

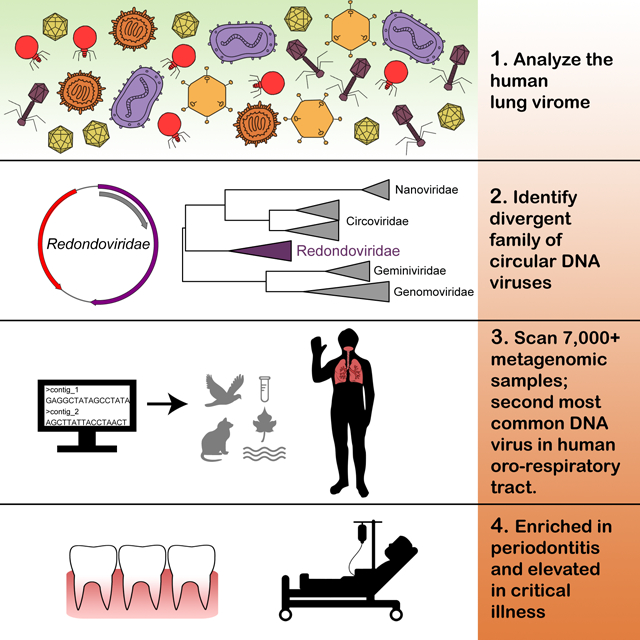
Summary:
The global virome is largely uncharacterized but is now being unveiled by metagenomic DNA sequencing. Exploring the human respiratory virome, in particular, can provide insights into oro-respiratory diseases. Here, we use metagenomics to identify a family of small, circular DNA viruses—named Redondoviridae—associated with human diseases. We first identified two redondovirus genomes from bronchoalveolar lavage samples from human lung donors. We then queried thousands of metagenomic samples and recovered 17 additional complete redondovirus genomes. Detections were exclusively in human samples and mostly from respiratory tract and oro-pharyngeal sites, where Redondoviridae was the second most prevalent eukaryotic DNA virus family.
Redondovirus sequences were associated with periodontal disease, and abundances decreased with treatment. Some critically ill patients in a medical intensive care unit were found to harbor high levels of redondoviruses in respiratory samples. These results suggest that redondoviruses colonize human oro-respiratory sites and can bloom in several human disorders.
Keywords: viral metagenomics, periodontitis, CRESS DNA virus, Redondoviridae, redondovirus, Brisavirus, Vientovirus
eTOC blurb
Abbas and Taylor et al. report the discovery and characterization of a family of circular DNA viruses subsequently named Redondoviridae. Redondoviruses are primarily found in the human oro-respiratory tract and reach high levels in subjects with periodontitis and critical illness.
Graphical Abstract
Introduction
Viruses are the most abundant biological entities on Earth, but global viral populations (the “virome”) are still mostly uncharacterized. Identifying novel viruses can be difficult if they have limited sequence homology to viral genomes in reference databases. Recent advances in sample preparation and sequencing techniques have uncovered a world of new viruses (Paez-Espino et al., 2016, Simmonds et al., 2017, Rosario and Breitbart, 2011, Minot et al., 2013, Minot et al., 2011). However the majority of reads in most studies remain unclassified (Aggarwala et al., 2017, Krishnamurthy and Wang, 2017), leaving our understanding of the virome incomplete. Here we describe the identification of a previously unstudied viral family, its localization in human oro-respiratory sites, and its association with disease states.
