

Abstract
Epigenetic alteration has been proposed to give rise to numerous classic hallmarks of cancer. Impaired DNA methylation plays a central role in the onset and progression of several types of malignancies, and DNA methylation is mediated by DNA methyltransferases (DNMTs) consisting of DNMT1, DNMT3A, and DNMT3B. DNMTs are frequently implicated in the pathogenesis and aggressiveness of acute myeloid leukaemia (AML) patients. In this review, we describe and discuss the oncogenic roles of DNMT1, DNMT3A, and DNMT3B in AML. The clinical response predictive roles of DNMTs in clinical trials utilising hypomethylating agents (azacitidine and decitabine) in AML patients are presented. Novel hypomethylating agent (guadecitabine) and experimental DNMT inhibitors in AML are also discussed. In summary, hypermethylation of tumour suppressors mediated by DNMT1 or DNMT3B contributes to the progression and severity of AML (except MLL-AF9 and inv(16)(p13;q22) AML for DNMT3B), while mutation affecting DNMT3A represents an early genetic lesion in the pathogenesis of AML. In clinical trials of AML patients, expression of DNMTs is downregulated by hypomethylating agents while the clinical response predictive roles of DNMT biomarkers remain unresolved. Finally, nucleoside hypomethylating agents have continued to show enhanced responses in clinical trials of AML patients, and novel non-nucleoside DNMT inhibitors have demonstrated cytotoxicity against AML cells in pre-clinical settings.
Keywords: acute myeloid leukaemia, DNMT1, DNMT3A, DNMT3B, azacitidine, decitabine, guadecitabine
Introduction
Epigenetic modifications are inherited changes in DNA that do not change the sequence itself. These include DNA methylation, histone deacetylation, and miRNA regulation.1 DNA methylation is one of the most extensively studied epigenetic changes and it regulates expression of genes in normal development of mammalian cells. Dysregulation in DNA methylation often plays a vital role in the onset of human diseases, and it has been proposed that epigenetic aberration potentially gives rise to several classic hallmarks of cancer.2 In particular, promoter hypermethylation of tumour suppressor genes resulting in the silencing of their expression is involved in driving cancer survival, growth, and metastasis.3,4
DNA methylation of cytosine bases is prevalent in mammalian and several other eukaryotic genomes. The addition of methyl groups is conducted by DNA methyltransferases (DNMTs) consisting of DNMT1, DNMT3A, and DNMT3B.5 Methylation occurs at the fifth carbon atom of cytosine bases in specific regions of CpG dinucleotides (Figure 1), and it is formed and preserved by DNMTs.5,6 Both DNMT3A and DNM3B catalyse de novo DNA methylation, while DNMT1 preserves such patterns of DNA methylation during cell division and preferentially methylates hemimethylated DNA.6,7
Figure 1.
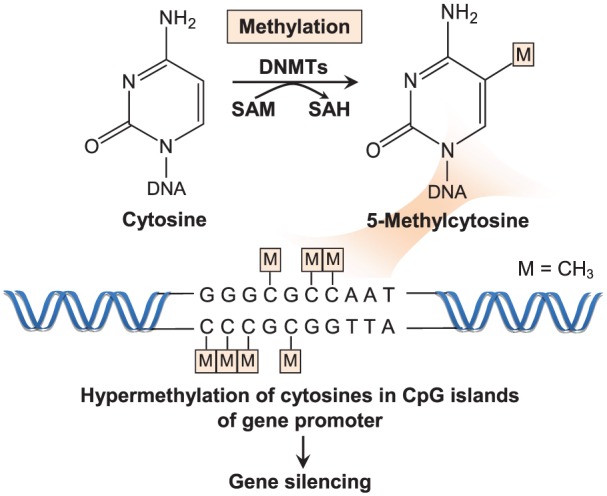
DNMTs catalyse the transfer of a methyl group from the methyl donor S-adenosylmethionine (SAM) to the fifth carbon of the cytosine base in the CpG islands of gene promoter. SAM is converted into S-adenosylhomocysteine (SAH) in the catalytic reaction. Hypermethylation of the cytosines suppresses transcription of the gene.8,9
Structurally, the domains of all DNMTs are distinguished by a variable N-terminal region consisting of regulatory domains while the C-terminal region contains the methyltransferase domain required for their enzymatic activities.6 The regulatory domains at N-terminal differ between DNMT1 and DNMT3 whereby the N-terminal region of DNMT1 harbours the DNA methyltransferase-associated protein (DMAP) charged-rich domain required for interaction with the transcriptional repressor DMAP1.10 In addition, DNMT1 contains the replication foci-targeting domain (RFD) that modulates anchoring to the replication fork, the zinc finger CXXC domain that recognises unmethylated CpG, and two bromo-adjacent homology (BAH) domains (ie, BAH1 and BAH2) proposed to act as protein-protein interaction module for the silencing of gene expression.10 DNMT3A and DNMT3B contain the Pro-Trp-Trp-Pro motif domain (PWWP; a methyl-lysine recognition motif) and the ATRX, DNMT3, DNMT3L (ADD) zinc finger domain (Figure 2). The C-terminal region of all three DNMTs consists of the methyltransferase domain required for their catalytic activities.6
Figure 2.

The domains of DNMT1 (RefSeq ID: NP_001124295.1), DNMT3A (RefSeq ID: NP_783328.1), and DNMT3B (RefSeq ID: NP_008823.1) proteins according to Pfam and PROSITE (for ADD domain of DNMT3A or DNMT3B) obtained from Ensembl database (https://www.ensembl.org/index.html). ADD indicates ATRX, DNMT3, DNMT3L zinc finger domain; BAH, bromo-adjacent homology domain; CXXC, CXXC zinc finger domain; DMAP, DNA methyltransferase-associated protein; PWWP, Pro-Trp-Trp-Pro motif domain; RFD, replication foci-targeting domain.
Acute myeloid leukaemia (AML) is a clonal disorder of haematopoiesis characterised by the expansion of undifferentiated myeloid precursors, leading to deregulated haematopoiesis and bone marrow failure.11 AML is the most common type of acute leukaemia with heterogeneous molecular profiles, clinical representations, response to therapies and outcomes. Although intensive chemotherapy and haematopoietic stem cell transplantation (HSCT) have improved the outcomes of AML patients, approximately 50% of younger patients and 80% of patients above the age of 60 years succumb to the disease due to refractory disease, relapse, or treatment-induced mortality.12 Older AML patients unable to receive intensive chemotherapy due to toxicities have a median survival of just 5-10 months.13
AML presents with fewer mutations than majority of other adult cancers and it is a highly heterogeneous disease,14 implying that other mechanisms such as epigenetics modifications play a role in the pathogenesis and outcomes of the disease. DNA hypermethylation at the CpG islands of tumour suppressors was reported in myelodysplastic syndrome (MDS) and AML nearly two decades ago.15 Deregulated genome-wide patterns of DNA methylation occur in AML and they are not associated with mutations of known epigenetic regulators.16
Apart from DNA methylation, alterations in DNA demethylation also contribute to leukaemogenesis and affect clinical outcomes in AML patients. The DNA demethylase ten-eleven translocation (TET) proteins (including TET1, TET2, and TET3) converts 5-methylcytosine into 5-hydroxymethylcytosine.17 TET1 is fused with the MLL gene in t(10;11)(q22;q23) AML,18 while TET2 mutations occur in 13% to 27% of AML patients with normal or intermediate-risk cytogenetics associated with unfavourable prognosis.19–21 Moreover, TET2 mutation status has been shown to predict higher response rate in AML and MDS patients.22
Findings in the past two decades demonstrating deregulated DNA methylation in the pathogenesis and aggressiveness of MDS and AML have led to the approval for the clinical use of pyrimidine analogues that inhibit DNMT methylating activities (ie, 5-azacitidine [azacitidine] and 5-aza-2′-deoxycytidine [decitabine]) in both diseases.23 These agents mimic cytosine and are able to trap DNMTs when incorporated into DNA in S phase of the replication cycle. The proteasome then degrades the trapped DNMTs leading to DNA hypomethylation and re-expression of tumour suppressor genes.24,25 However, azacitidine is usually administrated for older AML patients who are ineligible for HSCT and with low blasts count (20%-30% bone marrow blasts),26 while decitabine does not improve complete remission rates compared with supportive care and cytarabine in elderly AML patients.27 Hence, further understanding of the precise DNMT-mediated oncogenic mechanisms in AML is required to select for specific and potent novel DNMT inhibitors which is currently under intense investigation and discovery.28–30
In this review, we describe and discuss the oncogenic properties of DNMT1, DNMT3A, and DNMT3B in AML. We also describe the prognostic and predictive roles of DNMTs in clinical trials of AML patients with hypomethylating agents, as well as novel DNMT inhibitors that have been tested experimentally in AML cells.
DNMT1 in AML
DNMT1 is the most abundantly expressed DNMT in dividing cells and it represents a key therapeutic target in rapidly dividing cancer cells for methylation inhibition and re-expression of tumour suppressor genes.31 Several expression and mechanistic studies have shown DNMT1 to be a potential oncoprotein in AML.
