

Periodontitis, commonly referred to as gum disease, is a chronic inflammatory condition that affects a large proportion of the population. Porphyromonas gingivalis is a bacterium closely associated with periodontitis, although how and if it is a cause for the disease are not known. It has a formidable capacity to dampen the host's innate immune response, enabling its persistence in diseased sites and triggering microbial dysbiosis in animal models of infection. P. gingivalis is particularly adept at evading the host's TLR4-mediated innate immune response by modifying the structure of lipid A, the TLR4 ligand. In this paper, we report identification of the gene encoding lipid A deacylase, a key enzyme that modifies lipid A to TLR4-evasive structures.
KEYWORDS: Porphyromonas gingivalis, TLR4, deacylase, immune evasion, lipid A, transposon
ABSTRACT
Removal of one acyl chain from bacterial lipid A by deacylase activity is a mechanism used by many pathogenic bacteria to evade the host's Toll-like receptor 4 (TLR4)-mediated innate immune response. In Porphyromonas gingivalis, a periodontal pathogen, lipid A deacylase activity converts a majority of the initially synthesized penta-acylated lipid A, a TLR4 agonist, to tetra-acylated structures, which effectively evade TLR4 sensing by being either inert or antagonistic at TLR4. In this paper, we report successful identification of the gene that encodes the P. gingivalis lipid A deacylase enzyme. This gene, PGN_1123 in P. gingivalis 33277, is highly conserved within P. gingivalis, and putative orthologs are phylogenetically restricted to the Bacteroidetes phylum. Lipid A of ΔPGN_1123 mutants is penta-acylated and devoid of tetra-acylated structures, and the mutant strain provokes a strong TLR4-mediated proinflammatory response, in contrast to the negligible response elicited by wild-type P. gingivalis. Heterologous expression of PGN_1123 in Bacteroides thetaiotaomicron promoted lipid A deacylation, confirming that PGN_1123 encodes the lipid A deacylase enzyme.
IMPORTANCE Periodontitis, commonly referred to as gum disease, is a chronic inflammatory condition that affects a large proportion of the population. Porphyromonas gingivalis is a bacterium closely associated with periodontitis, although how and if it is a cause for the disease are not known. It has a formidable capacity to dampen the host's innate immune response, enabling its persistence in diseased sites and triggering microbial dysbiosis in animal models of infection. P. gingivalis is particularly adept at evading the host's TLR4-mediated innate immune response by modifying the structure of lipid A, the TLR4 ligand. In this paper, we report identification of the gene encoding lipid A deacylase, a key enzyme that modifies lipid A to TLR4-evasive structures.
INTRODUCTION
Porphyromonas gingivalis is an anaerobic Gram-negative bacterium found in subgingival plaque. It is closely associated with periodontitis, a chronic inflammatory disorder responsible for major tooth loss worldwide (1). P. gingivalis is found in higher abundance in diseased periodontal pockets than in healthy pockets (2, 3), indicating that it is adept at withstanding hostile immune conditions characteristic of periodontal diseased sites. The bacterium has been shown to express multiple virulence factors that enable it to evade, actively suppress, and modulate the host innate immune response (4–13), giving it the ability to persist under inflammatory conditions and to become a recurrent member of the bacterial community during disease. Furthermore, P. gingivalis infections in mouse and rabbit models have shown it to alter the composition of the subgingival microbiome and augment the bacterial load, marking it as a keystone pathogen (14–17) with the capacity to trigger microbial dysbiosis. A consequence of dysbiosis, commonly seen in chronic inflammatory disorders, is breakdown of the homeostatic bacterial host balance, leading to aggravated immune responses that, in the case of periodontitis, manifest in loss of supporting structures of the tooth.
P. gingivalis is unusually adept at evading the innate immune response mediated by Toll-like receptor 4 (TLR4) (1, 18, 19), a major bacterial clearance mechanism mounted by the host. TLR4 is a host innate immune receptor that, together with its coreceptor myeloid differentiation factor 2 (MD-2), recognizes lipopolysaccharide (LPS), an essential macromolecule that forms the outer layer of the outer membrane in Gram-negative bacteria (20–23). The TLR4 ligand specifically is lipid A, a hydrophobic glycolipid anchor of LPS. Lipid A satisfies the requirements first outlined by Medzhitov and Janeway for pattern recognition receptor ligands in that the structure is highly conserved, is essential for bacterial survival, and differs significantly from host (self) structures (24). Engagement of bacterial lipid A by the TLR4/MD-2 complex initiates a proinflammatory response, which in turn leads to production of costimulatory molecules required for adaptive immunity, culminating in clearance of local infection. The prototypical lipid A structure, first investigated in Escherichia coli, comprises a disaccharide, β-1′,6-glucosamine, to which is attached six C12 to C14 fatty acyl chains and two phosphate groups at the C-1 and C-4′ positions (25). E. coli lipid A is highly immunostimulatory, being capable of triggering endotoxic shock even when encountered in small quantities in the bloodstream.
In striking contrast, P. gingivalis lipid A is a poor activator of TLR4 (26–28). The bacterium exhibits a heterogeneous population of lipid A structures comprising penta- and tetra-acylated molecules that are bis-, mono-, or nonphosphorylated. The initial structure synthesized is penta-acylated and bisphosphorylated with C15 to C17 acyl chain lengths (Fig. 1) and is a moderate TLR4 agonist (29). Under standard growth conditions, it is subject to the action of three lipid A-modifying enzymes leading to a lipid A profile that is heavily biased toward a tetra-acylated, nonphosphorylated structure (29). The three distinct enzymatic activities that modify P. gingivalis lipid A are C-1-phosphatase, C-4′-phosphatase, and 3′-O-deacylase (Fig. 1). When P. gingivalis is grown under conditions with high hemin, hemin being an iron source required for growth, the C-1-phosphatase is inactive, leading to a shift of tetra-acylated nonphosphorylated lipid A to tetra-acylated C-1-phosphorylated lipid A (30). Notably, both tetra-acylated structures are TLR4 evasive at the TLR4/MD-2 receptor complex. Nonphosphorylated tetra-acylated lipid A is inert for TLR4 activation, whereas C-1-phosphorylated tetra-acylated lipid A is an antagonist of TLR4 activation (29, 31). Our group was the first to describe TLR4 antagonism by P. gingivalis lipid A in vitro, and we have been investigating the mechanistic process as well as clinical implications. Antagonism of TLR4 has the potential to have a community-wide effect, since it can dampen the host response to any Gram-negative member of the oral community. The two tetra-acylated structures are, in short, key to the ability of P. gingivalis to effectively evade the host TLR4 immune attack.
FIG 1.

Modification of the P. gingivalis lipid A structure. Penta-acylated bisphosphorylated lipid A (left), a moderate TLR4 agonist, is the initial lipid A structure synthesized by P. gingivalis. It is modified to tetra-acylated nonphosphorylated lipid A (right), which is silent for TLR4 activation, by the action of C-1- and C-4′-phosphatases and a 3′-O-deacylase. Inhibition of C-1-phosphatase activity leads to the formation of C-1-phosphorylated tetra-acylated lipid A, a TLR4 antagonist.
The C-1- and C-4′-phosphatases were identified by our group, by homology searches, to be encoded by PG1773 and PG1587, respectively (W83 designations) (29). Recently, PG0027 was identified to possess C-1-phosphatase activity as well (32). Deletion of the C-1-phosphatase, PG1773, led to accumulation of C-1-phosphorylated, tetra-acylated lipid A, the TLR4 antagonist. Deletion of the C-4′-phosphatase, PG1587, on the other hand, resulted in formation of C-4′-phosphorylated lipid A, but it was exclusively penta-acylated, suggesting that the C-4′-phosphate moiety needs to be removed for lipid A deacylation to occur. The contribution of lipid A deacylation to attenuation of TLR4-dependent proinflammatory responses, and for facilitating P. gingivalis persistence, has been experimentally demonstrated in vivo in a murine model of infection comparing wild-type (WT) versus ΔPG1587 mutant strains (33).
The gene encoding the lipid A deacylase, which is essential for the ability of P. gingivalis to evade TLR4 sensing, has, however, remained unidentified. Two known bacterial lipid A deacylase-encoding genes are pagL and lpxR, both of which were identified first in Salmonella enterica serovar Typhimurium (34, 35). These two deacylases are distinct from each other both in their primary sequence and in their modes of action, hydrolyzing a C-3 chain and C-3′ acyloxyacyl pair of acyl chains, respectively. A comprehensive search for homologs of pagL and lpxR by us and other groups failed to identify the lipid A deacylase gene in P. gingivalis (34–36). pagL and lpxR have orthologs in other bacteria, but, interestingly, are all restricted to the Proteobacteria phylum. P. gingivalis PG0027 was earlier reported to carry the lipid A deacylase gene (37). This possibility, however, was ruled out both by personal communications with the author, Eric Reynolds, and by the recent finding that PG0027 participates in lipid A C-1-phosphatase activity (32) and has no effect on deacylase function.
In this report, we demonstrate that the lipid A deacylase in P. gingivalis 33277 is encoded by PGN_1123. Identification of the gene began with a screen of a transposon mutant library, employing a functional assay developed for identifying novel mutants that elicit a strong host innate immune response. Using this approach, we identified a candidate mutant that demonstrated potent TLR4 activation, consistent with a lipid A deacylase phenotype. An affected gene in the transposon mutant was shown to be required for lipid A deacylation and identified to be PGN_1123. The encoded protein is highly conserved in P. gingivalis, bearing no significant homology to the two known lipid A deacylases or to other proteins of known function, indicating that PGN_1123 encodes a novel bacterial lipid A deacylase enzyme.
RESULTS
Development of a cell-based functional screen to identify P. gingivalis mutants that stimulate the human innate immune response.
Our goal was to identify novel genetic determinants that play a role in conferring to P. gingivalis its wide-ranging ability to evade immune recognition. We took the approach of screening a P. gingivalis mutant library for mutants that stimulate a high immune response. Monomac 6 (MM6) cells, a human monocyte cell line, were tested to serve as an in vitro model. Infection of MM6 cells with wild-type P. gingivalis 33277 did not induce secretion of the proinflammatory cytokine interleukin-6 (IL-6) in the supernatant, as measured by enzyme-linked immunosorbent assay (ELISA) (Fig. 2A). This is consistent with observations by us and others regarding the ability of P. gingivalis to effectively evade or dampen immune defense mechanisms (38–40). Infection with a 33277 ΔPG1587 mutant, which possesses TLR4-stimulating penta-acylated lipid A in its LPS, on the other hand, stimulated IL-6 secretion. This was in contrast to infection with a ΔPG1773 mutant, which harbors anti-inflammatory tetra-acylated antagonistic lipid A. The gingipain proteases Kgp and Rgp are also known to dampen innate immune responses by, for example, degrading CD14 (41, 42), a coreceptor of TLR2 and TLR4 signaling complexes. Infection of MM6 cells with a 33277 Δkgp mutant indeed led to a higher IL-6 response. The MM6/IL-6 assay hence was validated as a sensitive model system for detecting increased immune-stimulatory activity by P. gingivalis mutants that is conferred by functional loss of an immune-modulating gene.
