

The Tol-Pal complex plays a poorly defined role in envelope biogenesis. The data presented here show that DegP’s periplasmic protease activity becomes crucial in mutants lacking the intact Tol-Pal complex, but this requirement can be circumvented by suppressor mutations that activate the basal protease activity of a regulatory protease, DegS. These observations point to a critical role for periplasmic proteases when Tol-Pal-mediated envelope structure and/or functions are perturbed.
KEYWORDS: bacterial envelope, DegP protease, DegS, envelope stress, sigma E, Tol-Pal complex
ABSTRACT
In Escherichia coli, the periplasmic protease DegP plays a critical role in degrading misfolded outer membrane proteins (OMPs). Consequently, mutants lacking DegP display a temperature-sensitive growth defect, presumably due to the toxic accumulation of misfolded OMPs. The Tol-Pal complex plays a poorly defined but an important role in envelope biogenesis, since mutants defective in this complex display a classical periplasmic leakage phenotype. Double mutants lacking DegP and an intact Tol-Pal complex display exaggerated temperature-sensitive growth defects and the leaky phenotype. Two revertants that overcome the temperature-sensitive growth phenotype carry missense mutations in the degS gene, resulting in D102V and D320A substitutions. D320 and E317 of the PDZ domain of DegS make salt bridges with R178 of DegS’s protease domain to keep the protease in the inactive state. However, weakening of the tripartite interactions by D320A increases DegS’s basal protease activity. Although the D102V substitution is as effective as D320A in suppressing the temperature-sensitive growth phenotype, the molecular mechanism behind its effect on DegS’s protease activity is unclear. Our data suggest that the two DegS variants modestly activate RseA-controlled, σE-mediated envelope stress response pathway and elevate periplasmic protease activity to restore envelope homeostasis. Based on the release of a cytoplasmic enzyme in the culture supernatant, we conclude that the conditional lethal phenotype of ΔtolB ΔdegP mutants stems from a grossly destabilized envelope structure that causes excessive cell lysis. Together, the data point to a critical role for periplasmic proteases when the Tol-Pal complex-mediated envelope structure and/or functions are compromised.
IMPORTANCE The Tol-Pal complex plays a poorly defined role in envelope biogenesis. The data presented here show that DegP’s periplasmic protease activity becomes crucial in mutants lacking the intact Tol-Pal complex, but this requirement can be circumvented by suppressor mutations that activate the basal protease activity of a regulatory protease, DegS. These observations point to a critical role for periplasmic proteases when Tol-Pal-mediated envelope structure and/or functions are perturbed.
INTRODUCTION
Outer membrane (OM) biogenesis has been an active topic of investigation due to the important roles OM plays in the physiology and survival of Gram-negative bacteria (1, 2). The OM is populated with two main classes of proteins: β-barrels and lipoproteins. Whereas most β-barrel proteins form channels for nutrient uptake, lipoproteins participate in membrane biogenesis and structural integrity. Assembly of β-barrel proteins is mediated by a multiprotein complex called the β-barrel assembly machine (BAM [3–6]), while lipoproteins are targeted and assembled by the localization of lipoprotein complex (Lol) (7, 8). Interestingly, the BAM complex is composed of four lipoproteins, of which BamD is essential (9), and a single essential β-barrel protein, BamA (10–12).
Although BAM is critical for β-barrel outer membrane protein (OMP) assembly, it is not sufficient in vivo. This is because nascent amphipathic OMP assembly intermediates in the soluble milieu of the periplasm are prone to misfolding and aggregation. Two periplasmic chaperones, SurA and Skp, along with a major periplasmic protease, DegP, play critical roles in either keeping nascent OMPs assembly competent or degrading them to prevent their interference with normal OMP assembly (13). Expression of the BAM complex members and soluble periplasmic assembly factors is under the control of a major envelope stress response (ESR) pathway that controls σE activity (14). Normally, RseA, the inner membrane-localized anti-sigma factor, sequesters the majority of σE. However, when cells experience envelope stress due to defective BAM, absence of periplasmic assembly factors or expression of mutant OMPs (15), a two-step sequential cleavage of RseA by DegS and RseP proteases relieves σE from the membrane (16) to activate expression of the ESR genes (17). It is noteworthy that alleviation of stress under certain conditions requires simultaneous expression of degP from another ESR pathway composed of the CpxAR two-component system (18). Extensive studies have been carried out to identify ligands that activate DegS and trigger RseA proteolysis (19–23). Two important findings from these studies are as follows: first, the C-terminal OMP tripeptides act as ligands to activate DegS, and second, under the resting state the PDZ domain of DegS inhibits its protease domain, but this inhibition is reversed upon OMP binding to the PDZ domain.
We have taken genetic approaches to study OMP biogenesis through characterizing synthetic and conditionally lethal combination of mutations affecting OMPs and assembly factors (11, 15, 18, 24–29). One of the useful outcomes of conditional phenotypes has been the ability to isolate suppressor mutations under nonpermissive growth conditions. These mutations often map in genes whose products are also involved in the same pathway as the genes of the conditional lethal pair. For example, by employing the conditional lethal combination of bamB and bamE null mutations, we isolated novel suppressor mutations in bamA that rendered the BAM complex partially independent of BamB and BamE (28). Typas et al. (30) employed null mutations in 12 nonessential genes, encoding various envelope functions, to identify genetic interactions among them. Their work revealed several new interactions, including ones frequently involving a null mutation in the pal gene. Pal is an OM lipoprotein that forms the multiprotein Tol-Pal complex (31). Mutations in the tol-pal genes display two hallmark phenotypes: tolerance against colicins (32) and leakage of periplasmic proteins (33). Although tol mutants have reduced levels of certain OMPs (34) and TolAB reportedly interact with OMPs (35, 36), their physiological roles in OMP biogenesis have not been fully understood. Recent studies have implicated the Tol-Pal complex in OM invagination during cell septum formation (37) and polar localization of chemoreceptors (38).
Given our interest in OMP biogenesis, we sought to exploit the synthetic phenotype between mutations affecting tol-pal and a known OMP assembly component to gain insights into the cause of the synthetic phenotype and the means by which cells can overcome it. In particular, we focused on tolB and degP mutations, since we found a strong conditional growth defect ideal for suppressor isolation. This work describes characterization of novel suppressor mutations and the mechanism of suppression.
RESULTS AND DISCUSSION
Isolation of suppressor mutations in a ΔtolB ΔdegP strain.
