MDA-MB-231 cells, and myoglobin-expressing MDA-MB-231 cells. E, quantification of Western blots of the mitochondrial fusion and fission proteins was done by densitometry relative to β-actin expression. n = 3 per group. ****, p < 0.0001. F, representative Western blot and quantification by densitometry of the MFN1 protein expression relative to β-actin expression in MDA-MB-468 cells transfected with scrambled (SCR) or myoglobin (Mb −/−)-targeted siRNA. n = 4 per group. #, p < 0.05. G and H, representative Western blots (G) and proliferation analysis by crystal violet DNA staining (H) of WT MDA-MB-231 cells and myoglobin-expressing MDA-MB-231 cells transfected with scrambled (SCR), MFN1 (MFN1 −/−)-, or MFN2 (MFN2 −/−)-targeted siRNA. n = 6. ****, p < 0.0001. Error bars represent S.E.
Mitofusin 1–dependent fusion causes decreased proliferation in myoglobin-expressing cells
Mitochondrial dynamics are regulated by a number of small GTPases. Specifically, expression of MFN1 and -2 in the outer membrane mediates fusion, whereas recruitment of DRP1 to the mitochondrion from the cytosol catalyzes mitochondrial fission. To determine the mechanism of mitochondrial fusion in the myoglobin-expressing MDA-MB-231 cells, we measured the expression level of these fission and fusion proteins. Western blotting and densitometric analysis showed that myoglobin-containing MDA-MB-231 cells had a significant up-regulation of the mitochondrial fusion mediators MFN1 (3-fold; p < 0.001) and MFN2 (3-fold; p < 0.001) but not the mitochondrial fission mediator DRP1 compared with WT MDA-MB-231 cells (Fig. 3, D and E). The expression of MFN1, MFN2, and DRP1 in apomyoglobin-expressing cells was similar to control cells, indicating the necessity of the heme moiety for this effect (Fig. 3, D and E). Similarly, Western blotting and densitometric analysis showed that myoglobin-containing MCF7 cells had significant up-regulation of MFN1 and MFN2 protein expression compared with WT MCF7 cells (Fig. S1E). Consistent with this, genetic silencing of endogenous myoglobin in MDA-MB-468 cells resulted in decreased MFN1 expression compared with WT myoglobin-expressing MDA-MB-468 cells (Fig. 3F).
To determine whether this increased expression of MFN1 and -2 observed in myoglobin-expressing MDA-MB-231 cells was responsible for the decreased proliferation observed in these cells, MFN1 and MFN2 were silenced by siRNA transfection (Fig. 3G), and proliferation was measured by quantification of DNA content at 48 h. WT cells showed no change in proliferation rate in the absence of MFN1 or MFN2 expression, consistent with the low level of expression of these proteins in these cells (Fig. 3H). In contrast, myoglobin-expressing cells lacking MFN1 or MFN2 expression showed a significant increase in proliferation (p < 0.0001) with proliferation restored to the level of WT cells (Fig. 3H). These data demonstrate that increased mitofusin expression in myoglobin-expressing cells mediates decreased proliferation, likely through cell cycle arrest in these cells.
Myoglobin expression increases parkin oxidation and decreases parkin expression
We next sought to determine the mechanism by which myoglobin increases mitofusin expression. Mitofusin expression is predominantly regulated by parkin, an E3 ubiquitin ligase protein that is recruited to the mitochondrion and subsequently ubiquitinates mitofusin proteins, marking them for proteasomal degradation (23, 24). Thus, we measured parkin expression to determine whether decreased parkin expression was responsible for increased mitofusin expression in both myoglobin-expressing MDA-MB-231 and MCF7 cells. Parkin expression was significantly decreased in myoglobin-expressing cells when compared with the respective WT MDA-MB-231 (p < 0.05) and MCF7 cells (p < 0.01) (Figs. 4A and S1F). Similarly, genetic silencing of endogenous myoglobin in MDA-MB-468 cells showed an increase in parkin expression compared with WT myoglobin-expressing MDA-MB-468 cells (Fig. 4B).
Figure 4.

Myoglobin expression causes a reduction in parkin expression and increase in parkin oxidation. A, Western blot analysis (top) and densitometry quantification (bottom) of E3 ligase parkin expression in WT MDA-MB-231 cells and myoglobin-expressing MDA-MB-231 cells. n = 3. #, p < 0.05. B, representative Western blot and quantification by densitometry of the parkin protein expression relative to β-actin expression in MDA-MB-468 cells transfected with scrambled (SCR) or myoglobin (Mb −/−)-targeted siRNA. n = 4 per group. #, p < 0.05. C, flow cytometry analysis of total cellular ROS using CellROX marker in WT MDA-MB-231 cells, apomyoglobin-expressing MDA-MB-231 cells, and myoglobin-expressing MDA-MB-231 cells. n = 3. ****, p < 0.0001. D, quantification of total protein oxidation in WT MDA-MB-231 cells and myoglobin-expressing MDA-MB-231 cells using a carbonyl assay. n = 3. ****, p < 0.0001. E, proliferation rate of WT MDA-MB-231 and myoglobin-expressing MDA-MB-231 cells 48 h after ±10 μm TEMPOL treatment. Data are normalized to WT MDA-MB-231 no treatment (none). n = 5 per group. **, p < 0.01; ****, p < 0.0001. F, Western blot analysis of whole-cell lysates of oxidized proteins tagged with biotin in WT MDA-MB-231 cells and myoglobin-expressing MDA-MB-231 after biotin switch assay (left). Blots were also probed with parkin antibody to determine whether parkin was oxidized (right). Boxes indicate oxidized parkin protein. n ≥ 3. Error bars represent S.E.