FIG 2.

The J5-c5 transposon mutant induces a proinflammatory response. The amounts of IL-6 secreted by MM6 cells in response to exposure to 106 or 107 intact P. gingivalis 33277 bacteria versus the isogenic ΔPG1587, ΔPG1773, or Δkgp mutant (A) and the J5-c5 transposon mutant (B and C) are shown. The results are means ± standard deviations (SD) for triplicate samples from either one of two independent experiments (A) or from two separate experiments (B and C). Asterisks denote significant differences in amount of IL-6 secreted relative to that for the wild-type 33277 control (P < 0.01 by 2-tailed unpaired t tests).
Isolation of a P. gingivalis transposon mutant that stimulates a vigorous proinflammatory response.
We used a mariner transposon (Tn) isolated from Bacteroides thetaiotaomicron, a close relative of P. gingivalis, to create a P. gingivalis 33277 mutant library (43). Each round of mutagenesis yielded hundreds of mutants, obtained by selecting for erythromycin resistance conferred by the Tn-encoded erythromycin resistance cassette ermG. The Tn mutants were grown individually in 96-well plates for 2 days, at which point most had grown to visibly high turbidity. MM6 cells, also grown in 96-well plates, were infected with the mutant library and screened for IL-6 secretion by ELISA. The vast majority of ∼1,000 mutants screened exhibited nondetectable levels of IL-6 in the supernatant, similar to the case for the wild-type (WT) strain. A few mutants, ∼5 to 10%, exhibited a pronounced IL-6 response. A vast majority of the “positive-hit” mutants, however, were slow-growing strains, as was apparent by sight in the 96-well Tn mutant-holding plate. We speculated that slow-growing mutants were stimulating an increased inflammatory response due to low levels of expression of immune-suppressing proteases such as Kgp on the bacterial surface. We rescreened the putative positive-hit mutants and obtained one candidate, named J5-c5, which (i) displayed normal growth, similar to that of the WT, and (ii) stimulated IL-6 production, unlike the WT (Fig. 2B). These two features are characteristic phenotypes of ΔPG1587 and Δkgp mutants as well.
The J5-c5 mutant stimulates TLR4-dependent signaling and fails to produce tetra-acylated lipid A.
We investigated whether the proinflammatory response triggered by the J5-c5 Tn mutant was mediated by activation of either TLR2 or TLR4 innate immune receptors by utilizing a HEK293 luciferase reporter assay (44). Nonimmune HEK293 cells were transfected with either TLR2- or TLR4-expressing plasmids, followed by infection with wild-type 33277, J5-c5, or ΔPG1587 intact bacterial cells. As seen in Fig. 3A, J5-c5 stimulated TLR4 strongly relative to 33277, similarl to ΔPG1587 mutants, but did not increase levels of TLR2 stimulation (Fig. 3B). Since this assay measures fold change in activation, TLR4 activation by J5-c5 is highly significant.
FIG 3.

J5-c5 mutants stimulate TLR4 but not TLR2. (A and B) HEK293 cells expressing TLR4/MD-2 (A) or TLR2/mCD14 (B) were infected with wild-type 33277, ΔPG1587, or J5-c5 intact bacterial cells. (C) HEK293 cells expressing TLR4/MD-2 were exposed to LPS isolated from the wild type, ΔPG1587, or the J5-c5 transposon mutant. Fold NF-κB stimulation of infected cells relative to the unstimulated control is plotted on the y axis. The results are means ± SD for triplicate samples from one of two independent experiments. Asterisks denote a significant increase in fold NF-κB stimulation relative to that for the wild-type 33277 control (P < 0.05 by 2-tailed unpaired t tests).
The ability to stimulate TLR4 suggested that the lipid A structure in J5-c5 is different from that of the wild type, which was confirmed by subjecting purified LPS to a TLR4 assay. LPS from J5-c5, similar to LPS from ΔPG1587 mutants, stimulated TLR4 strongly relative to WT 33277 LPS (Fig. 3C), suggesting that the J5-c5 lipid A acylation status is shifted to a more penta-acylated profile. We ruled out the possibility that the PG1587 gene, whose C-4′-phosphatase activity was shown by us to be required for lipid A deacylation, is affected in J5-c5 by assaying for polymyxin B sensitivity. A characteristic feature of ΔPG1587 mutants is sensitivity to the cationic antimicrobial peptide polymyxin B, because of its penta-acylated lipid A being C-4′-phosphorylated (29). J5-c5 mutants, however, displayed polymyxin B resistance, similar to that of the wild-type bacterium, indicating that the PG1587 gene and the C-4′-phosphatase activity it encodes are intact in J5-c5.
We determined the J5-c5 mutant lipid A structure by matrix-assisted laser desorption ionization–time of flight (MALDI-TOF) mass spectrometry (MS). As seen in Fig. 4B, lipid A of the J5-c5 mutant displayed predominantly penta-acylated nonphosphorylated lipid A when viewed in positive-ion mode. This profile was strikingly different from that of the wild type, which displayed predominantly tetra-acylated nonphosphorylated lipid A (Fig. 4A). Tetra-acylated lipid A was abrogated to an undetectable level in J5-c5, indicating that deacylase function is absent or severely compromised.
FIG 4.

Lipid A of the J5-c5 mutant is predominantly penta-acylated. MALDI-TOF mass spectrometry of the lipid A structure was conducted in positive-ion (A and B) and negative-ion (C and D) modes, which detect nonphosphorylated and phosphorylated structures, respectively. Tetra-acylated nonphosphorylated lipid A is seen in wild-type 33277 (A) but not in the J5-c5 mutant (B), which instead exhibits penta-acylated nonphosphorylated lipid A. With growth under high-hemin conditions, tetra-acylated monophosphorylated antagonist is detected in the wild type (C) but not in the J5-c5 mutant (D), which instead displays penta-acylated lipid A that is either mono- or bisphosphorylated (m/z 1,769). Tetra-acylated lipid A is not detected under either condition in the J5-c5 mutant. Numbers refer to the m/z ratio of the predominant peak in each lipid A cluster.
The lipid A structure was also examined after growth of WT 33277 and the J5-c5 mutant under high-hemin conditions, a growth condition that abrogates C-1-phosphatase activity (30). In the WT, growth in high hemin leads to significant accumulation of antagonistic tetra-acylated C-1-phosphorylated lipid A, as can be viewed in negative-ion mode MALDI-TOF MS (Fig. 4C). This structure, however, was not detected in the largely penta-acylated J5-c5 lipid A profile (Fig. 4D), further confirming that deacylase function is severely compromised in this mutant.
Interestingly, we detected non-, mono-, and bisphosphorylated forms of penta-acylated lipid A in the J5-c5 mutant (Fig. 4B and D). The nonphosphorylated aspect accounts for resistance of J5-c5 to polymyxin B, while the penta-acylated feature accounts for high TLR4 stimulation. Nonphosphorylated penta-acylated lipid A is rarely observed in P. gingivalis 33277, but its production in J5-c5 indicates that both C-1- and C-4′-phosphatases are active, further validating that PG1587 C-4′-phosphatase has not been affected in J5-c5 and hence indicating disruption of a novel gene.
The J5-c5 mutant contains multiple transposon insertions.
Semirandom and nested PCR was used to identify the location of the transposon (Tn) insertion in J5-c5. An insertion was found to be located in an intergenic location, 122 bp downstream of PGN_0782 and 609 bp upstream of PGN_0783. Transposons, however, are capable of jumping to new locations until the replication-incompetent plasmid, which encodes both transposon and transposase, is lost from the population. A precise swap of the transposon with tetQ, a tetracycline resistance cassette, did not eliminate erythromycin resistance, indicating the presence of additional transposons in J5-c5, which was confirmed by PCR (data not shown).
We sequenced the entire J5-c5 genome on Illumina’s MiSeq platform to determine the number and locations of transposons in J5-c5. Analysis of the J5-c5 sequencing reads revealed approximately 5× coverage of Tn sequence relative to the 33277 RefSeq chromosomal sequence, suggesting the presence of five transposons in J5-c5. The insertion site of one of them, Tn-1, precisely matched the location identified by the PCR-based methods described above. The locations of the four other transposons are listed in Table 1.
TABLE 1.
Locations of the five transposon insertions in J5-c5
| Name | Chromosomal position in strain 33277 | Genetic location; protein |
|---|---|---|
| Tn1 | 854777 | Intergenic, 609 bp upstream of PGN_0783; histone-like DNA binding protein |
| Tn2 | 62040 | Intergenic, 52 bp upstream of PGN_0053; hypothetical protein, 408 aa |
| Tn3 | 85364 | In PGN_0081; matE family efflux transporter |
| Tn4 | 1252900 | In PGN_1124; Band 7 protein, 326 aa |
| Tn5 | 1653844 | Intergenic, 523 bp upstream of PGN_1476; T9SS C-terminal target domain-containing protein |
PGN_1123 encodes the lipid A deacylase.
Since it was not clear which one or combination of Tn insertions was responsible for the deacylase phenotype or, indeed, whether the phenotype was due to direct interruption of a deacylase gene or due to an indirect effect via gene regulation, we employed transcriptome sequencing (RNA-seq) to determine the genes that are differentially expressed between wild-type 33277 and the J5-c5 mutant. The top 20 genes displaying the most significant changes in log2 expression values in J5-c5 mutant relative to the wild type, as determined by utilizing edgeR with the default parameters described by Law et al. (45), are listed in Table 2. The statistical parameters of this package identified exactly one gene, PGN_1123, as significantly differentially expressed. PGN_1123 is located immediately downstream of the gene interrupted by Tn4, PGN_1124. Besides PGN_1123, the only genes that displayed >4-fold decreases in log2 expression values were located on one side of Tn1, i.e., PGN_0783 and PGN_784.
TABLE 2.