DNMT1 protein levels were higher in azacitidine-resistant AML cells (SKM1 azacitidine-sensitive and azacitidine-resistant clones), and reduced expression of anti-DNMT1 miRNAs (ie, targeted DNMT1 3′ untranslated region [UTR] for its reduction expression) was associated with azacitidine resistance in AML and high-risk MDS (HRMDS) patients.32 DNMT1 expression was increased in multi-drug resistant AML cells (HL60/ATRA), and knockdown of a drug resistance-related gene segment, HA117, decreased stem-like signature of the cells and blocked DNMT1 expression.33
A key pathogenic mechanism involving DNMT1 in AML is the DNMT1-mediated downregulation of the cyclin-dependent kinase inhibitor p15 (that encodes p15 protein, a tumour suppressor) expression in the disease. The expression of p15 is lost in approximately 80% of AML cases, and hypermethylation of its promoter is frequently associated with transformation of the disease to a more aggressive phenotype.34 DNMT1 transcripts were found to be upregulated (by 5.3-fold) in bone marrow cells from AML patients compared with bone marrow cells from healthy donors, and p15 was methylated in 72% of AML patients who had higher levels of DNMT1 expression, indicating the potential of DNMT1 to induce hypermethylation of tumour suppressors in AML.35
Subsequent studies have shown that treatment with receptor tyrosine kinase (RTK) inhibitor, nilotinib, reduced DNMT1 expression resulting in decreased global DNA methylation and upregulation of p15 expression via promoter hypomethylation in AML cells (MV4-11 and Kasumi-1) and patient blasts.36 Treatment with nilotinib led to apoptosis of AML leukaemia cell lines, leukaemia regression in mice (C1498 mouse AML cells injected into C57BL/6 mice), and impaired AML patient cell expansion ex vivo and in vivo through reduction of DNMT1. Also, p15 expression was increased through promoter hypomethylation. Moreover, treatment with harmine (a beta carboline alkaloid derivative of Peganum harmala, a type of herb originated from Asia) in AML cells (NB4) supressed their proliferation, decreased DNMT1 gene expression, and increased p15 promoter hypomethylation and reactivation.37
Interestingly, emerging evidence has shown an association between DNMT1 and lipid metabolism protein in the suppression of p15 expression in AML. Fatty acid-binding protein 4 (FABP4), a key regulator of lipid metabolism, is upregulated in AML cells and enhances their aggressiveness via DNMT1-dependent DNA methylation. Increased FABP4 expression induced IL-6 expression and STAT3 phosphorylation, causing DNMT1 overexpression and subsequent silencing of p15 expression while FABP4 silencing suppressed DNMT1-dependent DNA methylation that restored p15 expression in AML cells (C1498, MV4-11, and Kasumi-1).38 Similarly, inhibition of FABP4 by its selective inhibitor BMS309403 resulted in suppressed DNMT1 expression, a decrease in global DNA methylation, and re-expression of p15 through promoter DNA hypomethylation in AML cells (C1498, MV4-11, and Kasumi-1). Impairment of their growth in vitro, ex vivo (human and mouse AML primary cells), and in vivo (C1498 cells injected into C57BL/6 mice) was also reported.39
Synergistic inhibition of DNMT1 and other oncogenic proteins through the use of pharmacological inhibitors has been proposed to treat AML. MUC1C, a transmembrane oncoprotein expressed in AML stem-like cells, induced DNMT1 expression by activating NF-κB p65 pathway that drove DNMT1 mRNA transcription.40 Synergistic pharmacological inhibition of MUC1C (with GO-203) and DNMT1 (with decitabine) reduced DNMT1 levels followed by decreased AML cell survival in cell lines (THP-1 and MOML-14) and primary AML cells.40
DNMT1 has also been implicated in various other haematological malignancies. The protein is frequently expressed in diffuse large B-cell lymphoma (DLBCL) being highly associated with Ki-67 expression as demonstrated in our multi-centre series of DLBCL cases.41 We also showed that stable knockdown of DNMT1 in DLBCL cells upregulated expression of genes involved in the activation of cell cycles,42 similar with the DNMT1-p15 axis (p15 protein inhibits cell cycle progression) frequently reported in AML. Recent studies have also demonstrated its potentially oncogenic properties in acute lymphoblastic leukaemia,43 chronic myeloid leukaemia,44 multiple myeloma,45 and Burkitt’s lymphoma.46 Collectively, DNMT1 appears to be a promising target for therapeutic modulation in haematological malignancies.
DNMT3A in AML
Mutations affecting the loci of epigenetic regulators are commonplace in AML with DNMT3A being the most commonly mutated epigenetic regulator in the disease. Around 22%-33% of AML cases present with DNMT3A mutations.14,47–49
One of the vital findings pertaining to DNMT3A in AML is the identification of DNMT3A missense mutations that affect the encoding of arginine R882 (codon CGC), causing loss of methylation activity of DNMT3A.50 R882 mutation occurred in 13.2% (n = 37/281) of AML patients, and other types of mutations encompassing DNMT3A were found in 25 AML patients (8.9%).51 These mutations were significantly more frequent in patients with an intermediate-risk cytogenetic profile but absent in all patients with a favourable-risk cytogenetic profile (P < .001), and DNMT3A mutations conferred significantly shorter survival (P < .001). In a study of AML patients younger than 60 years of age, a similar proportion (14%, n = 58/415) of patients were found to harbour missense mutations of R882 and R882 mutations were significantly associated with inferior overall survival (OS; P = .018) and relapse-free survival (RFS; P = .029).52
Mutated DNMT3A is considered an early genetic lesion in the pathogenesis of leukaemia and DNMT3A mutation alone might be insufficient to cause malignancies as observed in asymptomatic older carriers,53–55 suggesting that additional genetic perturbation is required to transform into leukaemia cells. In line with this, functional studies in Dnmt3a knockout mice (C57BL/6-CD45.2 background) with Flt3 internal tandem duplication (Flt3-ITD) overexpression have shown the roles of DNMT3A mutation as a reservoir for the clonal expansion of HSCs until the acquirement of the additional genetic lesion Flt3-ITD, causing transformation into AML cells.56
An independent study also showed that mice (C57BL/6-CD45.2 background) with Flt3-ITD and inducible deletion of Dnmt3a developed a rapidly lethal, penetrant, and transplantable AML.57 The authors reported that single-cell assays identified clonogenic subpopulations expressing genes sensitive to methylation and responsive to Dnmt3a levels, and concluded that Dnmt3a haploinsufficiency transformed Flt3-ITD myeloproliferative neoplasms into AML by regulating methylation-sensitive gene expression. Likewise, a recent analysis of the mutational landscape of 85 AML patients with partial tandem duplication of MLL (MLL-PTD) suggested that DNMT3A (among others including IDH2, TET2, or U2AF1) mutations are clonal with early onset, and MLL-PTD likely occurs after these initial mutations while proliferative mutations involving FLT3 or RAS, that usually appear later, are largely subclonal.58
In conjunction with the aforementioned studies, several reports have also proposed the DNMT3Amut/Flt3-ITD axis as a promising therapeutic target for AML patients. AML patients with FLT3-ITD displayed inferior outcomes in AML patients after allogeneic HSCT due to higher risk of early relapse.59,60 Co-occurrence of DNMT3A mutations with FLT3-ITD comprised about 13% of AML patients (n = 210/1571),61 and in adult AML patients (n = 128), patients with DNMT3A R882 mutations positive for FLT3-ITD (DNMT3Amut/FLT3-ITDpos) had the worst OS (P = .025) and RFS (P = .011) compared with three other groups (DNMT3Awt/FLT3-ITDneg, DNMT3Awt/FLT3-ITDpos, DNMT3Amut/FLT3-ITDneg).62 In normal-karyotype (NK)-AML patients after allogeneic HSCT, patients with FLT3-ITDpos and DNMT3A R882mut had significantly worse survival compared with patients harbouring FLT3-ITDneg/DNMT3A R882wt, FLT3-ITDneg/DNMT3A R882mut, and FLT3-ITDpos/DNMT3A R882mut (all P < .05).63
There also appears to be less conventional findings pertaining to DNMT3A mutations in AML patients. The frequency of R882 mutation was much lower in Chinese patients where R882 mutations occurred in 6.6% (n = 12/182) of AML patients and in 7.8% (n = 4/51) of patients with MDS.64 None were found in patients with chronic myeloid leukaemia or myeloproliferative neoplasms in the same study. In AML patients with DNMT3A mutations (R882H/R882C), mutated DNMT3A transcript levels were higher in bone marrow than in the blood after induction and consolidation therapies but did not have association with endpoints remission duration and OS, although its levels were persistently high in remission.65 Interestingly, the molecular landscape of paediatric AML derived from nearly 1000 participants in the Children’s Oncology Group (COG) AML trials demonstrated that no mutations affecting the protein-coding regions of DNMT3A were found in paediatric AML, although these mutations were frequent in adults, implying that mutations in DNMT3A that promote leukaemogenesis occur in many apparently healthy adults but are rare in children.66
Other recent findings include the following: (1) DNMT3A was mutated in approximately 19% of AML cases with RUNX1 mutations that negatively impacted OS of the patients (P = .001)67; (2) methylation profiling studies on TCGA AML patients (n = 194) identified two distinct subgroups with profound hypomethylation signatures termed as DMP.1 and DMP.2. Mutations affecting DNMT3A and FLT3 were significantly enriched in the DMP.1 cases (P < .001).68 The DMP.1 group had the worst survival compared with DMP-negative and DMP.2 group, and immune response genes were enriched in the DMP.1 group, suggesting a link between DNMT3A mutations and altered immune response in AML.
