

Abstract
Mitochondrial dysfunction is a central protagonist of Alzheimer’s disease (AD) pathogenesis. Mitochondrial dysfunction stems from various factors including mitochondrial DNA damage and oxidative stress from reactive oxygen species, membrane and ionic gradient destabilization, and interaction with toxic proteins such as amyloid beta (Aβ). Therapeutic drugs such as cholinesterase and glutamate inhibitors have proven to improve synaptic neurotransmitters, but do not address mitochondrial dysfunction. Researchers have demonstrated that oxidative damage may be reduced by increasing endogenous antioxidants, and/or increasing exogenous antioxidants such as vitamin C & E, beta-carotene and glutathione. Nonetheless, as AD pathology intensifies, endogenous antioxidants are overwhelmed, and exogenous antioxidants are unable to reach neuronal mitochondria as they are blocked by the blood brain barrier. Current therapeutic methods however include novel usage of lipophilic phosphonium cation bound to antioxidants, to effect neuronal mitochondria targeted activity. Mitochondria targeted MitoQ, MitoVitE, MitoTempo, MitoPBN and MCAT concentrate within mitochondria where they scavenge free-radicals, and augment mitochondrial dysfunction. Additional molecules include Szeto-Schiller (SS) peptides which target stability of the inner mitochondrial membrane, and DDQ molecule capable of improving bioenergetics and reduce mitochondrial fragmentation. This article discusses advantages and disadvantages of small molecules, their ability to mitigate Aβ induced damage, and ability to ameliorate synaptic dysfunction and cognitive loss.
Keywords: Alzheimer’s Disease, Mitochondria-targeted molecules, Oxidative stress, Mitochondrial dysfunction, Huntington’s disease, Parkinson, disease, Aging, Mitophagy and Mitochondrial dynamics
Introduction
Alzheimer’s disease (AD), the most prevalent form of dementia, is a neurodegenerative disease which causes memory loss, cognitive impairment, and changes in personality (Hu, et al., 2017; Reiss et al., 2018; Reddy et al., 20182). AD has no cure, and affects over 50 million people worldwide (Reddy et al., 20182). AD is expected to double in prevalence every 20 years, reaching 75.6 million in 2030, and 131.5 million by 2050, already having an impact of $800 billion in 2015 alone (Reiss et al., 2018; Reddy et al., 20182).
Aβ deposits and neurofibrillary tangles (NFTs) in postmortem brains is indicative of AD (Hyman, et al., 2012). On a microscopic level, Aβ protein accumulation in neuronal cells, along with hyper-phosphorylation of tau (τ) protein are believed to cause dysfunction in mitochondria, the powerhouse of neuronal cells (Reddy et al., 20182; Hyman, et al., 2012) Subsequent loss of synapses and cholinergic fibers, and inflammatory response from astrocytes and microglia magnify the pathology of AD to cognitive impairment, and memory loss (Reddy et al., 20182; Hyman, et al., 2012; Wilkins & Swerdlow, 2016).
The purpose of this article is to highlight recent developments in mitochondria-targeted small molecules as therapeutic drugs for AD. This article also summarizes several aspects of Aβ and phosphorylated tau induced damage in AD, and how small molecules ameliorate synaptic dysfunction and cognitive loss.
Mitochondrial Cascade - Hypothesis Russell Swerdlow
AD can be described as a series of biomolecular interactions stemming from mitochondrial dysfunction. This origin of AD is described through the mitochondrial cascade hypothesis (MCH), which explains:
All persons have a genetically inherited predisposition for mitochondrial function and dysfunction (Swerdlow et al., 2013). Genetic contribution to mitochondrial function/dysfunction is mostly attributed to maternal mitochondrial DNA (mtDNA) though there is some contribution by paternal and maternal nuclear DNA (nDNA) (Swerdlow et al., 2013; Alexeyev et al., 2013; Blair et al., 2015).
Differences in expression of mtDNA are attributed to interaction between genetic and environmental factors (Swerdlow et al., 2013). Correlation between brain aging and mitochondrial dysfunction will intensify, but may be exacerbated or reduced by environmental factors (Swerdlow et al., 2013).
The MCH concludes that these interactions are the foundation for cognitive impairment and neurodegenerative diseases (Swerdlow et al., 2013). Furthermore, these factors determine the variant pathogenesis of the disease, such as early or late onset AD (Swerdlow et al., 2013).
The MCH provides the groundwork for understanding biomarkers indicating pathogenesis, and effectiveness of therapeutics, such as mitochondria targeted small molecules. (Swerdlow et al., 2013; Alexeyev et al., 2013; Blair et al., 2015; Reddy, 2011; Reddy, 2008; Reddy PH & Reddy TP, 2011).
Mitochondrial Dysfunction
Reactive Oxygen Species
Mitochondrial dysfunction is manifested in several ways. Reactive oxygen species (ROS) are toxic molecules which have the oxidant power to initiate a domino-effect of oxidation-reduction reactions (Reddy PH & Reddy TP, 2011; Wang et al., 2013). The oxygen molecule in these ROS’s are highly electronegative and reactive, and when left unmitigated cause damage to the cell by oxidizing nucleic acids, proteins, and lipids (Wang et al., 2013).
Mitochondria play a critical role as the source of 90% of endogenous ROS (Wang et al., 2013). In fact, the electron transport chain (ETC), particularly complexes I and III, provides a consistent supply of O2∙, H2O2, and other ROS despite mitigation by antioxidants (Reddy PH & Reddy TP, 2011; Wang et al., 2013). A steady baseline concentration of ROS is likely permitted due to ROS signaling properties, which function to indicate cellular homeostasis. Nonetheless, when mitochondrial and cellular antioxidants are overwhelmed by ROS, the result is oxidative stress, a precursor to aging (Reddy et al., 20172). Oxidative stress is one of the major sources of initial pathogenesis of AD (Wang et al., 2013; Cai & Tammineni, 2016; Du et al., 2010). The accumulation of ROS and oxidative stress also gives opportunity for additional toxic molecules to cause damage. These include 3-nitropropionic acid (3-NP), an endogenous toxin indicative of mitochondrial dysfunction in neurodegenerative disease, and tert-butyl hydroperoxide (t-BHP) also responsible for oxidative damage (Reddy, 2008; Lv et al., 2017; Chaturvedi & Beal, 2013). The molecules 3-NP and t-BHP both have lipophilic properties to permeate cells, and are capable of concentrating in cells up to 1000-fold in the inner mitochondrial membrane (Reddy, 2008).
The brain is especially susceptible to oxidative damage. This is due to its high demand on energy, high demand for and supply of oxygen, high supply of peroxidizable polyunsaturated fatty acids, high concentration of iron capable of ROS generation, and relative scarcity of antioxidants (Wang et al., 2013). Furthermore, neuronal mitochondria have longer half-lives than mitochondria in bodily tissues and less availability of glutathione, making them more susceptible to long-term nitro-oxidative damage (Hu et al., 2017).
Oxidative damage accumulates with age and as mitochondria become less efficient and more dysfunctional, fidelity of mtDNA is compromised (Swerdlow et al., 2013). Oxidative damage contributes to mtDNA and nDNA damage, mutation, and loss of mtDNA copy number (Swerdlow et al., 2013; Manczak et al., 2018; Kerr et al., 2017). These contribute to mitochondrial dysfunction, as well as mitophagy dysfunction, destruction of mitochondria in response to damage and compromised mtDNA (Swerdlow et al., 2013; Manczak et al., 2018; Kerr et al., 2017).
Toxic Proteins
Amyloid-Beta
Mitochondrial interaction with several toxic proteins assists with degeneration as well. The Aβ protein is a product of cleavage of the mutant amyloid precursor protein (mAPP) by beta- and gamma-secretases (Reddy et al., 20182). Aβ is able to interact with and open the mitochondrial permeability transition (MPT) pore, which affect an influx of calcium ion, gating of metabolites, destabilizing the membrane, causing loss of membrane potential (Reddy et al., 20182; Szeto, 2014). Aβ contributes to reduction in biogenesis, as well as increased oxidative stress in mitochondria (Reddy et al., 20182; Swerdlow et al., 2013). High concentration of Aβ in the brain contributes to Aβ deposition, which often accumulate to form polymers, oligomers, and eventual insoluble fibers, all capable of circulating through the cerebral spinal fluid and blood (Reiss et al., 2018; Reddy et al., 20182; Hyman et al., 2012). The Aβ also contributes to blockage in the synapse, preventing neurotransmission (Reddy & Beal, 2008). Aβ and mAPP also work to impede bioenergetics of the ETC by preventing proteins encoded by nDNA from reaching mitochondria, which further contributes to synaptic damage (Reddy & Beal, 2008).
Amyloid-Beta-Drp1 Interaction
Aβ also greatly affects mitochondrial dynamics, the organelle’s ability to divide and replicate (fission), fuse (fusion), and excise dysfunctional mitochondria (mitophagy) (Reddy & Beal, 2008). The GTPase-Drp1 activity, mitochondrial division protein, Drp1, interaction with Aβ causes an increase in GTPase activity (Manczak & Reddy, 20121; Manczak et al., 2011; Kuruva et al, 2017). This results in excessive mitochondrial fission, or fragmentation, due to the upregulation of fission proteins Drp1, and Fis1 (Reddy & Beal, 2008). Mitochondrial fragmentation and Aβ accumulation in the synapse also cause synapse inhibition (Reddy & Beal, 2008; Manczak & Reddy, 20121; Manczak et al., 2011; Kuruva et al, 2017). Drp1-Aβ interaction also greatly increases ROS production (Reddy & Beal, 2008; Manczak & Reddy, 20121; Manczak et al., 2011; Kuruva et al, 2017). Resultant synaptic damage is thought to be responsible for cognitive decline, the majority of which affects short term memory, rather than long term memory (Reddy & Beal, 2008).
Mitophagy, the means by which damaged mitochondria are excised from the cell, is initiated by the PTEN-induced putative kinase 1 (PINK1) protein, which signals E3 ligase activity by the ubiquitin ligase parkin (Angajala, et al., 2018; Zhu et al., 2013; Cai & Tammineni, 2016). Parkin causes perturbation of mitochondrial surface proteins, and an autophagosome which consumes the mitochondrion, subsequently fuses membranes with a lysosome, destroying it (Angajala, et al., 2018; Cai & Tammineni, 2016). Mitochondrial interaction with Aβ, overexpressed Drp1, and phosphorylated tau, affect functionality of PINK1 and parkin proteins, greatly reducing mitophagy activity (Kuruva et al., 2017; Angajala, et al., 2018; Cai & Tammineni, 2016).
Amyloid-Beta + Phosphorylated Tau
Tau particles associate with microtubules within the cytoplasm, and act as a conveyer belt for organelle motility (Reddy et al., 20182; Kerr et al., 2017). Stress, which accumulates with age, assists to phosphorylate these tau particles, causing a conformational change in the protein on the microtubule, which effectively slows organelle mobility and changes the cytoskeletal structure of the cell (Reddy et al., 20182; Kerr et al., 2017) For Mitochondria, this reduces their mobility along the axon, which starves axon and dendritic terminals from an energy source (Reddy et al., 20182; Kerr et al., 2017; Reddy & Beal, 2008; Zhu et al., 2013; Reddy, 2011). The end result is dysfunction at the synapse (Reddy et al., 20182; Kerr et al., 2017; Reddy & Beal, 2008; Reddy, 2011). Aβ and phosphorylated tau particles have also proven to contribute to one another’s proliferation, and induce mitochondrial and synapse dysfunction (Reddy et al., 20182; Kerr et al., 2017; Reddy & Beal, 2008; Manczak & Reddy, 20121).