Top 20 most significant changes in gene expression between the wild-type and J5-c5 transposon mutant strains as revealed by edgeR analysis
| Name | Product | Log2 fold change | P value |
|---|---|---|---|
| PGN_1123 | Hypothetical protein | −4.71590586 | 3.66E−06 |
| PGN_1281 | Conjugative transposon protein TraM | −2.276704451 | 0.000305862 |
| PGN_0783 | DNA binding protein | −7.281703828 | 0.00035261 |
| PGN_0090 | DNA binding protein | −2.304121237 | 0.000717639 |
| PGN_0066 | Transposase | −2.142286175 | 0.000941805 |
| PGN_0060 | Conjugative transposon protein TraM | −2.198185031 | 0.000882388 |
| PGN_1124 | Paraslipin | −1.690660121 | 0.001456315 |
| PGN_0084 | DNA topoisomerase I | −1.699090077 | 0.001497081 |
| PGN_0083 | Hypothetical protein | −1.703911427 | 0.001862623 |
| PGN_0065 | Transposase | −1.954910211 | 0.001886738 |
| PGN_1283 | Conjugal transfer protein TraO | −1.856001435 | 0.002120805 |
| PGN_0784 | Hypothetical protein | −5.588007472 | 0.002152355 |
| PGN_0061 | Transposase | −2.4521437 | 0.002448023 |
| PGN_0064 | Transposase | −1.800519514 | 0.002979058 |
| PGN_1282 | Conjugative transposon protein TraN | −1.636844785 | 0.003527579 |
| PGN_0063 | Conjugative transposon protein TraJ | −1.868293938 | 0.004728484 |
| PGN_0194 | Hypothetical protein | 3.227338786 | 0.004526265 |
| PGN_0074 | Hypothetical protein | −2.247462691 | 0.007013359 |
| PGN_0901 | 6,7-Dimethyl-8-ribityllumazine synthase | 2.462105689 | 0.00738567 |
| PGN_0067 | Transposase | −1.732652686 | 0.008098611 |
We targeted the PGN_1123, PGN_1124, and PGN_0783 genes for deletion analysis in P. gingivalis 33277. Deletion mutations were constructed by replacing each gene with a nonpolar erythromycin resistance cassette, ermF, using homologous recombination.
The deletion mutation in PGN_1123 was found to confer the lipid A deacylase phenotype, as demonstrated by structural and functional analyses. Lipid A of the ΔPGN_1123 mutant was devoid of tetra-acylated structures, and the mutant displayed predominantly penta-acylated nonphosphorylated lipid A (Fig. 5A), similar to the case for the J5-c5 Tn mutant, as determined by MALDI-TOF MS. On the other hand, both the ΔPGN_1124::ermF and ΔPGN_0783::ermF mutants displayed the wild-type tetra-acylated phenotype as seen in Fig. 4A (data not shown).
FIG 5.

The ΔPGN_1123 mutant is devoid of tetra-acylated lipid A. (A) Lipid A preparations of ΔPGN_1123::ermF mutants exhibit penta-acylated nonphosphorylated lipid A as detected by MALDI-TOF MS in positive-ion mode and a distinct absence of tetra-acylated lipid A. (B) ΔPGN_1123 mutants expressing the PGN_1123 gene in trans from pSJ836 display tetra-acylated nonphosphorylated lipid A, similar to that of the wild type, indicating complementation of lipid A deacylase function. Numbers refer to the m/z ratio of the predominant peak in each lipid A cluster.
We were successful in complementing the ΔPGN_1123::ermF deacylase phenotype by introducing a plasmid expressing PGN_1123 in trans. The PGN_1123 gene was cloned into pSJ46, a pT-COW-based plasmid vector capable of replicating in both E. coli and P. gingivalis, generating the plasmid pSJ836. Conjugation was used to mobilize pSJ836 from E. coli to ΔPGN_1123::ermF mutants, and expression of the PGN_1123 gene was confirmed by reverse transcription-PCR (RT-PCR) (data not shown). Analysis of ΔPGN_1123::ermF (pSJ836) lipid A demonstrated the presence of tetra-acylated nonphosphorylated structures, similar to the case for the wild type (Fig. 5B). Complementation of the ΔPGN_1123::ermF penta-acylated lipid A phenotype to tetra-acylated lipid A in ΔPGN_1123(pSJ836) confirmed that PGN_1123 is required for lipid A deacylation.
Consistent with an absence of tetra-acylated lipid A, infection of TLR4-expressing HEK293 cells with ΔPGN_1123::ermF mutant whole cells induced potent TLR4 stimulation (Fig. 6), similar to the case for J5-c5. ΔPGN_1124 mutants, on the other hand, displayed a nonactivating phenotype similar to that of the wild type, indicating that the gene interrupted by the Tn responsible for the deacylase phenotype due to polar effects on PGN_1123 itself does not play a role in TLR4 evasion. Introduction of pSJ836, the plasmid expressing PGN_1123, into ΔPGN_1123 mutants resulted in a substantial reduction of TLR4 stimulation, confirming complementation of the lipid A deacylase phenotype.
FIG 6.
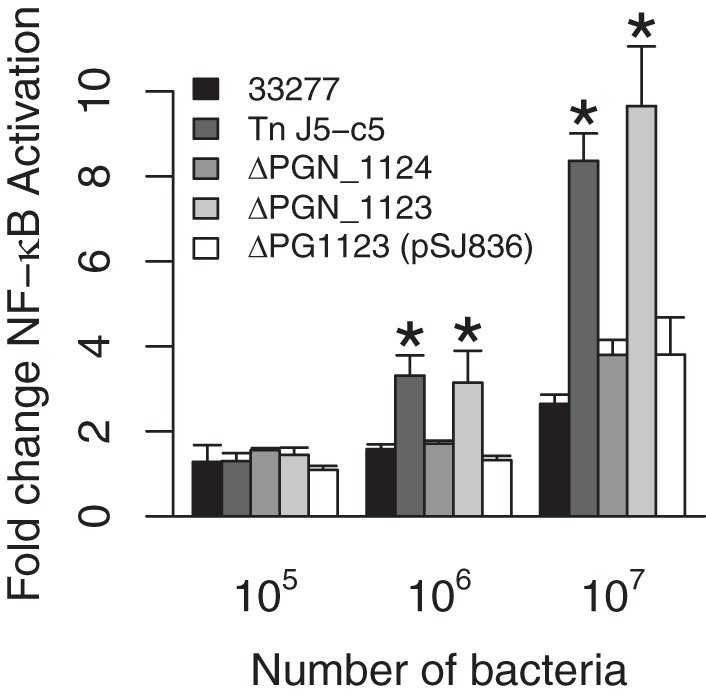
ΔPGN_1123::ermF mutants stimulate TLR4. HEK293 cells expressing TLR4/MD-2 were stimulated with wild-type 33277, J5-c5 transposon mutant, or isogenic ΔPGN_1124, ΔPGN_1123, or ΔPGN_1123(pSJ836) whole bacterial cells. The last sample, ΔPGN_1123 (pSJ836), expresses PGN_1123 from pSJ836 in trans. Fold NF-κB stimulation of infected cells relative to the unstimulated control is plotted on the y axis. The results are means ± SD for triplicate samples from one of three independent experiments. Asterisks denote a significant increase in fold NF-κB stimulation relative to that for the wild-type 33277 control (P < 0.05 by 2-tailed unpaired t tests).
Furthermore, replacement of Tn4 alone, out of the five Tns present in J5-c5, with a nonpolar tetracycline resistance cassette also restored lipid A deacylase activity (data not shown), confirming that Tn4 was the sole Tn in J5-c5 responsible for conferring the lipid A deacylase phenotype, specifically by exerting polar effects on downstream gene transcription.
PGN_1123 encodes a novel phylogenetically restricted deacylase and is part of an operon.
PGN_1123 is annotated to encode a conserved hypothetical protein comprising 399 amino acid residues. Homology searches did not reveal any obvious similarity to a protein of known function, and the protein does not display known motifs. PGN_1123 is highly conserved in P. gingivalis, with homologs displaying 92 to 98% identity present in all sequenced P. gingivalis strains. An ortholog with 92% identity was found in Porphyromonas gulae, after which identity to the next closest homolog fell to 43%, in P. crevioricanis. Orthologs with 30 to 35% identity were found in several bacteria, all belonging to the Bacteroidetes phylum, including Prevotella stercorea (35% identity), Tannerella forsythia (36% identity), Parabacteroides goldsteinii (35% identity), and Bacteroides fragilis (33% identity). Comparison of the PGN_1123 protein sequence with those of the known deacylases PagL and LpxR revealed low identity (<15%). Since PagL and LpxR are not orthologs of each other either and exhibit differences in their modes of action, PGN_1123 represents a third distinct class of bacterial lipid A deacylase enzymes.
Both PagL and LpxR have been shown to be outer membrane proteins with β-barrel structures (46, 47). Using several web-based tools, PGN_1123 is predicted to have a signal sequence, required for secretion across the inner membrane in Gram-negative bacteria, with a potential cleavage site located after the 19th amino acid (aa). One of several tertiary structure predictions of PGN_1123 using the I-TASSER server at the University of Michigan (48) revealed a β-barrel structure.
The start codon of the PGN_1123 gene overlaps the stop codon of the gene upstream, PGN_1124, suggesting that the two genes are translationally coupled. Indeed, it has been demonstrated that the homologs in P. gingivalis W83, PG1334 and PG1333, are cotranscribed together with the gene upstream, PG1335 (49), indicating that the three genes are organized in an operon. The first gene of the operon, PG1335 in W83, is 27 bp upstream of the second gene, PG1334. The three genes, PGN_1125, PGN_1124, and PGN_1123 (PG1335, PG1334, and PG1333 in W83), are all conserved in sequenced P. gingivalis strains.
Interestingly, PG1334, the middle gene of the operon in W83, was identified in an in vivo-induced antigen technology (IVIAT) study as a gene that is induced in vivo during human periodontitis (50). Subsequent quantitative RT-PCR studies by Walters et al. confirmed that the PG1334 transcript is detected more frequently in dental plaque samples from periodontal disease sites than from healthy sites after taking into account P. gingivalis abundance, consistent with induction of expression during disease (49).
We conducted semiquantitative RT-PCR (Fig. 7) to confirm that the three genes are cotranscribed in 33277 as well. We detected mRNA overlapping the PGN_1125 and PGN_1124 genes, PGN_1124 and PGN_1123, and, importantly, PGN_1125 and PGN_1123, the first and last genes, demonstrating that they form an operon. This strongly suggests that PGN_1123 is induced in vivo during disease.
FIG 7.
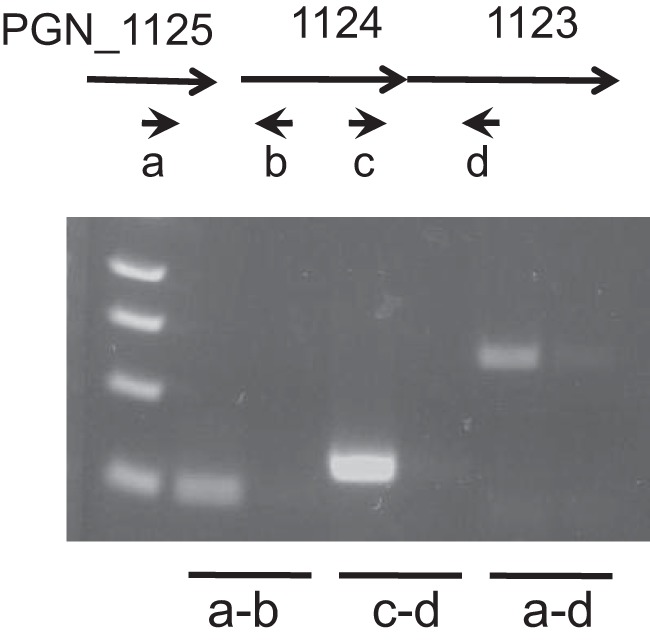
PGN_1123 is in an operon with the upstream PGN_1125 and PGN_1124 genes. RT-PCR of mRNA from wild-type 33277 using primers a and b overlapping PGN_1125 and PGN_1124 (lane 2), primers c and d overlapping PGN_1124 and PGN_1123 (lane 4), and primers a and d overlapping PGN_1125 PGN_1123 (lane 6) is shown. Reverse transcriptase is absent in lanes 3, 5, and 7, which use the same primer sets as for lanes 2, 4, and 6, respectively. A molecular weight ladder is in lane 1.