By employing a high-throughput genetic strategy and surveying synthetic or conditional lethal phenotypes, Typas et al. (30) identified functional interactions between many genes encoding envelope components. Among them, they revealed a negative interaction between degP and pal, as null mutations in these genes caused a growth defect at 37°C. The absence of pal also displayed a synthetic phenotype with null mutations in other genes, including surA and bamB, which encode envelope biogenesis functions. Due to a large number of synthetic interactions involving pal, the authors suggested that the Tol-Pal complex might be one of the central organizers of the Escherichia coli envelope. In this study, we investigated the cause of such synthetic interactions by isolating and characterizing suppressor mutations. Before commencing this exercise, we determined whether the absence of tolA and tolB, like pal, in the ΔdegP background would also confer a synthetically negative phenotype. Deletion alleles of tolA, tolB, and pal (replaced by the Kmr or Cmr gene) were recombined via P1 transduction into degP+ and ΔdegP backgrounds at 30°C. All resulting strains grew well at 30°C; however, at 37°C, double mutants displayed severe growth defects (Fig. 1A and B). These results indicated that the absence of TolA, TolB, and Pal produces a common envelope defect, which is exacerbated in the absence of the major periplasmic protease DegP.
FIG 1.

Effects of tol-pal mutations in degP+ and ΔdegP backgrounds. Bacterial growth on LBA was recorded after incubating petri plates at 30°C or 37°C for 24 h. (A and B) Bacterial strains used are as follows: 1, RAM1292 (wild type); 2, RAM2811 (ΔdegP); 3, RAM2806 (ΔtolA); 4, RAM2812 (ΔtolA ΔdegP); 5, RAM2807 (ΔtolB); 6, RAM2813 (ΔtolB ΔdegP); 7, RAM2808 (Δpal); and 8, RAM2814 (Δpal ΔdegP). (C and D) Bacterial strains used are as follows: 1, RAM1292 (wild type); 2, RAM2811 (ΔdegP); 3, RAM2807 (ΔtolB); 4, RAM2813 (ΔtolB ΔdegP); 5, RAM2817 (temperature-resistant revertant 1 of RAM2813); and 6, RAM2825 (temperature-resistant revertant 2 of RAM2813).
Although all three double mutant combinations produced acute temperature sensitivity, we initially isolated and characterized temperature-resistant revertants from the ΔtolB ΔdegP background. Subsequently, suppressor mutations were tested to see whether they could also reverse the temperature-sensitive phenotype of ΔtolA ΔdegP and Δpal ΔdegP double mutants. Temperature-resistant revertants were isolated as faster-growing colonies on rich medium after incubation of plates at 37°C for 24 h to 36 h. In this work, we conducted detailed characterization of two independently isolated revertants that consistently produced homogeneous colonies at 30°C and 37°C (Fig. 1C and D).
Characterization of suppressor mutations.
The Tol-Pal complex plays a poorly defined role in maintaining envelope integrity; consequently, in the absence of any one of the complex components, cells display a leaky phenotype resulting in the release of periplasmic contents in the culture supernatant (33). Unlike tol-pal, degP mutations are not reported to cause leakage of cellular contents. Thus, by analyzing cell-free culture supernatants, we hoped to gain an insight as to the effect of suppressor mutations on envelope integrity. Control and revertant strains were grown overnight at 30°C, the permissive temperature for the ΔtolB ΔdegP parental strain, after which cells from the growth medium were removed by centrifugation and then passaging of the supernatant through a 0.22-μm filter. Cell-free culture supernatants, normalized to cell density, were concentrated 10-fold and analyzed by SDS-PAGE, and the protein bands were visualized after staining the gel with Coomassie blue (Fig. 2A). As expected, culture supernatants of wild-type and ΔdegP strains showed no protein bands (Fig. 2A, lanes 1 and 2). In contrast, many proteins bands were readily detected from the culture supernatant of the ΔtolB strain (Fig. 2A, lane 3), and the intensity of these bands increased further in the ΔtolB ΔdegP sample (Fig. 2A, lane 4). Supernatants analyzed from the two revertants showed protein band intensities similar to that of the ΔtolB strain (Fig. 2A, compare lane 3 with lanes 5 and 6).
FIG 2.
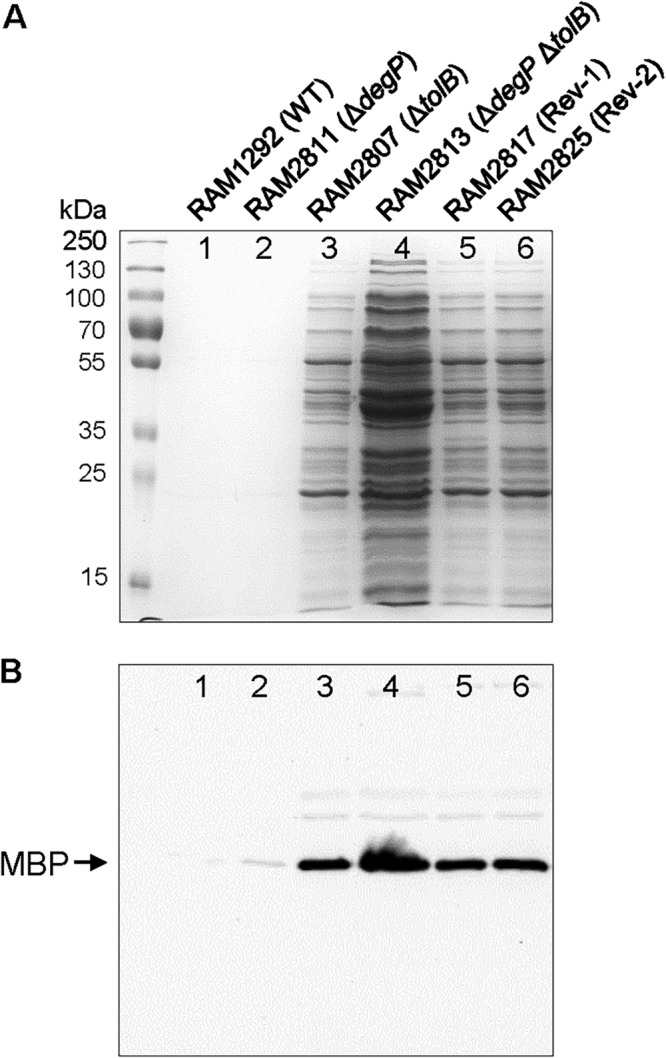
SDS-PAGE analysis of cell-free culture supernatants. Bacterial cultures were grown overnight at 30°C in a water bath with gentle shaking. Cells were pelleted and supernatants were passed through 0.22-μm filters. Cell-free supernatants, normalized to overnight culture OD600, were dried in a speed vacuum centrifuge and resuspended in 1/10 the original volume in a SDS buffer. (A) Protein bands were visualized after staining the gel with Coomassie blue. (B) Western blot analysis of the same samples as in panel A to detect maltose-binding protein (MBP). Positions of prestained protein markers are shown.