Methods for analyzing the virome are particularly efficient at recovering small circular DNA viruses. Metagenomic sample preparation commonly involves multiple displacement amplification (MDA) with a highly processive, strand-displacing DNA polymerase, which enriches for small, circular, single-stranded DNAs (ssDNA) (Rosario et al., 2012, Labonté and Suttle, 2013, Krupovic et al., 2016). Many ssDNA viruses encode a replication initiation protein (Rep)—thus this group is collectively known as circular Rep-encoding single-stranded DNA (CRESS) viruses (Rosario et al., 2012). Some aspects of genome architecture and functional domains of viral Rep and Capsid proteins are detectably conserved among CRESS viruses, though pairwise nucleotide identities are often low. A well-studied group of animal CRESS viruses is the Circovirus genus within the Circoviridae family, which includes pathogenic viruses of swine and birds (Ellis, 2014, Todd, 2000). The Circoviridae family also contains the genus Cyclovirus, which consists of viruses identified by metagenomic sequencing in samples from several mammalian species (Breitbart et al., 2017, Li et al., 2010), including some sporadically identified in human disease states (Phan et al., 2014, Smits et al., 2014). The recently identified Smacoviridae family has been detected in mammalian feces, though the definitive hosts are unknown (Varsani and Krupovic, 2018). Other CRESS families include viruses that infect plants, Geminiviridae and Nanoviridae (Harrison et al., 1977, Fauquet et al., 2005), fungi, Genomoviridae (Krupovic et al., 2016, Varsani and Krupovic, 2017), and additional apparent viruses detected as divergent sequences for which hosts are unknown (Simmonds et al., 2017).
We and others have previously investigated the human respiratory tract virome in health and disease. Typically anelloviruses, herpesviruses and bacteriophages dominate human respiratory tract samples (Willner et al., 2009, Abeles et al., 2015, Wylie et al., 2012, Pérez-Brocal and Moya, 2018, Young et al., 2015, Abbas et al., 2017, Abbas et al., 2018, Clarke et al., 2017a). Recently, we identified short sequence reads with limited homology to a swine-associated CRESS virus (Cheung et al., 2014) in bronchoalveolar lavage (BAL) from human organ donors (Abbas et al., 2018, Abbas et al., 2017), raising the possibility that we had detected a undescribed human virus.
Following up on this lead, we now report the identification of a group of CRESS viruses, highly divergent from other viral families, present in human respiratory and oral samples. These CRESS genomes are sufficiently different from previously described taxa that we propose establishing a viral family, which we name Redondoviridae (redondo—Spanish for “round”) containing the genera Vientovirus and Brisavirus (from the Spanish words for “wind” and “breeze”, alluding to their discovery in the respiratory tract). A recently described genome identified in upper respiratory secretions of a febrile individual (Cui et al., 2017) also classified as a redondovirus, and we propose it as the type species for the Brisavirus genus. Analysis of the distribution of redondoviruses showed that they were the second most prevalent virus in human respiratory samples, after anelloviruses, in samples from viral metagenomic studies. Analysis of redondovirus representation in numerous environmental and host-associated samples disclosed association of Redondoviridae with periodontal disease and acute critical illness.
Results
Initial Discovery of Redondoviruses in Human Bronchoalveolar Lavage Fluid
In two previous studies of lung transplant recipients (Abbas et al., 2017, Abbas et al., 2018), samples of BAL were enriched for viral particles, then RNA and DNA was purified and subjected to metagenomic sequence analysis. DNA fractions were amplified using MDA. Alignment of reads from two organ donor BAL samples to the NCBI viral genome database showed modest (14%) coverage of Porcine stool-associated circular virus 5 (PoSCV-5) isolate CP3 (GenBank: NC_023878) (Figure 1). PoSCV-5 is currently an unclassified and unstudied member of the Circoviridae family.
Figure 1: Discovery of Redondovirus Genomes in Metagenomic Samples.

A-Several hundred shotgun metagenomic reads from two organ donor BAL virome samples were identified as having limited homology to Porcine stool-associated circular virus 5 (PoSCV-5). Reads from these samples were assembled into two contigs, which were then cloned from multiple displacement amplified sample DNA using target specific primers and Sanger sequenced. The complete circular genomes were used to query additional internal and public microbial metagenomic datasets. Target-specific amplification, sequencing and genome assembly was repeated for additional samples with sequences homologous to these novel genomes if the original DNA was available. In cases where original samples were not available, metagenomic contigs were checked for circularity and completeness. A total of 19 complete genomes were recovered from 67 human samples (bottom). See also Figure S1.
B-The genomic architecture of redondoviruses shows ambisense open-reading frames (ORFs) encoding a conserved capsid, Replication associated protein (Rep) and unknown protein (ORF3). The average nucleotide identity of 20 Redondoviridae members (19 genomes discovered here and one genome previously reported (Cui et al., 2017)) is shown on the inside of the genome map as a heatmap. A putative origin of replication stem-loop structure with a conserved nonanucleotide motif is predicted to form in the 5’ end of the Rep coding region. The height of the letter in the motif represents its frequency.