Amyloid Beta and phosphorylated Tau Interaction with VDAC1
Furthermore, Reddy and Manczak were able to note, that voltage-dependent anion channel 1 protein (VDAC1) interact with both Aβ and phosphorylated τ particles, and block mitochondrial pores (Manczak & Reddy, 20122) Using transgenic 3XTg AD mice, the blockage caused by the interaction with VDAC1, Aβ and τ particles led to AD pathogenesis, particularly reduction in ATP production (Manczak & Reddy, 20122).
Amyloid Beta + ABAD
The protein amyloid binding alcohol dehydrogenase (ABAD) changes conformation due to Aβ, causing it to bind the coenzyme nicotinamide adenine dinucleotide (NAD+) (Yan & Stern, 2005). When Aβ associated ABAD binds with NAD+, ABAD permeability is changed, inhibiting its enzyme activity preventing it from removing toxic aldehydes, and compromising respiratory enzymes (Reiss et al., 2018; Yan & Stern, 2005; Blair et al., 2015).
Amyloid Beta + CypD
Aβ also interacts with cyclophilin D (CypD), a mitochondrial matrix protein essential for electron transport, reducing bioenergetics (Du et al., 2008). CypD protein has peptidyl-prolyl isomerase (PPI) activity, which enables it to change cis/trans conformation of peptide bonds around proline residues (Du et al., 2008; Blair et al., 2015). Aβ binds to CypD, forming a CypDAβ complex, which increases ROS-initiated oxidative damage, greatly contributing to mitochondrial degeneration in AD (Blair et al., 2015; Caspersen et al., 2005; Bernardi & Di Lisa, 2015).
There are many more means by which AD pathology is initiated by mitochondrial dysfunction, including Aβ interaction with sirtuin 4 which causes reduction in biogenesis (Guedes-Dias & Oliveira, 2013; Motyl et al., 2018; Li et al., 2018). Nonetheless, frequently prescribed therapeutics for AD, target dysfunction at the synapse, rather than the mitochondrion. These drugs are effective in mitigating AD pathological symptoms, but not necessarily effective in preventing mitochondrial dysfunction.
Alzheimer’s Disease Therapeutics
Table 1: Shows summary of small molecules for the treatment of AD
Table 1:
Summary of Small Molecules for the Treatment of Alzheimer’s disease
| Name | Molecular Weight |
Molecular Formula |
Properties | Molecular Structure | Reference |
|---|---|---|---|---|---|
| L-Ascorbic Acid Vitamin C (2R)-2-[(1S)-1,2- dihydroxyethyl]-3,4- dihydroxy-2H-furan- 5 - one | 176.12 g/mol |
C6H8O6 | Lowers prevalence and incidence of Alzheimer’s disease in elderly population | 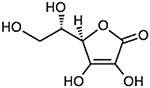 |
Reddy, 2006; Cragg & Pezzuto, 2015 |
| Beta Carotene 1,3,3-Trimethyl-2-
[3,7,12,16-tetramethyl- 18-(2,6,6- trimethylcyclohex-1 - en-1-yl)octadeca- 1,3,5,7,9,11,13,15,17- nonaen-1-yl]cyclohex- 1-ene |
536.8726 g/mol |
C40H56 | Isomer derivative of carotene; functions as antioxidant to reduce free radicals or is converted to retinol (vitamin A) by dioxygenase enzyme in body | 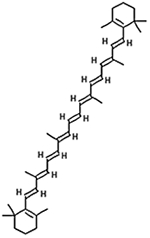 |
Reddy, 2006; Reddy, 2008; Strobel et al., 2007 |
| Catalase | 60–75kDa tetramer |
Antioxidant; porphyrin heme iron prosthetic group at core of redox activity; rapidly converts thousands of H2O2 to H2O and O2 per second |
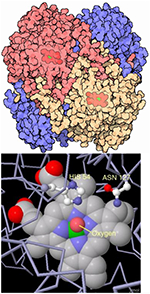 |
Chelikani et al., 2004; Imlay JA, 2003; Nicholls et al., 2001 | |
| Coumarins 1, 2-benzopyrone chromen-2-one |
146.143 g/mol |
C9H6O2 | Group of polyphenol benzopyrene anticarcinogen chemicals isolated from plants capable of reducing inflammation and oxidative stress, prevent cytochrome c leakage, t- BHP initiated NLRP3 inflammatory pathways, and apoptosis; increase signaling via the nuclear factor-erythroid 2-related factor 2 (Nrf2) and antioxidant response element pathway |
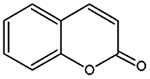 |
Apak et al.,
2007; Lv et al., 2016; Davatgaran-Taghipour et al., 2017 |
| Curcumin (1E,6E)-1,7-bis(4- hydroxy-3- methoxyphenyl)-1,6- heptadiene-3,5-dione |
368.38 g/mol |
C21H20O6 | Lipophilic character penetrates blood brain barrier; inhibits Aβ aggregation and Aβ fibril formation from Aβ monomer; disassembles fibril |
 |
2Reddy PH et al., 2018 |
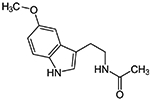
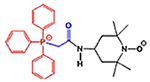
| DDQ Diethyl (3,4- dihydroxyphenethylami no) (quinolin-4- yl)methylphosphonate |
430.43 g/mol |
C22H27N27O5P | Dopamine derivative; Adheres to Aβ; prevents Aβ and Drp1 binding at the ser8 and Leu34 active sites of the Aβ polypeptide, and the ASN16 and Glu16 site of Drp1; disrupts Aβ interaction with mitochondria and synapses |
 |
Kuruva et al., 2017 |
| Donepezil 2-((1-Benzylpiperidin- 4-yl)methyl)-5,6- dimethoxy-2,3- dihydro-1H-inden-1- one |
379.50 g/mol |
C24H29NO3 | Prevents cholinesterase from breaking down acetylcholine (Ach), increasing its concentration in central nervous system; increases Ach regulated neurotransmission in memory centers of brain; mildly reduces memory loss; reversible cholinesterase inhibition |
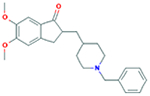 |
1,National Center for Biotechnology Information, 2019; Blanco-Silvente et al., 2017 |
| Glutathione
(GSH) γ-Glutamyl-Cysteinyl- Glycine γ-Glutamyl- Cysteinyl-Glycine (2S)-2-amino-4-{[(1R)- 1- [(carboxymethyl)carba moyl]-2- sulfanylethyl]carbamoy l}butanoic acid |
307.32 g/mol |
C10H17N3O6S | Endogenous antioxidant; balances protein sulfhydryl compounds; reduces sulfenic ion of the protein via covalent adduction; GSH to choline ester bond targets GSH to the mitochondrion, GSH reduces free radicals in mitochondria |
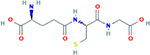 |
2,National Center for Biotechnology Information, 2019; Emanuele et al., 2018; Reddy, 2008 |
| Huperzine A (1R,9S,13E)- 1- Amino- 13-ethylidene- 11-methyl- 6- azatricyclo[7.3.1.02,7] trideca- 2(7),3,10- trien- 5-one |
242.32 g/mol |
C15H18N2O | Aromatic, cyclical, alkaloid with antioxidant activity; able to improve cognitive, non-cognitive, and activities of daily living |
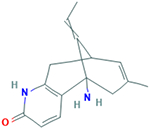 |
3,National Center for Biotechnology Information, 2019; Reddy, 2006 |
| MDivi1 3-(2,4-Dichloro-5- methoxyphenyl)-2- sulfanyl-4(3H)- quinazolinone |
353.217 g/mol |
C15H10Cl2N2O2S | Reduces H2O2
production and lipid peroxidation; increases cytochrome c oxidase activity, increases ATP production, and increases cell viability; inhibits Drp1 and Fis1 overexpression; increases mitochondrial fusion genes; inhibits mitochondrial degeneracy |
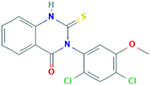 |
4,National Center for Biotechnology Information, 2019; Reddy et al., 20181; Manczak et al., 2018; Reddy et al., 2017 |
| Melatonin | 232.28 g/mol |
C13H16N2O2 | Decreases the accumulation of Aβ polypeptides in Tg2576 mouse brains; decreased abnormal protein nitration, and also extended lifespan of mice |
 |
Reddy, 2006; Man et al., 2014 |
| Memantine 3,5- dimethyladamantan-1- amine |
179.307 g/mol |
C12H21N | Blocks
N-methyl-Daspartate (NMDA) receptors (NMDAR), prevents glutamate from binding; reduces activation of caspase-3 and Bcl-2; reduces apoptotic effects caused by Aβ |
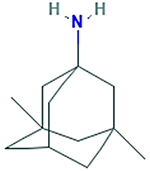 |
5,National Center for Biotechnology Information, 2019; Esposito et al., 2013 |
| MitoPBN [4-[4 (1,1-dimethylethyl) oxidoimino]- methyl]phenoxy]butyl] and triphenylphosphonium bromide |
590.542 g/mol |
C33H37BrN O2P |
Conjugated aromatic group (PBN) composed of antioxidant coenzyme Q (quinone) and phenyl tertbutylnitrone, fitted with mitochondria targeted TPP; prevents O-induced activation of uncoupled proteins; reduces free radicals |
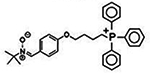 |
Kalogeris et
al., 2014; Apostolova &
Victor, 2015; Reddy,
2008 Reddy, 2006 |
| MitoQ [10-(6-ubiquinonyl) decyltriphenylphospho nium bromide] |
665.65 g/mol |
C37H46O4P Br |
Derivative of mitochondrial quinolone; fitted with mitochondria targeted TPP; may accumulate in high concentration in mitochondria, up to 5000- 6000; scavenges for and reduces free-radicals; reduces fragmentation, upregulate fusion of mitochondria; induce significant neurite growth; reduces mitochondrial fragmentation |
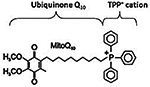 |
Apostolova & Victor, 2015; Yin et al., 2016; Reddy, 2008; Reddy, 2006 |
| MitoTEMPO (2-(2,2,6,6- Tetramethylpiperidin −1-oxyl-4-ylamino)-2- oxoethyl)triphenylpho sphonium chloride |
510.03 g/mol |
C29H35N2 O2P·Cl |
Contains a free radical electron capable of eliminating mitochondrial superoxide; fitted with mitochondria targeted TPP; to scavenges for ROS and mitigates ROS damage; reduces O2 production, Aβ induced lipid peroxidation, and overall oxidative stress; increases mtDNA fidelity and copy number |
 |
6,National Center for Biotechnology Information, 2019; Battogtokh G, et al., 2018; Hu & Li, 2016; Reddy, 2006 |
| MitoVitE [2–3,4dihydro-6- hydroxy-2,5,7,8tetramethyl- 2H-1- benzopyran-2-yl] and triphenylphosphonium bromide |
Vitamin E fitted with mitochondria targeted TPP; may accumulate in high concentration in mitochondria, up to 5000- 6000; Reduces H2O2 to less reactive O2 and H2O; able to prevent release of cytochrome c, caspase-3 activation and apoptosis; preserves glutathione peroxidase/glutathione reductase activity, gamma-glutamylcysteine synthetase activity, and overall glutathione concentration throughout the cell |
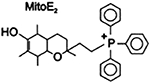 |
Apostolova & Victor, 2015; Reddy, 2006 | ||
| Rivastigmine Exelon (S)-N-ethyl-3-((1- dimethyl-amino)ethyl)- Nmethylphenylcarbamat e |
250.342 g/mol |
C14H22N2O2 | Prevents cholinesterase from breaking down acetylcholine (Ach), increasing its concentration in central nervous system; increases Ach regulated neurotransmission in memory centers of brain; mildly reduces memory loss; reversible cholinesterase inhibition |
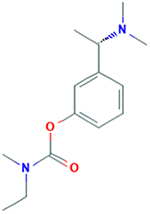 |
Blanco-Silvente et al., 2017; 7,National Center for Biotechnology Information, 2019 |
| SS-02 | Dmt-DArg- Phe- Lys-NH2 |
Mitochondria targeting; aromatic ring; positively charged cation; antioxidant properties |
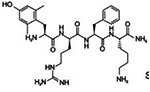 |
Reddy, 2008; Reddy, 2006; Szeto & Birk, 2014; Gruber et al., 2013 | |
| SS-19 | Dmt-DArg- PheatnDAP-NH2 |
Mitochondria targeting; aromatic ring; positively charged cation; antioxidant properties |
Reddy, 2006; Reddy, 2008; Szeto & Birk, 2014 | ||
| SS-20 | Phe-DArg- Phe- Lys-NH2 |