Prior studies have demonstrated that oxidative stress leads to oxidation of parkin and its subsequent degradation (25). Additionally, myoglobin is recognized to catalyze the production of cellular reactive oxygen species (ROS) (26–28). Thus, we sought to determine whether myoglobin expression increases cellular oxidant production, leading to parkin oxidation and degradation in the myoglobin-expressing MDA-MB-231 cells. To determine whether overall cellular oxidation was increased in the breast cancer cells expressing myoglobin compared with WT cells, we first measured total cellular ROS using the fluorescent dye CellROXTM Deep Red that predominantly detects superoxide anion (O2˙̄). We found an increased intensity of staining in the myoglobin-expressing cells compared with WT (p < 0.0001)- and apomyoglobin (p < 0.0001)-expressing cells, indicative of increased ROS in the myoglobin-expressing cells (Fig. 4C). To determine whether the increased ROS oxidized cellular proteins, we looked at total cellular protein carbonyls. Consistent with increased ROS production, we observed an increase in protein carbonyls in myoglobin-expressing cells compared with WT (p < 0.01)- and apomyoglobin (p < 0.0001)-expressing cells under normal growth conditions (Fig. 4D). To confirm that myoglobin-dependent increases in cellular oxidation were responsible for decreased cell proliferation, cells were treated with 10 μm TEMPOL (a synthetic antioxidant), and proliferation was measured. As expected, treatment with TEMPOL significantly increased the proliferation rate of the myoglobin-expressing MDA-MB-231 cells compared with the level of the untreated myoglobin-expressing MDA-MB-231 cells (p < 0.0028) (Fig. 4E). To specifically determine whether parkin was oxidized in the myoglobin-expressing cells, a modified biotin switch assay was employed to measure oxidative modifications on cellular proteins. Myoglobin-expressing cells showed an overall increase in the levels of oxidized proteins (Fig. 4F). Importantly, oxidation was increased in a band corresponding to the molecular weight of parkin in the myoglobin-overexpressing cells. Taken together, these data suggest that myoglobin-dependent oxidation of parkin potentially leads to its degradation in myoglobin-expressing breast cancer cells.
Myoglobin-expressing tumors have lower end tumor volume/weight and demonstrate mitochondrial fusion and cell cycle arrest
To determine whether myoglobin-mediated decreased cell proliferation was due to mitochondrial fusion in vivo, we utilized a murine xenograft model. Five NOD.CB17-Prkdcscid/J mice were injected in both mammary fat pads with either WT MDA-MB-231 or myoglobin-expressing MDA-MB-231 cells (3 × 106 cells/fat pad). All cells established tumor formation, and tumor growth was measured over 5 weeks. Tumors containing myoglobin-expressing cells showed a strong trend for lower tumor weight and significant decrease in tumor volume (p < 0.05) (Fig. 5, A and B), consistent with a decreased proliferative rate.
Figure 5.

Cellular changes due to myoglobin expression are recapitulated in murine tumors in vivo. A, final tumor weight from NOD.CB17-Prkdcscid/J mice injected with WT MDA-MB-231 cells or myoglobin-expressing MDA-MB-231 cells. n = 4 per group. B, change in tumor volume from NOD.CB17-Prkdcscid/J mice injected with WT MDA-MB-231 cells or myoglobin-expressing MDA-MB-231 cells over 25 days. n = 4 per group. #, p < 0.05. C, Western blot analysis (top) and densitometry quantification (bottom) of the protein expression of the G1–S phase cell cycle marker p21 in tumors injected with WT MDA-MB-231 cells or myoglobin-expressing MDA-MB-231 cells. n = 4. ***, p < 0.001. D, Western blot analysis (top) and densitometry quantification (bottom) of the protein expression of the G1–S phase cell cycle marker cyclin E in tumors injected with WT MDA-MB-231 cells or myoglobin-expressing MDA-MB-231 cells. n = 4. **, p < 0.005. E, EM analysis of mitochondrial morphology of tumors from WT MDA-MB-231 cells (left) and myoglobin-expressing MDA-MB-231 cells (right). Red arrows point to fused mitochondria. F, quantification of mitochondrial interconnectivity, represented as area over perimeter, of tumors from WT MDA-MB-231 cells and myoglobin-expressing MDA-MB-231 cells. n = 25. #, p < 0.05. G, Western blot analysis (left) and densitometry quantification (right) of the expression of the E3 ligase parkin in tumors from WT MDA-MB-231 cells and myoglobin-expressing MDA-MB-231 cells. β-Actin is used as a loading control. n ≥ 3. #, p < 0.05. Error bars represent S.E.
Consistent with our in vitro observations, the expression of p21 was about 2-fold higher (p < 0.001) and the expression of cyclin E was significantly lower (p < 0.01) in the myoglobin-expressing tumors compared with the tumors containing WT cells (Fig. 5, C and D), suggestive of cell cycle arrest. Furthermore, EM of excised tumors demonstrated increased mitochondrial fusion (p < 0.05) (Fig. 5, E and F) and significantly decreased parkin expression in tumors expressing myoglobin (p < 0.05) (Fig. 5G). These data demonstrate that myoglobin-dependent degradation of parkin is associated with mitochondrial fusion, cell cycle arrest, and attenuation of tumor growth in vivo.
Myoglobin-expressing human TNBCs have increased MFN1 expression
To determine whether our in vitro and murine in vivo observations are seen in human cancer tissues, we measured myoglobin, parkin, and MFN1 expression in tissue arrays from normal and TNBC tissues (Fig. 6A). Myoglobin protein expression in TNBC was significantly increased compared with normal breast tissue (2-fold, p < 0.0001) (Fig. 6B). Additionally, the expression of myoglobin in TNBC tumor tissue correlated significantly with the expression of MFN1 (p < 0.0001) (Fig. 6C). Taken together, these data are consistent with our hypothesis that myoglobin expression in breast cancer increases MFN1 expression.
Figure 6.

Expression of myoglobin in human TNBC tumors correlates with increased MFN1 expression in vivo. A, confocal image analysis of the expression of myoglobin (red), parkin (cyan), and MFN1 (green) from breast tissue arrays from one normal breast tissue (top) and two TNBC tissues (a and b; middle and bottom). n = 44 per group. B, increase in myoglobin expression in TNBC tumors compared with normal breast tissue. n = 44. #, p < 0.0001. C, increase in myoglobin expression in TNBC tumor arrays correlates with an increase in MFN1 expression. n = 44. p < 0.0001. Error bars represent S.E.
Discussion
Although it is known that myoglobin is expressed in invasive breast cancer tumors and this expression has been associated with better prognosis, the exact function of endogenous myoglobin in cancer cells remains unclear. In the current study, we demonstrate a novel function for myoglobin as a regulator of mitochondrial dynamics, leading to the inhibition of cell cycle progression and attenuation of breast cancer cell proliferation. Specifically, we show that myoglobin-dependent generation of oxidants oxidizes the E3 ligase parkin, leading to the up-regulation of MFN1 and -2, which catalyze mitochondrial fusion. Mitochondrial hyperfusion in turn inhibits cell cycle progression from G1 to S phase, leading to a decrease in cellular proliferation (Fig. 7). Furthermore, we show that this pathway is activated in a murine xenograft model in which myoglobin expression attenuates tumor volume and that myoglobin expression correlates with mitofusin expression in human TNBC tissue.
Figure 7.