Apart from the therapeutic potential, assessment of DNMT3A mutations might represent a diagnostic tool for AML. A recent study of women who were healthy at study baseline but were eventually diagnosed with AML (n = 212), somatic mutations of DNMT3A (among mutations of other genes) in peripheral blood DNA were found to predict an increased risk of developing AML years before diagnosis.69 Thus, screening peripheral blood for DNMT3A mutations in asymptomatic AML patients warrants expanded future research.
DNMT3B in AML
Unlike DNMT3A, DNMT3B mutation is a rare event in AML. Nonetheless, multiple lines of evidence have demonstrated that its expression in AML is associated with worse clinical outcomes:
Overexpression of DNMT3B was associated with inferior event-free survival (EFS; P = .006) and a trend towards worse OS (P = .056) in a panel of de novo AML patients (n = 191)70.
In an integrated analysis of the methylome and transcriptome of 151 paediatric AML patients, increased DNMT3B expression and reduced methylation were associated with poorer clinical outcomes (P ⩽ 10−5; q ⩽ 0.002), and higher DNMT3B expression was associated with worse minimal residual disease (MRD), higher rate of relapse or resistant disease, worse EFS, and higher genome-wide methylation burden (GWMB) in both the training and validation cohort (all P < .03 in both cohorts).71 Higher GWMB was also associated with worse MRD and EFS (all P < .05 in both cohorts), implying that DNMT3B-induced GWMB may play a role in the aggressiveness of paediatric AML.
Older adults (⩾60 years old) with primary, cytogenetically normal AML (n = 210) who expressed high DNMT3B transcript levels had significantly fewer complete remissions (P = .002), inferior disease-free survival (DFS; P = .02), and OS (P < .001), and all three characteristics remained significant (P < .05) in multivariable analyses.72 High DNMT3B levels in these patients were associated with gene expression profiles implicated in differentiation, proliferation, and survival pathways, but surprisingly high DNMT3B levels were not associated with DNA methylation changes, consistent with other reports demonstrating that DNMT3B expression did not influence the DNA methylation levels of leukaemic blasts derived from cytogenetically normal AML patients.73 These suggest that older AML patients with high DNMT3B expression might not respond to hypomethylation agents, and that DNMT3B might exert its effects through an additional, methylation-independent mechanism in older AML patients.
Myeloperoxidase (MPO), a microbicidal protein measured in leukaemia blasts by cytochemistry, is a biomarker for the diagnosis of AML and its expression is associated with better prognosis in AML patients. DNMT3B was found to have a significantly inverse relationship with MPO levels in CD34+ AML cells (P = .0283) without significant association with common mutations in AML (FLT3-ITD, CEBPA, or NPM1 mutations).74 This suggests that MPO gene transcription might be repressed through promoter methylation by DNMT3B, although this is subject to further proof by functional studies.
Furthermore, in a DNMT3B genotyping study on de novo AML patients (n = 317) and healthy control subjects (n = 406) of Chinese Han populations, the GG genotype of rs1569686 was most significantly associated with increased risk for AML (OR: 5.76; 95% confidence interval (CI): 2.60-12.73; P < .01), compared with the TT genotype, in contrast with the CC genotype of rs2424908 shown to have reduced AML risk (OR: 0.57; 95% CI: 0.36-0.91; P = .01) compared with the TT genotype, suggesting that DNMT3B gene polymorphisms could play roles in AML leukaemogenesis and as potential markers for AML.75 Moreover, DNMT3B overexpression leading to DNA hypermethylation has also been reported in T-cell acute lymphoblastic leukaemia and Burkitt’s lymphoma,76 illustrating the oncogenic role of DNMT3B through hypermethylation of tumour suppressor genes in haematological malignancies.
Despite of the association of DNMT3B expression with worse prognosis in AML patients, recent studies have also shown its potential tumour suppressive roles in two subtypes of AML, ie, MLL-AF9 AML, and inv(16)(p13;q22) AML.
In MLL-AF9 AML, one of most frequent MLL rearrangements in MLL-rearranged leukaemia, deletion of Dnmt3b in mice model increased their progression through enhanced stemness and cell cycle progression accompanied by upregulation of oncogenic gene sets, indicating a tumour suppressive role of Dnmt3b in MLL-AF9 AML.77 Moreover, the authors also showed that Dnmt3a/3b double-KO (DKO) AML cells had accelerated leukaemic development compared with control AML cells or deletion of either Dnmt3a or Dnmt3b gene, suggesting the synergistic involvement of DNMT3A and DNMT3B in suppressing MLL-AF9 leukaemia progression. These observations were in line with findings by an independent group in which Dnmt3b overexpression (inducible Dnmt3b-knock-in mice) slowed leukaemia development and deregulated leukaemia stem cell (LSC) function, and murine MLL-AF9 cells with Dnmt3b-knock-in demonstrated leukaemia development with prolonged survival compared with MLL-AF9 alone.78 These studies appear to tally with previous observations of the Dnmt3b tumour suppressive roles in lymphomas whereby Dnmt3b−/− mice with MYC-induced lymphomas demonstrated accelerated lymphomagenesis.79
The inv(16)(p13;q22) is one of the most common recurring chromosomal rearrangements in AML, and the transcriptional coactivator MN1, an oncogene in inv(16) AML, is known to be overexpressed in inv(16) AML.80 Recently, DNMT3B expression was found to be lower in inv(16) vs non-inv(16) paediatric AML patients, and knockdown of DNMT3B expression in AML cells (HL-60) led to decreased re-methylation efficiency of MN1 exon-1 locus that subsequently drove MN1 overexpresion in AML cells.81 This implies the involvement of DNMT3B in the leukaemogenesis of inv(16) AML and suggests a tumour suppressive role akin to those observed in MLL-AF9 AML.
miRNAs Implicated in the Regulation of DNMTs Expression
One of the pivotal findings in the regulation of DNMTs expression is through miR-29b-mediated suppression. Overexpression of miR-29b in AML cells (MV4-11 and Kasumi-1) reduced the expression of DNMT1, DNMT3A, and DNMT3B at both mRNA and protein levels, resulting in reduced global DNA methylation and re-expression of p15 through promoter DNA hypomethylation.82 The authors demonstrated that miR-29b directly targeted DNMT3A and DNMT3B in their 3′ UTRs, while miR-29b downregulated DNMT1 indirectly by targeting SP1, a transactivator of DNMT1 expression.
Treatment of AML cell lines (Kasumi-1 and NB4) and leukemic blasts from primary AML patients with a potent histone deacetylase 1 (HDAC1) inhibitor AR-42 resulted in downregulation of miR-29b targets including SP1, DNMT1, DNMT3A, and DNMT3B.83 Combination of AR-42 and decitabine yielded higher anti-leukemic activity both in vitro (Kasumi-1, NB4, and murine FDC-P1-KITmut) and in vivo (FDC-P1-KITmut cells injected into NOD/SCID mice).83 These findings suggest that miR-29b oligonucleotides might be effective hypomethylating therapeutic compounds by supressing DNMTs expression. Moreover, the levels of miR-29b, in addition to DNMTs, have since been evaluated as a potential predictive factor in decitabine clinical trials of AML patients as described in the next section.