Mitochondria targeting; aromatic ring; positively charged cation; antioxidant properties; protects mitochondrial cristae, prevent cytochrome c loss, promotes restoration of mitochondrial respiration after ischemic reperfusion, and preserve cardiac function |
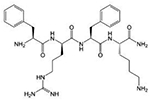 |
Reddy, 2006; Reddy, 2008; Szeto & Birk, 2014; Birk et al., 2015 | |
| SS-31 Elamipretide Bendavia |
639.802 g/mol |
D-Arg- Dmt-Lys- Phe-NH2 C32H49N9O5 |
Mitochondria targeting; aromatic ring; positively charged cation; antioxidant properties; binds to cardiolipin and localizes to IMM; protects mitochondrial cristae, prevent cytochrome c loss, promotes restoration of mitochondrial respiration after ischemic reperfusion, and preserve cardiac function |
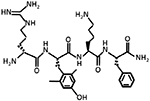 |
Reddy, 2006; Reddy, 2008; Szeto & Birk, 2014; Birk AV et al., 2015 |
| Tocopherol Vitamin E |
416.69 g/mol |
C28H48O2 | Lipophilic and membrane associated antioxidant; decreases Aβ and τ levels; decreases abnormal protein nitration |
Reddy, 2006 Cragg & Pezzuto, 2015 |
Cholinesterase Inhibitors and Glutamate Inhibitors
Cholinesterase Inhibitors
Cholinestrase inhibitors (CHEI) increase the number of acetylcholine (ACh) neurotransmitters in the Central Nervous System (CNS) (Blanco-Silvente et al., 2017). CHEI binds to the enzyme cholinesterase and effectively prevents the breakdown of the molecule (Blanco-Silvente et al., 2017). The resultant accumulation of ACh in the synapse, increases the signals between neurons, allowing for greater neuronal activity (Blanco-Silvente et al., 2017). ACh functions as the neurotransmitter between neurons responsible for learning. Examples of CHEI found effective for mildly reducing symptoms of memory loss include Donezepil and Rivastigmine, which both act reversibly (Blanco-Silvente et al., 2017). These drugs however only slow the neurodegenerative processes caused by AD, but have shown to allow AD patients to maintain autonomy and think independently (Blanco-Silvente et al., 2017). Still, the risks due to side effects such as nausea, diarrhea, vomiting, abdominal pain, weight loss, headache, insomnia, muscle cramps, bradycardia, syncope, and even hallucinations concurrently act to reduce quality of life (Blanco-Silvente et al., 2017).
Glutamate Inhibitors
Glutamate inhibitors (GluI) function by blocking N-methyl-D-aspartate (NMDA) receptors (NMDAR), and preventing glutamate from binding (Esposito et al., 2013). The calcium cation (Ca2+) passage through the NMDAR channel is necessary for learning and memory, and is dynamically regulated by glutamate, D-serine, or glycine (Esposito et al., 2013). Glutamate binding to NMDAR inhibits the flow of cations, including Ca2+ (Esposito et al., 2013). To counteract the deleterious effects to memory and learning caused by AD pathology, GluI’s are used to permit passage of Ca2+ and augment the capacity for memory and learning (Esposito et al., 2013). Memantine, a GluI is used for moderate to severe AD cases, and persons intolerant of CHEI, and enhances synaptic transmission (Esposito et al., 2013; Matsunaga et al., 2015). Memantine also has fewer adverse effects as CHEI’s, but may cause dizziness, headache, agitation, drowsiness (Matsunaga et al., 2015). Furthermore, the more AD pathology worsens, the less effective the therapeutic effects of Cholinesterase inhibitors and Glutamate Inhibitors (Blanco-Silvente et al., 2017; Esposito et al., 2013). These therapeutic methods may aid proliferation of neurotransmitters, but oxidative damage will persist, leading to mitochondrial dysfunction, and inevitable apoptosis.
Endogenous Antioxidants
Antioxidants target the oxidative damage, pervasive throughout AD pathology, and experimentally have been very effective in vitro. Antioxidants reduce the ability of ROS to oxidize. Endogenous antioxidants, such as superoxide dismutase (SOD), reduce ROS to less reactive molecules, minimizing their threat. UCP2 also reduces mitochondrial ROS by shifting the movement of protons into the matrix rather than the intermembrane space, effectively behaving like an antioxidant (Angajala et al., 2018). Mitochondrial ubiquinone (Q) in the ETC also serves as an in-house antioxidant for mitochondria (Reddy, 2006). Brain derived neurotrophic factor (BDNF) initiate biomolecular pathways which inhibit the neurotoxic effects of 3-NP (Chen et al., 2017; Tanila, 2016).
Loss of mtDNA copy number has been shown to be prevented by natural antioxidants such as coenzyme Q, vitamin E, melatonin, and mtDNA re-synthesis processes (Alexeyev et al., 2013). Experimentally, intra-gastric introduction of ethanol resulted in loss of approximately half of mtDNA copies in all organs sampled, including heart, liver, brain, and skeletal musles of mice, and antioxidant administration was able to prevent loss (Mansouri et al., 2001).
Glutathione
Glutathione (GSH) is a tripeptide (γ-Glutamyl-Cysteinyl-Glycine with Glu-Cys gamma peptide bond) that exhibits antioxidant properties, and is endogenously produced by cells (Emanuele et al., 2018). It plays a role in the balance of protein sulfhydryl compounds in eukaryotic cells, which undergo sulfenylation (oxidation to sulfenic ion) (Emanuele et al., 2018). GSH acts as an antioxidant by reducing the sulfenic ion of the protein via covalent adduction (Emanuele et al., 2018).
An ester bond of GSH to choline targets GSH to the mitochondrion, and the resultant glutathione choline ester and N-acetyl-l-cysteine choline ester have both been effective in reducing free radicals in mitochondria (Reddy, 2008). Glutathione has proven to stave off oxidative damage in mitochondria of AD cells or neurons affected by mutant APP, effectively alleviating protein oxidation, and preventing Aβ peptide prompted DNA fragmentation (Reddy, 2008).
Nonetheless, with age, as mitochondrial dysfunction persists and proliferates, endogenous antioxidants become less effective in mitigating oxidative damage (Kristilis et al., 2018). However, a diet rich with antioxidants is an effective method at reducing oxidative damage.
Exogenous Antioxidant: Antioxidant Rich Diet
There are several natural products such as green tea, vitamin C, vitamin E, curcumin, Gingko biloba, rosemary, sage, ginseng, and β-carotene (Reddy et al., 20182). In several studies, Vitamin E, including when supplemented with Vitamin C, was shown to correlate with lower prevalence and incidence of AD in elderly population (Reddy, 2006). Vitamin E, also known as Tocopherol, is a lipophilic and membrane associated antioxidant, may be a beneficial supplement for AD patients (Reddy, 2006).
There is evidence that antioxidants may reduce or even reverse the toxic effects of Aβ or τ particles. Vitamin E was shown to decrease Aβ and τ levels in Tg2576 mice, and curcumin, another antioxidant present in food seasoning like cumin and curry, may also reduce oxidative damage as well as Aβ deposition (Reddy, 2006). Melatonin, a sleep inducing agent, decreased the accumulation of Aβ polypeptides in Tg2576 mouse brains, decreased abnormal protein nitration, and also extended their lifespan (Reddy, 2006).
Huperzine A
Huperzine A, an aromatic, cyclical, alkaloid with antioxidant activity was investigated for ability to treat AD patients with doses of 300mg/day for the first 2–3 weeks, and 400mg/day for the subsequent 3–12 weeks (Reddy, 2006). Overall, the AD patients showed improvement in their cognitive, non-cognitive and ADL functions (Reddy, 2006). ADL or “Activities of Daily Living, are considered to be: eating, bathing, getting dressed, grooming toileting/continence, and transferring/ambulating (Mlinac & Feng, 2016).
Vitamin C
Vitamin C, also known as ascorbic acid, in its anion form ascorbate, was shown to ameliorate the dysfunction of a mutated yeast cell (Saccharomyces cerevisiae) lacking Cu-Zn (copper, zinc) superoxide dismutase (SOD) (Reddy, 2006). Without the Cu-Zn SOD, the yeast were hypersensitive to oxygen and vulnerable to oxidation, but the supplementation of ascorbate prolonged the lifespan the yeast cell, showing the benefits of not only Vitamin C, but the impact of functional SOD as antioxidants (Reddy, 2006). Additional enzymes Mn-SOD (manganese superoxide dismutase), and methionine sulfoxide reductase, which target mitochondria, were also shown to agonistically subsidize the extension of the life of Drosophila fruit flies via reduction in ROS (Reddy, 2006). Studies have shown that Mn-SOD, mitoquinone (MitoQ, a derivative of mitochondrial quinoline), Euk-8, and Euk-134, were all shown to alleviate the dysfunction caused by oxidative stress in experiments with Drosophila, but not in wild-type Drosophila (Reddy, 2006). In fact, there were dose-dependent increases in toxicity in wild-type flies (Reddy, 2006). Nonetheless, the studies showed that administration of exogenous antioxidants and beneficence leading to extended life of the antioxidant deficient flies was dose dependent, sex-specific, and age-specific (Reddy, 2006).
Coumarin
Coumarins, a group of polyphenol benzopyrene chemicals isolated from plants are capable of reducing inflammation and oxidative stress, and fighting cancer (Lv et al., 2016; Davatgaran-Taghipour et al., 2017). They function by increasing signaling via the nuclear factor-erythroid 2-related factor 2 (Nrf2) and antioxidant response element pathway (Johnson et al., 2008). The Nrf2/ARE pathway defends the cell by mitigating oxidative stress which signal apoptosis (Lv et al., 2017). Daphnetin, a type of Coumarin, was proven by researchers to reduce oxidative damage, cyto-toxicity, and resultant apoptosis (Lv et al., 2017). Significantly, Daphentin is capable of preventing loss of membrane potential and cytochrome c leakage in mitochondria, and also prevents t-BHP initiated NLRP3 inflammatory pathways (Lv et al., 2017).
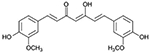
Curcumin
Curcumin, isolated from the Asian spice turmeric, sourced from the rhizome of Curcuma longa has been proven to exhibit anti-amyloid properties in AD (Reddy et al., 20182). The lipophilic character of curcumin allows it to diffuse the blood-brain barrier as well as bind to Aβ (Reddy et al., 20182). No adverse effects have been noted regarding curcumin, and thus it may be used for clinical trials (Reddy et al., 20182).