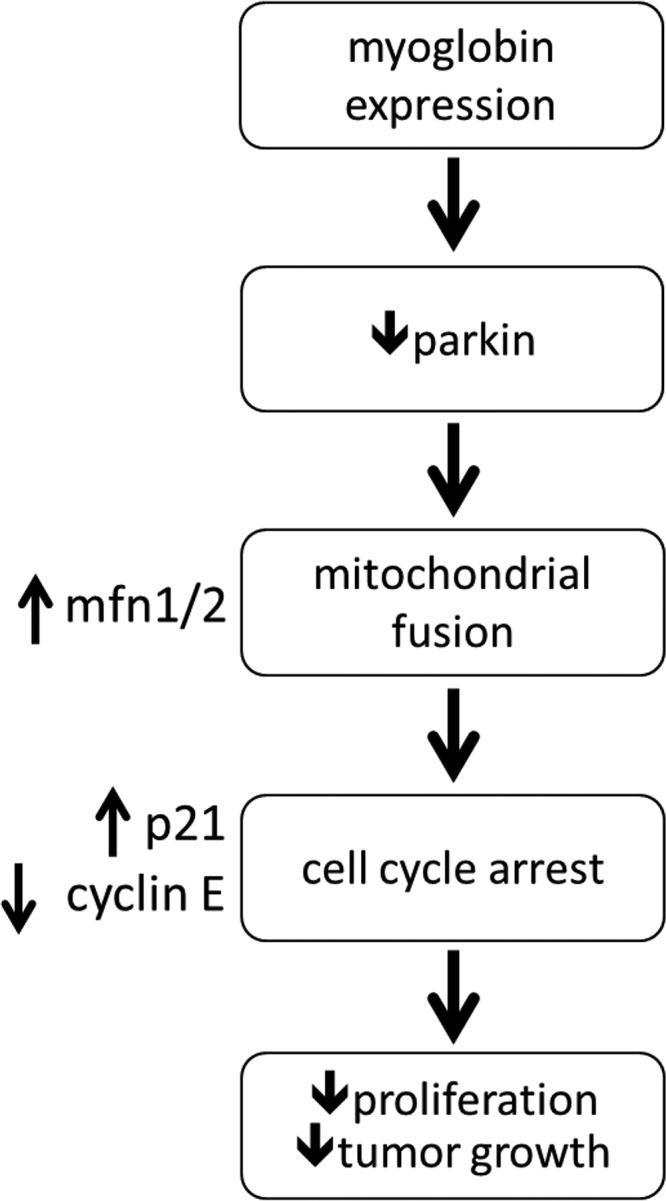
Schematic diagram of the pathway by which myoglobin expression decreases tumor growth and proliferation in breast cancer cells. We propose that expression of myoglobin in breast cancer cells causes an increase in oxidant damage to parkin, resulting in parkin's degradation. This decrease in parkin allows for stabilization of MFN1/2 and increased mitochondrial hyperfusion, leading to cell cycle arrest through increased p21 and decreased cyclin E expression, ultimately culminating in inhibition of cellular proliferation and tumor growth.
The conventional role of myoglobin is the storage of oxygen and facilitated delivery to mitochondria in hypoxic conditions. However, the very low micromolar concentration at which myoglobin is estimated to be endogenously expressed in cancer cells seems to preclude this function (20). Consistent with this, Kristiansen et al. (1) confirmed through respirometry studies that myoglobin does not play a significant oxygen buffering role in breast cancer cells and may have other tumor suppressor functions. Myoglobin has been shown to bind fatty acids, and a role for myoglobin-dependent fatty acid transport has been postulated due to the colocalization of myoglobin and fatty acid synthase in cancer cells as well as studies in which inhibition of fatty acid synthase decreases myoglobin expression in these cells (20). The data presented in the current study suggest an additional novel tumor suppressor function by which myoglobin (through ROS production) increases mitochondrial fusion to inhibit cell proliferation. Importantly, our studies demonstrate that this pathway is operant even with low (<1 μm) concentrations of myoglobin expression. Notably, both myoglobin expression and mitochondrial fusion have been associated with increased fatty acid oxidation in other cell types, potentially linking our findings to prior studies (29–32). Although we did not investigate fatty acid metabolism in this study, it is interesting to speculate that myoglobin-dependent mitochondrial fusion may not only regulate cellular proliferation but be part of an adaptive response to increased fatty acid synthesis that results in both improved transport and metabolism.
Our data confirm that the heme moiety within myoglobin is required for its antiproliferative effects in vitro as expression of the apomyoglobin protein does not modulate cell proliferation. Additionally, apo-Mb expression does not increase cellular oxidant production, unlike WT myoglobin. This is consistent with the catalytic role of the heme iron in the pseudoperoxidase activity of myoglobin that leads to lipid peroxidation and oxidant production (33), which potentially leads to the oxidation of parkin.
Beyond the generation of oxidants, the reduced heme in myoglobin can interact with reactive nitrogen species generated in the cancer cell. NO has been shown to potentiate tumor cell proliferation as well as cancer stem cell conversion in several studies (34, 35). Rapid oxidation of NO to nitrate by the heme center of myoglobin may prevent these detrimental effects. Additionally, the reduced heme iron of myoglobin also functions as a nitrite reductase in deoxygenated conditions, which can result in production of free NO or S-nitrosation of critical cysteine residues on proteins (36, 37). Recent studies show that S-nitrosation of parkin in dopaminergic neurons inhibited its E3 ligase activity, similar to oxidation. Thus, it is possible that in more hypoxic, nitrite-rich areas of a tumor, myoglobin shifts from an oxidant generator to a nitrite reductase, and both these functions result in inhibition of parkin activity, which increases expression of MFN1 and -2.
Although myoglobin is known to regulate mitochondrial function and expression of mitochondrial proteins such as cytochrome c oxidase (38, 39), this is to our knowledge the first demonstration of myoglobin-dependent regulation of mitochondrial dynamics. We show that myoglobin expression promotes mitochondrial fusion through the expression of mitofusins. In cancer, emphasis has been placed on the role of mitochondrial dynamics in regulating cell proliferation. Westrate et al. (40) showed that cells in cell cycle arrest have hyperfused mitochondria, whereas Mitra et al. (16) demonstrated that mitochondrial fragmentation occurs during mitotic progression. Moreover, several studies show that mitofusins are down-regulated in cancer cells and when overexpressed can have a tumor suppressor function (15, 41–43). Our findings are consistent with the antiproliferative effects of mitofusins in cancer cells. Notably, we saw no difference between β-gal production between myoglobin-expressing and WT cells (data not shown). Furthermore, silencing of myoglobin in the nonproliferative myoglobin-expressing cells reinstated proliferation. These data are consistent with a myoglobin-dependent quiescent state rather than irreversible senescence. However, more work must be done to truly understand the permanence of cell cycle arrest.