Growing evidence has shown the potential mechanisms involving DNMT3B-mediated hypermethylation in the promotion of leukaemogenesis and aggressiveness of AML, particularly involving miRNAs.82,84,85 miR-375 has been demonstrated to be a tumour suppressive miRNA in various cancers; its expression conferred better OS (P = .007) and DFS (P = .015) in AML patients (n = 102), and DNA hypermethylation of the promoter of precursor-miR-375 (pre-miR-375) caused lower miR-375 expression in AML.85 In the same study, it was reported that overexpression of miR-375 suppressed proliferation and colony formation of AML cells and reduced tumour size with prolonged survival in leukaemia xenograft mouse model (HL-60 cells in nude mice). More importantly, HOXB3 (a homeobox protein) induced DNMT3B expression to bind pre-miR-375 promoter that led to increased DNA hypermethylation of pre-miR-375, causing lower expression of miR-375 in AML cells (HL-60 and THP1), and the authors proposed a novel miR-375-HOXB3-CDCA3/DNMT3B regulatory circuitry in AML.85
Predictive Roles of DNMTs in AML Clinical Trials
In clinical trials of AML patients with and without hypomethylating agents that have assessed changes in DNMTs, the clinical response predictive role of DNMT markers remains unresolved. In a phase I study of decitabine alone or in combination with valproic acid, DNMT1 protein was detected in eight of 14 (57.1%) AML patients. DNMT1 was significantly (P = .02) decreased by decitabine treatment in the patients but the depletion was not associated with clinical response.86 In another phase I trial of decitabine plus bortezomib, bone marrow samples from five patients showed that DNMT1, DNMT3A, and DNMT3B were downregulated while miR-29b and estrogen receptor (ESR) were upregulated post-treatment (day 26) but none of the genes reached statistical significance.87
In terms of decitabine treatment alone, a phase II study of untreated older (⩾60 years old) AML patients receiving low-dose decitabine showed that responders (n = 14) had a significantly higher pre-treatment level of miR-29b vs non-responders (n = 9) (P = .02).88 In the same study, a trend for lower DNMT3A (P = .06) was observed in responders vs non-responders, and no significant differences in the levels of DNMT1, DNMT3B, or ESR1. The COG conducted a trial on eight young adults or children with refractory/relapsed (R/R) AML to receive low-dose decitabine alone.89 Three bone marrow samples were available in the study for expression analysis that showed increased miR-29b expression associated with a decrease in DNMT1 expression post-treatment. Furthermore, a phase I/II trial of adult R/R AML patients (n = 122) receiving guadecitabine showed that global DNA demethylation was strongly associated with clinical response, and DNMT3B (but not DNMT1 and DNMT3A) expression contributed >5% in predicting clinical response. FLT3-ITD and NPM1 mutations did not predict response in this cohort of AML patients.90
Although DNMT1-mediated suppression of p15 is common in AML, the expression of p15 in clinical trials also does not always correlate with response or prognosis. In a phase I/II study of 5-azacitidine in combination with valproic acid and all-trans retinoic acid, p15 mRNA expression was significantly upregulated after 7 days of treatment (P = .02) but it was not associated with clinical response.91 Likewise, in a separate phase I study, no association was observed between p15 methylation status at baseline or after therapy and response to decitabine in R/R AML patients.92 Conversely, p15 mRNA levels were increased in five of six (83.3%) assessable AML patients treated with decitabine, and global DNA hypomethylation occurred in 7 of 12 (58.3%) patients.86 Changes in DNMT1, DNMT3A, or DNMT3B expression and response predictive roles of each biomarker in clinical trials of AML patients treated with DNMT inhibitors are summarised in Table 1.
Table 1.
Changes in DNMT1, DNMT3A, or DNMT3B expression in AML patients treated with DNMT inhibitors in clinical trials.
| Treatment | Phase and ID | Patients | Regimen | Type of samplesa | Changes in expression | Predictive of response |
|---|---|---|---|---|---|---|
| Decitabine or decitabine + valproic acid (VA)86
Decitabine + bortezomib87 Decitabine89 Decitabine88 Guadecitabine90 |
Phase I ID: NCT 00079378 Phase I ID: NCT 00703300 N/A Phase II ID: NC T00492401 Phase I/II ID: NCT 01261312 |
Adult relapsed AML (n = 13) and older (>60 y/o) untreated AML (n = 12) Untreated poor-risk older (⩾65 y/o) AML patients (n = 19) Young adults or children R/R AML (n = 8) Untreated older (⩾60 y/o) AML patients (n = 53) Adult R/R AML (n = 122) |
Decitabine alone for OBD (days 1-10; n = 14) or decitabine for 10 days + VA for MTD (days 5-21) Decitabine (days 1-10) with cycles repeated every 28 days + bortezomib (days 5 and 8, or days 5, 8, 12, and 15) Decitabine (days 1-10) at ~4-week intervals. Subsequent courses shortened to 5 days in responders Low-dose decitabine (days 1-10) Guadecitabine for 5 (daily ×5) or 10 days (daily ×10) up to 4 cycles followed by daily ×5 cycles. All regimens dosed with a 28-day treatment cycle |
Unselected bone marrow mononuclear cells (n = 14) Serial bone marrow samples (n = 5) Sequential bone marrow samples (n = 3) Unselected diagnostic bone marrow samples (n = 23) Type of samples not stated (n = 122) |
Post-treatment: ↑p15, ER, ER promoter demethylation; ↓DNMT1 protein, global hypomethylation Post-treatment: ↑miR-29b, ESR; ↓DNMT1, DNMT3A, DNMT3B Post-treatment: ↑miR-29b; ↓DNMT1 Responders: ↑miR-29b, a trend for lower DNMT3A. No differences in DNMT1, DNMT3B, or ESR1 N/A |
ER expression N/A N/A miR-29b expression Global DNA demethylation; DNMT3B contributed >5% |
For the primary outcomes (eg, response rate, survival) of the actual clinical trial, readers are directed to the individual reference for each study.
Abbreviaitons: MTD, maximum-tolerated dose; N/A, not available; OBD, optimal biological dose.
Denotes the type of samples used to examine the changes in the levels of DNMTs and other genes, and the number of available samples.
Novel DNMT Inhibitors and Combination Therapies in Clinical Trials of AML
Complete tumour responses are infrequent in AML and MDS patients treated with azacitidine or decitabine (a deoxy derivative of azacitidine). This is thought to be due to degradation of both agents by cytidine-deaminase (CDA) present in the liver, spleen, and intestinal epithelium, resulting in their short plasma half-life.93–95 Guadecitabine (SGI-110), a dinucleotide of decitabine, demonstrates improved stability in plasma due to CDA resistance,96,97 and it is under active assessment in several AML clinical trials. In a multicenter phase I study of R/R AML (n = 74) and MDS patients (n = 19), the dosage 60 mg/m2 daily in a 5-day schedule and 28-day cycle administered subcutaneously (60 mg/m2/d SC 5-day/28) was shown to be well-tolerated in both patient populations.98
The clinical trial was followed by a phase II study of elderly (⩾65 years) treatment-naïve AML patients (n = 107) to receive guadecitabine at different doses and treatment schedules.99 Over 50% of the patients achieved a composite complete response at all doses and schedules of guadecitabine administration, and 60 mg/m2/d SC 5-day/28 maintained as the recommended regimen. Similar findings were reported by another multicenter phase II study of R/R AML and MDS (n = 55) where eight (14.3%) patients responded to guadecitabine (60 mg/m2/d SC 5-day/28) with prolonged survival of 17.9 months vs median survival of 7.1 months.100 In the study, patients with no or few somatic mutations (mutated DNMT3A was present in 20% of the study cohort) was the only factor that significantly predicted clinical response (P = .035), and high rate of demethylation in blood was significantly associated with longer survival (P = .03).
Ongoing clinical trials involving DNMT inhibitors (azacitidine, decitabine, and guadecitabine) in AML patients are summarised in Table 2. In particular, a multicenter phase III randomised clinical trial of guadecitabine vs treatment of choice in 404 previously treated AML patients is currently underway (ClinicalTrials.gov ID: NCT02920008). Interestingly, combination of decitabine with talazoparib, a novel inhibitor recently approved by the Federal Drug Administration (FDA) in October 2018 for the treatment of BRCA+ breast cancer, is also under investigation by a phase I/II study in untreated AML and R/R AML patients (n = 171; ID: NCT02878785) (Table 2).
Table 2.
Ongoing clinical trials of DNMT inhibitors in AML patients.