In vitro inoculation of curcumin was shown to inhibit Aβ aggregation, inhibit Aβ fibril formation from Aβ monomer, and was also able to disassemble fibril form of Aβ, indicating curcumin neuroprotective activity against Aβ toxicity (Reddy et al., 20182). Curcumin was able to disaggregate Aβ deposits in vivo as well, prevent the accumulation of new Aβ deposits, as well as reduce the size of remaining Aβ aggregations (Reddy et al., 20182). In mice brains with mutant APP which were inoculated with curcumin, Aβ was reduced by 40% and Aβ deposits were reduced by 43%, and at high concentrations curcumin may even prevent self-assembly (Reddy et al., 20182). Curcumin may also destabilize Aβ40 and Aβ42 (Reddy et al., 20182). Isoxazole and pyrazoles derived from curcumin were able to bind to Aβ and inhibit the proteolytic metabolism of APP (Reddy et al., 20182). Curcumin inhibits Aβ induced oxidative stress in PC12 cells and normal human umbilical endothelial cells, and is shown to decrease concentration of oxidized proteins and the inflammatory producing interleukin 1B in the brains of APP mice (Reddy et al., 20182). Heightened uptake of Aβ by macrophages in AD patients is induced by curcumin, denoting a plausible immunotherapy approach to AD patients (Reddy et al., 20182). Curcumin also prevents activity of peroxidase and reduces cyto-pathologies in AD patients (Reddy et al., 20182). Furthermore, curcumin may bind to redox cofactor metals such as iron and copper to reduce inflammatory damaged caused by nuclear factor κB (Reddy et al., 20182).
In a recent study by Reddy’s group (Reddy et al., 2016) the neuroprotective effects of curcumin with respect to Aβ propagation, mitochondrial and synaptic toxicities were quantitatively and qualitatively studied. Immortalized SHSY5Y human neuroblastoma Cells were used, and treated with Aβ for 4 hours, or Aβ for 4 hours and curcumin for 24 hours (Reddy et al., 2016). Aβ was shown to reduce biogenesis and synaptic proteins as well as reduce mitochondrial function (Reddy et al., 2016). With treatment from curcumin, there was enhanced mitochondrial fusion activity, reduced fission machinery, increased biogenesis as well as synaptic proteins (Reddy et al., 2016). Furthermore, there was elevated mitochondrial function and cell viability, and pre-treatment of the cell with curcumin had greater advantageous effects than post-treatment (Reddy et al., 2016). Thus, curcumin was found to work better as preventative to Aβ induced pathology than treatment in AD like neurons (Reddy et al., 2016). Nonetheless, this study demonstrated short term results, mostly from notable changes in concentration of transcriptions of mRNA and translations of proteins. A longitudinal study would better demonstrate the ability of curcumin to circumvent the cycles of mitochondrial and synaptic health over long time period, and its ability to extend the lifespan of the experimental cell, which is the ultimate goal of an AD cure. Important pathways to note would be ability for curcumin to maintain fission/fusion balance, long term.
Mitochondria Targeted Molecules
The aforementioned studies demonstrate that free-radicals and reactive oxygen species directly impact lifespan and function of mitochondria, and treatment with antioxidant molecules and enzymes play a critical role in extension of lifespan and supporting healthy mitochondrial function (Reddy, 2006). Nonetheless, many of these antioxidants are incapable of ameliorating oxidative stress specifically in the mitochondria. Hydrophilic antioxidants are often excreted quickly, or undergo first or second pass metabolism giving them a short half-life, and removed from the body before reaching the brain (Reddy, 2008; Szeto, 2006; Apostolova & Victor, 2015). Given antioxidant effectiveness however, it would behoove researchers to focus on mitochondrially targeted antioxidant small molecules which ameliorate the effects of mitochondrial dysfunction (Reddy, 2006; Reddy, 2008; Szeto, 2006; Apostolova & Victor, 2015).
Enzymes and compounds of interest include catalase targeting mitochondria (MCAT), MitoQ, MitoVitE, MitoPBN, glutathione peroxidase and additional mitochondrially targeted peptides which reduce H2O2 to less reactive O2 and H2O (Reddy, 2006). Three main vehicles used for targeting antioxidants to mitochondria are triphenylphosphonium, amino acid, and peptide moieties (Reddy, 2006). Due to the negatively charge potential gradient in mitochondria, caused by proton-motive force of the ETC, Murphy and colleagues developed small molecules that contained lipophilic cation moieties, to traverse the lipophilic membrane of mitochondria, and direct the small molecules antioxidants via attraction to the opposite charge (Reddy, 2006). The following reviews studies that detail mitochondria-targeted molecules’ ability to accumulate within mitochondria, and scavenge for free radicals.
Exogenous Mitochondria-Targeted Antioxidants and Small Molecules
Mitochondrial Division Inhibitor 1
In Huntington’s disease (HD) there is degeneration of bioenergetics due to defective enzyme complexes II, III, and IV in mitochondria just as with AD (Manczak et al., 2016; Reddy et al., 2010). Also similar to AD, in HD, there is an increased permeability of the mitochondrial membrane due to calcium cation imbalance (Manczak et al., 2016). Furthermore, between HD and AD structural deformation of highly conserved regions of the GTPase Drp1 and Fis1 mitochondrial fission and Mfn1, Mfn2, and Opa1 GTPase mitochondrial fusion enzymes cause mitochondrial anomalies (Manczak et al., 2016; Reddy et al., 2010; Scott & Youle, 2010). The long-term effect of these anomalies is the excessive fragmentation of mitochondria, and the inhibition of bioenergetics (Manczak et al., 2016). This fragmentation of mitochondria is a characteristic of AD cells induced particularly by Aβ particles (Reddy et al., 2017).
Mitochondrial division inhibitor 1 (Mdivi 1) has been shown to subsidize the health of healthy neurons, while increasing mitochondrial fusion genes, while decreasing mitochondrial fission genes (Manczak et al., 2016; Scott & Youle, 2010). While used to alleviate the pathology of HD, Mdivi 1 has shown to be effective in inhibiting the mitochondrial degeneration is AD as well (Manczak et al., 2016; Reddy et al., 2017; Manczak et al., 2018). Quite novel about Mdivi 1 is that AD neuronal cells treated with Mdivi 1 showed reduction in mitochondrial degeneration, but cells treated with Mdivi 1 prior to induction of AD pathology, showed even less mitochondrial degeneracy than the cells treated after AD pathology (Reddy et al., 2017; Manczak et al., 2018). Mdivi 1 treatment in mouse neuroblastoma cells (N2a) with Aβ toxicity lowered Aβ42 levels, whether the N2a cells were treated with Mdivi 1 before or after Aβ42 inoculation (Reddy et al., 2017). This indicates that Mdivi1 likely functions as a preventative molecule to AD pathology (Manczak et al., 2016; Reddy et al., 2017; Manczak et al., 2018).
Mdivi 1 treatment prior and post Aβ42 inoculation in N2a cells demonstrated similar effects reducing toxicity caused by cytochrome c, ATP, and cell viability reduction, by increased cytochrome c oxidase activity, increased ATP production, and increased cell viability, mitochondrial biogenesis and synaptic activity (Reddy et al., 2017). Mdivi 1 treatment also reduced H2O2 production and lipid peroxidation, whether pre or post inoculation with Aβ42 toxic protein (Reddy et al., 2017). The method by which Mdivi 1 accomplishes therapeutic effects likely is related to the inhibition of Drp1 and Fis1 overexpression, as lowered concentration of Drp1 and Fis1 were noted in N2a cells treated with Mdivi 1 (Reddy et al., 2017).
Drp1 and Fis1 are proteins which regulate mitochondrial division by way of fission, and are upregulated to the point of mitochondrial fragmentation in cells where Aβ accumulation has propagated (Manczak et al., 2016; Reddy et al., 2010; Scott & Youle, 2010; Reddy et al., 2017; Manczak et al., 2018). Mdivi1 blocks the self-assembly of Drp1, GTPase dependent mitochondrial fission regulator, which leaves mitochondrial fusion factors uninhibited to promote mitochondrial elongation (Manczak et al., 2016; Scott & Youle, 2010; Reddy et al., 2017; Manczak et al., 2018). Nonetheless, Drp1 and Fis1 as aforementioned are critical to normal, healthy mitochondrial function, and healthy neuronal and neuronal synaptic function, particularly as it counterbalances the effects of mitochondrial fusion factors (Reddy et al., 2010). In an experiment conducted by Shields’ research team knockout mice with deletion of Drp1 functionality were able to survive with functional neurons, but with greatly diminished neuronal activity (Shields et al., 2015). The mitochondria in the synapses were able to support synaptic activity, but minimally, as they were swollen in size, but within the first year of growth, unable to multiply to sufficient numbers (Shields et al., 2015). Mdivi 1 may suppress the excessive fragmentation initiated by Aβ aggregation. In particular, Mdivi 1 may improve regulation of mitophagy, the mitochondrial network’s ability to self-propagate via fission, amalgamation via fusion, and phagocytosis of degenerate mitochondria. Nonetheless, excessive fragmentation of mitochondria may be a characteristic of AD pathology, but the root cause of mitochondria dysregulation may not simply be Drp1 and Fis1 overexpression. Furthermore, aforementioned studies lack longitudinal analysis, to investigate ability of Mdivi 1 to extend lifespan of cells (Manczak et al., 2018).
Mito-Q
Oxidative damage to the mitochondria is a source of bioenergetics degeneracy and mtDNA mutation. An antioxidant which targets oxidative damage in the mitochondria has the ability to alleviate the effects of damage caused by ROS. Effects of neuron degeneration include excessive influx of Ca2+ in mitochondria, which lead to excessive ROS, causing mitochondrial dysfunction and subsequent neuronal cell death (Reddy, 2006; Mao et al., 2013). Mitochondrial CoQ10 (MitoQ) is similar in function to coenzyme Q10, but MitoQ differs in conjugation to triphenylphosphonium cation, a lipophilic cation (Reddy, 2006; Mao et al., 2013). In both animal and human cells under AD conditions, MitoQ has been shown to induce the uptake of CoQ10 into the mitochondrion, to ameliorate from ROS damage (Reddy, 2006; Mao et al., 2013).
The mechanism of MitoQ in the mitochondrion is profound and effective. Overall, MitoQ reduces neuronal axon loss, via reduction in neuronal inflammation and demyelination (Reddy, 2006; Mao et al., 2013). MitoQ maintains mitochondrial ATP, improves remyelination, allows for rapid cognition, and thus reduction in demyelination, protects against additional loss of mental function including memory loss.
The structure of MitoQ consists of a triphenylphosphonium (TPP), a novel lipophilic cation, attached to ubiquinone via a ten carbon alkane (together the triphenyl-phosponium and decyl group are abbreviated DecylTPP) (Mao et al., 2013). MitoQ antioxidant contiains two redox forms of ubiquinone, reduced mitoquinol and reduced mitoquinone (Reddy, 2006). The overall molecular formula of MitoQ is C37H46O4PBr, and its molecular weight is 665.65, and is a red, oily solid that may be stored in −20°C cryopreservation in the dark (Reddy, 2006). Incubation in 95% ethanol fully oxidizes MitoQ, and it may dissolve in DMSO a known agent for maintenance of cell viability during cryopreservation (Reddy, 2006).
The antioxidant component of MitoQ, ubiquinone, is a derivative of ubiquinone of the ETC where it more frequently remains in its reduced form of ubiquinol (Reddy, 2006). In this form, it serves as an antioxidant, and a promoter of bioenergetics (Reddy, 2006). Its highly conjugated structure, as well as its stable aromatic highly substituted hydroquinone moiety allows it to readily donate a hydrogen from one of its hydroxyl groups to lipid peroxyl radicals (Reddy, 2006). The unstable semi-ubiquinone radical formed during the process transfers the unstable radical to others resulting in ubiquinone or ubiquinol (Reddy, 2006). The continuous transfer of electrons in the ETC quickly reduces ubiquinone to ubiquinol, for it to readily act as an antioxidant again (Reddy, 2006).