Our results have implications beyond modulation of proliferation. Mitochondrial fusion is tightly linked with bioenergetic function and redox signaling as well as mitochondrial quality control. For example, mitochondrial fusion has been shown to shift metabolism in some cancer cell types from glycolysis to increased oxidative phosphorylation (44, 45). Therefore, additional studies are needed to determine whether myoglobin-dependent stimulation of mitochondrial fusion may result in greater respiratory efficiency (46) or attenuates the mitophagic pathway (47). Investigation of these pathways is potentially important in cancer cells as well as skeletal and cardiac muscle cells, which express micromolar levels of myoglobin.
A number of clinical and preclinical trials testing the efficacy of antioxidants, such as β-carotene, in attenuating tumor progression have failed (48, 49). Moreover, oxidative stress has been shown to have beneficial effects in some cancer models, including in the inhibition of metastasis (50). Our data further support a protective role for oxidants as we demonstrate that antioxidant treatment propagates cancer cell proliferation in myoglobin-expressing cells. Further study is required to determine whether myoglobin-dependent oxidant generation modulates tumor cell metastasis in addition to proliferation.
Our data demonstrate that expression of myoglobin in tumors attenuates tumor volume in a murine xenograft model. We also show up-regulation of myoglobin and mitochondrial fusion proteins in triple-negative breast cancer as proof of concept that our mechanistic findings may hold true in human models where myoglobin expression is known to correlate with improved outcome. This work highlights the necessity for additional studies to examine the temporal expression of myoglobin across distinct breast cancer subtypes. Quantification of the degree of myoglobin and MFN1 expression across a spectra of tumor samples and sizes would provide additional insight to the intratumoral effects of myoglobin-induced mitochondrial fusion, and further studies are warranted. Additional studies are also required to explore whether the heme moiety of myoglobin or parkin could be viable therapeutic targets for the treatment of TNBC.
In summary, we have described a novel pathway by which myoglobin stimulates mitochondrial fusion. We show that myoglobin-stimulated fusion leads to cell cycle arrest and attenuation of breast cancer cell proliferation. These data expand myoglobin's role from a modulator of bioenergetic function to a regulator of mitochondrial dynamics and structure. Furthermore, this study advances the understanding of the protective effect of myoglobin in breast cancer.
Experimental procedures
All reagents were purchased from Sigma-Aldrich unless otherwise noted. MDA-MB-231 (catalog number HTB-26), MDA-MB-468 (catalog number HTB-132), and MCF7 (catalog number HTB-22) cells were purchased from ATCC.
MDA-MB-231, myoglobin-expressing MDA-MB-231, and MDA-MB-468 cells were cultured in Dulbecco's modified Eagle's medium with 4.5 g/liter glucose (catalog number 11965-118, Invitrogen) supplemented with 10% fetal bovine serum (catalog number 10438026, Thermo Fisher Scientific), 1% penicillin-streptomycin (catalog number 17-602E, Lonza, Basel, Switzerland), and 1% Geneticin (catalog number 10131035, Thermo Fisher Scientific). MCF7 and myoglobin-expressing MCF7 cells were cultured in Dulbecco's modified Eagle's medium with 1 g/liter glucose (Invitrogen) supplemented with 10% fetal bovine serum, 1% penicillin-streptomycin, 1% insulin (catalog number I0516, Sigma-Aldrich), and 1% Geneticin. Cells were stored in an incubator maintained at 37 °C, 21% O2, and 5% CO2.
GFP-tagged human Mb was purchased from Origene (catalog number RG212352). Bacterial transformation was performed using XL1-Blue competent cells (catalog number 200249, Agilent) following the manufacturer's instructions. 200 μl of the transformed cells was spread on agar plates containing 100 μg/μl ampicillin and incubated overnight at 37 °C. Viable colonies were chosen, and glycerol stocks were prepared. DNA was purified using a Qiagen plasmid kit (catalog number 12123; Venlo, Netherlands). Cells were transfected with Mb DNA using Lipofectamine 3000 (catalog number L3000008, Life Technologies). Approximately 48 h after transfection, cells were passaged in media containing geneticin to select only for cells resistant to the antibiotic. Single cells from resistant colonies were transferred to 96-well plates to confirm they were able to yield antibiotic-resistant colonies, which were then expanded.
Site-directed mutagenesis was performed on the myoglobin-GFP plasmid where histidine 94 was mutated to phenylalanine using the QuikChange XL Site-Directed Mutagenesis kit (catalog number 200516, Agilent) with custom forward primer 5′-ttgtgcttggtggcaaacgactgtgccagggg-3′ and reverse primer 5′-cccctggcacagtcgtttgccaccaagcacaa-3′ (Integrated DNA Technologies). Apo-Mb plasmid amplification was confirmed on a 1% agarose gel, and bacterial transformation was performed as detailed above. Sequence was confirmed by GENEWIZ. Cells were transfected with apo-Mb DNA, and colonies were selected as detailed above.
Ten 4–5-week-old female NOD.CB17-Prkdcscid/J mice were purchased from The Jackson Laboratory (catalog number 001303). The animal studies were approved by the University of Pittsburgh Institutional Animal Care and Use Committee (protocol number 12030315B-7). Following a 3-day acclimation period, mice were placed on irradiated AIN-76A diet for the duration of the study (catalog number CA.170481, Harlan-Teklad). After 1 week on the diet, both inguinal mammary fat pads of the mice were orthotopically injected with 3 × 106 of either control MDA-MB-231 or myoglobin-expressing MDA-MB-231 cells, which were previously cultured to ∼40–60% confluence in culture dishes. Dishes were washed with PBS, trypsinized, centrifuged at 1100 rpm for 5 min, and diluted to 108 cells/ml of serum-free media before injection into the mice. Tumor growth was measured using Vernier calipers and recorded each week beginning when tumors became measurable. Tumor volume was determined using the following equation.
| (Eq. 1) |
The study was terminated when the average tumor volume in the control group approached 1.5 cm3. Tumors were harvested from each animal, and weights, length, and width were recorded.
For [3H]thymidine incorporation, cells were plated in a 6-well plate and incubated with media containing [3H]thymidine (5 μCi/ml) for 24 h. To determine the amount of [3H]thymidine incorporation, cells were washed with 1 ml of PBS and incubated in 5% TCA at 4 °C for 30 min. Cells were washed once with 5% TCA and PBS and incubated in 0.2 n NaOH at room temperature for 30 min to precipitate DNA. Samples were added to scintillation tubes containing 4 ml of scintillation fluid (catalog number 6013326, PerkinElmer Life Sciences) and read in a scintillation counter.