Western blot analysis was carried out to detect maltose binding protein (MBP), a bona fide periplasmic protein, from culture supernatants to assess whether MBP levels correlate with total supernatant protein levels observed in different strains (Fig. 2B). While no MBP was detected from the wild-type strain (Fig. 2B, lane 1), a small amount could be seen in the ΔdegP strain (Fig. 2B, lane 2). In contrast, MBP was readily detected from the culture supernatant of the ΔtolB strain and its level increased severalfold in the ΔtolB ΔdegP sample (Fig. 2B, lanes 3 and 4). Therefore, for control strains, MBP levels correlated well with total supernatant protein levels. Samples from the two revertants showed the same level of MBP as the ΔtolB strain, and this is consistent with the observation that these three strains have the same level of total proteins in the culture supernatant (Fig. 2A and B). These data showed that the two suppressor mutations reverse the ΔdegP-associated growth defect and the exacerbated leaky phenotype without correcting the intrinsic leaky phenotype associated with the loss of TolB.
Identification of suppressor mutations.
The whole-genome sequence analysis was carried out to locate the suppressor mutations in two revertants. In both strains, a single base pair substitution was found in the degS protease gene, resulting in D320A (RAM2817) and D102V (RAM2825) substitutions. The presence of these mutations was confirmed by Sanger sequencing and by transducing the mutant degS alleles into a fresh ΔtolB ΔdegP background using a linked yhcA::Kmr marker. The presence of the mutant degS alleles in the newly constructed strains was also able to reverse the temperature-sensitive growth phenotype, thus confirming that the mutant degS alleles are solely responsible for suppression.
We asked whether the mutant degS alleles could suppress the temperature-sensitive phenotype of the ΔtolA ΔdegP and Δpal ΔdegP strains. The degS alleles were transduced into double mutants using a linked marker, and the presence of the degS mutation was confirmed by Sanger sequencing of the PCR-amplified degS gene. Growth results showed that the presence of either degS allele reverses the temperature-sensitive phenotype of the ΔtolA ΔdegP and Δpal ΔdegP strains (Fig. 3A and B). These data are consistent with the notion that the absence of any one component of the Tol-Pal complex in the ΔdegP background produces a common envelope defect that can be partly reversed by the same suppressor mutation.
FIG 3.

Effects of degS suppressor mutations in various conditional lethal genetic backgrounds. Bacterial growth on LBA was recorded after incubation of petri plates at 30°C, 37°C, or 42°C for 24 h. Bacterial strains used for panels A and B are as follows: 1, RAM3065 (ΔtolA ΔdegP); 2, RAM3066 (ΔtolA ΔdegP degS-D102V); 3, RAM3067 (ΔtolA ΔdegP degS-D320A); 4, RAM3068 (Δpal ΔdegP); 5, RAM3069 (Δpal ΔdegP degS-D102A); and 6, RAM3070 (Δpal ΔdegP degS-D320A). Bacterial strains used for panels C and D are as follows: 1 and 2, RAM3061 (ΔbamB ΔdegP); 3 and 4, RAM3062 (ΔbamB ΔdegP degS-D102V); and 5 and 6, RAM3063 (ΔbamB ΔdegP degS-D320A). Bacterial strains used for panels E and F are as follows: 1, RAM3108 (parent); 2, RAM3109 (ΔdegP); 3, RAM3110 (ΔdegP degS-D102V); and 4, RAM3111 (ΔdegP degS-D320A).
We also tested whether degS alleles can reverse the temperature-sensitive growth defect in other genetic backgrounds, including ΔbamB ΔdegP and ΔdegP. BamB is a nonessential component of the BAM complex (9, 12). The absence of BamB alone causes a relatively minor growth and OMP assembly defects (26, 39); however, the absence of BamB together with other BAM complex members (28, 29, 40) or the DegP protease (26, 41) confers a severe growth defect. Data presented in Fig. 3C and D showed that the mutant degS alleles were able to partially correct the growth defect the ΔbamB ΔdegP strain, indicating that the suppression mechanism does not appear to involve correcting a tol-pal-specific defect. Consistent with this notion, we found that the mutant degS alleles fully reversed the high-temperature (42°C) growth defect of a ΔdegP strain (Fig. 3E and F). Together, these observations show that the mutant degS alleles offset the loss of a general periplasmic protease.
Mechanism of suppression by the mutant degS alleles.
The DegS protease plays a key role in controlling the activation of the σE-mediated envelope stress pathway (14, 16, 42). It does so by initiating degradation of RseA, a membrane-bound anti-sigma factor that sequesters σE to the inner membrane (16), thus preventing it from binding to RNA polymerase and activating transcription of the ESR genes (17). The protease activity of DegS is stimulated upon binding of the C-terminal OMP fragments to its PDZ domain, thus relieving the allosteric inhibition of the PDZ domain on its protease domain (23). It is possible that without DegP, fragments of misfolded OMP are not generated in sufficient quantities to fully activate DegS protease and, thus, the σE pathway. If so, the degS suppressor alleles may constitutively activate DegS protease, leading to elevated degradation of RseA and activation of the σE pathway.
We first tested the status of the σE pathway by employing a σE-regulated lacZ transcription fusion construct, yfgC::lacZ. In a ΔrseA background, constitutive activation of σE increases yfgC::lacZ activity 10-fold (Fig. 4A). Complementation of ΔrseA by a plasmid-carried copy of rseA reduced yfgC::lacZ expression back to the basal rseA+ level (Fig. 4A). These data are consistent with an earlier report (17) and show that yfgC is a member of the RseA-σE regulon. Because σE activity is regulated in a growth phase-dependent manner (43), yfgC::lacZ expression in an rseA+ background was determined from cultures grown to the early log (optical density at 600 nm [OD600] = 0.2 to 0.25), mid-log (OD600 = 0.55 to 0.65), late log (OD600 = 0.9 to 1.0), and stationary (overnight; OD600 ≥ 1.8) phases. In the wild-type degS background, expression of yfgC::lacZ from early- and mid-log-phase-grown cultures was 4- and 5-fold lower than from cultures grown to the late log and stationary phases (Fig. 4B). These data are consistent with the previous report of elevated σE activity during the late log and stationary growth phases (43). Interestingly, the presence of the two mutant degS alleles elevated yfgC::lacZ expression by about 50%, regardless of the culture growth phase (Fig. 4B), indicating that the mutant degS alleles affect expression of the σE-regulated yfgC gene intrinsically and independent of factors, such as ppGpp, which influence the σE regulon based on growth phase (43).
FIG 4.