With the exogenous MitoQ compound, the positive charge on the phosphorus cation is stabilized by the aromaticity of the three phenyl groups which also serve a physical barrier from nucleophilic attack (Reddy, 2006). The MitoQ is drawn to the neuronal cell membrane due to its negative membrane potential, and cross the membrane due to its hydrophobicity and lipophilicity, gathering up in the cytoplasm of the cell (Mao et al., 2013). Then the positive charge on the TPP is what attracts the MitoQ to the mitochondrion which is negatively charged, due to the charge difference (Mao et al., 2013). It too crosses the mitochondrial membrane due to its hydrophobicity and lipophilicity of the decyl-TPP group, and going against the proton gradient, crosses the inner mitochondrial membrane (IMM) and accumulates in the mitochondrial matrix by the hundreds (Mao et al., 2013). From this point, the MitoQ attack ROS in the mitochondrion effectively defending it from damage due to oxidative stress (Mao et al., 2013). MitoQ reduction of free radicals and oxidative species such as H2O2 and ultimately protects the neuron from age related dysfunction caused by mitochondrial degeneracy (Reddy, 2006).
In practice, MitoQ has been very effective in protection in against oxidative stress in in-vitro studies. In cells cultured from Friedreich Ataxia (rare autosomal recessive neuronal disease) (FRDA) patients, MitoQ prevented cell death by reducing endogenous oxidative stress (Reddy, 2006). In bovine aortic endothelial cells treated with MitoQ, there was inhibition of cytochromec release from the IMM and caspase 3 activation (signals for apoptosis or cell death), and DNA fragmentation (a cause for DNA mutation, which may eventually cause dysfunction and/or apoptosis), and MitoQ did perform these inhibitions better than Vitamin E (Reddy, 2006).
In an animal study, MitoQ fed laboratory mice were shown to successfully accumulate MitoQ within all tissues and organs assessed by researchers, Smith et al., which included the liver, heart, brain and kidney (Smith et al., 1999). The accumulation of MitoQ was more pronounced when aided with MitoVitE, and the MitoQ was easily cleared, along with MitoVitE, once oral administration ceased (Reddy, 2006). Adam et al. was able to demonstrate MitoQ’s antioxidant properties via cardiac ischemia-reperfusion injury (mitochondrial oxidative damage) rat model, where rats fed MitoQ had a significant reduction in heart dysfunction and mitochondrial damage (Reddy, 2006).
In clinical studies however, MitoQ are toxic to neuronal cells above 0.3 μM concentration, and thus optimal dosage for treatment of oxidative damage is critical for neurodegenerative disease (Reddy, 2008).In a study conducted by Reddy’s lab, the N2a cells, mouse neuroblastoma cells, given 0.3 μM of MitoQ showed increased growth, but above this threshold demonstrated toxicity (Reddy, 2008).
MitoQ taken up by N2a cells expressing mutant APP was able to induce significant neurite growth (axon and dendrite outgrowths), reduced transcription of mitochondrial fission mRNA, and also were able to maintain longer cell viability (Manczak et al., 2010). MitoQ treatment of cells increased PGC1α expression and prevented mitochondrial dysfunction, including preventing membrane depolarization, reducing free radicals, and increasing cytochrome oxidase activity (Manczak et al., 2010). Furthermore, MitoQ and SS31 protected the cells from Aβ toxicity more effectively than untargeted antioxidants, such as resveratrol (Manczak et al., 2010).
It should be noted that MitoQ also has strong potential for working synergistically with other small molecules for therapy of mitochondrial degeneracy in neurodegenerative diseases. In a 2016 study by Yin, Manczak and Reddy, when Mito Q was used alongside SS31 peptide, was able to reduce fragmentation, upregulate fusion of mitochondria in cells induced with disease via mutant Huntingtin protein (Yin et al., 2016). This effect was much greater than MitoQ on its own, and given the similarity to mitochondrial degeneracy in AD, such a combination should prove therapeutic for AD, and other similar neurodegenerative diseases as well (Yin et al., 2016).
MitoVitE
MitoVitE, a mitochondrially targeted Vitamin E, acts as an antioxidant and contains an aromatic head with two ring structures [2–3,4dihydro-6-hydroxy-2,5,7,8tetra-methyl-2H-1-benzopyran-2-yl], and a tiriphenylphosphonium bromide group (TPPB) which dissociates in solution to form cationic and hydrophobic TPP group (Reddy, 2008). Similar to the mode with which MitoQ is taken up by mitochondria, MitoVitE is quickly attracted to mitochondrial negative charge, and taken up quickly through its membranes due to its hydrophobic nature (Reddy, 2008). Also like MitoQ, MitoVitE may accumulate in high concentration in mitochondria, up to 5000–6000 units as demonstrated by Smith’s research team in 1999 by incubating mitochondria with 20 μM MitoVitE (Reddy, 2008; Smith et al., 1999).
MitoVitE is able to protect mitochondria and their cells from oxidative damage and apoptosis, by reducing H2O2 and inhibiting caspase activation and stave off apoptosis (Reddy, 2008; Smith et al., 1999). Likewise, MitoVitE was able to inhibit cell death due to oxidative stress in vitro fibroblasts from FRDA patients (Reddy, 2008; Smith et al., 1999). During this same study of MitoVitE on FRDA fibroblasts, MitoVitE was noted to be 350 times more potent than its analog Trolox, which is a more hydrophilic soluble analog of Vitamin E sans aliphatic tail and with an additional carboxylic (Reddy, 2008; Smith et al., 1999). Additionally, MitoVitE of 1μM concentration was shown to prevent the release of cytochrome c, inhibiting caspase-3 activation and thus staving off apoptosis, in bovine aortic epithelial cells, rejuvenating the membrane potential necessary for effective bioenergetics, and ubiquitin initiated proteasome activity (Reddy, 2008; Smith et al., 1999). Furthermore, MitoVitE was able to ameliorate intracellular ROS accumulation induced ethanol in upwards of 1600mg/dL concentration, with as little as 1nM concentration of MitoVitE (Reddy, 2008; Smith et al., 1999). Here, MitoVitE preserved glutathione peroxidase/glutathione reductase activity, gamma-glutamylcysteine synthetase activity, and overall glutathione concentration throughout the cell (Reddy, 2008; Smith et al., 1999).
MitoPBN
MitoPBN is composed of the antioxidant coenzyme Q (quinone) and phenyl tertbutylnitrone, a conjugated aromatic group abbreviated as PBN, attached to positively charged lipophilic triphenylphosphonium bromide (Kalogeris et al., 2014). Its overall structure is [4-[4 (1,1-dimethylethyl) oxidoimino]—methyl]phenoxy]butyl] and triphenylphosphonium bromide, and similar to MitoQ and MitoVitE, is subjective to quick uptake by the mitochondria due to both its lipophilic and cationic character, even up to concentrations of 2.2 to 4.0 mM (Reddy, 2008). MitoPBN is believed to prevent O-induced activation of uncoupled proteins (Reddy, 2008).
LPBNAH, which is analogous to MitoPBN, and derived from α-phenyl-N-tert-butyl nitrone, also acts as an antioxidant, inhibiting the damage of ROS damage originating from the mitochondrion, effectively ameliorating neural damage (Reddy, 2008). LPBNAH (whose nomenclature is N-[4-(octa-O-acetylactobionamidomethylene) benzylidene]-N-[1,1-dimethyl-2-(N-octanoyl) amido]-ethylamine N-oxide), is capable of reducing free radicals in rotifers, and increase their lifespan, and was much more effective than PBN from which it was derived (Reddy, 2008).
MitoTEMPO
Similar to the aforementioned mitochondria targeted antioxidants, MitoTEMPO is the 2,2,6,6-Tetramethylpiperidin-1-yl)oxyl (TEMPO) compound, fitted with TPP (Hu & Li, 2016). TEMPO, though a stable compound, contains a free radical electron capable of eliminating mitochondrial superoxide (Hu & Li, 2016). In studies, MitoTEMPO has shown to scavenge for ROS and mitigate damage (Hu & Li, 2016). Experimental results include reduction of O2∙production, without reduction of SOD2 expression, reduction of Aβ induced lipid peroxidation, mtDNA fidelity and copy number, and reduce overall oxidative stress (Hu & Li, 2016). Overall bioenergetics of mitochondria in cells treated with MitoTEMPO improved as well (Hu & Li, 2016). Toxicity of MitoTEMPO is of question, in addition to other mitochondria targeted compounds with TPP, nonetheless.
Catalase
In mice, catalase targeted toward mitochondria decreased H2O2 and reduced ROS damage in mitochondria (Reddy, 2006). There was a measured 25% reduction in H2O2 production by mitochondria in cardiac muscles, and these cardiac muscles also showed significantly less arteriosclerosis and myopathy compared to wild-type control (Schriner et al., 2005). This correlated with additional studies which show correlation between mouse aging process and oxidative stress and DNA damage (which likely too, results from ROS species) (Schriner et al., 2005). Furthermore, compared to wild-type mice, the mice which overexpressed human mitochondrial catalase (MCAT) averaged lifespans of 5.5 months longer, about a 20% increase in longevity (Schriner et al., 2005). Important to note is that nuclear catalase (NCAT) expression did not change the median lifespan or maximal lifespan of the mice (Schriner et al., 2005).
MCAT×APP Mice
Additional studies with MCAT in mouse models demonstrated effectiveness with increasing the lifespan of mice expressing toxic Aβ protein (Mao et al., 2012). In a recent 2012 study, mAPP/Aβ and MCAT mice lived 4 months longer if crossed with MCAT (Mao et al., 2012). Furthermore, the double mutant mice (APP×MCAT) demonstrated reduced 8-hydroxy-2-deoxyguanosine (8-OHdG) markers than Aβ mice left untreated (Mao et al., 2012). This is important as 8-OHdG DNA damage is most prevalent form of damage for mtDNA and nDNA, believed to be responsible for aging, and neurodegeneration (Alexeyev et al., 2013; Reddy & Beal, 2008; Mao et al., 2012). The MCAT induced reduction in oxidative damage from free radicals and reactive oxygen species, is thus likely for the reduction in DNA damage in the mice brains, and reduction of mutant proteins mAPP and Aβ in the Aβ expressing mice (Mao et al., 2012). MCAT was also noted to reduce beta site amyloid precursor protein cleaving enzyme 1 (BACE1) expression, which is also responsible for reduction in Aβ proteins (Mao et al., 2012). Overall, there was a reduction in cognitive decline for Aβ mice treated with MCAT, which was demonstrated enhanced short term/working memory (mouse ability to poke nose for food acquisition) (Mao et al., 2012).
Szeto-Schiller Peptides with Antioxidant Activity
Szeto-Schiller (SS) peptides share similar composition in being small molecules, of four amino acids alternating with aromatic groups, one of which is a 2’, 6’-dimethyltyrosine residue, and a cation exhibiting positive charge (Reddy, 2008; Szeto & Birk, 2014). SS compounds are tetrapeptides that exhibit antioxidant characteristic in a similar fashion to 3,5-dimethylphenols, and target the mitochondrion due to its positive charge and lipophilic properties of aliphatic chains and hydrophobic aromatic rings (Reddy, 2008; Szeto & Birk, 2014). The following SS compounds developed have the aforementioned characteristics, particularly antioxidant properties: SS-02 (Dmt-D-Arg-Phe-Lys-NH2), SS-20 (Phe-D-Arg-Phe-Lys-NH2), SS-31 (D-Arg-Dmt-Lys-Phe-NH2), and SS-19 (Dmt-d-Arg-Phe-atnDAP-NH2) (Reddy, 2008; Szeto & Birk, 2014).