PGN_1123 deacylates Bacteroides thetaiotaomicron lipid A.
In order to obtain definitive evidence that PGN_1123 is a structural gene encoding the lipid A deacylase, we expressed PGN_1123 heterologously in B. thetaiotaomicron, a phylogenetically close relative of P. gingivalis; both belong to the order Bacteroidales in the phylum Bacteroidetes. Lipid A from B. thetaiotaomicron is penta-acylated and C-1-phosphorylated (Fig. 8A, m/z 1,675), structurally similar to the penta-acylated, C-4′-phosphorylated lipid A cluster seen in P. gingivalis. To our knowledge, tetra-acylated lipid A has not been observed before in B. thetaiotaomicron, nor does the bacterium contain a PGN_1123 homolog when searched with BLAST tools, which is in marked contrast to the case for B. fragilis. B. thetaiotaomicron LPS is a strong activator of TLR4 (stronger than P. gingivalis LPS) due to location of the phosphate group at the C-1 position instead of the C-4′ position (51). Strong TLR4 activation by B. thetaiotaomicron LPS, and by whole bacteria, suggests that, unlike in P. gingivalis, there are no attenuating tetra-acylated lipid A structures capable of dampening TLR4 activation.
FIG 8.

B. thetaiotaomicron lipid A is deacylated by PGN_1123. (A and B) Lipid A structure of B. thetaiotaomicron with empty plasmid vector pSJ46 (A) or PGN_1123-expressing plasmid pSJ836 (B) as detected by MALDI-TOF MS in negative-ion mode. (C) HEK293 cells expressing TLR4/MD-2 were stimulated with B. thetaiotaomicron(pSJ46) or B. thetaiotaomicron(pSJ836) intact bacterial cells. Fold NF-κB stimulation of infected cells relative to an unstimulated control is plotted on the y axis. The results are means ± SD for triplicate samples from one of two independent experiments. Asterisks denote a significant decrease in fold NF-κB stimulation relative to that for B. thetaiotaomicron(pSJ46) (P < 0.01 by 2-tailed unpaired t tests).
Consistent with PGN_1123 encoding deacylase activity, B. thetaiotaomicron harboring pSJ836 in trans led to deacylation of penta-acylated monophosphorylated lipid A to tetra-acylated monophosphorylated lipid A (m/z 1,420), as revealed by MALDI-TOF MS in the negative-ion mode (Fig. 8B). This marks the first time we have observed tetra-acylated lipid A in B. thetaiotaomicron. A HEK293 TLR4 assay revealed that TLR4 activation by B. thetaiotaomicron(pSJ836) intact bacteria was substantially lower than that by B. thetaiotaomicron containing vector alone (Fig. 8C), providing novel evidence that expression of the PGN_1123 gene product is sufficient to confer a TLR4-evasive phenotype in B. thetaiotaomicron. Lipid A deacylation in B. thetaiotaomicron by heterologous expression of PGN_1123 confirms that PGN_1123 encodes a lipid A deacylase.
DISCUSSION
We report identification of PGN_1123 as the lipid A deacylase-encoding gene in P. gingivalis, a gene that plays a pivotal role in evasion and suppression of the TLR4-mediated host innate immune response. Identification of this gene has been sought since TLR4 antagonism by tetra-acylated lipid A in P. gingivalis was first described, over 2 decades ago. The reason for the inability to identify this gene by homology searches is now clear, since PGN_1123 does not display homology to the two known deacylases, PagL and LpxR. Additionally, the sequence of PGN_1123 is unique in that it does not have close homologs outside P. gingivalis (with the exception of P. gulae), nor does it contain motifs that could inform us of its function. In this report, we have shown that PGN_1123 is required for lipid A deacylation by structural and functional studies and have established that it encodes the lipid A deacylase enzyme by demonstrating deacylation following heterologous expression in B. thetaiotaomicron.
Lipid A is a fundamental component of the bacterial outer membrane. Remodeling of the membrane by lipid A deacylation has been shown to play a significant role in pathogenesis in several bacteria on account of its moderating effect on the immune response. Lipid A deacylation in B. thetaiotaomicron as mediated by PGN_1123 did indeed attenuate TLR4 activation, leading us to speculate on the reason for the absence of the genetic capability for deacylation in this intestinal commensal bacterium, which results in synthesis of a lipid A structure that is an unequivocal stimulator of TLR4. B. fragilis, belonging to the same genus and considered an opportunistic pathogen, on the other hand, possesses an ortholog of the PGN_1123 protein, suggesting that it has the capacity to undergo lipid A deacylation. This bacterium, however, has not been shown to display deacylated lipid A when grown in vitro. Further studies are warranted to more fully understand the expression of putative deacylase genes in Bacteroides spp. Notably, PGN_1123 belongs to a third distinct class of bacterial lipid A deacylase enzymes, and it remains feasible that there are other novel classes that remain unidentified.
Many pathogens possess the ability to disrupt TLR4 signaling by lipid A deacylation. S. Typhimurium, for example, undergoes pagL-dependent lipid A deacylation in response to low pH and low Ca2+/Mg2+ concentrations, as would be encountered in phagolysosomes, suggesting that deacylation is important for pathogenesis (52). Pseudomonas aeruginosa lipid A was shown to undergo deacylation in isolates from cystic fibrosis patients by upregulation of pagL expression, which was not seen in environmental isolates (53). A striking example of deacylated lipid A contributing to pathogenesis is seen in Yersinia pestis, the etiologic agent of bubonic plague, which shuttles between a flea vector and humans. When incubated at 27°C, the environmental temperature of its flea vector, lipid A is composed of predominantly hexa-acylated lipid A structures, but when incubated at 37°C, the mammalian host temperature, a shift to a tetra-acylated form was observed (54, 55). The temperature-dependent shift in lipid A structure was shown to occur in Yersinia enterocolitica and Yersinia pseudotuberculosis as well, and lpxR activity was demonstrated to remove the 3′-acyl-oxy-acyl chain in Y. enterocolitica (56). Tetra-acylated lipid A in Y. pestis, which is considered to occur due to decreased acyl transferase activity, rendered the bacterium silent to TLR4 recognition and reduced the inflammatory response, hence potentially allowing the pathogen to proliferate undetected in the bloodstream during early stages of infection. Interestingly, genetic modification of tetra-acylated lipid A to a hexa-acylated structure led to potent TLR4 stimulation but rendered the strain avirulent in a mouse model of infection (57), highlighting the important role of a reduced number of acyl chains in pathogenesis.
P. gingivalis is a unique member of the subgingival plaque microbial community in that it has the distinct capacity to foil TLR4 signaling, even antagonizing it, giving it the capacity to protect itself and, potentially, bystander bacteria in the plaque community as well. This is consistent with the observation that P. gingivalis is observed in high abundance in diseased plaque sites and is also associated with an increase in microbial load and an alteration in plaque microbial composition in animal models of infection. P. gingivalis hence has pathogenic properties that can be significantly attributed to TLR4 evasion by tetra-acylated lipid A. The phenotype of P. gingivalis ΔPGN_1123 mutants in vivo with respect to colonization, instigation of dysbiosis, and chronic inflammation remains to be tested. Mutants displaying penta-acylated lipid A due to deletion of the PG1587 gene were shown to stimulate a proinflammatory response and displayed reduced survival in a mouse macrophage model of infection (33). ΔPG1587 mutants were shown to be defective for colonization in a rabbit model of infection as well (17), indicating that lipid A deacylation is required for pathogenesis. The distinction, if any, between the penta-acylated lipid A mutants, the ΔPGN_1123 and ΔPG1587 mutants, which possess largely nonphosphorylated lipid A versus exclusively C-4′-phosphorylated lipid A, respectively, will become clear from future in vivo studies.
Expression of the PGN_1123 gene is predicted to be upregulated during disease as inferred from upregulation of the cotranscribed PGN_1124 gene during disease. Gene regulation has been reported for both pagL and lpxR deacylases, by several molecular mechanisms and in response to environmental conditions. pagL transcription in S. Typhimurium, for example, is activated by the two-component regulator PhoPQ in response to different host microenvironments that include acidic pH, cationic antimicrobial peptides, and depletion of Ca2+ and Mg2+ (34). Noncoding small RNA (sRNA) molecules are known to modulate expression of outer membrane proteins and have been shown to regulate both pagL and lpxR. MicF is an sRNA that binds to lpxR transcripts in Salmonella, increasing degradation of the mRNA by exposing regions that are susceptible to RNase E (58). A recent study identified an sRNA, Sr006, as a positive regulator of pagL expression in P. aeruginosa, leading to a reduction in the proinflammatory response (59). Another recent study in enterohemorrhagic E. coli (EHEC) identified two virulence regulators, Ler and Pch, as positive regulators of lpxR (60).
Increased expression of PGN_1123 during disease would correlate with enhanced survival under inflammatory conditions. The cotranscribed PGN_1124, shown to be expressed prominently during disease, has a motif indicating it belongs to the Band 7 family of proteins, a family conserved from protozoa to mammals. Also called the slipin (stomatin-like integral membrane protein) family, it is implicated in regulating membrane protein turnover in stomatins and other membrane-associated proteins. It is often cotranscribed with a gene encoding a membrane protease, which in P. gingivalis is predicted to be encoded by PGN_1125, the first gene of the operon. Genes in an operon often are part of the same functional pathway, but since a deletion mutation in PGN_1124 did not affect lipid A deacylase function (Fig. 6) or structure (data not shown), the reason for these three genes to be in an operon is not clear. Interestingly, the transcript overlapping PGN_1124 and PGN_1123, the second and third genes of the operon, was more abundant than that overlapping the first two genes or all three genes (Fig. 7), indicating an additional level of transcriptional control for PGN_1123 expression independent of the operon.
In addition to TLR4 evasion, it is becoming established that lipid A deacylation contributes to pathogenesis in bacteria by facilitating the formation of outer membrane vesicles (OMVs) (61–63). The presence of fewer acyl chains in the lipid A moiety leads to a reduction in the hydrophobic area of the molecule. The associated change in membrane structure, from a cone to either a cylinder or an inverted cone, has been proposed to facilitate increased membrane curvature, the first step toward outer membrane blebbing and OMV formation (64).