For crystal violet staining, cells were grown in a 96-well plate for the required time. Media were aspirated, 0.25% crystal violet was added to cover the bottom of the plate, and the plate was put on a shaker for 30 min. The plate was rinsed twice in water and allowed to dry overnight to remove crystal violet. Crystal violet was dissolved with 1% SDS. Absorbance was measured in a Synergy plate reader (BioTek Instrument Inc.), which is correlated to DNA content.
For immunofluorescence measurement of fusion, cells were stained with rabbit anti-TOM20 (catalog number sc-11415, Santa Cruz Biotechnology) diluted 1:1000 in 50% LI-COR® Biosciences PBS solution overnight at 4 °C and imaged with a confocal microscope using a 60× objective and 3× optical zoom at the University of Pittsburgh Center for Biologic Imaging as described previously (51). Bright field images were taken with a Zeiss Axiovert 40 CFL microscope and analyzed with AxioVision software Rel. 4.8 (Carl Zeiss Microscopy).
Western blotting was performed as described previously in lysed cells or homogenized and lysed tissue (52). Antibodies used included MFN1 (catalog number sc-50330, Santa Cruz Biotechnology), DRP1 (catalog number sc-32898, Santa Cruz Biotechnology), parkin (catalog number 2132, Cell Signaling Technology), p21 (catalog number 556430, BD Pharmingen), and cyclin E (catalog number sc-481, Santa Cruz Biotechnology). After blots were treated with the appropriate fluorophore-conjugated secondary antibody, blots were imaged with a LI-COR Biosciences Odyssey imaging system and analyzed using LI-COR Biosciences Odyssey IR imaging software version 3.0. To normalize protein concentration to total protein, blots were reprobed with antibodies against either α-tubulin (catalog number CP06, Calbiochem) or β-actin (catalog number A5316, Sigma-Aldrich).
For transmission electron microscopy, cells and tissues were imaged after fixation with 2.5% glutaraldehyde to assess mitochondrial fission and fusion as described previously (53). Once fixed and embedded, all samples were imaged using a JEOL JEM 1011 transmission electron microscope (JEOL Ltd., Tokyo, Japan).
For flow cytometry analysis of cell cycle arrest, cells were plated and allowed to grow to 90% confluence. Dishes were washed with PBS, trypsinized, and centrifuged at 1200 rpm for 5 min, and flow tubes were prepared with a concentration of 1 × 106 cells/ml. Cells were permeabilized with ice-cold 70% ethanol for 30 min at 4 °C and then washed twice with 1× PBS. Cells were treated with 100 μg/ml RNase (catalog number EN0531, Thermo Fisher Scientific) for 5 min, then stained with propidium iodide (catalog number P4170, Sigma-Aldrich) for a 50 μg/ml final stain concentration, and incubated at 37 °C for 30 min protected from light. Samples were analyzed at the analytical cytometry core facility on our campus using the BD LSRFortessaTM with excitation/emission 496/578 nm and flow cytometer laser line at 405 nm. Data were processed using FlowJo acquisition and analysis software.
For siRNA transfections, cells were plated and allowed to grow to 70% confluence. Cells were washed with PBS and media was changed to Opti-MEM® reduced serum medium (#31985070, Thermo Fischer Scientific). Cells were transfected with human myoglobin (#sc-35995, Santa Cruz Biotechnology), MFN1 (#sc-43927, Santa Cruz Biotechnology), or MFN2 (#sc-43928, Santa Cruz Biotechnology) targeted siRNA using Santa Cruz siRNA Transfection Reagent (#sc-29528, Santa Cruz Biotechnology) according to the manufacturer's instructions.
To determine Oxidative Stress, cells were plated and allowed to grow to 70% confluence. Dishes were washed with PBS, trypsinized, centrifuged at 1100 RPM for 5 min and flow tubes were prepared with each containing a concentration of 1 × 106 cells/ml. CellROX® Deep Red Reagent (#C10422, Thermo Fisher Scientific) was added for a final of 2 μm stain concentration, and incubated at 37 °C for 30 min protected from light. Samples were washed three times with PBS, and analyzed at the analytical cytometry core facility on our campus using the BD LSRFortessaTM with excitation/emission 644/665 nm. Data were processed using FlowJo acquisition and analysis software.
A Protein Carbonyl Colormetric Assay kit (catalog number 10005020, Cayman Chemical) was used according to manufacturer's instructions for the carbonyl assay. To determine antioxidant effects on the cells, 5000 cells/well were grown in a 96-well plate for 24 h. Media were aspirated and replaced with media ± 10 μm TEMPOL (catalog number 581500, Sigma-Aldrich), and cells were grown for the required time before crystal violet staining was performed as described above.
For protein oxidation detection using a modified biotin switch assay, cells were plated and allowed to grow to 90% confluence. Cells were washed twice with PBS and lysed with 100 μl of lysis buffer (pH 7.4; PBS, 1% SDS, 0.5% Triton X-100, and 0.1 mm neocuprione) for 30 min on ice, vortexing every few minutes. Lysates were centrifuged for 10 min at 16,000 × g, and protein concentrations of the supernatants were measured using a BCA assay. Three aliquots of 250 μg of protein (one for sample, one for positive control, and one for negative control) were made. An equal volume of blocking reagent with N-ethylmaleimide (NEM) (20 mm NEM, 5% SDS, 20 mm Tris, pH 7.4, 1 mm EDTA, and 0.2 nm neocuprione) was added to the sample and negative control, and blocking reagent without NEM (5% SDS, 20 mm Tris, pH 7.4, 1 mm EDTA, and 0.2 nm neocuprione) was added to the positive control and incubated in the dark for 30 min at 50 °C, vortexing every 10 min. 20 μl of labeling reagent (4 mm HPDP-Biotin stock dissolved in DMSO) and 4 μl of 10 mm tris(2-carboxyethyl)phosphine (in Tris buffer, pH 7.4) were added to the sample and positive control and 20 μl of labeling reagent was added to the negative control and incubated at 40 °C for 2 h. Proteins were analyzed by Western blotting.