SS-02 reduces H2O2 to O2 and H2O, and prevent the oxidation of linoleic acid and low-density lipoproteins (LDL) (Reddy, 2008; Szeto & Birk, 2014). In experiment with Caco-2 cells (derived from human colorectal adenocarcinoma, epithelial cancer cells) it was shown to have greater uptake within the cell, as much as 10 times greater than dissolution in extracellular matrix (Reddy, 2008; Szeto & Birk, 2014). From there, SS-02 may be rapidly taken up by mitochondria, as demonstrated with isolated mouse mitochondria, up to 105-fold, and SS-19 too was able to demonstrate the same selective uptake (Reddy, 2008; Szeto & Birk, 2014). SS-02 treated with digitonin showed
SS-19, like SS-02 is shown to be selectively rapidly taken up by mitochondria (Reddy, 2008). Via experiment, where incubation with mitochondrial uncoupler FCCP was shown to reduce SS-19 uptake by 20%, suggesting that selective uptake of SS-19 is partially dependent on mitochondrial membrane potential (Reddy, 2008; Szeto & Birk, 2014).
It has been shown that cardiolipin is key for the stability of the inner mitochondrial membrane (IMM), particularly for constructing of the many folds to increase the surface area of the membrane, and thus the overall functionality of the ETC (Szeto & Birk, 2014). When Aβ disruption of the IMM causes loss of cardiolipin, the folds of the IMM are compromised, as well as the efficiency of the ETC pathway (Szeto & Birk, 2014). SS-31 peptide sequence as well as SS-20 bind to cardiolipin and restore IMM structural integrity (Szeto & Birk, 2014).
SS-31
SS-31 has a similar function as SS-02 due to it having the same amino acids with different arrangement, but has greater antioxidant characteristics (Reddy, 2008; Szeto, 2014; Szeto & Birk, 2014; Calkins et al., 2011). In addition, researchers found that after mitochondrial damage, Cytochrome C is capable of continuing its function in the electron transport if SS-31 binds to cardiolipin in the inner mitochondrial membrane (Szeto, 2014). SS-31 may also inhibit peroxidase activity of cytochrome c in mitochondria, which would reduce ROS production and aid reversal of mitochondrial dysfunction (Szeto, 2014). SS-31 was proven to prevent lipid peroxidation and hydrogen peroxide scavenging (Szeto, 2014; Reddy et al., 20181). In studies involving amyotrophic lateral sclerosis (ALS) and myocardial infarction in rat models, SS-31 was proven to reduce ROS production (Reddy et al., 20181) SS-31, in vitro in ischaemic tissues, protects mitochondrial cristae, facilitates ATP recovery, and prevents peroxidase activity of CytC with an effective concentration 50, EC50, of under 1μM (Szeto, 2014).
SS-31 has the benefit of being localized to the mitochondrion, specifically targeting the IMM rather than the mitochondrial matrix. Its uptake is linear and independent of receptor mediated processes or transporter mediated processes (Szeto, 2014). It can be taken up readily by different cell types, such as: endothelial, renal and intestinal epithelial cells, myotubes, cardiomyocytes, macrophages, and especially neurons (Szeto, 2014). Furthermore, SS-31 does not have an effect on normal mitochondria, which only aids its profile as safe to use (Szeto, 2014).
In mouse neuroblastoma N2a cells grown with mutant APP, and incubated with Aβ (25–35 length) peptide, treatment with SS31 was shown to increase neurite growth from the N2a cells and prevent Aβ induced mitochondrial dysfunction by maintaining membrane potential (Manczak et al., 2010). SS31 was able to reduce mitochondrial fission, evidenced by reduction in mitochondrial fission protein, while greatly increasing neuroprotective genes PGC1α (2.5 fold increase), FOXO1 (14.9 fold increase), and NMDA receptor (8.8 fold increase) (Manczak et al., 2010). SS31 treatment also increased Prxs protein levels induced with Aβ peptide, prevented Aβ dysfunction of cytochrome oxidase, enhanced ATP production after Aβ toxicity, support overall cell viability (Manczak et al., 2010). In addition, mutant APP primary neuron cells treated with SS-31 were able to improve and increase mitochondrial function and synaptic activities (Manczak et al., 2011; Manczak et al., 2010).
In an experiment accomplished with transgenic mice (Tg2576) expressing Aβ precursor protein, SS-31 demonstrated additional therapeutic effects for mitochondria and synapse (Calkins et al., 2011). Tg2576 mice accumulated Aβ in neurons, demonstrated inhibition of anterograde or forward mitochondrial transport from neuronal cell body to synapse, concentration of Aβ in mitochondria, increased mitochondrial fission, and reduced mitochondrial and synaptic gene expression and protein translation (Calkins et al., 2011). There was also notable increase in H2O2 production, reduced cytochrome c oxidase and ATP levels, indicative of known oxidative damage and reduction in bioenergetics typical for Aβ induced AD pathology (Calkins et al., 2011). Tg2576 mice treated with SS-31 however demonstrated recovery of mitochondrial axonal transport, and mitochondrial numbers and sizes returned to similar size and number of the control/wild-type (Calkins et al., 2011). The SS-31 peptide was shown to target the inner mitochondrial membrane and also reduce ROS damage (Calkins et al., 2011). SS-31’s dimethyl-tyrosine residue demonstrated oxyradical scavenging properties and inhibit linoleic acid low density lipoprotein oxidation (Calkins et al., 2011).
SS-31 has proven to be an agonist of the therapeutic effects of other small molecules used to treat AD induced degeneracy and works in a synergistic effect. In an experiment with Mdivi1, N2A cells were transfected via lipofectamine to express cDNA with mutant APP for 24 hours (Reddy et al., 20181). SS-31 treatment and Mdivi1 treatment were for 6 hours, and separately, the small molecules were capable of reducing mitochondrial dysfunction, increase mtDNA copy number and cell survival, levels of Aβ40 and Aβ42 were reduced (Reddy et al., 20181). Synergistically, the combined treatment proved to be more beneficial as a therapeutic strategy for AD (Reddy et al., 20181).
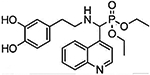
DDQ
Molecular structure of DDQ
Diethyl (3,4-dihydroxyphenethylamino) (quinolin-4-yl) methylphosphonate, or DDQ, is a compound specifically designed to inhibit the interaction of mutant proteins such as Aβ, with mitochondrial fission protein Drp1 and reduce mitochondrial toxicity in AD cells. (Kuruva et al., 2017). DDQ and the other 81 similar compounds were derived from dopamine, 4-(2-Aminoethyle) benzene-1, 2-diol, a neurotransmitter created naturally in the human body, by removing a carboxyl group from its precursor L-Dopa (3,4-Dihydroxyphenethylamine) (Kuruva et al., 2017). Dopamine is essential for the brain’s regulation of reward and pleasure, and deficiency may result greater susceptibility to addiction, and development of Parkinson’s disease (Kuruva et al., 2017). Excessive amounts of dopamine may result in allergic reactions such as hives, respiratory inhibition, swelling of face, lips, tongue or throat (Kuruva et al., 2017). Pharmacologically, exogenous dopamine, aka intropin, may be used to treat very low blood pressure and heart rate, resultant from shock from myocardial infarction, heart failure, trauma, surgery, renal failure, or a number of other conditions (Kuruva et al., 2017).
Dopamine’s structure is already advantageous because of its ability to cross the BBB and target synapses. Manipulation of its structure led to the development of 82 molecule designs that were fitted with scaffold structures to adhere to Aβ and disrupt its interaction with mitochondria and synapses (Kuruva et al., 2017). Of the molecules, DDQ was chosen particularly for its adherence to Aβ and its ability to inhibit the dysregulation of Drp1, which is known to cause fragmentation of mitochondria in AD cells (Kuruva et al., 2017). Specifically, DDQ prevents Aβ and Drp1 binding at the ser8 and Leu34 active sites of the Aβ polypeptide, and the ASN16 and Glu16 site of Drp1 (Kuruva et al., 2017).
In a study with immortalized human neuroblastoma SHSY5Y cells that exhibited AD pathology, application of DDQ was able to disrupt Aβ’s toxic interaction with Drp1 (Kuruva et al., 2017). In the SHSY5Y cells DDQ was able to reduce Aβ concentration and Drp1 overproduction, and in APPSWE/IND cells DDQ was able to reduce concentration of soluble Aβ42 as well (Kuruva et al., 2017). Overall, measurements of mRNA levels of mitochondrial dynamics Drp1, Fis1, Mfn1, Mfn2, mitochondrial biogenesis (PGC1x, Nrf1, Nrf2, and TFAM), and synaptic genes (PSD95, synaptophysin, synapsin 1, synapsin 2, synaptobrevin 1, synaptobrevin 2, synaptopodin, and GAP43) were used to determine the therapeutic nature of DDQ (Kuruva et al., 2017). DDQ’s ability to induce improvement of mitochondrial dynamics resulted in reduction of mitochondrial fragmentation, improvement of mitochondrial fusion, and resulting in improved biogenesis and synaptic activity (Kuruva et al., 2017).
Conclusions and Future Directions
Cholinesterase Inhibitors and glutamate inhibitors improve synaptic function but are not therapeutic for mitochondrial degradation. Natural antioxidants are unable to specifically target mitochondria, and are unable to cross blood brain barrier. The small molecules of topic however have antioxidant, and restorative properties, with moieties lipophilic and positively charged in nature. Mitochondria-targeted molecules protect cells from mutant proteins’ induced mitochondrial and cellular toxicities, and also protect neurons from age-related oxidative stress. Cultured cells induced with neurodegenerative disease demonstrate recovery at the synapse once treated with mitochondria targeted molecules. Mice induced with neurodegenerative disease and treated with mitochondria targeted molecules exhibit longer lifespan.
Interestingly, some of these molecules such as SS31 has shorter lifespan and metabolize quickly than other molecules such as MitoQ and Mdivi-1. Thus far, SS31 did not show any adverse effects in cell culture and animal models. Mitochondria-targeted molecules exhibit promising potential for increasing lifespan of persons with neurodegenerative diseases.
It is interesting to note that mitochondria-targeted small molecules have been tested in cell culture and animal models of neurodegenerative diseases, such as AD, Huntington’s, Parkinson’s, ALS, multiple sclerosis and other human diseases. However, there are no clinical trials reported, particularly for Mdivi 1, SS31 and DDQ to determine their efficacies for human applications. It is important to focus on small molecules for preclinical models and also for human clinical studies.
Figure 1:

Exhibiting difference between normal brain and AD brain, including factors leading to late-onset and early on-set AD.
Figure 2:

Factors contributing to neuronal cellular changes in AD.
Figure 3:

Demonstration of healthy mitochondrial dynamics, including fission and fusion.
Figure 4:

Demonstration of dysfunctional mitochondrial fission and fusion.
Figure 5:

Therapeutic effects of natural and mitochondria targeted antioxidants in mitochondria.
Figure 6:

Exhibition of mitochondria targeted antioxidant and SS-31 mechanism of action.
Highlights.
Mitochondria are power houses of cells and important for cell survival and cell death.
Mitochondrial dysfunction has been implicated in aging and age-related neurodegenerative diseases.
Natural antioxidants such as vitamin C & E, beta-carotene and glutathione have protective properties with limited blood brain barrier capacity.