P. gingivalis is known to release a large number of OMVs, which may be a function of its largely tetra-acylated lipid A profile. OMVs from P. gingivalis mediate coaggregation with other bacteria and heme acquisition and, furthermore, are known to invade host epithelial tissue themselves with, as it turns out, a large arsenal of virulence proteins (62, 63). The proteomic and LPS contents of P. gingivalis OMVs have been shown to be different from those of the outer membrane by having (i) elevated levels of proteins secreted by the type IX secretion system (T9SS), which include gingipain proteases and other virulence factors, and (ii) an increased proportion of anionic A-LPS relative to neutral O-LPS (65, 66). Colocalization of type IX secreted proteins and A-LPS is consistent with the finding that T9SS proteins use A-LPS as an anchor for attachment to the outer membrane (67–69). Interestingly, many bacteria have been shown to pack a preferential cargo of proteins into their OMVs (70–75), suggesting that membrane remodeling occurs prior to, and may facilitate, vesiculation.
The lipid A content of P. gingivalis OMVs was also shown to be different from that of the outer membrane by being nearly devoid of penta-acylated lipid A structures and instead composed of tetra- and/or triacylated lipid A, leading the authors to conclude that lipid A in OMVs is more heavily deacylated than lipid A in outer membranes (32, 65). Interestingly, a recent study on an S. Typhimurium strain expressing recombinant pagL deacylase demonstrated lipid A deacylation from hexa- to penta-acylated structures as expected, but this, notably, led to increased vesiculation (61). Moreover, the deacylated (penta-acyl) lipid A was detected exclusively in OMVs and not in the bacterial outer membrane, suggesting that PagL-mediated changes in membrane structure contributed to membrane curvature and to formation of OMVs.
We recently reported in a collaborative study that nondeacylated (penta-acyl) lipid A from P. gingivalis ΔPG1587 mutants is not only an agonist for TLR4 stimulation but also an agonist of inflammasome activation (33), an immune pathway that targets intracellular cytosolic bacteria for elimination by pyroptosis. Bacterial lipid A has been shown to bind murine caspase-11 and its homolog in humans, caspase-4/5, resulting in inflammasome activation by the noncanonical pathway (76, 77). Underacylated lipid A, on the other hand, specifically tetra-acylated forms, was shown to bind caspases but failed to induce activation. Hence, lipid A structural modifications that abrogate TLR4 activation also abrogate inflammasome activation, as has been shown by us with P. gingivalis and by others in bacteria such as Francisella novicida.
In summary, lipid A deacylation has been shown to be involved in a wide range of functions that each contributes to pathogenesis. Identification of PGN_1123 as the deacylase-encoding gene in P. gingivalis will now enable us to gain further insights into the precise role of deacylation in immune evasion and virulence factor expression and, furthermore, the potential contribution in vivo toward survival, dysbiosis, and chronic inflammation. Identification also lays the foundation for investigating the use of deacylase gene expression as a potential diagnostic tool, and inhibition of the enzyme as a potential therapeutic, for periodontitis.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
P. gingivalis 33277, Bacteroides thetaiotaomicron ATCC 29148, and E. coli DH10b were obtained from our culture collection. E. coli SM10λpir(pSAM_Bt) was obtained from Andrew Goodman’s laboratory (43). P. gingivalis strains were grown on blood agar plates containing 5% sheep’s blood and in TYHK broth (30 g/liter Trypticase soy broth, 5 g/liter yeast extract, and 1 mg/liter vitamin K3). Following sterilization by autoclaving, filter-sterilized hemin was added to TYHK broth, just prior to inoculation, to a final concentration of 1 μg/ml. Hemin was added at 10 μg/ml for growth under high-hemin conditions. TYHK agar plates were used for growth of P. gingivalis on solid medium as well. Hemin (1 μg/ml) and antibiotics were added following sterilization. Antibiotics were added to the following concentrations: erythromycin (Erm), 5 μg/ml; tetracycline (Tet), 0.5 μg/ml. E. coli strains were grown in Luria broth (10 g/liter tryptone, 5 g/liter yeast extract, 5 g/liter NaCl); 100 μg/ml ampicillin was added when required for selection. B. thetaiotaomicron was grown on TYHK agar plates and in TYHK broth containing 1 μg/ml hemin. All anaerobic strains were grown in an anaerobic growth chamber (5% H2, 5% CO2, 90% N2) at 37°C.
Transposon mutagenesis.
Conjugation was used to transfer the mariner transposon-containing plasmid pSAM_Bt from E. coli SM10λpir to P. gingivalis 33277. pSAM_Bt encodes the (i) RP4 origin of transfer, which is required for mobilization by biparental mating, (ii) the mariner transposon, encompassing an ermG cassette that confers erythromycin resistance, and (iii) the transposase encoding gene, which is required for transposition. P. gingivalis 33277 (100 ml) and SM10λpir(pSAM_Bt) (25 ml) were grown to mid-log phase to optical densities at 600 nm (OD600) of ∼1.0 and ∼0.5, respectively. After pelleting and resuspending each culture in 1 ml TYK broth, the two cultures were mixed, and 100-μl aliquots were spotted on blood agar plates and incubated overnight aerobically. The bacterial mixes from 8 to 10 spots were transferred using sterile swabs into 10 ml phosphate-buffered saline (PBS), spun, and resuspended in 1.5 ml TYK broth. Aliquots (100 μl) were plated on TYHK plates containing 5 μg/ml erythromycin for selection of transposon-containing 33277 strains and containing 50 μg/ml gentamicin for counterselection of E. coli. Transposon-containing erythromycin- and gentamicin-resistant 33277 colonies were seen after 4 to 5 days of anaerobic incubation.
Transposon mutant candidate colonies were patched on TYHK-Erm-Gm plates at 48 colonies per plate. After 3 to 4 days of growth, they were inoculated into 150 μl TYHK-Erm medium in 96-well plates. Two days later, they were used for infection of MM6 cells.
IL-6 production from MM6 cells.
Monomac 6 (MM6) cells were grown in medium comprising RPMI 1640 supplemented with 10% heat-inactivated fetal bovine serum, 1% penicillin-streptomycin, 1% nonessential amino acids, and 1% OPI medium supplement, and then 2.5 × 105 cells were seeded in 96-well plates in 200-μl aliquots. Twenty-four hours later, the cells were infected with 20 μl of each Tn mutant that had been grown for 2 days in the 96-well holding plate. At 24 h after infection, the cells were spun down in the 96-well plate, and the amount of IL-6 in 100 μl supernatant was measured by ELISA according to the manufacturer’s instructions (eBioscience).
Preparation of LPS and isolation of lipid A.
Bacteria were cultured for 48 h in TYHK medium containing 1 μg/ml hemin or 10 μg/ml hemin (for growth under high-hemin conditions). LPS was isolated from 150 ml of culture using the Tri-reagent protocol as previously described (44). To generate lipid A, dried LPS samples were resuspended in 10 mM sodium acetate (pH 4.5) containing 1% sodium dodecyl sulfate. The solution was heated for 1 h at 100°C, followed by lyophilization overnight. The resulting dried lipid A was resuspended in ice-cold 95% ethanol containing 0.02 N HCl, spun, washed three times in 95% ethanol, and subjected to a final extraction with 1,160 μl of chloroform-methanol-water (1:1:0.9), a biphasic solution that separates residual carbohydrate contaminants into the aqueous phase. The chloroform layer containing the lipid A was dried in a fume hood and used for MALDI-TOF MS analyses.
MALDI-TOF MS.
Lipid A samples were dissolved in 10 μl norharmane matrix, which was prepared by adding 10 mg norharmane to 1 ml of a 1:1 chloroform-methanol solution and mixing well. One microliter of each sample was analyzed in both positive- and negative-ion modes on an AutoFlex II Analyzer (Bruker Daltonics). Data were acquired with a 50-Hz repletion rate, and up to 3,000 shots were accumulated for each spectrum. Instrument calibration and all other tuning parameters were optimized using HP Calmix (Sigma-Aldrich). Data were acquired and processed using Flex Analysis software (Bruker Daltonics).
HEK293 TLR2 and TLR4 assays.
The HEK293 TLR2 and TLR4 assays were performed as previously described (44). Briefly, HEK293 cells were plated in 96-well plates and transfected the following day with plasmids encoding human TLRs, NF-κB-dependent firefly luciferase reporter, and β-actin promoter-dependent Renilla luciferase reporter. In the case of human TLR4, 0.002 μg plasmid encoding human TLR4 was cotransfected with 0.0025 μg plasmid encoding human MD-2. In the case of human TLR2, 0.001 μg plasmid encoding TLR2 was cotransfected with 0.002 μg plasmid encoding human mCD14. At 18 to 20 h posttransfection, test wells were stimulated in triplicate for 4 h at 37°C with various doses of sample, which were suspended in Dulbecco’s modified Eagle medium (DMEM) containing 10% human serum. For stimulation with intact bacteria, 1-ml cultures of the indicated strains were first washed with TYHK, and their concentration was estimated by measuring the optical density at 600 nm. Luciferase activity was assayed using a dual-luciferase assay reporter system (Promega, Madison, WI). NF-κB activity was measured as the ratio of NF-κB-dependent firefly luciferase activity to β-actin promoter-dependent Renilla luciferase activity, which served as an internal standard. The data were plotted as the fold difference between the NF-κB activity of the sample and that of the unstimulated control.
Nested PCR to identify transposon insertion sites.
Semirandom primer Arb1 (78) was used with a primer reading outwards from the mariner transposon, SamSeq2 (43), to amplify the transposon-chromosome junction by PCR. Several fragments obtained were gel extracted together and used as the template for a nested PCR product using primers Arb2 (same as Arb1 but lacking the semirandom N nucleotides) and SamSeq2, a primer reading outwards and closer to the edge of the transposon than SamSeq1. A prominent 650-bp PCR product was obtained, which was cleaned and sent for Sanger sequencing at Genewiz.
Whole-genome sequencing and analysis.
Genomic DNA was purified from P. gingivalis cultures using the DNeasy blood and tissue kit (Qiagen). Genomic DNAs from wild-type 33277 and the transposon mutant J5-c5 were subjected to paired-end, whole-genome sequencing on Illumina’s MiSeq platform.
Transposon insertion sites in J5-c5 were identified by aligning reads spanning junction sites between the genomic sequence and the termini of the transposon. The reads were first trimmed and filtered based on quality analysis by FastQC. Next, they were aligned against the P. gingivalis 33277 reference genome using Bowtie2, and unalignable reads were aligned against the sequence of the mariner transposon. The ratio of whole-genome coverage to mariner Tn coverage in J5-c5 was 4.4, suggesting that there were at least four, and perhaps five, transposon insertions. Reads that could not be aligned strictly within the 33277 reference genome or mariner Tn sequence were assumed to be enriched in junction spanners. These reads were searched for the termini of the mariner Tn, and reads confirmed to be positive for the Tn terminus were subjected to BLAST analysis, from which genomic sequences flanking the insertion could be inferred. The precise insertion sites inferred by this method were verified by amplifying the junction site using PCR and sequencing the PCR product.
RNA-seq.