The breast adjacent normal tissue array (catalog number BRN801a) and cancer tissue array (catalog number BR1201) were purchased from US Biomax Inc. Arrays were deparaffinized and hydrated as follows: 10 min in xylenes, 10 min in 100% ethanol, 10 min in 95% ethanol, 10 min in 70% ethanol, and 10 min in in water. The arrays were immersed in 10 mm citric acid buffer, pH 6.0, and heated at full power in a microwave until boiling. Upon boiling, the microwave was set at 30% power, and the arrays were boiled for 6 min. The arrays were cooled to room temperature for 10 min and washed in water. Arrays were blocked with 20% donkey serum (catalog number 566460-5ML, Millipore) in PBS + 0.5% BSA (PBB) for 45 min and washed five times with PBB. All the primary antibodies (rabbit anti-MFN1, mouse anti-parkin, and goat anti-myoglobin) were diluted to 10 μg/ml and incubated on arrays overnight at 4 °C. The next day the arrays were washed five times with PBB followed by incubation with the secondary antibodies (donkey anti-rabbit Alexa Fluor 488 (catalog number A-21202, Invitrogen), donkey anti-mouse Cy5 (catalog number 715-605-150, Jackson ImmunoResearch Laboratories), and donkey anti-goat Cy3 (catalog number 705-165-174, Jackson ImmunoResearch Laboratories) for 1 h. Arrays were washed five times with PBB followed by five washes with PBS. Arrays were incubated with Hoechst stain for 1 min (catalog number B-2883, Sigma) and washed three times with PBS. Arrays were mounted with Gelvatol and imaged on a Nikon A1 confocal microscope using a 60× objective at the University of Pittsburgh Center for Biologic Imaging as described previously (51).
All values are expresses at mean ± S.E. Single comparisons were tested for significance by a two-tailed Student's t test. Analysis of variance was used when comparing multiple groups with a Bonferroni post hoc analysis to correct for multiple comparisons. Differences were considered significant when p values were <0.05.
Author contributions
A. B., K. Q., J. B., C. R., Y. W., M. J., C. S. C., J. S., and S. V. S. data curation; A. B., K. Q., J. B., C. R., Y. W., C. S. C., and J. S. formal analysis; A. B., K. Q., and S. V. S. investigation; A. B., Y. W., C. S. C., S. V. S., and S. S. methodology; A. B., K. Q., and S. S. writing-original draft; Y. W. and S. S. conceptualization; S. V. S. and S. S. resources; S. V. S. and S. S. writing-review and editing; S. S. supervision; S. S. project administration.
Supplementary Material
Acknowledgment
We thank Su Kim for help with determining the appropriate tissue arrays for this study.
This work was supported by National Institutes of Health Grants R01 HL133003-01A1 and 1R01GM113816-01 (to S. S.) and T32 HL110849 Training in Translational Research and Entrepreneurship in Pulmonary Vascular Biology (to A. B.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Fig. S1.
- MFN
- mitofusin
- DRP1
- dynamin-related protein 1
- TNBC
- triple-negative breast cancer
- ROS
- reactive oxygen species
- Mb
- myoglobin
- TEMPOL
- 1-oxyl-2,2,6,6-tetramethyl-4-hydroxypiperidine
- NEM
- N-ethylmaleimide.
References
- 1. Kristiansen G., Hu J., Wichmann D., Stiehl D. P., Rose M., Gerhardt J., Bohnert A., ten Haaf A., Moch H., Raleigh J., Varia M. A., Subarsky P., Scandurra F. M., Gnaiger E., Gleixner E., et al. (2011) Endogenous myoglobin in breast cancer is hypoxia-inducible by alternative transcription and functions to impair mitochondrial activity: a role in tumor suppression? J. Biol. Chem. 286, 43417–43428 10.1074/jbc.M111.227553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Qiu Y., Sutton L., and Riggs A. F. (1998) Identification of myoglobin in human smooth muscle. J. Biol. Chem. 273, 23426–23432 10.1074/jbc.273.36.23426 [DOI] [PubMed] [Google Scholar]
- 3. Wittenberg B. A., and Wittenberg J. B. (1987) Myoglobin-mediated oxygen delivery to mitochondria of isolated cardiac myocytes. Proc. Natl. Acad. Sci. U.S.A. 84, 7503–7507 10.1073/pnas.84.21.7503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wittenberg B. A., Wittenberg J. B., and Caldwell P. R. (1975) Role of myoglobin in the oxygen supply to red skeletal muscle. J. Biol. Chem. 250, 9038–9043 [PubMed] [Google Scholar]
- 5. Rayner B. S., Hua S., Sabaretnam T., and Witting P. K. (2009) Nitric oxide stimulates myoglobin gene and protein expression in vascular smooth muscle. Biochem. J. 423, 169–177 10.1042/BJ20090716 [DOI] [PubMed] [Google Scholar]
- 6. Totzeck M., Hendgen-Cotta U. B., Luedike P., Berenbrink M., Klare J. P., Steinhoff H. J., Semmler D., Shiva S., Williams D., Kipar A., Gladwin M. T., Schrader J., Kelm M., Cossins A. R., and Rassaf T. (2012) Nitrite regulates hypoxic vasodilation via myoglobin-dependent nitric oxide generation. Circulation 126, 325–334 10.1161/CIRCULATIONAHA.111.087155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kamga C., Krishnamurthy S., and Shiva S. (2012) Myoglobin and mitochondria: a relationship bound by oxygen and nitric oxide. Nitric Oxide 26, 251–258 10.1016/j.niox.2012.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Higdon A. N., Benavides G. A., Chacko B. K., Ouyang X., Johnson M. S., Landar A., Zhang J., and Darley-Usmar V. M. (2012) Hemin causes mitochondrial dysfunction in endothelial cells through promoting lipid peroxidation: the protective role of autophagy. Am. J. Physiol. Heart Circ. Physiol. 302, H1394–H1409 10.1152/ajpheart.00584.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cole R. P., Sukanek P. C., Wittenberg J. B., and Wittenberg B. A. (1982) Mitochondrial function in the presence of myoglobin. J. Appl. Physiol. Respir. Environ. Exerc. Physiol. 53, 1116–1124 [DOI] [PubMed] [Google Scholar]
- 10. Wittenberg J. B., and Wittenberg B. A. (2007) Myoglobin-enhanced oxygen delivery to isolated cardiac mitochondria. J. Exp. Biol. 210, 2082–2090 10.1242/jeb.003947 [DOI] [PubMed] [Google Scholar]
- 11. Jue T., Simond G., Wright T. J., Shih L., Chung Y., Sriram R., Kreutzer U., and Davis R. W. (2016) Effect of fatty acid interaction on myoglobin oxygen affinity and triglyceride metabolism. J. Physiol. Biochem. 73, 359–370 10.1007/s13105-017-0559-z [DOI] [PubMed] [Google Scholar]