| Treatment | Phase and ID | Patients | Primary outcome measures | Regimen | Primary completion |
|---|---|---|---|---|---|
| Guadecitabine vs treatment of choice (TC)101
Guadecitabine102 Guadecitabine103 Guadecitabine104 Decitabine + talazoparib105 Azacitidine or decitabine + chemotherapy106 Guadecitabine vs guadecitabine + idarubicin107 |
Phase III ID: NCT 02920008 Phase I ID: NCT 02293993 Phase II ID: NCT 03603964 Phase II ID: NCT 03454984 Phase I/II ID: NCT 02878785 Phase II ID: NCT 03164057 Phase II ID: NCT 02096055 |
Previously treated AML (n = 404) Relapsed or not eligible for chemotherapy AML (n = 21) AML and MDS who participated in a previous guadecitabine clinical study (n = 250) Previously treated AML and MDS after allogeneic stem cell transplantation (n = 40) Untreated AML and relapsed/refractory AML (n = 171) Untreated AML (n = 200) Untreated elderly AML (n = 44) |
Overall survival (OS) Dose-limiting toxicity (DLT) when administered subcutaneously Incidence of adverse events (AEs) Disease-free survival (DFS) Dose finding and efficacy Safety, change in genome-wide methylation and its association with event-free survival (EFS) Complete remission (CR) and toxicity |
Guadecitabine or TC in a 1:1 ratio. Guadecitabine is given in 28-day cycles. TC includes either high- or low-intensity regimen, or best supportive care. Patients divided into four cohorts each receiving different dose once daily for 5 consecutive days followed by a 23-day non-dosing period, or once daily for 10 days followed by a 16-day non-dosing period Guadecitabine at the same dose received in the last cycle of their prior study or at a different dose. Treatment is continued as long as the patient continues to benefit based on investigator’s judgement Guadecitabine for 5 days in a total of 10 cycles (cycle = 28 days) Phase I: decitabine daily for 5 days every 28 days. Talazoparib orally daily days 1-28; Phase II: decitabine for 5 days every 28 days. Talazoparib orally daily days 1-28 Azacitidine or decitabine for 5 days prior to induction chemotherapy followed by intensification chemotherapy. Objective is to evaluate safety and efficacy of epigenetic priming prior to chemotherapy Group A: guadecitabine for 5 days; Group B: guadecitabine plus idarubicin for 5 days |
September 2019 December 2019 July 2020 September 2020 December 2020 June 2025 April 2026 |
Talazoparib inhibits the activities of poly (ADP-ribose) polymerase (PARP) that subsequently induces DNA damage and cell death of BRCA+ breast cancer cells through synthetic lethality (ie, simultaneous disruption of multiple genes).108–110 PARP-BRCA represents the first synthetic lethality approach that has successfully translated into the clinic,111 and similar synthetic lethality interaction is thought to occur for PARP-DNMT. The PARP inhibitor olaparib has been shown to synergise with decitabine whereby treatment with both inhibitors caused synthetic lethality with increased apoptosis and DNA damage in a panel of AML cell lines (HL60, K562, Mv4-11, KG1a, and PL21).112
Likewise, treatment of AML cell lines (KBM3/Bu2506 and MOLM14) with combinations of decitabine (DNMT inhibitor), niraparib (PARP inhibitor), and romidepsin or panobinostat (both HDAC inhibitors) suppressed their proliferation through trapping of PARP1 and DNMT1 to chromatin, leading to increased double-strand DNA breaks and cell death.113 These findings support the ongoing or future clinical trials of PARP-DNMT inhibitors combination in AML patients.
Experimental DNMT Inhibitors Targeting AML
Approved nucleoside analogues for AML treatment (azacitidine and decitabine) are relatively non-specific with low chemical stability, confer significant toxicities, and require incorporation into DNA to exert their effects as covalent inhibitors.114 This intensifies the identification and characterisation of novel, non-nucleoside DNMT inhibitors with various chemical scaffolds, and multiple of such inhibitors have recently been assessed in AML cells.
A recent structure-based virtual screening in combination with biological assays identified two compounds, termed as compounds 40 and 40_3, that inhibited DNMT3A activities with IC50 of 46.5 and 41 µM, respectively.29 The authors demonstrated that both compounds were more cytotoxic against AML cell lines (Kasumi-1, KG-1, MV4-11, and THP-1) than the cervical cancer HeLa cells, and both compounds showed similar growth inhibitory effects. Compounds 40 and 40_3 at 50 µM concentration conferred over 50% inhibition in MV4-11 AML cells.
NSC-319745 is an inhibitor of DNMT1 originally identified through docking-based virtual screening and enzymatic assays, although it contains relatively low potency against DNMT1.115 Recently, an independent research group synthesised hydroxamic acid derivatives of NSC-319745, resulting in compounds potent against DNMTs and HDACs.116 One of the inhibitors termed as compound 15a showed higher DNMT1 inhibitory potency than NSC-319745 (% inhibition at 100 µM: 69.88 ± 1.97) as well as inhibiting HDAC1 (IC50: 57 nM) and HDAC6 (IC50: 17 nM). Compound 15a was cytotoxic against AML (U937; IC50: 1.06 ± 0.09 µM) and chronic myeloid leukaemia (CML) (K562; IC50: 2.85 ± 0.12 µM) cells, and induced expression of p16 through its CpG islands demethylation and histones acetylation. The compound also demonstrated inhibition, albeit lower than DNMT1, against DNMT3A (% inhibition at 100 µM: 23.39 ± 4.12) and DNMT3B (% inhibition at 100 µM: 48.53 ± 2.38).116
Harmine, a type of β-carboline alkaloid originally isolated from the seeds of the herbal plant Peganum harmala, decreased DNMT1 expression and re-activated p15 expression through promoter hypomethylation. In AML cells (NB4), the compound significantly suppressed cell proliferation (P < .05) in a dose- and time-dependent manner and arrested the cells in G0/G1 phase.37 In addition, an analogue of procaine (a non-nucleoside DNMT inhibitor) termed as derivative 3b was capable of inducing the demethylation of chromosomal satellite repeats in HL60 AML cell line, although it was not cytotoxic against the cells.117
Conclusions
DNMT1 is rarely mutated in AML and its high expression levels in AML imply the requirement of its increased functions, through overexpression, for the survival of AML cells. DNMT1 frequently targets the promoter region of p15 for expression downregulation. Hence, assessment of increased p15 levels serves as a recommended parameter to measure the impaired activity of DNMT1 in AML, and this is applicable in future research for the identification and characterisation of novel inhibitors specific against DNMT1 which has been a subject of active investigations in recent years.28,118,119
A wide body of reports have demonstrated the high frequency of DNMT3A mutations occurring in AML. Mutations of DNMT3A locus is regarded as one of the prerequisite early genetic lesions in the pathogenesis of AML before the acquisition of additional mutations such as FLT3-ITD that transform a pre-malignant clone into AML, while R882 mutations confer loss-of-function of the encoded protein. DNMT3A-specific therapeutic modalities might require genetic editing for the restoration of wild-type DNMT3A protein expression.
The roles of DNMT3B in AML might be subtype specific as it appears to be oncogenic in AML xenograft mouse models as well as cell lines. Paradoxically, it may also play a tumour suppressive role in MLL-AF9 AML, and inv(16)(p13;q22) AML. Its oncogenic properties have been frequently shown to be associated with HOX genes, particularly the oncoprotein HOXB3 in AML as well as breast cancer,120 and DNMT3B expression is positively associated with those of HOX family genes in AML.71 Disruption of the expression or functions of DNMT3B as a potential treatment for AML patients might thus require assessment for the expression levels of HOX genes.
The expression of DNMTs was not predictive of response in most clinical trials of hypomethylating agents in AML. However, there are limitations to the interpretation of predictive values whereby most clinical trials had limited number of samples for expression studies pre- and post-treatment. Furthermore, different patient populations (young or older AML patients) or regimens (hypomethylation monotherapy or combination treatments) might contribute to differences in their DNMTs expression or prediction of response results. Nonetheless, majority of the studies reported downregulation of DNMTs expression accompanied with increased expression of their suppressor mir-29b post-treatment (Table 1).
In conclusion, increased hypermethylation of tumour suppressor genes in AML is attributable to the aberrant activities of DNMT proteins whose expression is primarily regulated by miR-29b. Expression of DNMTs is downregulated in clinical trials of AML patients treated with hypomethylating agents targeting DNMTs. Recent clinical trials have shown the novel hypomethylating drug guadecitabine conferring enhanced response in AML patients. Multiple phase II and III clinical trials are currently underway to test the efficacy of azacitidine, decitabine, or guadecitabine in combination with other agents (eg, PARP inhibitor or chemotherapy regimens) for AML patients. Collectively, these imply that nucleoside-based agents might still be the mainstay for AML hypomethylation therapy in the upcoming years. Novel non-nucleoside DNMT inhibitors have demonstrated cytotoxicity in pre-clinical experimental settings against AML cells, and their translation into the clinic remains to be elucidated.
Footnotes
Funding:The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work has been supported by the Bridging Grant (304.PPSP.6316332), Universiti Sains Malaysia, awarded to KKW.
Declaration of conflicting interests:The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions: KKW conceived and designed the manuscript, conducted literature searches, prepared the figures, and wrote the manuscript. CHL and TMG critically revised and edited the manuscript.
ORCID iD: Kah Keng Wong  https://orcid.org/0000-0001-7359-6202
https://orcid.org/0000-0001-7359-6202
References
- 1. Perri F, Longo F, Giuliano M, et al. Epigenetic control of gene expression: potential implications for cancer treatment. Crit Rev Oncol Hematol. 2017;111:166–172. [DOI] [PubMed] [Google Scholar]
- 2. Flavahan WA, Gaskell E, Bernstein BE. Epigenetic plasticity and the hallmarks of cancer. Science. 2017;357:eaal2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. He W, Li X, Xu S, et al. Aberrant methylation and loss of CADM2 tumor suppressor expression is associated with human renal cell carcinoma tumor progression. Biochem Biophys Res Commun. 2013;435:526–532. [DOI] [PubMed] [Google Scholar]
- 4. Xia L, Huang W, Bellani M, et al. CHD4 has oncogenic functions in initiating and maintaining epigenetic suppression of multiple tumor suppressor genes. Cancer Cell. 2017;31:653.e7–668.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Smith ZD, Meissner A. DNA methylation: roles in mammalian development. Nat Rev Genet. 2013;14:204–220. [DOI] [PubMed] [Google Scholar]
- 6. Ambrosi C, Manzo M, Baubec T. Dynamics and context-dependent roles of DNA methylation. J Mol Biol. 2017;429:1459–1475. [DOI] [PubMed] [Google Scholar]
- 7. Edwards JR, Yarychkivska O, Boulard M, Bestor TH. DNA methylation and DNA methyltransferases. Epigenet Chromat. 2017;10:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Li Y, Tollefsbol TO. Impact on DNA methylation in cancer prevention and therapy by bioactive dietary components. Curr Med Chem. 2010;17:2141–2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. May O. DNA methylation: fingerprints of the (epi)genome. 2010. Website. https://www.caymanchem.com/article/2153. Accessed March 29, 2019.