RNA was purified from P. gingivalis cultures that were treated with TRIzol (Invitrogen) to facilitate cell lysis. Briefly, a fully grown 3-ml culture was spun, the pellet was brought up in 900 μl TRIzol and mixed with 200 μl chloroform, and the sample was spun for 15 min. The upper aqueous layer was transferred to a new tube, and an equal volume of isopropanol was added, mixed well, and frozen in a −80°C freezer. The next day, the samples were thawed and spun, and the pellet was washed with 70% ethanol and resuspended in water. Twenty micrograms of this sample, comprising crude RNA, was subjected to DNase treatment using RQ1 DNase (Promega). The sample was again subjected to TRIzol and chloroform treatment and spun, and the upper aqueous layer was concentrated using the Clean and Concentrator kit (Zymo Research) according to the manufacturer’s instructions. RNA-seq was performed on three independent biological samples of 33277 and J5-c5 RNA using Illumina’s HiSeq platform at the University of Washington Center for Precision Diagnostics.
RNA-seq data were analyzed by closely following the guidance outlined by Law et al. (45).
Construction of P. gingivalis 33277 mutants.
Deletion mutations were constructed in P. gingivalis 33277 by replacing the gene of interest with either an erythromycin resistance cassette, ermF, or a tetracycline resistance cassette, tetQ. Flanking sequences of the gene to be deleted, ∼500 to 700 bp upstream and 500 to 700 bp downstream, respectively, were amplified by PCR using genomic DNA from P. gingivalis 33277 as the template. The primers used for the upstream flanking sequence, a and b, and for the downstream flanking sequence, c and d, are listed in Table 3 for ΔPGN_1123::erm, for ΔPGN_1124::erm and Δkgp::erm deletion constructs, and for replacement of Tn1 with tetQ in J5-c5. The b and c primers were designed to be overlapping primers that are reverse complements of each other. In general, the forward primer c comprised 15 nucleotides specific to the end of the upstream flanking sequence, an XbaI restriction site, and 15 nucleotides specific to the beginning of the downstream flanking sequence. The upstream and downstream flanking fragments were first amplified separately using a-b and c-d primer sets, respectively. The two products were cleaned and mixed together, and outer primers a-d were used to amplify the aligned product that comprised both flanks. The amplified flanks were ligated by TA cloning into pGEM-T Easy, a narrow-host-range vector that can propagate in E. coli but is a suicide vector in P. gingivalis. Finally, an ermF or tetQ cassette was introduced between the flanking sequences into the XbaI site to complete each deletion construct.
TABLE 3.
Primers used in this study
| Oligonucleotide | Specificity | Orientation | Sequencea |
|---|---|---|---|
| Δ1124-a | PGN_1124 upstream flanking sequence | Forward | AGACTCAATAGCAGATCGACG |
| Δ1124-b | PGN_1124 upstream flanking sequence | Reverse | TTCATACCGCCCAAGgatCCTGTTGCTGTCATA |
| Δ1124-c | PGN_1124 downstream flanking sequence | Forward | TATGACAGCAACAGGatcCTTGGGCGGTATGAA |
| Δ1124-d | PGN_1124 downstream flanking sequence | Reverse | GACCCAAACATCCATTCGG |
| Δ1124-confirm | PGN_1124 | Reverse | TCTCATACATGGCACGCAT |
| Δ1123-a | PGN_1123 upstream flanking sequence | Forward | ATGCGTGCCATGTATGAGA |
| Δ1123-b | PGN_1123 upstream flanking sequence | Reverse | GGACTGTCAAAGGAGgatcCTAAGCACAGCAGG |
| Δ1123-c | PGN_1123 downstream flanking sequence | Forward | CCTGCTGTGCTTAGgatcCTCCTTTGACAGTCC |
| Δ1123-d | PGN_1123 downstream flanking sequence | Reverse | CCGTAACCGGGTACGAT |
| Δ1123-confirm | PGN_1123 | Reverse | GACCCAAACATCCATTCGG |
| Δkgp-a | kgp upstream flanking sequence | Forward | atccatggACGCCCGATACCCATACTC |
| Δkgp-b | kgp upstream flanking sequence | Reverse | CACAAAGTCTCCGAGTggatccATTCAGAGAACCACG |
| Δkgp-c | kgp downstream flanking sequence | Forward | CGTGGTTCTCTGAATggatccACTCGGAGACTTTGTG |
| Δkgp-d | kgp downstream flanking sequence | Reverse | tatgtcgacAACAGGTTGTCCGTCAGC |
| Δkgp-confirm | kgp | Reverse | AACTTCCTAACTGCTGGCAC |
| ΔTn1-a | Tn1 upstream flanking sequence | Forward | AAGGCTTGATGCTGAAGACC |
| ΔTn1-b | Tn1 upstream flanking sequence | Reverse | GTCATTTCTTATTAAGAATAggatccTATTTACGTTGCGAGC |
| ΔTn1-c | Tn1 downstream flanking sequence | Forward | GCTCGCAACGTAAATAggatccTATTCTTAATAAGAAATGAC |
| ΔTn1-d | Tn1 downstream flanking sequence | Reverse | TCTTTGCCGGCATCTTTGC |
| 1123-start | PGN_1123 | Forward | ataggcctATGAGATTCTCTGCCATTATTATCG |
| 1123-stop | PGN_1123 | Reverse | attctaGAGGAATCCACTCGCAAATATC |
| SJ59 | ermF promoter | Forward | ggaattcggccgCCGATAGCTTCCGCTATTG |
| SJ60 | ermF promoter | Reverse | taggtaccaggcctGTAACTTCTTACAGGTGAATAC |
Lowercase indicates bases that add or complete a restriction site.
P. gingivalis 33277 deletion mutants were generated by introducing each deletion plasmid into P. gingivalis 33277 by natural transformation. Briefly, 0.5 × 109 bacteria were mixed with 1 μg of the deletion plasmid, incubated overnight in a loosely fastened screw-cap Eppendorf tube in an anaerobic chamber, and plated the next morning on TYHK agar plates containing the appropriate antibiotic. Putative mutant colonies that arose by homologous recombination, entailing a double-crossover of the flanking regions into the chromosome, were seen 5 to 6 days later. Loss of the targeted gene in the deletion mutant was confirmed by PCR analysis using primers designed to detect the coding sequences (Table 3).
Expression of PGN_1123 in trans in P. gingivalis and B. thetaiotaomicron.
The PGN_1123 gene was amplified using primers 1123-start and 1123-stop (Table 3) by PCR. The 1.2-kb product was cloned into pGem-T-EZ by TA cloning, generating pSJ831. It was next moved into a pT-COW-based plasmid, which is a broad-host-range vector capable of replicating in both E. coli and Bacteroidetes. pT-COW carries tetQ, conferring tetracycline resistance in Porphyromonas and Bacteroides. This vector was modified into an expression vector, pSJ46, by removing the tetC gene, encoding tetracycline resistance in E. coli, and replacing it with the promoter of the ermF gene, obtained from the ermFA erythromycin resistant cassette, and a multiple-cloning site (MCS) immediately downstream of the promoter. The majority of the tetC gene was removed by digesting pT-COW with EagI-HinDIII and replacing it with the ermF promoter plus MCS. The ermF promoter was first cloned into pSU20 using primers SJ59 andSJ60 (Table 3) on an EcoRI-KpnI fragment and was moved from there on an EagI-HinDIII fragment to the EagI-HinDIII sites of the tetC-deficient pT-COW vector. The newly introduced EagI-HinDIII fragment contained both the ermF promoter and the multiple-cloning site of pSU20.
We were at first unable to introduce PGN_1123 under control of the ermF promoter in pSJ46. We were, however, successful in cloning the gene in the reverse orientation by digesting PGN_1123 out of pSJ831 on an SphI-XbaI fragment and introducing it into the same sites of pSJ46, generating the plasmid pSJ836. The PGN_1123-containing pSJ836 was moved to ΔPGN_1123 mutants by conjugation. Triparental mating was used to mobilize plasmids from E. coli to P. gingivalis ΔPGN_1123 or to B. thetaiotaomicron with the help of the broad-host-range conjugative helper plasmid pRK231. Briefly, E. coli donor and helper strains were subcultured with no antibiotics and grown to an OD600 of ∼0.5. The recipient ΔPGN_1123 strain was grown to stationary phase, to an OD600 of >2.0. Fifty milliliters of recipient was spun and resuspended in 5 ml of TYK medium, of which 750 μl was mixed with 250 μl donor and 250 μl helper E. coli strains. The mixture was spun down, and the pellet brought up in 100 μl TYK broth and spotted on a TYHK agar plate. After aerobic incubation overnight at 37˚C, the spotted mixture was transferred by swab to 1 ml TYK broth and mixed well by vortexing, and 100 μl of the resuspension was plated on TYHK medium containing 0.5 μg/ml tetracycline and incubated anaerobically. Tetracycline-resistant exconjugants were obtained 4 to 5 days later. We confirmed that PGN_1123 was expressed from pSJ836 in ΔPGN1123::erm mutants by RT-PCR, using purified RNA.
pSJ836 was similarly transferred by triparental mating to B. thetaiotaomicron. The method was similar to that with P. gingivalis except that B. thetaiotaomicron was grown to an OD600 of ∼0.2, and 700 μl was mixed with 150 μl E. coli donor and helper strains, also grown to an OD600 of ∼0.2.
Accession number(s).
RNA-seq data obtained by next-generation sequencing of wild-type strain 33277 and the J5-c5 transposon mutant, performed on three biological replicates, are available in the NCBI GEO database under accession number GSE122289.
ACKNOWLEDGMENTS
We sincerely thank Andrew Goodman for providing the mariner transposon-containing E. coli strain SM10λpir(pSAM_Bt). We are grateful to Gabrielle Juliette Sisco and Anusha Etikala for providing technical support during construction of the transposon mutant library and analysis of the J5-c5 mutant. We also thank the Center for Precision Diagnostics at the University of Washington for advice offered and for conducting RNA-seq on a HiSeq platform.
This study was supported by NIH grant R21DE026344 to S.J. and R.P.D.
We note that Roger W. Kramer's work on analysis of next generation sequencing reads was unconnected to his affiliation with the Institute of Systems Biology.
Footnotes
For a commentary on this article, see https://doi.org/10.1128/JB.00146-19.