After assembling reads from these samples into contigs, we found that sequences matching PoSCV-5 were present in circular contigs of approximately 3,000 base pairs (bp). Thus, whole viral genomes were present in the initial BAL samples, but only a small region of these genomes resembled PoSCV-5. Several sets of nested primers (Table S1 and Figure S1) were used to amplify overlapping fragments from the original BAL samples. These fragments were sequenced using the Sanger method and assembled to construct two circular genomes of 3,026 bp (Human lung-associated brisavirus RC; accession MK059757) and 3,056 bp (Human lung-associated vientovirus FB; accession MK059763) (Figure S1).
Contigs assembled from shotgun metagenomic reads of other BAL samples processed by our group were then queried for DNA sequence similarity to the two genomes described above. In total, seven complete Redondoviridae genomes were discovered and cloned from independent BAL samples from organ donors and patients with sarcoidosis (Figure S1). These genomes were then used as alignment targets to interrogate publicly available datasets. Twelve more samples had sufficient coverage of redondovirus sequences to allow assembly, yielding 19 complete genomes (Figure 1A, Table S2).
A danger is that small circular viruses may be derived from environmental contamination in clinical or laboratory reagents (Naccache et al., 2013, Salter et al., 2014). We queried 144 contamination controls from seven studies analyzed by shotgun metagenomics, six of which were from our laboratory (most viral metagenomic data sets in databases do not include sequenced contamination controls). None of these negative controls had any reads aligning to redondoviruses. This included 24 bronchoscopes prewashes, which consist of a sterile saline solution passed through bronchoscopes before insertion into a patient. These were processed at our site in parallel with virome preps of multiple positive BAL samples; control samples were subjected to MDA, library preparation, and shotgun sequencing all in parallel (Clarke et al., 2017a). In further tests, we used a qPCR assay targeting redondovirus genomes to check the 24 bronchoscope prewashes and two additional DNA extraction controls subjected to MDA. All were negative by qPCR analysis. As positive controls, we detected robust qPCR signals in MDA-amplified DNA extracted from the original acellular BAL samples from which these genomes were cloned (Figure S1C).
To strengthen the notion that Redondoviridae are of eukaryotic origin, we investigated whether they showed sequence signatures of bacteriophages. The presence of prokaryotic ribosomal binding sites (RBS) upstream of viral open reading frames (ORFs) can provide evidence for a prokaryotic host (Krishnamurthy and Wang, 2018) . We implemented the algorithm described in Krishnamurthy and Wang and identified no prokaryotic RBS proximal to any redondovirus protein coding sequence. These data support the idea that redondovirus sequences were not derived from environmental contamination and are not bacteriophages.
Redondovirus Genomes Contain Conserved Features of CRESS Viruses and a third distinct ORF
Redondoviruses share some genomic features with other CRESS DNA viruses, but display several unique characteristics (Table 1). Redondovirus genomes contain ambisense ORFs (Figure 1B) encoding a 334–363 amino acid Rep and a 449–531 amino acid capsid (Cp), which are only 10–15% identical to those of porcine circovirus-1 and −2, and 40–55% for PoSCV-5 , which provided the initial database “hit”. All redondovirus genomes also contain a third ORF (ORF3) overlapping the capsid gene, which is not found in either porcine circoviruses or in PoSCV-5. ORF3 has no homology to any described protein family. Thus, while PoSCV-5 is the most closely related known virus to the redondoviruses, PoSCV-5 is markedly divergent in genome architecture and protein identity and does not meet criteria for a member of the Redondoviridae family.