- 10. Maresca A, Zaffagnini M, Caporali L, Carelli V, Zanna C. DNA methyltransferase 1 mutations and mitochondrial pathology: is mtDNA methylated? Front Genet. 2015;6:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Weinberg OK, Sohani AR, Bhargava P, Nardi V. Diagnostic work-up of acute myeloid leukemia. Am J Hematol. 2017;92:317–321. [DOI] [PubMed] [Google Scholar]
- 12. Burnett A, Wetzler M, Lowenberg B. Therapeutic advances in acute myeloid leukemia. J Clin Oncol. 2011;29:487–494. [DOI] [PubMed] [Google Scholar]
- 13. Dohner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukemia. N Engl J Med. 2015;373:1136–1152. [DOI] [PubMed] [Google Scholar]
- 14. Cancer Genome Atlas Research Network, Ley TJ, Miller C, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368:2059–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Christiansen DH, Andersen MK, Pedersen-Bjergaard J. Methylation of p15INK4B is common, is associated with deletion of genes on chromosome arm 7q and predicts a poor prognosis in therapy-related myelodysplasia and acute myeloid leukemia. Leukemia. 2003;17:1813–1819. [DOI] [PubMed] [Google Scholar]
- 16. Figueroa ME, Lugthart S, Li Y, et al. DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer Cell. 2010;17:13–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tahiliani M, Koh KP, Shen Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lorsbach RB, Moore J, Mathew S, Raimondi SC, Mukatira ST, Downing JR. TET1, a member of a novel protein family, is fused to MLL in acute myeloid leukemia containing the t(10;11)(q22;q23). Leukemia. 2003;17:637–641. [DOI] [PubMed] [Google Scholar]
- 19. Chou WC, Chou SC, Liu CY, et al. TET2 mutation is an unfavorable prognostic factor in acute myeloid leukemia patients with intermediate-risk cytogenetics. Blood. 2011;118:3803–3810. [DOI] [PubMed] [Google Scholar]
- 20. Metzeler KH, Maharry K, Radmacher MD, et al. TET2 mutations improve the new European LeukemiaNet risk classification of acute myeloid leukemia: a Cancer and Leukemia Group B study. J Clin Oncol. 2011;29:1373–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Weissmann S, Alpermann T, Grossmann V, et al. Landscape of TET2 mutations in acute myeloid leukemia. Leukemia. 2012;26:934–942. [DOI] [PubMed] [Google Scholar]
- 22. Itzykson R, Kosmider O, Cluzeau T, et al. Impact of TET2 mutations on response rate to azacitidine in myelodysplastic syndromes and low blast count acute myeloid leukemias. Leukemia. 2011;25:1147–1152. [DOI] [PubMed] [Google Scholar]
- 23. Wouters BJ, Delwel R. Epigenetics and approaches to targeted epigenetic therapy in acute myeloid leukemia. Blood. 2016;127:42–52. [DOI] [PubMed] [Google Scholar]
- 24. Kelly TK, De Carvalho DD, Jones PA. Epigenetic modifications as therapeutic targets. Nat Biotechnol. 2010;28:1069–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lund K, Cole JJ, VanderKraats ND, et al. DNMT inhibitors reverse a specific signature of aberrant promoter DNA methylation and associated gene silencing in AML. Genome Biol. 2014;15:406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pleyer L, Dohner H, Dombret H, et al. Azacitidine for front-line therapy of patients with AML: reproducible efficacy established by direct comparison of international phase 3 trial data with registry data from the Austrian Azacitidine Registry of the AGMT study group. Int J Mol Sci. 2017;18:E415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Malik P, Cashen AF. Decitabine in the treatment of acute myeloid leukemia in elderly patients. Cancer Manag Res. 2014;6:53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Krishna S, Shukla S, Lakra AD, Meeran SM, Siddiqi MI. Identification of potent inhibitors of DNA methyltransferase 1 (DNMT1) through a pharmacophore-based virtual screening approach. J Mol Graph Model. 2017;75:174–188. [DOI] [PubMed] [Google Scholar]
- 29. Shao Z, Xu P, Xu W, et al. Discovery of novel DNA methyltransferase 3A inhibitors via structure-based virtual screening and biological assays. Bioorg Med Chem Lett. 2017;27:342–346. [DOI] [PubMed] [Google Scholar]
- 30. Miletic V, Odorcic I, Nikolic P, Svedruzic ZM. In silico design of the first DNA-independent mechanism-based inhibitor of mammalian DNA methyltransferase Dnmt1. PLoS ONE. 2017;12:e0174410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Singh V, Sharma P, Capalash N. DNA methyltransferase-1 inhibitors as epigenetic therapy for cancer. Curr Cancer Drug Targets. 2013;13:379–399. [DOI] [PubMed] [Google Scholar]
- 32. Solly F, Koering C, Mohamed AM, et al. An miRNA-DNMT1 axis is involved in azacitidine resistance and predicts survival in higher-risk myelodysplastic syndrome and low blast count acute myeloid leukemia. Clin Cancer Res. 2017;23:3025–3034. [DOI] [PubMed] [Google Scholar]
- 33. Li S, Jin X, Wu H, et al. HA117 endows HL60 cells with a stem-like signature by inhibiting the degradation of DNMT1 via its ability to down-regulate expression of the GGL domain of RGS6. PLoS ONE. 2017;12:e0180142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fares J, Wolff L, Bies J. p15INK4b, a tumor suppressor in acute myeloid leukemia. In: Koschmieder S, Krug U, eds. Myeloid Leukemia – Basic Mechanisms of Leukemogenesis. London, England: IntechOpen; 2011:289–312. [Google Scholar]
- 35. Mizuno S, Chijiwa T, Okamura T, et al. Expression of DNA methyltransferases DNMT1, 3A, and 3B in normal hematopoiesis and in acute and chronic myelogenous leukemia. Blood. 2001;97:1172–1179. [DOI] [PubMed] [Google Scholar]
- 36. Shen N, Yan F, Pang J, et al. Inactivation of receptor tyrosine kinases reverts aberrant DNA methylation in acute myeloid leukemia. Clin Cancer Res. 2017;23:6254–6266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Oodi A, Norouzi H, Amirizadeh N, Nikougoftar M, Vafaie Z. Harmine, a novel DNA methyltransferase 1 inhibitor in the leukemia cell line. Indian J Hematol Blood Transfus. 2017;33:509–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yan F, Shen N, Pang JX, et al. Fatty acid-binding protein FABP4 mechanistically links obesity with aggressive AML by enhancing aberrant DNA methylation in AML cells. Leukemia. 2017;31:1434–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yan F, Shen N, Pang JX, et al. A vicious loop of fatty acid-binding protein 4 and DNA methyltransferase 1 promotes acute myeloid leukemia and acts as a therapeutic target. Leukemia. 2018;32:865–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tagde A, Rajabi H, Stroopinsky D, et al. MUC1-C induces DNA methyltransferase 1 and represses tumor suppressor genes in acute myeloid leukemia. Oncotarget. 2016;7:38974–38987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Loo SK, Ch’ng ES, Lawrie CH, et al. DNMT1 is predictive of survival and associated with Ki-67 expression in R-CHOP-treated diffuse large B-cell lymphomas. Pathology. 2017;49:731–739. [DOI] [PubMed] [Google Scholar]
- 42. Loo SK, Ab Hamid SS, Musa M, Wong KK. DNMT1 is associated with cell cycle and DNA replication gene sets in diffuse large B-cell lymphoma. Pathol Res Pract. 2018;214:134–143. [DOI] [PubMed] [Google Scholar]
- 43. Shu Y, Zhou X, Qi X, et al. β-Arrestin1 promotes the self-renewal of the leukemia-initiating cell-enriched subpopulation in B-lineage acute lymphoblastic leukemia related to DNMT1 activity. Cancer Lett. 2015;357:170–178. [DOI] [PubMed] [Google Scholar]
- 44. Wang J, Hua L, Guo M, et al. Notable roles of EZH2 and DNMT1 in epigenetic dormancy of the SHP1 gene during the progression of chronic myeloid leukaemia. Oncol Lett. 2017;13:4979–4985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Harada T, Ohguchi H, Grondin Y, et al. HDAC3 regulates DNMT1 expression in multiple myeloma: therapeutic implications. Leukemia. 2017;31:2670–2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Robaina MC, Mazzoccoli L, Arruda VO, et al. Deregulation of DNMT1, DNMT3B and miR-29s in Burkitt lymphoma suggests novel contribution for disease pathogenesis. Exp Mol Pathol. 2015;98:200–207. [DOI] [PubMed] [Google Scholar]
- 47. Baer C, Pohlkamp C, Haferlach C, Kern W, Haferlach T. Molecular patterns in cytopenia patients with or without evidence of myeloid neoplasm-a comparison of 756 cases. Leukemia. 2018;32:2295–2298. [DOI] [PubMed] [Google Scholar]