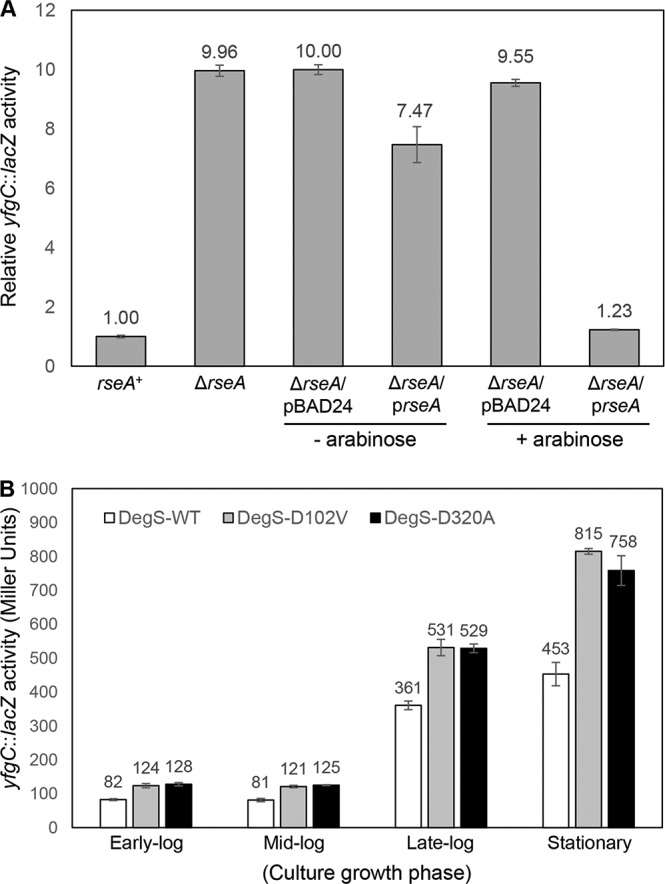
(A) RseA/σE-mediated control yfgC::lacZ expression. β-Galactosidase activities were measured from mid-log-phase-grown cultures. Expression of the plasmid-borne rseA gene was induced for 3 h with 0.2% arabinose. (B) Effects of degS suppressor mutations on σE activation were assessed by examining activities of the σE-regulated yfgC::lacZ fusion construct (yfgC is also known as bepA). β-Galactosidase assays were carried out from three independent cultures, grown to various growth phases as shown, in duplicate. Error bars indicate SDs.
We then tested whether elevated σE activity in the two degS mutants coincides with reduced RseA levels. Cells from overnight-grown cultures, adjusted to equal optical densities, were analyzed by SDS-PAGE, and RseA was detected by Western blotting using polyclonal RseA antibodies (Fig. 5A). As a control, MBP was also probed from same cell extracts (Fig. 5B). Quantification of RseA bands relative to an unknown protein band migrating above RseA (Fig. 5A) or MBP (Fig. 5B) showed that RseA levels were reduced approximately 30% and 50% in strains expressing DegSD102V and DegSD320A substitutions, respectively. These data support the hypothesis of elevated DegS protease and σE activities in the two DegS mutants.
FIG 5.

Effects of degS mutations on RseA and AcrAL222Q levels. (A and B) RseA and MBP (control) were detected by Western blot analysis. Bacterial cultures were grown overnight at 37°C in a roller drum. Samples from two independent cultures, per strain, normalized to OD600, were boiled in a SDS buffer and analyzed by SDS-PAGE. Membrane filters were probed with antibodies against RseA (A) and MBP (B). Relevant genotypes of the strains, as well as positions of RseA, MBP and prestained protein markers are shown. (C and D) Effects of degS mutations on AcrAL222Q levels. Western blot analysis was conducted to observe the effects of degS suppressor mutations on a labile AcrA protein carrying an L222Q substitution. Bacterial cultures were grown overnight at 37°C in a roller drum. Samples from two independent cultures, per strain, normalized to OD600, were boiled in an SDS buffer and analyzed by SDS-PAGE. Membrane filters, carrying samples from the degS-D102V (C) or degS-D320A (D) background, were probed with antibodies against AcrA. Relevant genotypes of the strains used are shown, except that all strains had deletions of tolC and expressed the mutant allele of acrA-L222Q from the chromosome.
As a second way of assessing elevated envelope protease activity, we used a mutant AcrA protein, AcrAL222Q, which, without its complex partners AcrB and TolC, is degraded in a DegP-dependent manner (27, 44, 45). If restoration of envelope homeostasis involves activation of an envelope protease, we may observe increased degradation of AcrAL222Q in the mutant degS backgrounds lacking DegP. Mutant degS alleles were transduced into a ΔdegP ΔtolC acrAL222Q strain and AcrA levels were examined by Western blotting (Fig. 5C and D). In a degP+ strain, AcrAL222Q levels were 3- to 5-fold lower than in a ΔdegP strain expressing wild-type degS (Fig. 5C and D), confirming the previous data of DegP-dependent degradation of the mutant AcrA protein. The presence of mutant degS alleles reduced AcrAL222Q levels by about half in a ΔdegP background, indicating elevated protease activity in DegSD102V and DegSD320A backgrounds. These experiments do not show that the DegS variants themselves are responsible for degrading AcrAL222Q. We have previously shown that a mutant allele of σE (rpoE3) also reverses the temperature-sensitive growth defect of a degP mutant and increases AcrAL222Q proteolysis (27). These data suggest that another periplasmic protease of the DegS-RseA-σE pathway may be responsible for the effects of DegSD102V and DegSD320A on AcrAL222Q. YfgC/BepA, an σE-controlled protease, is not involved in degrading AcrAL222Q (27). Regardless of the identity of a specific protease, reduced levels of RseA and AcrAL222Q in the DegSD102V and DegSD320A backgrounds indicate elevated periplasmic protease activity, which, together with other σE regulon members, compensates for the loss of the major periplasmic protease DegP and restores envelope homeostasis.
Structure of DegS explains the suppressor phenotype of DegSD320A.