Table 1:
Comparison of genomic features between Redondoviridae and other CRESS-DNA viruses
| Feature | Redondoviridae | Circoviridae | Nanoviridae | Geminiviridae | Genomoviridae | Smacoviridae |
|---|---|---|---|---|---|---|
| Size (kb) | 3.0–3.1 | 1.7–2.0 | 1.0 * 6 segments | 2.5–3.0 | 2.1–2.2 | 2.6–2.9 |
| ORFs | Cp, Rep, ORF3 | Cp, Rep, ORF3/4 | Cp, Rep, others | Cp, Rep, others | Cp, Rep | Cp, Rep |
| ORF orientation | Ambisense | Ambisense | Segmented | Ambisense (or segmented) | Ambisense | Ambisense |
| Origin sequence | TATTATTAT | TAGTATTAC | TATTATTAC | TAATATTAC | TAATATTAT | NAGTATTAC |
| Origin location | Noncoding (upstream) / in Rep | Noncoding (upstream) / in Rep | Noncoding (upstream) | Noncoding (upstream) | Noncoding (upstream) | Noncoding (downstream) |
Redondoviruses display considerable sequence divergence when comparing their Cp and Rep proteins. The range of pairwise amino acid identities of capsid is 67.5–99.6% (median 82.3%) while the range of Rep amino acid identity is 36.6–99.7% (median 54%) (Figure 1B). Surprisingly, Cp is more conserved than Rep. One might have expected that the capsid protein, which is presumably recognized by host antibodies, would be under stronger diversifying (positive) selection. Part of Cp overlaps ORF3, and so could be constrained in sequence drift for that reason, but even in the non-overlapping carboxy-terminal coding region (Figure 1B) the variability is still lower than in Rep.
To clarify the phylogenetic relationships between viral proteins within the Redondoviridae and other CRESS virus families, we built maximum-likelihood phylogenetic trees of Rep and Cp protein sequences. Redondoviruses are more similar to each other than to other CRESS families by protein identity and genome organization (Figure 2, Table 1). The capsid and Rep protein phylogenies show different relationships between the isolates, suggesting that recombination is common in redondoviruses, as in other circular ssDNA viruses (Ma et al., 2007, Lefeuvre et al., 2009, Fahsbender et al., 2017, Leppik et al., 2007). Based on previous definitions (Varsani and Krupovic, 2018) and analysis of the diversity of viral Rep proteins, redondovirus genomes can be grouped in two genera, demarcated by 50% Rep protein identity, which we propose to call Vientovirus and Brisavirus (Table S2).
Figure 2: Redondoviridae is a Distinct Virus Family Based on Capsid and Rep Identities.

Phylogenetic trees of redondovirus Rep (A) and capsid (B) proteins from CRESS DNA viruses. Collapsed viral genera or families are indicated by grey triangles. Branch likelihood, determined by approximate likelihood ratio test, is shown by colored circles at each node and the scale shows amino acid substitutions per site. The sample type of origin for each redondovirus is shown as colored boxes next to each virus’ name, which is colored to reflect genus designation. See also Table S2.
The redondovirus Rep protein (Figure 3B) contains two domains found in many small DNA and RNA viruses: one involved in rolling-circle replication (Pfam: PF00799) and a second helicase domain within the P-loop NTPase superfamily (Pfam: PF00910) (Ilyina and Koonin, 1992, Gorbalenya et al., 1990).
Figure 3: Redondovirus Genomes Contain Conserved Motifs Implicated in Rolling-circle Replication.

A-The sequence and predicted structure of the putative replication origin of Human lung-associated brisavirus AA is shown in the top left. The inverted repeat forming the stem is shown in orange, the nonanucleotide motif within the loop is shown in green, and an imperfect 6 bp direct repeat sequence is shown in purple. Individual predicted stem loop sequences (threshold for stability: ΔG°<−5 kcal/mol) are shown to the right of the folded sequence. The calculated ΔG° of melting for the predicted stem-loops ranges from −5.0 to −9.45 kcal/mol.
B-Conserved rolling circle replication and superfamily 3 (SF3) helicase motifs were found in redondoviruses. The positions for the motifs are given using the Human lung-associated brisavirus AA genome sequence (Accession MK059754). The height of each letter represents its frequency. Amino acid positions identified as possibly under positive selection pressure are marked by a red star.
C-The putative redondovirus capsid protein contains a basic amino-terminus and a predicted virus coat protein-like fold. The positions for the motifs correspond to Human lung-associated brisavirus AA, as above. Amino acid positions identified as possibly under positive selection pressure are marked by a red star.
Redondovirus capsids, like those of other ssDNA viruses, contain a basic amino-terminus. Protein modeling by PHYRE2 (Kelley et al., 2015) weakly predicted folds similar to coat proteins of ssRNA viruses that infect plants (Figure 3C; 58% confidence over 7% of sequence).