- 12. Sriram R., Kreutzer U., Shih L., and Jue T. (2008) Interaction of fatty acid with myoglobin. FEBS Lett. 582, 3643–3649 10.1016/j.febslet.2008.09.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li D., Li X., Guan Y., and Guo X. (2015) Mitofusin-2-mediated tethering of mitochondria and endoplasmic reticulum promotes cell cycle arrest of vascular smooth muscle cells in G0/G1 phase. Acta Biochim. Biophys. Sin 47, 441–450 10.1093/abbs/gmv035 [DOI] [PubMed] [Google Scholar]
- 14. Taguchi N., Ishihara N., Jofuku A., Oka T., and Mihara K. (2007) Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J. Biol. Chem. 282, 11521–11529 10.1074/jbc.M607279200 [DOI] [PubMed] [Google Scholar]
- 15. Lou Y., Li R., Liu J., Zhang Y., Zhang X., Jin B., Liu Y., Wang Z., Zhong H., Wen S., and Han B. (2015) Mitofusin-2 over-expresses and leads to dysregulation of cell cycle and cell invasion in lung adenocarcinoma. Med. Oncol. 32, 132 10.1007/s12032-015-0515-0 [DOI] [PubMed] [Google Scholar]
- 16. Mitra K., Wunder C., Roysam B., Lin G., and Lippincott-Schwartz J. (2009) A hyperfused mitochondrial state achieved at G1-S regulates cyclin E buildup and entry into S phase. Proc. Natl. Acad. Sci. U.S.A. 106, 11960–11965 10.1073/pnas.0904875106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Xia Y., Wu Y., He X., Gong J., and Qiu F. (2008) Effects of mitofusin-2 gene on cell proliferation and chemotherapy sensitivity of MCF-7. J. Huazhong Univ. Sci. Technolog. Med. Sci. 28, 185–189 10.1007/s11596-008-0218-2 [DOI] [PubMed] [Google Scholar]
- 18. Flonta S. E., Arena S., Pisacane A., Michieli P., and Bardelli A. (2009) Expression and functional regulation of myoglobin in epithelial cancers. Am. J. Pathol. 175, 201–206 10.2353/ajpath.2009.081124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bicker A., Brahmer A. M., Meller S., Kristiansen G., Gorr T. A., and Hankeln T. (2015) The distinct gene regulatory network of myoglobin in prostate and breast cancer. PLoS One 10, e0142662 10.1371/journal.pone.0142662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kristiansen G., Rose M., Geisler C., Fritzsche F. R., Gerhardt J., Lüke C., Ladhoff A. M., Knüchel R., Dietel M., Moch H., Varga Z., Theurillat J. P., Gorr T. A., and Dahl E. (2010) Endogenous myoglobin in human breast cancer is a hallmark of luminal cancer phenotype. Br. J. Cancer 102, 1736–1745 10.1038/sj.bjc.6605702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Oleksiewicz U., Daskoulidou N., Liloglou T., Tasopoulou K., Bryan J., Gosney J. R., Field J. K., and Xinarianos G. (2011) Neuroglobin and myoglobin in non-small cell lung cancer: expression, regulation and prognosis. Lung Cancer 74, 411–418 10.1016/j.lungcan.2011.05.001 [DOI] [PubMed] [Google Scholar]
- 22. Meller S., Bicker A., Montani M., Ikenberg K., Rostamzadeh B., Sailer V., Wild P., Dietrich D., Uhl B., Sulser T., Moch H., Gorr T. A., Stephan C., Jung K., Hankeln T., et al. (2014) Myoglobin expression in prostate cancer is correlated to androgen receptor expression and markers of tumor hypoxia. Virchows Arch. 465, 419–427 10.1007/s00428-014-1646-y [DOI] [PubMed] [Google Scholar]
- 23. Metzger M. B., Pruneda J. N., Klevit R. E., and Weissman A. M. (2014) RING-type E3 ligases: master manipulators of E2 ubiquitin-conjugating enzymes and ubiquitination. Biochim. Biophys. Acta 1843, 47–60 10.1016/j.bbamcr.2013.05.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lim K. L., Ng X. H., Grace L. G., and Yao T. P. (2012) Mitochondrial dynamics and Parkinson's disease: focus on parkin. Antioxid. Redox Signal. 16, 935–949 10.1089/ars.2011.4105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Meng F., Yao D., Shi Y., Kabakoff J., Wu W., Reicher J., Ma Y., Moosmann B., Masliah E., Lipton S. A., and Gu Z. (2011) Oxidation of the cysteine-rich regions of parkin perturbs its E3 ligase activity and contributes to protein aggregation. Mol. Neurodegener. 6, 34 10.1186/1750-1326-6-34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jourd'heuil D., Mills L., Miles A. M., and Grisham M. B. (1998) Effect of nitric oxide on hemoprotein-catalyzed oxidative reactions. Nitric Oxide 2, 37–44 10.1006/niox.1998.0167 [DOI] [PubMed] [Google Scholar]
- 27. Richards M. P. (2013) Redox reactions of myoglobin. Antioxid. Redox Signal. 18, 2342–2351 10.1089/ars.2012.4887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Reeder B. J., Svistunenko D. A., Cooper C. E., and Wilson M. T. (2004) The radical and redox chemistry of myoglobin and hemoglobin: from in vitro studies to human pathology. Antioxid. Redox Signal. 6, 954–966 10.1089/ars.2004.6.954 [DOI] [PubMed] [Google Scholar]
- 29. Buck M. D., O'Sullivan D., Klein Geltink R. I., Curtis J. D., Chang C. H., Sanin D. E., Qiu J., Kretz O., Braas D., van der Windt G. J., Chen Q., Huang S. C., O'Neill C. M., Edelson B. T., Pearce E. J., et al. (2016) Mitochondrial dynamics controls T cell fate through metabolic programming. Cell 166, 63–76 10.1016/j.cell.2016.05.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hendgen-Cotta U. B., Esfeld S., Coman C., Ahrends R., Klein-Hitpass L., Flögel U., Rassaf T., and Totzeck M. (2017) A novel physiological role for cardiac myoglobin in lipid metabolism. Sci. Rep. 7, 43219 10.1038/srep43219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rambold A. S., Cohen S., and Lippincott-Schwartz J. (2015) Fatty acid trafficking in starved cells: regulation by lipid droplet lipolysis, autophagy, and mitochondrial fusion dynamics. Dev. Cell 32, 678–692 10.1016/j.devcel.2015.01.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rao S. I., Wilks A., Hamberg M., and Ortiz de Montellano P. R. (1994) The lipoxygenase activity of myoglobin. Oxidation of linoleic acid by the ferryl oxygen rather than protein radical. J. Biol. Chem. 269, 7210–7216 [PubMed] [Google Scholar]
- 33. Newman E. S., Rice-Evans C. A., and Davies M. J. (1991) Identification of initiating agents in myoglobin-induced lipid peroxidation. Biochem. Biophys. Res. Commun. 179, 1414–1419 10.1016/0006-291X(91)91730-Z [DOI] [PubMed] [Google Scholar]