Once activated, DegS degrades RseA to release σE from the inner membrane. A key activator of DegS is the exposed C-terminal end of a misfolded β-barrel OMP, which binds at the interface of the PDZ and protease domains of DegS and stabilizes its protease domain (19). Structural, biochemical, and mutagenesis work has provided valuable insights into DegS’s activation mechanism and identified key residues in the PDZ and protease domains that play an important role in the enzyme’s equilibrium between the active and inactive states (20, 21, 23). These studies revealed that D320 and E317 of the PDZ domain make critical salt bridges with R178 of the protease domain when DegS is not bound to its ligand, thus favoring the inactive state (Fig. 6). Interestingly, a D320A substitution, which was also isolated in this study as one of the suppressors, was shown to increase the basal protease activity of DegS in vitro (23). This is consistent with our genetic data and suggests a plausible mechanism of suppression. The two mutant degS alleles reduce RseA levels and activate σE pathway (Fig. 4 and 5). Although σE activation in the mutant degS backgrounds is modest (50% increase) compared to its full activation (10-fold) in the ΔrseA background (Fig. 4A), this σE activation by the two degS alleles is sufficient to overcome envelope stress. Consistent with these data, the mutant rpoE3 allele, which fully reversed the temperature-sensitive growth defect of ΔdegP, also only modestly activated σE-mediated gene expression (27). It is worth noting that overexpression of wild-type DegS was reported not to complement the temperature-sensitive growth phenotype of a ΔdegP allele (46). However, work carried out in this study clearly showed that chromosomally expressed DegSD320A fully reverses the temperature-sensitive phenotype of a degP null allele (Fig. 3E and F). We think that this difference is due to the fact wild-type DegS without its ligand is largely inactive, while D320A partially disengages the PDZ domain of DegS from its protease domain to increase its basal activity (23) to compensate for ΔdegP. Unlike the D320A substitution, it is not apparent how D102A enhances DegS protease activity. The D102 residue is present in the protease domain and its side chain is oriented away from the catalytic triad (H96, D126, and S201) or the DegS subunit interface (Fig. 6). Based on available DegS crystal structures, we could not identify any interacting partners of D102. The positive effect of D102V on DegS activity may be indirect and driven through an effect on enzyme’s folding/conformation or altered interactions by an unidentified modulator.
FIG 6.

Cartoon showing X-ray structure of ligand-free DegS (PDB accession number 1te0 [21]). Shown are key side chain interactions between the PDZ domain (wheat) and the protease domain (cyan), as well as three active-site residues. The two suppressor alterations isolated in this study affect D102 and D320 of the protease and PDZ domains, respectively.
Reasons for the conditional lethal phenotype of ΔtolB ΔdegP.
Strains lacking tolB or degP produce distinct phenotypes: the ΔtolB strain displays a classical leaky phenotype, while the ΔdegP strain is unable to grow at 42°C on rich media. The combination of ΔtolB and ΔdegP mutations greatly exacerbates both the temperature-sensitive growth and leaky phenotypes (Fig. 1 and 2), indicating a drastically compromised envelope structure and perturbed envelope homeostasis processes. It is conceivable that excessive leakage of proteins and metabolites out of the cell compromises critical cellular functions, including nutrient transport and macromolecular synthesis, resulting in a severe growth defect. We employed a σE-regulated rpoHP3::lacZ reporter construct (47) to monitor envelope stress and possible leakage of cytoplasmic contents in the double mutant. At 30°C, the absence of DegP or TolB caused a 15% or 32% increase, respectively, in rpoHP3::lacZ expression over the parent strain (Table 1). In the double mutant, rpoHP3::lacZ expression increased by 50%, reflecting a further elevation in envelope stress. We then measured rpoHP3::lacZ expression from the cell-free culture supernatant to gauge any signs of the cytoplasmic membrane disruption that would release cytoplasmic contents, including LacZ, into the medium. Around 1% of the total LacZ activity was present in the culture supernatant of the parent and degP strains. However, in the tolB and tolB degP mutants, the LacZ activity in the culture supernatant accounted for 5% and 10%, respectively, of the total LacZ activity, indicating a significant increase in cell lysis (Table 1).
TABLE 1.
Determination of rpoHP3::lacZ activities in different genetic backgrounds from cultures grown at 30°C and 35°Ca
| Relevant genotype |
rpoHP3::lacZ activity at 30°C |
rpoHP3::lacZ activity at 35°C |
||||
|---|---|---|---|---|---|---|
| Culture | Supernatant | % Sup/cul | Culture | Supernatant | % Sup/cul | |
| WT | 340 ± 23 | 4.8 ± 0.5 | 1.40 | 390 ± 22 | 5.5 ± 1.3 | 1.41 |
| ΔdegP | 372 ± 16 | 5.0 ± 0.7 | 1.34 | 488 ± 27 | 9.0 ± 1.1 | 1.84 |
| ΔtolB | 451 ± 15 | 23 ± 3.7 | 5.10 | 597 ± 32 | 32.0 ± 1.8 | 5.36 |
| ΔtolB ΔdegP | 515 ± 9 | 51 ± 4.3 | 9.90 | 1,572 ± 94 | 890.0 ± 12 | 56.62 |
LacZ activities (Miller units) were determined from four independent overnight-grown cultures. Cul, culture; Sup, cell-free culture supernatant. Values following the “±” sign are SDs obtained from four independent biological samples. WT, wild type.
We also measured rpoHP3::lacZ expression from cultures grown at 35°C, at which the double mutant can grow, albeit poorly compared to the single mutants. The overall pattern of rpoHP3::lacZ expression in the parent strain and single mutants was similar to that observed at 30°C (Table 1). In contrast, the cell culture and cell-free supernatant of the double mutant showed a dramatic increase in the rpoHP3::lacZ activity (Table 1), reflecting significantly heightened envelope stress and cell lysis. Together, these observations indicate that the ΔtolB ΔdegP mutant experiences substantial damage in the envelope structure in a temperature-dependent manner that results in cell lysis and death. It is worth noting that the leaky phenotype of the tolB mutant is traditionally considered to reflect the release of the periplasmic and not the cytoplasmic proteins (33). Yet we consistently found as much as 5% of the total LacZ activity in the cell-free culture supernatant of the tolB strain. One reason for this difference could be that we assayed LacZ activity from overnight-grown cultures, while Lopes et al. (33) used freshly grown cells.
In summary, we described the isolation of two suppressor mutations in the degS gene that reverse the acute temperature-sensitive growth phenotype of the strain simultaneously lacking DegP, a major periplasmic protease and a component of the Tol-Pal complex. Our data suggest that suppressor mutations in degS overcome the synthetic lethal phenotype by enhancing DegS’s protease activity, which through the activation of the σE pathway compensates for the loss of DegP. Without DegP or the two mutant DegS proteins described here, cells with the destabilized Tol-Pal complex experience substantially elevated envelope stress and damage, causing cell lysis and death at growth temperatures of 35°C and above. We do not know exactly why cell lysis is intensified when DegP and TolB are simultaneously absent, but we speculate that it is due to the cumulative effect of defects in cell division, envelope biogenesis, OMP assembly, and envelope homeostasis processes.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
Escherichia coli K-12 strains used in this study are listed in Table 2. For bacterial growth, Difco LB broth or agar (LBA) was used and, when desired, supplemented with kanamycin (25 μg/ml), chloramphenicol (12.5 μg/ml), or tetracycline (10 μg/ml). Bacteria on LBA were grown for 24 to 36 h at 30°C, 37°C, or 42°C. The genetic transfer of various null alleles marked by the kanamycin (Kmr), chloramphenicol (Cmr), or tetracycline resistance gene was carried out by P1 transduction. To avoid antibiotic resistance incompatibility, in some instances Kmr or Cmr genes were scarred out by the method of Datsenko and Wanner (48). In some instances, lacZ was recombined at the deletion site by the method of Ellermeier et al. (49). Temperature-resistant revertants of a ΔdegP ΔtolB strain were isolated by plating 5 × 108 cells on LBA plates and incubating for 24 to 36 h at 37°C. Revertants that consistently formed homogeneous (same-size) colonies at 37°C were further characterized.