Circovirus genomes typically contain a conserved motif (“NANTATTAC”) within a stem-loop structure followed by short direct repeats, located in the intergenic region at the 5’ end of Cp and Rep. Such sequences are candidates for the origin of replication (Mankertz et al., 1997) where the viral-encoded Rep binds and cleaves, mediating replication by host polymerases. Such stem-loops are found in other CRESS virus families. In the one previous report of a single redondovirus genome (Cui et al., 2017), the authors suggested a hairpin in the large intergenic region as the origin of replication. However, analysis of all 20 genomes showed that a conserved, stable stem loop structure is predicted to form in the second smaller intergenic region, partially overlapping Rep. Although the length of the stem, size of the loops, and presence of downstream direct repeats vary, most redondovirus genomes contain a nonanucleotide motif (“TATTATTAT”) (Figure 1B) similar but not identical to that of other CRESS viruses. This structure is highly conserved among redondoviruses (Figure 3A), while the sequence of the alternative intergenic hairpin is not, suggesting that this is a more likely candidate for the replication origin.
Redondovirus Genomes Identified in Shotgun Metagenomic Data
To investigate redondovirus distribution in the biosphere, we surveyed metagenomic sequence datasets for homology to redondoviruses (Table S3 presents the datasets queried). Studies were favored for analysis if they 1) biochemically enriched for viral nucleic acids, 2) used MDA, which enriches for small circular viral genomes (Kim and Bae, 2011, Kim et al., 2008), 3) reported detection of Circovirus-like sequences, and/or 4) included a diverse range of sample types. In total, we queried 7,581 samples from 173 datasets covering 51 organisms or environments. Within human metagenomes, 18 body sites or fluids were examined. A positive hit was defined as 25% coverage of any redondovirus genome.
Redondoviruses were detected in metagenomic sequences from human oral cavity (3.8% of samples), lung (3.3%), nasopharynx (0.95%), and gut (0.59%). The most frequent sites of detection were the mouth and respiratory tract (Figure 4A). Redondovirus sequences were rare in human gut (17 detections total). Redondoviruses were not found in other animals, freshwater, marine, or soil samples (1,087 non-human biological samples), nor in laboratory reagents (144 contamination control samples). Importantly, 24 of the contamination controls analyzed were prepared side-by-side with redondovirus-positive samples and consist of saline bronchoscope pre-washes performed immediately prior to BAL sampling, which reflects the entire pipeline of specimen acquisition and nucleic acid processing. We thus conclude that redondoviruses are authentically present in the human oro-respiratory tract. Whether infrequent detection in gut samples reflects an authentic site of replication or transient passage after swallowing is uncertain. We cannot rule out that redondoviruses colonize other animal species, although thus far we have only identified hits in human samples.
Figure 4: Frequency of Redondovirus Detection and Co-occurrence with Human DNA Viruses.

A-Reads from 173 metagenomic datasets encompassing different human and non-human sample types were aligned to redondovirus genomes. A positive hit was determined based on 25% coverage of any redondovirus genome by short-read alignment. The percentage of samples that were positive is plotted on the y-axis and human body sites and other sample types are shown on the x-axis. The total number of samples analyzed in each category is annotated above and the total number of positive samples is indicated within the bar. See also Table S3.
B-The clinical status breakdown, if available, of redondovirus-positive samples is shown.
C-Reads from a subset of 20 datasets across 9 body sites (n = 2,675) were analyzed for homology to 20 redondovirus genomes and 133 animal-cell DNA viruses from six viral families. The height of each column represents the total number of samples that had detections of multiple viral families (rows). The viral families included in the co-detections are depicted as filled dots connected with lines below. The length of the bars on the left represents the total number of samples in which that viral family was detected. Cases where redondoviruses were detected are indicated in blue. See also Figure S2 and S3.