- 48. Silva P, Neumann M, Schroeder MP, et al. Acute myeloid leukemia in the elderly is characterized by a distinct genetic and epigenetic landscape. Leukemia. 2017;31:1640–1644. [DOI] [PubMed] [Google Scholar]
- 49. Renaud L, Nibourel O, Marceau-Renaut A, et al. Comprehensive molecular landscape in patients older than 80 years old diagnosed with acute myeloid leukemia: a study of the French Hauts-de-France AML observatory. Am J Hematol. 2019;94:E24–E27. [DOI] [PubMed] [Google Scholar]
- 50. Yamashita Y, Yuan J, Suetake I, et al. Array-based genomic resequencing of human leukemia. Oncogene. 2010;29:3723–3731. [DOI] [PubMed] [Google Scholar]
- 51. Ley TJ, Ding L, Walter MJ, et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med. 2010;363:2424–2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ribeiro AF, Pratcorona M, Erpelinck-Verschueren C, et al. Mutant DNMT3A: a marker of poor prognosis in acute myeloid leukemia. Blood. 2012;119:5824–5831. [DOI] [PubMed] [Google Scholar]
- 53. Genovese G, Kahler AK, Handsaker RE, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med. 2014;371:2477–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Jaiswal S, Fontanillas P, Flannick J, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371:2488–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Balasubramanian SK, Aly M, Nagata Y, et al. Distinct clinical and biological implications of various DNMT3A mutations in myeloid neoplasms. Leukemia. 2018;32:550–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yang L, Rodriguez B, Mayle A, et al. DNMT3A loss drives enhancer hypomethylation in FLT3-ITD-associated leukemias. Cancer Cell. 2016;29:922–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Meyer SE, Qin T, Muench DE, et al. DNMT3A haploinsufficiency transforms FLT3ITD myeloproliferative disease into a rapid, spontaneous, and fully penetrant acute myeloid leukemia. Cancer Discov. 2016;6:501–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sun QY, Ding LW, Tan KT, et al. Ordering of mutations in acute myeloid leukemia with partial tandem duplication of MLL (MLL-PTD). Leukemia. 2017;31:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sengsayadeth SM, Jagasia M, Engelhardt BG, et al. Allo-SCT for high-risk AML-CR1 in the molecular era: impact of FLT3/ITD outweighs the conventional markers. Bone Marrow Transplant. 2012;47:1535–1537. [DOI] [PubMed] [Google Scholar]
- 60. Brunet S, Labopin M, Esteve J, et al. Impact of FLT3 internal tandem duplication on the outcome of related and unrelated hematopoietic transplantation for adult acute myeloid leukemia in first remission: a retrospective analysis. J Clin Oncol. 2012;30:735–741. [DOI] [PubMed] [Google Scholar]
- 61. Rose D, Haferlach T, Schnittger S, Perglerova K, Kern W, Haferlach C. Subtype-specific patterns of molecular mutations in acute myeloid leukemia. Leukemia. 2017;31:11–17. [DOI] [PubMed] [Google Scholar]
- 62. Ardestani MT, Kazemi A, Chahardouli B, et al. FLT3-ITD compared with DNMT3A R882 mutation is a more powerful independent inferior prognostic factor in adult acute myeloid leukemia patients after allogeneic hematopoietic stem cell transplantation: a retrospective cohort study. Turk J Haematol. 2018;35:158–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ahn JS, Kim HJ, Kim YK, et al. DNMT3A R882 mutation with FLT3-ITD positivity is an extremely poor prognostic factor in patients with normal-karyotype acute myeloid leukemia after allogeneic hematopoietic cell transplantation. Biol Blood Marrow Transplant. 2016;22:61–70. [DOI] [PubMed] [Google Scholar]
- 64. Lin J, Yao DM, Qian J, et al. Recurrent DNMT3A R882 mutations in Chinese patients with acute myeloid leukemia and myelodysplastic syndrome. PLoS ONE. 2011;6:e26906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Gaidzik VI, Weber D, Paschka P, et al. DNMT3A mutant transcript levels persist in remission and do not predict outcome in patients with acute myeloid leukemia. Leukemia. 2018;32:30–37. [DOI] [PubMed] [Google Scholar]
- 66. Ho PA, Kutny MA, Alonzo TA, et al. Leukemic mutations in the methylation-associated genes DNMT3A and IDH2 are rare events in pediatric AML: a report from the Children’s Oncology Group. Pediatr Blood Cancer. 2011;57:204–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Stengel A, Kern W, Meggendorfer M, et al. Number of RUNX1 mutations, wild-type allele loss and additional mutations impact on prognosis in adult RUNX1-mutated AML. Leukemia. 2018;32:295–302. [DOI] [PubMed] [Google Scholar]
- 68. Kelly AD, Madzo J, Madireddi P, et al. Demethylator phenotypes in acute myeloid leukemia. Leukemia. 2018;32:2178–2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Desai P, Mencia-Trinchant N, Savenkov O, et al. Somatic mutations precede acute myeloid leukemia years before diagnosis. Nat Med. 2018;24:1015–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Hayette S, Thomas X, Jallades L, et al. High DNA methyltransferase DNMT3B levels: a poor prognostic marker in acute myeloid leukemia. PLoS ONE. 2012;7:e51527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Lamba JK, Cao X, Raimondi SC, et al. Integrated epigenetic and genetic analysis identifies markers of prognostic significance in pediatric acute myeloid leukemia. Oncotarget. 2018;9:26711–26723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Niederwieser C, Kohlschmidt J, Volinia S, et al. Prognostic and biologic significance of DNMT3B expression in older patients with cytogenetically normal primary acute myeloid leukemia. Leukemia. 2015;29:567–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Russler-Germain DA, Spencer DH, Young MA, et al. The R882H DNMT3A mutation associated with AML dominantly inhibits wild-type DNMT3A by blocking its ability to form active tetramers. Cancer Cell. 2014;25:442–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Itonaga H, Imanishi D, Wong YF, et al. Expression of myeloperoxidase in acute myeloid leukemia blasts mirrors the distinct DNA methylation pattern involving the downregulation of DNA methyltransferase DNMT3B. Leukemia. 2014;28:1459–1466. [DOI] [PubMed] [Google Scholar]
- 75. Zheng Q, Zeng TT, Chen J, Liu H, Zhang H, Su J. Association between DNA methyltransferases 3B gene polymorphisms and the susceptibility to acute myeloid leukemia in Chinese Han population. PLoS ONE. 2013;8:e74626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Poole CJ, Zheng W, Lodh A, et al. DNMT3B overexpression contributes to aberrant DNA methylation and MYC-driven tumor maintenance in T-ALL and Burkitt’s lymphoma. Oncotarget. 2017;8:76898–76920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Zheng Y, Zhang H, Wang Y, et al. Loss of Dnmt3b accelerates MLL-AF9 leukemia progression. Leukemia. 2016;30:2373–2384. [DOI] [PubMed] [Google Scholar]
- 78. Schulze I, Rohde C, Scheller-Wendorff M, et al. Increased DNA methylation of Dnmt3b targets impairs leukemogenesis. Blood. 2016;127:1575–1586. [DOI] [PubMed] [Google Scholar]
- 79. Hlady RA, Novakova S, Opavska J, et al. Loss of Dnmt3b function upregulates the tumor modifier Ment and accelerates mouse lymphomagenesis. J Clin Invest. 2012;122:163–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Grosveld GC. MN1, a novel player in human AML. Blood Cells Mol Dis. 2007;39:336–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Larmonie NSD, Arentsen-Peters T, Obulkasim A, et al. MN1 overexpression is driven by loss of DNMT3B methylation activity in inv(16) pediatric AML. Oncogene. 2018;37:107–115. [DOI] [PubMed] [Google Scholar]
- 82. Garzon R, Liu S, Fabbri M, et al. MicroRNA-29b induces global DNA hypomethylation and tumor suppressor gene reexpression in acute myeloid leukemia by targeting directly DNMT3A and 3B and indirectly DNMT1. Blood. 2009;113:6411–6418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Mims A, Walker AR, Huang X, et al. Increased anti-leukemic activity of decitabine via AR-42-induced upregulation of miR-29b: a novel epigenetic-targeting approach in acute myeloid leukemia. Leukemia. 2013;27:871–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Memari F, Joneidi Z, Taheri B, Aval SF, Roointan A, Zarghami N. Epigenetics and Epi-miRNAs: potential markers/therapeutics in leukemia. Biomed Pharmacother. 2018;106:1668–1677. [DOI] [PubMed] [Google Scholar]