TABLE 2.
Escherichia coli K-12 strains used in this study
| Strain name | Relevant genotype | Reference or source |
|---|---|---|
| RAM1292 | MC4100 Δara714 | 11 |
| RAM2051 | CAG45114 rpoHP3::lacZ | 47 |
| RAM2498 | RAM2051 degP::Tn10 | This study |
| RAM2806 | RAM1292 ΔtolA::scar | This study |
| RAM2807 | RAM1292 ΔtolB::scar | This study |
| RAM2808 | RAM1292 Δpal::scar | This study |
| RAM2811 | RAM1292 ΔdegP::Kmr | This study |
| RAM2812 | RAM2806 ΔdegP::Kmr | This study |
| RAM2813 | RAM2807 ΔdegP::Kmr | This study |
| RAM2814 | RAM2808 ΔdegP::Kmr | This study |
| RAM2817 | RAM2813 degS-D320A (revertant 1) | This study |
| RAM2825 | RAM2813 degS-D102V (revertant 2) | This study |
| RAM2831 | RAM2051 ΔtolB::Kmr | This study |
| RAM2832 | RAM2498 ΔtolB::Kmr | This study |
| RAM3061 | RAM1292 ΔyhcA::scar degP::Tn10 | This study |
| RAM3062 | RAM1292 ΔyhcA::scar degS-D102V degP::Tn10 | This study |
| RAM3063 | RAM1292 ΔyhcA::scar degS-D320A degP::Tn10 | This study |
| RAM3065 | RAM3061 ΔtolA::Cmr | This study |
| RAM3066 | RAM3062 ΔtolA::Cmr | This study |
| RAM3067 | RAM3063 ΔtolA::Cmr | This study |
| RAM3068 | RAM3061 Δpal::Kmr | This study |
| RAM3069 | RAM3062 Δpal::Kmr | This study |
| RAM3070 | RAM3063 Δpal::Kmr | This study |
| RAM3075 | RAM2051 yhcA::Kmr | This study |
| RAM3077 | RAM2051 yhcA::Kmr degS-D102V | This study |
| RAM3079 | RAM2051 yhcA::Kmr degS-D320A | This study |
| RAM3108 | RAM1292 ΔyhcA::Kmr | This study |
| RAM3109 | RAM1292 ΔyhcA::Kmr degP::Tn10 | This study |
| RAM3110 | RAM1292 ΔyhcA::Kmr degS-D102V degP::Tn10 | This study |
| RAM3111 | RAM1292 ΔyhcA::Kmr degS-D320A degP::Tn10 | This study |
| RAM3112 | RAM1292 ΔyhcA::scar yfgC::lacZ-Kmr | This study |
| RAM3113 | RAM1292 ΔyhcA::scar degS-D102V yfgC::lacZ-Kmr | This study |
| RAM3114 | RAM1292 ΔyhcA::scar degS-D320A yfgC::lacZ-Kmr | This study |
| RAM3115 | RAM1292 ΔtolC::Cmr acrA-L222Q-Tn10 | 44 |
| RAM3116 | RAM3115 ΔyhcA::Kmr | This study |
| RAM3117 | RAM3115 ΔyhcA::Kmr degS-D102V | This study |
| RAM3118 | RAM3116 ΔyhcA::Kmr degS-D320A | This study |
| RAM3119 | RAM1292 ΔtolC::Cmr acrA-L222Q-Tn10 ΔdegP::scar | 44 |
| RAM3120 | RAM3119 ΔyhcA::Kmr | This study |
| RAM3121 | RAM3119 ΔyhcA::Kmr degS-D102V | This study |
| RAM3122 | RAM3119 ΔyhcA::Kmr degS-D320A | This study |
| RAM3144 | RAM1292 yfgC::lacZ-Kmr | This study |
| RAM3145 | RAM1292 yfgC::lacZ-Kmr ΔrseA::scar | This study |
| RAM3146 | RAM1292 pBAD24 | This study |
| RAM3147 | RAM1292 pBAD24-rseA | This study |
β-Galactosidase assays.
β-Galactosidase assays were carried out to measure gene expression and determine leakage of the cytoplasmic contents in the culture supernatant. β-Galactosidase activities were measured from four to six independent cultures in duplicate by the method described by Miller (50). To determine leakage of cytoplasmic proteins, cells were removed from the growth medium by centrifugation in a microcentrifuge at 16,000 × g for 5 min. The supernatant was then passed through a 0.22-μm filter and the presence of viable cells in the filtrate was determined by the serial dilution method. β-Galactosidase activity from the cell-free supernatant was determined without the addition of chloroform or SDS, which is normally done to permeabilize cells prior to adding the β-galactosidase substrate.
Protein and DNA analyses.
Proteins were analyzed on SDS-polyacrylamide (11%) gels and visualized after staining by Coomassie blue. For detection of specific proteins, Western blot analysis was carried out as described previously (15). Primary antibodies (dilutions in parentheses) used were raised against AcrA (1:16,000), MBP (1:16,000), and RseA (1:5,000).
Whole-genome sequence analysis was carried out to determine the location of suppressor mutations. The bacterial chromosome was isolated using a DNeasy blood and tissue kit from Qiagen and subjected to sequencing by Illumina’s MiSeq system. Whole-genome sequencing reads for each sample were quality checked using FastQC v0.10.1 and aligned to the Escherichia coli K-12 MC4100 assembly from the NCBI Database (https://www.ncbi.nlm.nih.gov/assembly/GCF_000499485.1/) using the Burrows-Wheeler short-read alignment tool, BWA version 0.7.15. After alignment, single nucleotide polymorphisms (SNPs) and indels were discovered following the GATK Best Practices workflow of Germline short variant discovery (https://gatkforums.broadinstitute.org/gatk/discussion/11145/germline-short-variant-discovery-snps-indels). Raw mapped reads were preprocessed by adding read groups, indexing, marking duplicates, sorting, and recalibrating base quality scores. Then variants were called by HaplotypeCaller. Per-base genome coverage was computed by bedtools genomecov. All regions with zero coverage were reported. Structural variations were identified by BreakDancer and LUMPY 0.2.13. The presence of nonsynonymous mutations was confirmed by Sanger sequencing using PCR-amplified fragments of the targeted region.
ACKNOWLEDGMENTS
We thank Carol Gross for antibodies against RseA and a bacterial strain (CAG45114) containing rpoHP3::lacZ. We are also thankful to Tom Silhavy and his colleagues for sharing their degS mutant data with us prior to publication.