REFERENCES
- 1.Darveau RP. 2010. Periodontitis: a polymicrobial disruption of host homeostasis. Nat Rev Microbiol 8:481–490. doi: 10.1038/nrmicro2337. [DOI] [PubMed] [Google Scholar]
- 2.Socransky SS, Haffajee AD. 1994. Evidence of bacterial etiology: a historical perspective. Periodontol 2000 5:7–25. [DOI] [PubMed] [Google Scholar]
- 3.Rafiei M, Kiani F, Sayehmiri K, Sayehmiri F, Tavirani M, Dousti M, Sheikhi A. 2018. Prevalence of anaerobic bacteria (P. gingivalis) as major microbial agent in the incidence of periodontal diseases by meta-analysis. J Dent (Shiraz) 19:232–242. [PMC free article] [PubMed] [Google Scholar]
- 4.Hajishengallis G, Lamont RJ. 2014. Breaking bad: manipulation of the host response by Porphyromonas gingivalis. Eur J Immunol 44:328–338. doi: 10.1002/eji.201344202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zenobia C, Hajishengallis G. 2015. Porphyromonas gingivalis virulence factors involved in subversion of leukocytes and microbial dysbiosis. Virulence 6:236–243. doi: 10.1080/21505594.2014.999567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Darveau RP, Belton CM, Reife RA, Lamont RJ. 1998. Local chemokine paralysis, a novel pathogenic mechanism for Porphyromonas gingivalis. Infect Immun 66:1660–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hasegawa Y, Tribble GD, Baker HV, Mans JJ, Handfield M, Lamont RJ. 2008. Role of Porphyromonas gingivalis SerB in gingival epithelial cell cytoskeletal remodeling and cytokine production. Infect Immun 76:2420–2427. doi: 10.1128/IAI.00156-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kadowaki T, Takii R, Yamatake K, Kawakubo T, Tsukuba T, Yamamoto K. 2007. A role for gingipains in cellular responses and bacterial survival in Porphyromonas gingivalis-infected cells. Front Biosci 12:4800–4809. [DOI] [PubMed] [Google Scholar]
- 9.Curtis MA, Aduse-Opoku J, Rangarajan M. 2001. Cysteine proteases of Porphyromonas gingivalis. Crit Rev Oral Biol Med 12:192–216. [DOI] [PubMed] [Google Scholar]
- 10.Wang M, Shakhatreh M-AK, James D, Liang S, Nishiyama S-I, Yoshimura F, Demuth DR, Hajishengallis G. 2007. Fimbrial proteins of Porphyromonas gingivalis mediate in vivo virulence and exploit TLR2 and complement receptor 3 to persist in macrophages. J Immunol 179:2349–2358. doi: 10.4049/jimmunol.179.4.2349. [DOI] [PubMed] [Google Scholar]
- 11.Hajishengallis G, Wang M, Liang S, Shakhatreh MA, James D, Nishiyama S, Yoshimura F, Demuth DR. 2008. Subversion of innate immunity by periodontopathic bacteria via exploitation of complement receptor-3. Adv Exp Med Biol 632:203–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bielecka E, Scavenius C, Kantyka T, Jusko M, Mizgalska D, Szmigielski B, Potempa B, Enghild JJ, Prossnitz ER, Blom AM, Potempa J. 2014. Peptidyl arginine deiminase from Porphyromonas gingivalis abolishes anaphylatoxin C5a activity. J Biol Chem 289:32481–32487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lasica AM, Ksiazek M, Madej M, Potempa J. 2017. The type IX secretion system (T9SS): highlights and recent insights into its structure and function. Front Cell Infect Microbiol 7:215. doi: 10.3389/fcimb.2017.00215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Darveau RP, Hajishengallis G, Curtis MA. 2012. Porphyromonas gingivalis as a potential community activist for disease. J Dent Res 91:816–820. doi: 10.1177/0022034512453589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hajishengallis G, Darveau RP, Curtis MA. 2012. The keystone-pathogen hypothesis. Nat Rev Microbiol 10:717–725. doi: 10.1038/nrmicro2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hajishengallis G, Liang S, Payne MA, Hashim A, Jotwani R, Eskan MA, McIntosh ML, Alsam A, Kirkwood KL, Lambris JD, Darveau RP, Curtis MA. 2011. Low-abundance biofilm species orchestrates inflammatory periodontal disease through the commensal microbiota and complement. Cell Host Microbe 10:497–506. doi: 10.1016/j.chom.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zenobia C, Hasturk H, Nguyen D, Van Dyke TE, Kantarci A, Darveau RP. 2014. Porphyromonas gingivalis lipid A phosphatase activity is critical for colonization and increasing the commensal load in the rabbit ligature model. Infect Immun 82:650–659. doi: 10.1128/IAI.01136-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ogawa T, Asai Y, Makimura Y, Tamai R. 2007. Chemical structure and immunobiological activity of Porphyromonas gingivalis lipid A. Front Biosci 12:3795–3812. [DOI] [PubMed] [Google Scholar]
- 19.Bainbridge BW, Darveau RP. 2001. Porphyromonas gingivalis lipopolysaccharide: an unusual pattern recognition receptor ligand for the innate host defense system. Acta Odontol Scand 59:131–138. [DOI] [PubMed] [Google Scholar]
- 20.Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, Freudenberg M, Ricciardi-Castagnoli P, Layton B, Beutler B. 1998. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 282:2085–2088. [DOI] [PubMed] [Google Scholar]
- 21.Hoshino K, Takeuchi O, Kawai T, Sanjo H, Ogawa T, Takeda Y, Takeda K, Akira S. 1999. Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J Immunol 162:3749–3752. [PubMed] [Google Scholar]
- 22.Shimazu R, Akashi S, Ogata H, Nagai Y, Fukudome K, Miyake K, Kimoto M. 1999. MD-2, a molecule that confers lipopolysaccharide responsiveness on Toll-like receptor 4. J Exp Med 189:1777–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Coats SR, Pham TT, Bainbridge BW, Reife RA, Darveau RP. 2005. MD-2 mediates the ability of tetra-acylated and penta-acylated lipopolysaccharides to antagonize Escherichia coli lipopolysaccharide at the TLR4 signaling complex. J Immunol 175:4490–4498. [DOI] [PubMed] [Google Scholar]
- 24.Medzhitov R, Janeway C Jr.. 2000. The Toll receptor family and microbial recognition. Trends Microbiol 8:452–456. [DOI] [PubMed] [Google Scholar]
- 25.Raetz CR, Whitfield C. 2002. Lipopolysaccharide endotoxins. Annu Rev Biochem 71:635–700. doi: 10.1146/annurev.biochem.71.110601.135414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jain S, Darveau RP. 2010. Contribution of Porphyromonas gingivalis lipopolysaccharide to periodontitis. Periodontol 2000 54:53–70. doi: 10.1111/j.1600-0757.2009.00333.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kumada H, Haishima Y, Watanabe K, Hasegawa C, Tsuchiya T, Tanamoto K, Umemoto T. 2008. Biological properties of the native and synthetic lipid A of Porphyromonas gingivalis lipopolysaccharide. Oral Microbiol Immunol 23:60–69. doi: 10.1111/j.1399-302X.2007.00392.x. [DOI] [PubMed] [Google Scholar]
- 28.Zhang Y, Gaekwad J, Wolfert MA, Boons GJ. 2008. Synthetic tetra-acylated derivatives of lipid A from Porphyromonas gingivalis are antagonists of human TLR4. Org Biomol Chem 6:3371–3381. doi: 10.1039/b809090d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Coats SR, Jones JW, Do CT, Braham PH, Bainbridge BW, To TT, Goodlett DR, Ernst RK, Darveau RP. 2009. Human Toll-like receptor 4 responses to P. gingivalis are regulated by lipid A 1- and 4′-phosphatase activities. Cell Microbiol 11:1587–1599. doi: 10.1111/j.1462-5822.2009.01349.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Al-Qutub MN, Braham PH, Karimi-Naser LM, Liu X, Genco CA, Darveau RP. 2006. Hemin-dependent modulation of the lipid A structure of Porphyromonas gingivalis lipopolysaccharide. Infect Immun 74:4474–4485. doi: 10.1128/IAI.01924-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reife RA, Coats SR, Al-Qutub M, Dixon DM, Braham PA, Billharz RJ, Howald WN, Darveau RP. 2006. Porphyromonas gingivalis lipopolysaccharide lipid A heterogeneity: differential activities of tetra- and penta-acylated lipid A structures on E-selectin expression and TLR4 recognition. Cell Microbiol 8:857–868. doi: 10.1111/j.1462-5822.2005.00672.x. [DOI] [PubMed] [Google Scholar]
- 32.Rangarajan M, Aduse-Opoku J, Hashim A, McPhail G, Luklinska Z, Haurat MF, Feldman MF, Curtis MA. 2017. LptO (PG0027) is required for lipid A 1-phosphatase activity in Porphyromonas gingivalis W50. J Bacteriol 199:e00751-16. doi: 10.1128/JB.00751-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Slocum C, Coats SR, Hua N, Kramer C, Papadopoulos G, Weinberg EO, Gudino CV, Hamilton JA, Darveau RP, Genco CA. 2014. Distinct lipid a moieties contribute to pathogen-induced site-specific vascular inflammation. PLoS Pathog 10:e1004215. doi: 10.1371/journal.ppat.1004215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Trent MS, Pabich W, Raetz CR, Miller SI. 2001. A PhoP/PhoQ-induced lipase (PagL) that catalyzes 3-O-deacylation of lipid A precursors in membranes of Salmonella typhimurium. J Biol Chem 276:9083–9092. doi: 10.1074/jbc.M010730200. [DOI] [PubMed] [Google Scholar]
- 35.Reynolds CM, Ribeiro AA, McGrath SC, Cotter RJ, Raetz CRH, Trent MS. 2006. An outer membrane enzyme encoded by Salmonella typhimurium lpxR that removes the 3′-acyloxyacyl moiety of lipid A. J Biol Chem 281:21974–21987. doi: 10.1074/jbc.M603527200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Geurtsen J, Steeghs L, Hove JT, van der Ley P, Tommassen J. 2005. Dissemination of lipid A deacylases (pagL) among gram-negative bacteria: identification of active-site histidine and serine residues. J Biol Chem 280:8248–8259. doi: 10.1074/jbc.M414235200. [DOI] [PubMed] [Google Scholar]
- 37.Chen Y-Y, Peng B, Yang Q, Glew MD, Veith PD, Cross KJ, Goldie KN, Chen D, O'Brien-Simpson N, Dashper SG, Reynolds EC. 2011. The outer membrane protein LptO is essential for the O-deacylation of LPS and the co-ordinated secretion and attachment of A-LPS and CTD proteins in Porphyromonas gingivalis. Mol Microbiol 79:1380–1401. doi: 10.1111/j.1365-2958.2010.07530.x. [DOI] [PubMed] [Google Scholar]
- 38.Barth K, Remick DG, Genco CA. 2013. Disruption of immune regulation by microbial pathogens and resulting chronic inflammation. J Cell Physiol 228:1413–1422. doi: 10.1002/jcp.24299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Palm E, Khalaf H, Bengtsson T. 2013. Porphyromonas gingivalis downregulates the immune response of fibroblasts. BMC Microbiol 13:155. doi: 10.1186/1471-2180-13-155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hajishengallis G. 2011. Immune evasion strategies of Porphyromonas gingivalis. J Oral Biosci 53:233–240. doi: 10.2330/joralbiosci.53.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Duncan L, Yoshioka M, Chandad F, Grenier D. 2004. Loss of lipopolysaccharide receptor CD14 from the surface of human macrophage-like cells mediated by Porphyromonas gingivalis outer membrane vesicles. Microb Pathog 36:319–325. doi: 10.1016/j.micpath.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 42.Sugawara S, Nemoto E, Tada H, Miyake K, Imamura T, Takada H. 2000. Proteolysis of human monocyte CD14 by cysteine proteinases (gingipains) from Porphyromonas gingivalis leading to lipopolysaccharide hyporesponsiveness. J Immunol 165:411–418. [DOI] [PubMed] [Google Scholar]