- 85. Bi L, Zhou B, Li H, et al. A novel miR-375-HOXB3-CDCA3/DNMT3B regulatory circuitry contributes to leukemogenesis in acute myeloid leukemia. BMC Cancer. 2018;18:182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Blum W, Klisovic RB, Hackanson B, et al. Phase I study of decitabine alone or in combination with valproic acid in acute myeloid leukemia. J Clin Oncol. 2007;25:3884–3891. [DOI] [PubMed] [Google Scholar]
- 87. Blum W, Schwind S, Tarighat SS, et al. Clinical and pharmacodynamic activity of bortezomib and decitabine in acute myeloid leukemia. Blood. 2012;119:6025–6031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Blum W, Garzon R, Klisovic RB, et al. Clinical response and miR-29b predictive significance in older AML patients treated with a 10-day schedule of decitabine. Proc Natl Acad Sci U S A. 2010;107:7473–7478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Phillips CL, Davies SM, McMasters R, et al. Low dose decitabine in very high risk relapsed or refractory acute myeloid leukaemia in children and young adults. Br J Haematol. 2013;161:406–410. [DOI] [PubMed] [Google Scholar]
- 90. Gottipati S, Rohatagi S, Chung W, et al. Multivariate analysis identifies significant correlations between baseline biomarkers, DNA demethylation and clinical responses in 122 patients treated in a phase 1/2, study of guadecitabine (SGI-110), in the treatment of relapsed/refractory acute myeloid leukemia (r/r AML). Blood. 2015;126:2594. [Google Scholar]
- 91. Soriano AO, Yang H, Faderl S, et al. Safety and clinical activity of the combination of 5-azacytidine, valproic acid, and all-trans retinoic acid in acute myeloid leukemia and myelodysplastic syndrome. Blood. 2007;110:2302–2308. [DOI] [PubMed] [Google Scholar]
- 92. Issa JP, Garcia-Manero G, Giles FJ, et al. Phase 1 study of low-dose prolonged exposure schedules of the hypomethylating agent 5-aza-2’-deoxycytidine (decitabine) in hematopoietic malignancies. Blood. 2004;103:1635–1640. [DOI] [PubMed] [Google Scholar]
- 93. Estey EH. Epigenetics in clinical practice: the examples of azacitidine and decitabine in myelodysplasia and acute myeloid leukemia. Leukemia. 2013;27:1803–1812. [DOI] [PubMed] [Google Scholar]
- 94. Navada SC, Steinmann J, Lubbert M, Silverman LR. Clinical development of demethylating agents in hematology. J Clin Invest. 2014;124:40–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Bohl SR, Bullinger L, Rucker FG. Epigenetic therapy: azacytidine and decitabine in acute myeloid leukemia. Expert Rev Hematol. 2018;11:361–371. [DOI] [PubMed] [Google Scholar]
- 96. Yoo CB, Jeong S, Egger G, et al. Delivery of 5-aza-2′-deoxycytidine to cells using oligodeoxynucleotides. Cancer Res. 2007;67:6400–6408. [DOI] [PubMed] [Google Scholar]
- 97. Roboz GJ, Kantarjian HM, Yee KWL, et al. Dose, schedule, safety, and efficacy of guadecitabine in relapsed or refractory acute myeloid leukemia. Cancer. 2018;124:325–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Issa JJ, Roboz G, Rizzieri D, et al. Safety and tolerability of guadecitabine (SGI-110) in patients with myelodysplastic syndrome and acute myeloid leukaemia: a multicentre, randomised, dose-escalation phase 1 study. Lancet Oncol. 2015;16:1099–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Kantarjian HM, Roboz GJ, Kropf PL, et al. Guadecitabine (SGI-110) in treatment-naive patients with acute myeloid leukaemia: phase 2 results from a multicentre, randomised, phase 1/2 trial. Lancet Oncol. 2017;18:1317–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Sebert M, Renneville A, Bally C, et al. A phase II study of guadecitabine in higher-risk myelodysplastic syndrome and low blast count acute myeloid leukemia after azacitidine failure [published online ahead of print February 7, 2019]. Haematologica. doi: 10.3324/haematol.2018.207118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. ClinicalTrials.gov. Phase 3 randomized, open-label study of guadecitabine vs treatment choice in previously treated acute myeloid leukemia. 2016. Website. https://clinicaltrials.gov/ct2/show/NCT02920008. Accessed March 24, 2019.
- 102. ClinicalTrials.gov. Phase 1 study of SGI-110 in patients with acute myeloid leukemia. 2014. Website. https://clinicaltrials.gov/ct2/show/NCT02293993. Accessed March 24, 2019.
- 103. ClinicalTrials.gov. Guadecitabine extension study. 2018. Website. https://clinicaltrials.gov/ct2/show/NCT03603964. Accessedarch 24, 2019.
- 104. ClinicalTrials.gov. SGI-110 and donor lymphocyte infusions (DLI) after allogeneic stem cell transplantation. 2018. Website. https://clinicaltrials.gov/ct2/show/NCT03454984. Accessed March 24, 2019.
- 105. ClinicalTrials.gov. Decitabine and talazoparib in untreated AML and R/R AML (1565GCC). 2019. Website. https://clinicaltrials.gov/ct2/show/NCT02878785. Accessed March 25, 2019.
- 106. ClinicalTrials.gov. A trial of epigenetic priming in patients with newly diagnosed acute myeloid leukemia. 2017. Website. https://clinicaltrials.gov/ct2/show/NCT03164057. Accessed March 24, 2019.
- 107. ClinicalTrials.gov. Study of SGI-110 in elderly acute myeloid leukemia (AML). 2014. Website. https://clinicaltrials.gov/ct2/show/NCT02096055. Accessed March 24, 2019.
- 108. Shen Y, Rehman FL, Feng Y, et al. BMN 673, a novel and highly potent PARP1/2 inhibitor for the treatment of human cancers with DNA repair deficiency. Clin Cancer Res. 2013;19:5003–5015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Sonnenblick A, de Azambuja E, Azim HA, Jr, Piccart M. An update on PARP inhibitors-moving to the adjuvant setting. Nat Rev Clin Oncol. 2015;12:27–41. [DOI] [PubMed] [Google Scholar]
- 110. Nur Husna SM, Tan HT, Mohamud R, Dyhl-Polk A, Wong KK. Inhibitors targeting CDK4/6, PARP and PI3K in breast cancer: a review. Ther Adv Med Oncol. 2018;10:1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Helleday T. The underlying mechanism for the PARP and BRCA synthetic lethality: clearing up the misunderstandings. Mol Oncol. 2011;5:387–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Orta ML, Hoglund A, Calderon-Montano JM, et al. The PARP inhibitor Olaparib disrupts base excision repair of 5-aza-2’-deoxycytidine lesions. Nucleic Acids Res. 2014;42:9108–9120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Valdez BC, Li Y, Murray D, et al. Combination of a hypomethylating agent and inhibitors of PARP and HDAC traps PARP1 and DNMT1 to chromatin, acetylates DNA repair proteins, down-regulates NuRD and induces apoptosis in human leukemia and lymphoma cells. Oncotarget. 2018;9:3908–3921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Stresemann C, Lyko F. Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. Int J Cancer. 2008;123:8–13. [DOI] [PubMed] [Google Scholar]
- 115. Kuck D, Singh N, Lyko F, Medina-Franco JL. Novel and selective DNA methyltransferase inhibitors: docking-based virtual screening and experimental evaluation. Bioorg Med Chem. 2010;18:822–829. [DOI] [PubMed] [Google Scholar]
- 116. Yuan Z, Sun Q, Li D, et al. Design, synthesis and anticancer potential of NSC-319745 hydroxamic acid derivatives as DNMT and HDAC inhibitors. Eur J Med Chem. 2017;134:281–292. [DOI] [PubMed] [Google Scholar]
- 117. Castellano S, Kuck D, Sala M, Novellino E, Lyko F, Sbardella G. Constrained analogues of procaine as novel small molecule inhibitors of DNA methyltransferase-1. J Med Chem. 2008;51:2321–2325. [DOI] [PubMed] [Google Scholar]
- 118. Florean C, Schnekenburger M, Lee JY, et al. Discovery and characterization of Isofistularin-3, a marine brominated alkaloid, as a new DNA demethylating agent inducing cell cycle arrest and sensitization to TRAIL in cancer cells. Oncotarget. 2016;7:24027–24049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Joshi M, Rajpathak SN, Narwade SC, Deobagkar D. Ensemble-based virtual screening and experimental validation of inhibitors targeting a novel site of human DNMT1. Chem Biol Drug Des. 2016;88:5–16. [DOI] [PubMed] [Google Scholar]
- 120. Palakurthy RK, Wajapeyee N, Santra MK, et al. Epigenetic silencing of the RASSF1A tumor suppressor gene through HOXB3-mediated induction of DNMT3B expression. Mol Cell. 2009;36:219–230. [DOI] [PMC free article] [PubMed] [Google Scholar]