Footnotes
For a companion article on this topic, see https://doi.org/10.1128/JB.00745-18.
REFERENCES
- 1.Nikaido H. 2003. Molecular basis of bacterial outer membrane revisited. Microbiol Mol Biol Rev 67:593–656. doi: 10.1128/MMBR.67.4.593-656.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Silhavy TJ, Kahne D, Walker S. 2010. The bacterial cell envelope. Cold Spring Harb Perspect Biol 2:a000414. doi: 10.1101/cshperspect.a000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hagan CL, Silhavy TJ, Kahne D. 2011. β-Barrel membrane protein assembly by the Bam complex. Annu Rev Biochem 80:189–210. doi: 10.1146/annurev-biochem-061408-144611. [DOI] [PubMed] [Google Scholar]
- 4.Misra R. 2012. Assembly of the β-barrel outer membrane proteins in Gram-negative bacteria, mitochondria, and chloroplasts. ISRN Mol Biol 2012:708203. doi: 10.5402/2012/708203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Selkrig J, Leyton DL, Webb CT, Lithgow T. 2014. Assembly of β-barrel proteins into bacterial outer membranes. Biochim Biophys Acta 1843:1542–1550. doi: 10.1016/j.bbamcr.2013.10.009. [DOI] [PubMed] [Google Scholar]
- 6.Noinaj N, Gumbart JC, Buchanan SK. 2017. The β-barrel assembly machine in motion. Nat Rev Microbiol 15:197–204. doi: 10.1038/nrmicro.2016.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tokuda H, Matsuyama S-I. 2004. Sorting of lipoproteins to the outer membrane of E. coli. Biochim Biophys Acta 1693:5–13. doi: 10.1016/j.bbamcr.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 8.Okuda S, Tokuda H. 2011. Lipoprotein sorting in bacteria. Annu Rev Microbiol 65:239–259. doi: 10.1146/annurev-micro-090110-102859. [DOI] [PubMed] [Google Scholar]
- 9.Malinverni JC, Werner J, Kim S, Sklar JG, Kahne D, Misra R, Silhavy J. 2006. YfiO stabilizes the YaeT complex and is essential for outer membrane protein assembly in Escherichia coli. Mol Microbiol 61:151–164. doi: 10.1111/j.1365-2958.2006.05211.x. [DOI] [PubMed] [Google Scholar]
- 10.Doerrler WT, Raetz C. 2005. Loss of outer membrane proteins without inhibition of lipid export in an Escherichia coli YaeT mutant. J Biol Chem 280:27679–27687. doi: 10.1074/jbc.M504796200. [DOI] [PubMed] [Google Scholar]
- 11.Werner J, Misra R. 2005. YaeT affects the assembly of lipid-dependent and lipid-independent outer membrane proteins of Escherichia coli. Mol Microbiol 57:1450–1459. doi: 10.1111/j.1365-2958.2005.04775.x. [DOI] [PubMed] [Google Scholar]
- 12.Wu T, Malinverni J, Ruiz N, Kim S, Silhavy TJ, Kahne D. 2005. Identification of a multicomponent complex required for outer membrane biogenesis in Escherichia coli. Cell 121:235–245. doi: 10.1016/j.cell.2005.02.015. [DOI] [PubMed] [Google Scholar]
- 13.Sklar JG, Wu T, Kahne D, Silhavy TJ. 2007. Defining the roles of periplasmic chaperones SurA, Skp, and DegP in Escherichia coli. Genes Dev 21:2473–2484. doi: 10.1101/gad.1581007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ades SE, Connolly LE, Alba BM, Gross CA. 1999. The Escherichia coli σE-dependent extracytoplasmic stress response is controlled by the regulated proteolysis of anti-sigma factor. Genes Dev 13:2449–2461. doi: 10.1101/gad.13.18.2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bennion D, Charlson ES, Coon E, Misra R. 2010. Dissection of β-barrel outer membrane protein assembly pathways through characterizing BamA POTRA 1 mutants of Escherichia coli. Mol Microbiol 77:1153–1171. doi: 10.1111/j.1365-2958.2010.07280.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alba BM, Leeds JA, Onufryk C, Lu CZ, Gross CA. 2002. DegS and YaeL participate sequentially in the cleavage of RseA to activate the σE-dependent extracytoplasmic stress response. Genes Dev 16:2156–2168. doi: 10.1101/gad.1008902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rhodius VA, Suh WC, Nonaka G, West J, Gross CA. 2006. Conserved and variable functions of the σE stress response in related genomes. PLoS Biol 4:43–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gerken H, Leiser OP, Bennion D, Misra R. 2010. Involvement and necessity of the Cpx regulon in the event of aberrant β‐barrel outer membrane protein assembly. Mol Microbiol 75:1033–1046. doi: 10.1111/j.1365-2958.2009.07042.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Walsh NP, Alba BM, Bose B, Gross CA, Sauer RT. 2003. OMP peptide signals initiate the envelope-stress response by activating DegS protease via relief of inhibition mediated by its PDZ domain. Cell 113:61–71. doi: 10.1016/S0092-8674(03)00203-4. [DOI] [PubMed] [Google Scholar]
- 20.Wilken C, Kitzing K, Kurzbauer R, Ehrmann M, Clausen T. 2004. Crystal structure of the DegS stress sensor: how a PDZ domain recognizes misfolded protein and activates a protease. Cell 117:483–494. doi: 10.1016/S0092-8674(04)00454-4. [DOI] [PubMed] [Google Scholar]
- 21.Zeth K. 2004. Structure analysis of DegS, a stress sensor of the bacterial periplasm. FEBS Lett 569:351–358. doi: 10.1016/j.febslet.2004.06.012. [DOI] [PubMed] [Google Scholar]
- 22.Hasselblatt H, Kurzbauer R, Wilken C, Krojer T, Sawa J, Kurt J, Kirk R, Hasenbein S, Ehrmann M, Clausen T. 2007. Regulation of the σE stress response by DegS: how the PDZ domain keeps the protease domain inactive in the resting state and allows integration of different OMP-derived signals upon folding stress. Genes Dev 21:2659–2670. doi: 10.1101/gad.445307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sohn J, Grant RA, Sauer RT. 2007. Allosteric activation of DegS, a stress sensor PDZ protease. Cell 131:572–583. doi: 10.1016/j.cell.2007.08.044. [DOI] [PubMed] [Google Scholar]