- 43.Goodman AL, McNulty NP, Zhao Y, Leip D, Mitra RD, Lozupone CA, Knight R, Gordon JI. 2009. Identifying genetic determinants needed to establish a human gut symbiont in its habitat. Cell Host Microbe 6:279–289. doi: 10.1016/j.chom.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jain S, Coats SR, Chang AM, Darveau RP. 2013. A novel class of lipoprotein lipase-sensitive molecules mediates Toll-like receptor 2 activation by Porphyromonas gingivalis. Infect Immun 81:1277–1286. doi: 10.1128/IAI.01036-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Law CW, Alhamdoosh M, Su S, Smyth GK, Ritchie ME. 2016. RNA-seq analysis is easy as 1-2-3 with limma, Glimma and edgeR. F1000Res 5:1408. doi: 10.12688/f1000research.9005.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rutten L, Geurtsen J, Lambert W, Smolenaers JJM, Bonvin AM, de Haan A, van der Ley P, Egmond MR, Gros P, Tommassen J. 2006. Crystal structure and catalytic mechanism of the LPS 3-O-deacylase PagL from Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 103:7071–7076. doi: 10.1073/pnas.0509392103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rutten L, Mannie J-PBA, Stead CM, Raetz CRH, Reynolds CM, Bonvin AMJJ, Tommassen JP, Egmond MR, Trent MS, Gros P. 2009. Active-site architecture and catalytic mechanism of the lipid A deacylase LpxR of Salmonella typhimurium. Proc Natl Acad Sci U S A 106:1960–1964. doi: 10.1073/pnas.0813064106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y. 2015. The I-TASSER Suite: protein structure and function prediction. Nat Methods 12:7–8. doi: 10.1038/nmeth.3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Walters S, Rodrigues P, Bélanger M, Whitlock J, Progulske-Fox A. 2009. Analysis of a band 7/MEC-2 family gene of Porphyromonas gingivalis. J Dent Res 88:34–38. doi: 10.1177/0022034508328381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Song Y-H, Kozarov EV, Walters SM, Cao SL, Handfield M, Hillman JD, Progulske-Fox A. 2002. Genes of periodontopathogens expressed during human disease. Ann Periodontol 7:38–42. doi: 10.1902/annals.2002.7.1.38. [DOI] [PubMed] [Google Scholar]
- 51.Coats SR, Berezow AB, To TT, Jain S, Bainbridge BW, Banani KP, Darveau RP. 2011. The lipid A phosphate position determines differential host Toll-like receptor 4 responses to phylogenetically related symbiotic and pathogenic bacteria. Infect Immun 79:203–210. doi: 10.1128/IAI.00937-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kawasaki K, Ernst RK, Miller SI. 2004. 3-O-deacylation of lipid A by PagL, a PhoP/PhoQ-regulated deacylase of Salmonella typhimurium, modulates signaling through Toll-like receptor 4. J Biol Chem 279:20044–20048. doi: 10.1074/jbc.M401275200. [DOI] [PubMed] [Google Scholar]
- 53.Ernst RK, Adams KN, Moskowitz SM, Kraig GM, Kawasaki K, Stead CM, Trent MS, Miller SI. 2006. The Pseudomonas aeruginosa lipid A deacylase: selection for expression and loss within the cystic fibrosis airway. J Bacteriol 188:191–201. doi: 10.1128/JB.188.1.191-201.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kawahara K, Tsukano H, Watanabe H, Lindner B, Matsuura M. 2002. Modification of the structure and activity of lipid A in Yersinia pestis lipopolysaccharide by growth temperature. Infect Immun 70:4092–4098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rebeil R, Ernst RK, Gowen BB, Miller SI, Hinnebusch BJ. 2004. Variation in lipid A structure in the pathogenic yersiniae. Mol Microbiol 52:1363–1373. doi: 10.1111/j.1365-2958.2004.04059.x. [DOI] [PubMed] [Google Scholar]
- 56.Reinés M, Llobet E, Dahlström KM, Pérez-Gutiérrez C, Llompart CM, Torrecabota N, Salminen TA, Bengoechea JA. 2012. Deciphering the acylation pattern of Yersinia enterocolitica lipid A. PLoS Pathog 8:e1002978. doi: 10.1371/journal.ppat.1002978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Montminy SW, Khan N, McGrath S, Walkowicz MJ, Sharp F, Conlon JE, Fukase K, Kusumoto S, Sweet C, Miyake K, Akira S, Cotter RJ, Goguen JD, Lien E. 2006. Virulence factors of Yersinia pestis are overcome by a strong lipopolysaccharide response. Nat Immunol 7:1066–1073. doi: 10.1038/ni1386. [DOI] [PubMed] [Google Scholar]
- 58.Corcoran CP, Podkaminski D, Papenfort K, Urban JH, Hinton JCD, Vogel J. 2012. Superfolder GFP reporters validate diverse new mRNA targets of the classic porin regulator, MicF RNA. Mol Microbiol 84:428–445. doi: 10.1111/j.1365-2958.2012.08031.x. [DOI] [PubMed] [Google Scholar]
- 59.Zhang Y-F, Han K, Chandler CE, Tjaden B, Ernst RK, Lory S. 2017. Probing the sRNA regulatory landscape of P. aeruginosa: post-transcriptional control of determinants of pathogenicity and antibiotic susceptibility. Mol Microbiol 106:919–937. doi: 10.1111/mmi.13857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ogawa R, Yen H, Kawasaki K, Tobe T. 2018. Activation of lpxR gene through enterohaemorrhagic Escherichia coli virulence regulators mediates lipid A modification to attenuate innate immune response. Cell Microbiol 20. doi: 10.1111/cmi.12806. [DOI] [PubMed] [Google Scholar]
- 61.Elhenawy W, Bording-Jorgensen M, Valguarnera E, Haurat MF, Wine E, Feldman MF. 2016. LPS remodeling triggers formation of outer membrane vesicles in Salmonella. mBio 7:e00940-16. doi: 10.1128/mBio.00940-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gui MJ, Dashper SG, Slakeski N, Chen Y-Y, Reynolds EC. 2016. Spheres of influence: Porphyromonas gingivalis outer membrane vesicles. Mol Oral Microbiol 31:365–378. doi: 10.1111/omi.12134. [DOI] [PubMed] [Google Scholar]
- 63.Xie H. 2015. Biogenesis and function of Porphyromonas gingivalis outer membrane vesicles. Future Microbiol 10:1517–1527. doi: 10.2217/fmb.15.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schromm AB, Brandenburg K, Loppnow H, Moran AP, Koch MH, Rietschel ET, Seydel U. 2000. Biological activities of lipopolysaccharides are determined by the shape of their lipid A portion. Eur J Biochem 267:2008–2013. [DOI] [PubMed] [Google Scholar]
- 65.Haurat MF, Aduse-Opoku J, Rangarajan M, Dorobantu L, Gray MR, Curtis MA, Feldman MF. 2011. Selective sorting of cargo proteins into bacterial membrane vesicles. J Biol Chem 286:1269–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Veith PD, Chen Y-Y, Gorasia DG, Chen D, Glew MD, O’Brien-Simpson NM, Cecil JD, Holden JA, Reynolds EC. 2014. Porphyromonas gingivalis outer membrane vesicles exclusively contain outer membrane and periplasmic proteins and carry a cargo enriched with virulence factors. J Proteome Res 13:2420–2432. doi: 10.1021/pr401227e. [DOI] [PubMed] [Google Scholar]
- 67.Gabarrini G, Heida R, van Ieperen N, Curtis MA, van Winkelhoff AJ, van Dijl JM. 2018. Dropping anchor: attachment of peptidylarginine deiminase via A-LPS to secreted outer membrane vesicles of Porphyromonas gingivalis. Sci Rep 8:8949. doi: 10.1038/s41598-018-27223-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gorasia DG, Veith PD, Chen D, Seers CA, Mitchell HA, Chen Y-Y, Glew MD, Dashper SG, Reynolds EC. 2015. Porphyromonas gingivalis type IX secretion substrates are cleaved and modified by a sortase-like mechanism. PLoS Pathog 11:e1005152. doi: 10.1371/journal.ppat.1005152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Veith PD, Glew MD, Gorasia DG, Reynolds EC. 2017. Type IX secretion: the generation of bacterial cell surface coatings involved in virulence, gliding motility and the degradation of complex biopolymers. Mol Microbiol 106:35–53. doi: 10.1111/mmi.13752. [DOI] [PubMed] [Google Scholar]
- 70.Haurat MF, Elhenawy W, Feldman MF. 2015. Prokaryotic membrane vesicles: new insights on biogenesis and biological roles. Biol Chem 396:95–109. doi: 10.1515/hsz-2014-0183. [DOI] [PubMed] [Google Scholar]
- 71.Lappann M, Otto A, Becher D, Vogel U. 2013. Comparative proteome analysis of spontaneous outer membrane vesicles and purified outer membranes of Neisseria meningitidis. J Bacteriol 195:4425–4435. doi: 10.1128/JB.00625-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sidhu VK, Vorhölter F-J, Niehaus K, Watt SA. 2008. Analysis of outer membrane vesicle associated proteins isolated from the plant pathogenic bacterium Xanthomonas campestris pv. campestris. BMC Microbiol 8:87. doi: 10.1186/1471-2180-8-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Galka F, Wai SN, Kusch H, Engelmann S, Hecker M, Schmeck B, Hippenstiel S, Uhlin BE, Steinert M. 2008. Proteomic characterization of the whole secretome of Legionella pneumophila and functional analysis of outer membrane vesicles. Infect Immun 76:1825–1836. doi: 10.1128/IAI.01396-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kato S, Kowashi Y, Demuth DR. 2002. Outer membrane-like vesicles secreted by Actinobacillus actinomycetemcomitans are enriched in leukotoxin. Microb Pathog 32:1–13. doi: 10.1006/mpat.2001.0474. [DOI] [PubMed] [Google Scholar]
- 75.Elhenawy W, Debelyy MO, Feldman MF. 2014. Preferential packing of acidic glycosidases and proteases into Bacteroides outer membrane vesicles. mBio 5:e00909-14. doi: 10.1128/mBio.00909-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, Hu L, Shao F. 2014. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 514:187–192. doi: 10.1038/nature13683. [DOI] [PubMed] [Google Scholar]
- 77.Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA. 2013. Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science 341:1250–1253. doi: 10.1126/science.1240988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Klein BA, Tenorio EL, Lazinski DW, Camilli A, Duncan MJ, Hu LT. 2012. Identification of essential genes of the periodontal pathogen Porphyromonas gingivalis. BMC Genomics 13:578. doi: 10.1186/1471-2164-13-578. [DOI] [PMC free article] [PubMed] [Google Scholar]