- 34. Kim J. K., Jeon H. M., Jeon H. Y., Oh S. Y., Kim E. J., Jin X., Kim S. H., Kim S. H., Jin X., and Kim H. (2018) Conversion of glioma cells to glioma stem-like cells by angiocrine factors. Biochem. Biophys. Res. Commun. 496, 1013–1018 10.1016/j.bbrc.2017.02.076 [DOI] [PubMed] [Google Scholar]
- 35. Salimian Rizi B., Caneba C., Nowicka A., Nabiyar A. W., Liu X., Chen K., Klopp A., and Nagrath D. (2015) Nitric oxide mediates metabolic coupling of omentum-derived adipose stroma to ovarian and endometrial cancer cells. Cancer Res. 75, 456–471 10.1158/0008-5472.CAN-14-1337 [DOI] [PubMed] [Google Scholar]
- 36. Hendgen-Cotta U. B., Merx M. W., Shiva S., Schmitz J., Becher S., Klare J. P., Steinhoff H. J., Goedecke A., Schrader J., Gladwin M. T., Kelm M., and Rassaf T. (2008) Nitrite reductase activity of myoglobin regulates respiration and cellular viability in myocardial ischemia-reperfusion injury. Proc. Natl. Acad. Sci. U.S.A. 105, 10256–10261 10.1073/pnas.0801336105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shiva S., Sack M. N., Greer J. J., Duranski M., Ringwood L. A., Burwell L., Wang X., MacArthur P. H., Shoja A., Raghavachari N., Calvert J. W., Brookes P. S., Lefer D. J., and Gladwin M. T. (2007) Nitrite augments tolerance to ischemia/reperfusion injury via the modulation of mitochondrial electron transfer. J. Exp. Med. 204, 2089–2102 10.1084/jem.20070198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yamada T., Furuichi Y., Takakura H., Hashimoto T., Hanai Y., Jue T., and Masuda K. (2013) Interaction between myoglobin and mitochondria in rat skeletal muscle. J. Appl. Physiol. 114, 490–497 10.1152/japplphysiol.00789.2012 [DOI] [PubMed] [Google Scholar]
- 39. Yamada T., Takakura H., Jue T., Hashimoto T., Ishizawa R., Furuichi Y., Kato Y., Iwanaka N., and Masuda K. (2016) Myoglobin and the regulation of mitochondrial respiratory chain complex IV. J. Physiol. 594, 483–495 10.1113/JP270824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Westrate L. M., Sayfie A. D., Burgenske D. M., and MacKeigan J. P. (2014) Persistent mitochondrial hyperfusion promotes G2/M accumulation and caspase-dependent cell death. PLoS One 9, e91911 10.1371/journal.pone.0091911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ma L. I., Chang Y., Yu L., He W., and Liu Y. (2015) Pro-apoptotic and anti-proliferative effects of mitofusin-2 via PI3K/Akt signaling in breast cancer cells. Oncol. Lett. 10, 3816–3822 10.3892/ol.2015.3748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhang G. E., Jin H. L., Lin X. K., Chen C., Liu X. S., Zhang Q., and Yu J. R. (2013) Anti-tumor effects of Mfn2 in gastric cancer. Int. J. Mol. Sci. 14, 13005–13021 10.3390/ijms140713005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Xu K., Chen G., Li X., Wu X., Chang Z., Xu J., Zhu Y., Yin P., Liang X., and Dong L. (2017) MFN2 suppresses cancer progression through inhibition of mTORC2/Akt signaling. Sci. Rep. 7, 41718 10.1038/srep41718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mourier A., Motori E., Brandt T., Lagouge M., Atanassov I., Galinier A., Rappl G., Brodesser S., Hultenby K., Dieterich C., and Larsson N. G. (2015) Mitofusin 2 is required to maintain mitochondrial coenzyme Q levels. J. Cell Biol. 208, 429–442 10.1083/jcb.201411100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Fang D., Yan S., Yu Q., Chen D., and Yan S. S. (2016) Mfn2 is required for mitochondrial development and synapse formation in human induced pluripotent stem cells/hiPSC derived cortical neurons. Sci. Rep. 6, 31462 10.1038/srep31462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Boutant M., Kulkarni S. S., Joffraud M., Ratajczak J., Valera-Alberni M., Combe R., Zorzano A., and Cantó C. (2017) Mfn2 is critical for brown adipose tissue thermogenic function. EMBO J. 36, 1543–1558 10.15252/embj.201694914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Song M., Mihara K., Chen Y., Scorrano L., and Dorn G. W. 2nd. (2015) Mitochondrial fission and fusion factors reciprocally orchestrate mitophagic culling in mouse hearts and cultured fibroblasts. Cell Metab. 21, 273–286 10.1016/j.cmet.2014.12.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Touvier M., Kesse E., Clavel-Chapelon F., and Boutron-Ruault M. C. (2005) Dual association of β-carotene with risk of tobacco-related cancers in a cohort of French women. J. Natl. Cancer Inst. 97, 1338–1344 10.1093/jnci/dji276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Le Gal K., Ibrahim M. X., Wiel C., Sayin V. I., Akula M. K., Karlsson C., Dalin M. G., Akyürek L. M., Lindahl P., Nilsson J., and Bergo M. O. (2015) Antioxidants can increase melanoma metastasis in mice. Sci. Transl. Med. 7, 308re8 10.1126/scitranslmed.aad3740 [DOI] [PubMed] [Google Scholar]
- 50. Piskounova E., Agathocleous M., Murphy M. M., Hu Z., Huddlestun S. E., Zhao Z., Leitch A. M., Johnson T. M., DeBerardinis R. J., and Morrison S. J. (2015) Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 527, 186–191 10.1038/nature15726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kamga Pride C., Mo L., Quesnelle K., Dagda R. K., Murillo D., Geary L., Corey C., Portella R., Zharikov S., St Croix C., Maniar S., Chu C. T., Khoo N. K., and Shiva S. (2014) Nitrite activates protein kinase A in normoxia to mediate mitochondrial fusion and tolerance to ischaemia/reperfusion. Cardiovasc. Res. 101, 57–68 10.1093/cvr/cvt224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Khoo N. K., Rudolph V., Cole M. P., Golin-Bisello F., Schopfer F. J., Woodcock S. R., Batthyany C., and Freeman B. A. (2010) Activation of vascular endothelial nitric oxide synthase and heme oxygenase-1 expression by electrophilic nitro-fatty acids. Free Radic. Biol. Med. 48, 230–239 10.1016/j.freeradbiomed.2009.10.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Stolz D. B., Ross M. A., Salem H. M., Mars W. M., Michalopoulos G. K., and Enomoto K. (1999) Cationic colloidal silica membrane perturbation as a means of examining changes at the sinusoidal surface during liver regeneration. Am. J. Pathol. 155, 1487–1498 10.1016/S0002-9440(10)65464-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.