- 24.Misra R, CastilloKeller M, Deng M. 2000. Overexpression of protease deficient DegPS210A rescues the lethal phenotype of Escherichia coli OmpF assembly mutants in a degP background. J Bacteriol 182:4882–4888. doi: 10.1128/JB.182.17.4882-4888.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.CastilloKeller M, Misra R. 2003. Protease-deficient DegP suppresses lethal effects of a mutant OmpC protein by its capture. J Bacteriol 185:148–154. doi: 10.1128/JB.185.1.148-154.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Charlson ES, Werner JN, Misra R. 2006. Differential effects of yfgL mutation on the outer membrane proteins and lipopolysaccharide. J Bacteriol 188:7186–7194. doi: 10.1128/JB.00571-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leiser OP, Charlson ES, Gerken H, Misra R. 2012. Reversal of the ΔdegP phenotypes by a novel rpoE allele of Escherichia coli. PLoS One 7:e33979. doi: 10.1371/journal.pone.0033979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tellez R Jr, Misra R. 2012. Substitutions in the BamA β-barrel domain overcome the conditional lethal phenotype of a ΔbamB ΔbamE strain of Escherichia coli. J Bacteriol 194:317–324. doi: 10.1128/JB.06192-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Misra R, Stikeleather R, Gabriele R. 2015. In vivo roles of BamA, BamB and BamE in the biogenesis of BamA, a core protein of the β-barrel assembly machine of Escherichia coli. J Mol Biol 427:1061–1074. doi: 10.1016/j.jmb.2014.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Typas A, Nichols RJ, Siegele DA, Shales M, Collins SR, Lim B, Braberg H, Yamamoto N, Takeuchi R, Wanner BL, Mori H, Weissman JS, Krogan NJ, Gross CA. 2008. High-throughput, quantitative analyses of genetic interactions in E. coli. Nat Methods 5:781–787. doi: 10.1038/nmeth.1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lazzaroni JC, Germon P, Ray MC, Vianney A. 1999. The Tol proteins of Escherichia coli and their involvement in the uptake of biomolecules and outer membrane stability. FEMS Microbiol Lett 177:191–197. doi: 10.1111/j.1574-6968.1999.tb13731.x. [DOI] [PubMed] [Google Scholar]
- 32.Nagel de Zwaig R, Luria SE. 1967. Genetics and physiology of colicin-tolerant mutants of Escherichia coli. J Bacteriol 94:1112–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lopes J, Gottfried S, Rothfield L. 1972. Leakage of periplasmic enzymes by mutants of Escherichia coli and Salmonella typhimurium: isolation of “periplasmic leaky” mutants. J Bacteriol 109:520–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lazzaroni JC, Fognini-Lefebvre N, Portalier RC. 1986. Effects of lky mutations on the expression of ompF, ompC and lamB porin structural genes in Escherichia coli K-12. FEMS Microbiol Lett 33:235–239. doi: 10.1111/j.1574-6968.1986.tb01278.x. [DOI] [Google Scholar]
- 35.Derouiche R, Gavioli M, Benédétti H, Prilipov A, Lazdunski C, Lloubès R. 1996. TolA central domain interacts with Escherichia coli porins. EMBO J 15:6408–6415. doi: 10.1002/j.1460-2075.1996.tb01032.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rigal A, Bouveret E, Lloubes R, Lazdunski C, Benedetti H. 1997. The TolB protein interacts with the porins of Escherichia coli. J Bacteriol 179:7274–7279. doi: 10.1128/jb.179.23.7274-7279.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gerding MA, Ogata Y, Pecora ND, Niki H, de Boer PA. 2007. The trans-envelope Tol-Pal complex is part of the cell division machinery and required for proper outer membrane invagination during cell constriction in E. coli. Mol Microbiol 63:1008–1025. doi: 10.1111/j.1365-2958.2006.05571.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Santos TMA, Lin T-Y, Rajendran M, Anderson SM, Weibel DB. 2014. Polar localization of Escherichia coli chemoreceptors requires an intact Tol-Pal complex. Mol Microbiol 92:985–1004. doi: 10.1111/mmi.12609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vuong P, Bennion D, Mantei J, Frost D, Misra R. 2008. Analysis of YfgL and YaeT interactions through bioinformatics, mutagenesis, and biochemistry. J Bacteriol 190:1507–1517. doi: 10.1128/JB.01477-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Onufryk C, Marie-Laure C, Fang FC, Gross CA. 2005. Characterization of the six lipoproteins in the σE regulon. J Bacteriol 187:4552–4561. doi: 10.1128/JB.187.13.4552-4561.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gerken H, Charlson ES, Cicirelli EM, Kenney LJ, Misra R. 2009. MzrA: a novel modulator of the EnvZ-OmpR two-component regulon. Mol Microbiol 72:1408–1422. doi: 10.1111/j.1365-2958.2009.06728.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Alba BM, Zhong HJ, Pelayo JC, Gross CA. 2001. degS (hhoB) is an essential Escherichia coli gene whose indispensable function is to provide σE activity. Mol Microbiol 40:1323–1333. doi: 10.1046/j.1365-2958.2001.02475.x. [DOI] [PubMed] [Google Scholar]
- 43.Costanzo A, Ades SE. 2006. Growth phase-dependent regulation of the extracytoplasmic stress factor, σE, by guanosine 3′,5′-bispyrophosphate (ppGpp). J Bacteriol 188:4627–4634. doi: 10.1128/JB.01981-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gerken H, Misra R. 2004. Genetic evidence for functional interactions between TolC and AcrA proteins of a major antibiotic efflux pump of Escherichia coli. Mol Microbiol 54:620–631. doi: 10.1111/j.1365-2958.2004.04301.x. [DOI] [PubMed] [Google Scholar]
- 45.Weeks JW, Celaya-Kolb T, Pecora S, Misra R. 2010. AcrA suppressor alterations reverse the drug hypersensitivity phenotype of a TolC mutant by inducing TolC aperture opening. Mol Microbiol 75:1468–1483. doi: 10.1111/j.1365-2958.2010.07068.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Waller PRH, Sauer RT. 1996. Characterization of degQ and degS, Escherichia coli genes encoding homologs of the DegP protease. J Bacteriol 178:1146–1153. doi: 10.1128/jb.178.4.1146-1153.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mecsas J, Rouviere PE, Erickson JW, Donohue TI, Gross CA. 1993. The activity of σE, an Escherichia coli heat-inducible σ-factor, is modulated by expression of outer membrane proteins. Genes Dev 7:2618–2628. doi: 10.1101/gad.7.12b.2618. [DOI] [PubMed] [Google Scholar]
- 48.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ellermeier CD, Janakiraman A, Slauch JM. 2002. Construction of targeted single copy lac fusions using lambda Red and FLP-mediated site-specific recombination in bacteria. Gene 290:153–161. [DOI] [PubMed] [Google Scholar]
- 50.Miller JH. 1992. A short course in bacterial genetics: a laboratory manual and handbook for Escherichia coli and related bacteria, p 71–74. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]