Mitochondria targeted molecules such as MitoQ, MitoVitE, MitoTempo, MitoPBN and SS31 concentrate within mitochondria where they scavenge free-radicals, and augment mitochondrial dysfunction.
Acknowledgements
Work presented in this article is supported by NIH grants R01AG042178, R01AG047812, R01NS105473, and R41AG060836, the Garrison Family Foundation, CH Foundation and Alzheimer’s Association SAGA grant (to PHR).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict Interest Statement
None declared
References
- Alexeyev M, Shokolenko I, Wilson G, & LeDoux S (2013) The Maintenance of Mitochondrial DNA Integrity—Critical Analysis and Update. Cold Spring Harbor Perspectives in Biology. 5(5): a012641. doi: 10.1101/cshperspect.a012641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angajala A, Lim S, Phillips JB, Kim J-H, Yates C, You Z, & Tan M (2018) Diverse Roles of Mitochondria in Immune Responses: Novel Insights Into Immuno-Metabolism. Frontiers in Immunology, 9, 1605. doi: 10.3389/fimmu.2018.01605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apak R, Guclu K, Demirata B, Ozyurek M, Celik SE, Bektasoglu B, Berker KI, Ozyurt (2007) Comparative evaluation of various total antioxidant capacity assays applied to phenolic compounds with the CUPRAC assay. Molecules. 12(7):1496–1547 doi: 10.3390/12071496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apostolova N, & Victor VM (2015). Molecular strategies for targeting antioxidants to mitochondria: therapeutic implications. Antioxidants & redox signaling, 22(8), 686–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battogtokh G, Choi YS, Kang DS, Park SJ (2018) Mitochondria-targeting drug conjugates for cytotoxic, anti-oxidizing and sensing purposes: current strategies and future perspectives. Acta Pharmaceutica Sinica B 2018. 8(6):862–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi P, & Di Lisa F (2015) The mitochondrial permeability transition pore: molecular nature and role as a target in cardioprotection. Journal of molecular and cellular cardiology, 78,100–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birk AV, Chao WM, Liu S, Soong Y, Szeto HH (2015) Disruption of cytochrome c heme coordination is responsible for mitochondrial injury during ischemia. Biochimica et Biophysica Acta (BBA) – Bioenergetics. 1847(10):1075–1084 10.1016/j.bbabio.2015.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blair LJ, Baker JD, Sabbagh JJ, & Dickey CA (2015) The emerging role of peptidyl-prolyl isomerase chaperones in tau oligomerization, amyloid processing, and Alzheimer’s disease. Journal of neurochemistry, 133(1), 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco-Silvente L, Castells X, Saez M, Barceló MA, Garre-Olmo J, Vilalta-Franch J, & Capellà D (2017). Discontinuation, Efficacy, and Safety of Cholinesterase Inhibitors for Alzheimer’s Disease: a Meta-Analysis and Meta-Regression of 43 Randomized Clinical Trials Enrolling 16 106 Patients. International Journal of Neuropsychopharmacology, 20(7), 519–528. 10.1093/ijnp/pyx012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Q, & Tammineni P (2016) Alterations in Mitochondrial Quality Control in Alzheimer’s Disease. Frontiers in cellular neuroscience, 10, 24. doi: 10.3389/fncel.2016.00024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calkins MJ, Manczak M, Mao P, Shirendeb U, & Reddy PH (2011). Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer’s disease. Human molecular genetics, 20(23), 4515–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspersen C, Wang N, Yao J, Sosunov A, Chen X, Lustbader JW, Xu HW, Stern D, McKhann G, Yan SD (2005) Mitochondrial Abeta: a potential focal point for neuronal metabolic dysfunction in Alzheimer’s disease. FASEB journal. 19(14): 2040–2041 [DOI] [PubMed] [Google Scholar]
- Chaturvedi RK & Beal MF (2013) Mitochondrial Diseases of the Brain. Free Radical Biology and Medicine. (63): 1–29. 10.1016/j.freeradbiomed.2013.03.018 [DOI] [PubMed] [Google Scholar]
- Chelikani P, Fita I, Loewen PC (2004) Diversity of structures and properties among catalases. Cellular and Molecular Life Sciences 61: 192–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SD, Wu CL, Hwang WC, & Yang DI (2017). More Insight into BDNF against Neurodegeneration: Anti-Apoptosis, Anti-Oxidation, and Suppression ofAutophagy. International journal of molecular sciences, 18(3), 545. doi: 10.3390/ijms18030545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cragg GM, Pezzuto JM (2015) Natural Products as a Vital Source for the Discovery of Cancer Chemotherapeutic and Chemopreventative Agents. Medical Principles and Practice 2016. 25(suppl 2):41–59 DOI: 10.1159/000443404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davatgaran-Taghipour Y, Masoomzadeh S, Farzaei MH, Bahramsoltani R, Karimi-Soureh Z, Rahimi R, & Abdollahi M (2017). Polyphenol nanoformulations for cancer therapy: experimental evidence and clinical perspective. International journal of nanomedicine, 12, 2689–2702. doi: 10.2147/IJN.S131973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du H, Guo L, Yan S, Sosunov AA, McKhann GM, Yan SS (2010) Early deficits in synaptic mitochondria in an Alzheimer’s disease mouse model. Proceedings of the National Academy of Sciences. 107(43): 18670–18675; DOI: 10.1073/pnas.1006586107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du H, Guo L, Fang F, Chen D, Sosunov AA, McKhann GM, Yan Y, Wang C, Zhang H, Molkentin JD, Gunn-Moore FJ, Vonsattel JP, Arancio O, Chen JX, … Yan SD (2008). Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nature medicine, 14(10), 1097–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emanuele S, D’Anneo A, Calvaruso G, Cernigliaro C, Giuliano M, & Lauricella M (2018) The Double-Edged Sword Profile of Redox Signaling: Oxidative Events As Molecular Switches in the Balance between Cell Physiology and Cancer. Chemical Research in Toxicology. 31(4): 201–210. doi: 10.1021/acs.chemrestox.7b00311 [DOI] [PubMed] [Google Scholar]
- Esposito Z, Belli L, Toniolo S, Giuseppe S, Claudio B, Martorana A (2013) Amyloid β, Glutamate, Excitotoxicity in Alzheimer’s Disease: Are We on the Right Track? CNS Neuroscience & Therapeutics. (19)8: 549–555 10.1111/cns.12095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber J, Fong S, Chen C, Yoong S, Pastorin G, Schaffer S, Cheah I, Halliwell B (2013) Mitochondria-targeted antioxidants and metabolic modulators as pharmacological interventions to slow ageing. Biotechnology Advances. 31(5): 563–592. 10.1016/j.biotechadv.2012.09.005 [DOI] [PubMed] [Google Scholar]
- Guedes-Dias P, Oliveira JMA (2013) Lysine deacetylases and mitochondrial dynamics in neurodegeneration. Biochimica et Biophysica Acta. (1832)8: 1345–1359. doi: 10.1016/j.bbadis.2013.04.005 [DOI] [PubMed] [Google Scholar]
- Hu H, Tan CC, Tan L, and Yu JT (2017) A Mitocentric View of Alzheimer’s disease. Molecular Neurobiology. 54(8): 6046–6060. doi: 10.1007/s12035-016-0117-7 [DOI] [PubMed] [Google Scholar]
- Hu H, Li M (2016) Mitochondria-targeted antioxidant mitotempo protects mitochondrial function against amyloid beta toxicity in primary cultured mouse neurons. Biochemical and Biophysical Research Communications. 478(1): 174–180. 10.1016/j.bbrc.2016.07.071 [DOI] [PubMed] [Google Scholar]
- Hyman BT, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Carrillo MC, Dickson DW, Duyckaerts C, Frosch MP, Masliah E, Mirra SS, Nelson PT, Schneider JA, Thal DR, Thies B, Trojanowski JQ, Vinters HV, … Montine TJ (2012) National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimer’s & dementia: the journal of the Alzheimer’s Association, 8(1), 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imlay JA (2003) Pathways of oxidative damage. Annual Review of Microbiology 57: 395–418. [DOI] [PubMed] [Google Scholar]
- Johnson JA, Johnson DA, Kraft AD, Calkins MJ, Jakel RJ, Vargas MR, & Chen PC (2008). The Nrf2-ARE pathway: an indicator and modulator of oxidative stress in neurodegeneration. Annals of the New York Academy of Sciences, 1147, 61–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalogeris T, Bao Y, & Korthuis RJ (2014). Mitochondrial reactive oxygen species: a double edged sword in ischemia/reperfusion vs preconditioning. Redox biology, 2, 702–14. doi: 10.1016/j.redox.2014.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr JS, Adriaanse BA, Greig NH, Mattson MP, Cader MZ, Bohr VA, & Fang EF (2017) Mitophagy and Alzheimer’s disease: cellular and molecular mechanisms. Trends in Neurosciences. 40(3): 151–166. doi: 10.1016/j.tins.2017.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kritsilis M, V Rizou S, Koutsoudaki PN, Evangelou K, Gorgoulis VG, & Papadopoulos D (2018) Ageing, Cellular Senescence and Neurodegenerative Disease. International journal of molecular sciences, 19(10), 2937. doi: 10.3390/ijms19102937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuruva CS, Manczak M, Yin X, Ogunmokun G, Reddy AP, & Reddy PH (2017) Aqua-soluble DDQ reduces the levels of Drp1 and Aβ and inhibits abnormal interactions between Aβ and Drp1 and protects Alzheimer’s disease neurons from Aβ- and Drp1-induced mitochondrial and synaptic toxicities. Human Molecular Genetics. 26(17): 3375–3395. doi: 10.1093/hmg/ddx226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Zhou Y, Wang F, Chen X, Wang C, Wang J, Liu T, Li Y, He B (2018) SIRT4 is the last puzzle of mitochondrial sirtuins. Bioorganic & Medicinal Chemistry. 26(14): 3861–3865. [DOI] [PubMed] [Google Scholar]
- Lv H, Liu Q, Zhou J, Tan G, Deng X, Ci X (2017) Daphnetin-mediated Nrf2 antioxidant signaling pathways ameliorate tert-butyl hydroperoxide (t-BHP)-induced mitochondrial dysfunction and cell death. Free Radical Biology and Medicine. (106): 38–52; DOI: 10.1016/j.freeradbiomed.2017.02.016 [DOI] [PubMed] [Google Scholar]
- Man GC, Wang WW, Yim AP, Wong JH, Ng TB, Lam TP, Lee SK, Ng BK, Wang CC, Qiu Y, Cheng CY (2014). A review of pinealectomy-induced melatonin-deficient animal models for the study of etiopathogenesis of adolescent idiopathic scoliosis. International journal of molecular sciences, 15(9), 16484–99. doi: 10.3390/ijms150916484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manczak M, Kandimalla R, Yin X, Reddy PH (2018) Hippocampal mutant APP and amyloid beta-induced cognitive decline, dendritic spine loss, defective autophagy, mitophagy and mitochondrial abnormalities in a mouse model of Alzheimer’s disease. Human Molecular Genetics 27(8): 1332–1342. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Manczak M, Kandimalla R, Fry D, Sesaki H, & Reddy PH (2016) Protective effects of reduced dynamin-related protein 1 against amyloid beta-induced mitochondrial dysfunction and synaptic damage in Alzheimer’s disease. Human molecular genetics, 25(23), 5148–5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manczak M, & Reddy PH (2012). Abnormal interaction of VDAC1 with amyloid beta and phosphorylated tau causes mitochondrial dysfunction in Alzheimer’s disease. Human molecular genetics, 21(23), 5131–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manczak M, & Reddy PH (2012) Abnormal interaction between the mitochondrial fission protein Drp1 and hyperphosphorylated tau in Alzheimer’s disease neurons: implications for mitochondrial dysfunction and neuronal damage. Human molecular genetics, 21(11), 2538–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manczak M, Calkins MJ, & Reddy PH (2011) Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: implications for neuronal damage. Human molecular genetics, 20(13), 2495–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manczak M, Mao P, Calkins M, Cornea A, Arubala RP, Murphy MP, Szeto HH, Park B, Reddy PH (2010) Mitochondria-targeted antioxidants protect against Abeta toxicity in Alzheimer’s disease neurons. Journal of Alzheimer’s Disease : JAD. 20(Suppl 2): S609–S631. doi: 10.3233/JAD-2010-100564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao P, Manczak M, Shirendeb UP, & Reddy PH (2013) MitoQ, a mitochondria-targeted antioxidant, delays disease progression and alleviates pathogenesis in an experimental autoimmune encephalomyelitis mouse model of multiple sclerosis. Biochimica et Biophysica Acta, 1832(12): 2322–2331. 10.1016/j.bbadis.2013.09.005. doi: 10.1016/j.bbadis.2013.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao P, Manczak M, Calkins MJ, Truong Q, Reddy TP, Reddy AP, Shirendeb U, Lo HH, Rabinovitch PS, Reddy PH (2012) Mitochondria-targeted catalase reduces abnormal APP processing, amyloid β production and BACE1 in a mouse model of Alzheimer’s disease: implications for neuroprotection and lifespan extension. Human Molecular Genetics. 21(13): 2973–2990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansouri A, Demeilliers C, Amsellem S, Pessayre D, & Fromenty B (2001) Acute Ethanol Administration Oxidatively Damages and Depletes Mitochondrial DNA in Mouse Liver, Brain, Heart, and Skeletal Muscles: Protective Effects of Antioxidants. The Journal of Pharmacology and Experimental Therapeutics. 298(2): 737–743. [PubMed] [Google Scholar]
- Matsunaga S, Kishi T, & Iwata N (2015). Memantine Monotherapy for Alzheimer’s Disease: A Systematic Review and Meta-Analysis. PLoS ONE, 10(4), e0123289 10.1371/journal.pone.0123289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mlinac ME, Feng MC (2016) Assessment of Activities of Daily Living, Self-Care, and Independence. Archives of Clinical Neuropsychology. 31(6): 506–516. 10.1093/arclin/acw049 [DOI] [PubMed] [Google Scholar]
- Motyl J, Wencel PL, Cieślik M, Strosznajder RP, & Strosznajder JB (2018) Alpha-synuclein alters differently gene expression of Sirts, PARPs and other stress response proteins: implications for neurodegenerative disorders. Molecular Neurobiology. 55(1): 727–740. doi: 10.1007/s12035-016-0317-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1.National Center for Biotechnology Information (2019) PubChem Compound Database; CID=3152, https://pubchem.ncbi.nlm.nih.gov/compound/3152 (accessed Jan. 23, 2019). [Google Scholar]
- 2.National Center for Biotechnology Information (2019) PubChem Compound Database; CID=124886, https://pubchem.ncbi.nlm.nih.gov/compound/124886 (accessed Jan. 23, 2019). [Google Scholar]
- 3.National Center for Biotechnology Information (2019) PubChem Compound Database; CID=1253, https://pubchem.ncbi.nlm.nih.gov/compound/1253 (accessed Jan. 23, 2019). [Google Scholar]
- 4.National Center for Biotechnology Information (2019) PubChem Compound Database; CID=3825829, https://pubchem.ncbi.nlm.nih.gov/compound/3825829 (accessed Jan. 23, 2019). [Google Scholar]
- 5.National Center for Biotechnology Information (2019) PubChem Compound Database; CID=4054, https://pubchem.ncbi.nlm.nih.gov/compound/4054 (accessed Jan. 23, 2019). [Google Scholar]
- 6.National Center for Biotechnology Information (2019) PubChem Substance Database; SID=332864229, https://pubchem.ncbi.nlm.nih.gov/substance/332864229 (accessed Jan. 23, 2019). [Google Scholar]
- 7.National Center for Biotechnology Information (2019) PubChem Compound Database; CID=77991, https://pubchem.ncbi.nlm.nih.gov/compound/77991 (accessed Jan. 23, 2019). [Google Scholar]
- Nicholls P, Fita I, Loewen PC (2001) Enzymology and structure of catalases. Advances in Inorganic Chemistry 51: 51–106 [Google Scholar]
- 1.Reddy PH, Manczak M, Yin X, & Reddy AP (2018). Synergistic Protective Effects of Mitochondrial Division Inhibitor 1 and Mitochondria-Targeted Small Peptide SS31 in Alzheimer’s Disease. Journal of Alzheimer ’s disease : JAD, 62(4), 1549–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reddy PH, Manczak M, Yin X, Grady MC, Mitchell A, Tonk S, Kuruva CS, Bhatti JS, Kandimalla R, Vijayan M, Kumar S, Wang R, Pradeepkiran JA, Ogunmokun G, Thamarai K, Quesada K, Boles A, Reddy AP (2018) Protective Effects of Indian Spice Curcumin Against Amyloid Beta in Alzheimer’s Disease. Journal of Alzheimer’s Disease : JAD, 61(3), 843–866. doi: 10.3233/JAD-170512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy PH, Manczak M, & Yin X (2017). Mitochondria-Division Inhibitor 1 Protects Against Amyloid-β induced Mitochondrial Fragmentation and Synaptic Damage in Alzheimer’s Disease. Journal of Alzheimer’s disease : JAD, 58(1), 147–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy PH, Williams J, Smith F, Bhatti JS, Kumar S, Vijayan M, Kandimalla R, Kuruva CS, Wang R, Manczak M, Yin X, Reddy AP (2017) Chapter Five – MicroRNAs, Aging, Cellular Senescence, and Alzheimer’s Disease. Progress in Molecular Biology and Translational Science. 146(2017): 127–171 [DOI] [PubMed] [Google Scholar]
- Reddy PH, Manczak M, Yin X, Grady MC, Mitchell A, Kandimalla R, & Kuruva CS (2016). Protective effects of a natural product, curcumin, against amyloid β induced mitochondrial and synaptic toxicities in Alzheimer’s disease. Journal of investigative medicine : the official publication of the American Federation for Clinical Research, 64(8), 1220–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy PH (2011). Abnormal tau, mitochondrial dysfunction, impaired axonal transport of mitochondria, and synaptic deprivation in Alzheimer’s disease. Brain research, 1415, 136–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy PH, & Reddy TP (2011). Mitochondria as a therapeutic target for aging and neurodegenerative diseases. Current Alzheimer research, 8(4), 393–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy PH, Reddy TP, Manczak M, Calkins MJ, Shirendeb U, & Mao P (2010). Dynamin-related protein 1 and mitochondrial fragmentation in neurodegenerative diseases. Brain research reviews, 67(1–2), 103–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy PH (2008) Mitochondrial Medicine for Aging and Neurodegenerative Diseases. Neuromolecular Medicine. 10(4): 291–315. doi: 10.1007/s12017-008-8044-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy PH, & Beal MF (2008). Amyloid beta, mitochondrial dysfunction and synaptic damage: implications for cognitive decline in aging and Alzheimer’s disease. Trends in molecular medicine, 14(2), 45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy PH (2006) Mitochondrial Oxidative Damage in Aging and Alzheimer’s Disease: Implications for Mitochondrially Targeted Antioxidant Therapeutics. Journal of Biomedicine and Biotechnology. 2006(3): 31372. doi: 10.1155/JBB/2006/31372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiss AB, Arain HA, Stecker MM, Siegart NM, Kasselman LJ (2018) Amyloid toxicity in Alzheimer’s disease. Reviews in Neurosciences. 29(6):613–627. doi: 10.1515/revneuro-2017-0063 [DOI] [PubMed] [Google Scholar]
- Schriner SE, Linford NJ, Martin GM, Treuting P, Ogburn CE, Emond M, Coskun PE, Ladiges W, Wolf N, Van Remmen H, Wallace DC, Rabinovitch PS (2005) Extension of murine life span by overexpression of catalase targeted to mitochondria. Science. 308(5730):1909–1911 DOI: 10.1126/science.1106653 [DOI] [PubMed] [Google Scholar]
- Scott I, & Youle RJ (2010) Mitochondrial fission and fusion. Essays in Biochemistry. 47: 85–98. doi: 10.1042/bse0470085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shields LY, Kim H, Zhu L, Haddad D, Berthet A, Pathak D, Lam M, Ponnusamy R, Diaz-Ramirez LG, Gill TM, Sesaki H, Mucke L, … Nakamura K (2015). Dynamin-related protein 1 is required for normal mitochondrial bioenergetic and synaptic function in CA1 hippocampal neurons. Cell death & disease, 6(4), e1725. doi: 10.1038/cddis.2015.94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RA, Porteous CM, Coulter CV, Murphy MP (1999) Selective targeting of an antioxidant to mitochondria. European Journal of Biochemistry. 263(3): 709–716. [DOI] [PubMed] [Google Scholar]
- Strobel M, Tinz J, Biesalski HK (2007) The importance of β-carotene as a source of vitamin A with special regard to pregnant and breastfeeding women. European Journal of Nutrition. 46[Supplement1]:1–20 10.1007/s00394-007-1001-z [DOI] [PubMed] [Google Scholar]
- Swerdlow RH, Burns JM, & Khan SM (2013) The Alzheimer’s disease mitochondrial cascade hypothesis: progress and perspectives. Biochimica et biophysica acta, 1842(8), 1219–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szeto HH (2014) First-in-class cardiolipin-protective compound as a therapeutic agent to restore mitochondrial bioenergetics. British Journal of Pharmacology. 171(8): 2029–2050. doi: 10.1111/bph.12461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szeto H, & Birk A (2014) Serendipity and the Discovery of Novel Compounds That Restore Mitochondrial Plasticity. Clinical Pharmacology and Therapeutics. 96(6): 672–683. doi: 10.1038/clpt.2014.174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szeto HH (2006). Mitochondria-targeted peptide antioxidants: novel neuroprotective agents. The AAPS journal, 8(3), E521–31. doi: 10.1208/aapsj080362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanila H (2016) The role of BDNF in Alzheimer’s disease. Neurobiology of Disease. 97(2017): 114–118. 10.1016/j.nbd.2016.05.008 [DOI] [PubMed] [Google Scholar]
- Wang X, Wang W, Li L, Perry G, Lee HG, & Zhu X (2013). Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochimica et biophysica acta, 1842(8), 1240–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkins HM, & Swerdlow RH (2016) Relationships Between Mitochondria and Neuroinflammation: Implications for Alzheimer’s Disease. Current topics in medicinal chemistry, 16(8), 849–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan SD, & Stern DM (2005) Mitochondrial dysfunction and Alzheimer’s disease: role of amyloid-beta peptide alcohol dehydrogenase (ABAD). International journal of experimental pathology, 86(3), 161–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin X, Manczak M, & Reddy PH (2016). Mitochondria-targeted molecules MitoQ and SS31 reduce mutant huntingtin-induced mitochondrial toxicity and synaptic damage in Huntington’s disease. Human molecular genetics, 25(9), 1739–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Perry G, Smith MA, & Wang X (2013) Abnormal mitochondrial dynamics in the pathogenesis of Alzheimer’s disease. Journal of Alzheimer’s disease : JAD, 33 Suppl 1(01), S253–62. [DOI] [PMC free article] [PubMed] [Google Scholar]