IBD, inflammatory bowel disease
Redondovirus Co-occurrence with Human DNA Viruses
Adeno-associated virus, a ssDNA virus of the Parvoviridae family, also encodes capsid and Rep proteins, and is known to require coinfection with a helper virus such as adenovirus to replicate. We thus asked whether redondoviruses co-occurred with any other eukaryotic viral family suggestive of a helper virus. We analyzed a subset (20) of the 173 datasets we previously screened for redondoviruses for the presence of common human DNA virus families (Adenoviridae, Anelloviridae, Herpesviridae, Papillomaviridae, Parvoviridae and Polyomaviridae). Redondoviridae was the second-most frequent human DNA virus family detected, exceeded only by Anelloviridae, which are known to be ubiquitous in humans (Spandole et al., 2015)—this high frequency is likely affected by use of MDA for virome preps, which enriches for small circular DNAs. Figure 4C shows the representation of additional human DNA viruses that co-occurred with redondoviruses in metagenomic datasets. Only anelloviruses were found to co-occur significantly with redondoviruses (Figure 4C, p=5.7×10−7, Fisher’s Exact Test with Bonferroni correction). Anelloviruses are small ssDNA viruses that seem unlikely to contribute helper functions to redondovirus replication. We speculate that the inflammatory milieu known to favor anellovirus replication (Maggi et al., 2001, Mariscal et al., 2002) may be similarly favorable for redondoviruses. Alternatively, given the ubiquitous nature of anelloviruses in humans, this association may reflect the fact that MDA enriches for both anelloviruses and redondoviruses, resulting in their co-detection. Rarely, other human viruses were found in redondovirus positive samples; these included Human mastadenovirus C and Epstein-Barr virus.
Redondoviruses in the Respiratory Tract are Elevated in Abundance in Critical Illness
Several sample sets were further queried using metagenomic analysis and qPCR to assess redondovirus abundance in the respiratory tract. We investigated 916 selected oro-respiratory samples using metagenomic analysis of datasets described above, reflecting a mixture of health and disease states, and found that redondoviruses were still the second-most frequent DNA virus detected, after anelloviruses (Figure S3).
To investigate the presence of redondovirus in healthy subjects further, we tested DNA isolated from oropharyngeal swabs from 60 adults using qPCR (Charlson et al., 2010). DNA was subjected to selective whole genome amplification (SWGA) (Clarke et al., 2017b) to enrich for redondovirus sequences over the human genome background, followed by redondovirus qPCR. Nine of 60 healthy subjects were positive (15%), although quantities even following SWGA amplification showed generally modest levels (Figure 5A).
Figure 5: Redondoviruses in the Oro-Respiratory Tract in Humans with Critical Illness and Periodontitis.

A. Quantification of redondovirus genome sequences in post-SWGA DNA from oropharyngeal swabs (oropharynx) from 60 healthy volunteers, and oropharyngeal swabs and endotracheal aspirates (lung secretions) from 69 critically ill subjects. The average cycle of threshold (Ct) value of technical replicates is plotted on the y-axis. Samples with undetermined (i.e: no amplification) value in all 3 replicates are assigned an arbitrary value above the Ct value of the limit of resolution of the assay (37) which corresponds to 11 target copies per reaction. Samples below this value are counted as authentic detections. Negative controls included extraction blanks, reagent blanks and no template controls. Positive controls represent replicates of 104 copies of Human lung-associated brisavirus RC spiked into DNA extracted from a redondovirus-negative lung sample, subjected to SWGA, and assayed by qPCR.
B qPCR was used for redondovirus detection in respiratory and/or stool samples from 69 subjects in the medical intensive care unit (ICU). Six total subjects, 3 males and 3 females, were positive for redondoviruses. The time point and type of sample surveyed for these six subjects is shown on the x- and y-axis, respectively. Positive samples are indicated by a filled-circle and negative samples by an open circle.
C-Number of reads mapping to a redondovirus in periodontitis samples from (Shi et al., 2015). Each point represents the average of all samples from a particular individual either before treatment (red) or after disease resolution (blue). Points from the same subject are connected by grey lines. The horizontal black line indicates the median. The Wilcoxon signed-rank test was used to test for paired differences between groups. D-Each point represents the number of reads mapping to a redondovirus in samples from (Califf et al., 2017) from subjects with periodontitis whose disease either did (blue) or did not (red) improve during the study. The horizontal black line indicates the median. The Wilcoxon rank-sum test was used to test for differences between groups. See also Table S4.
We then tested samples from 69 critically ill individuals (44 male, 25 female) using SWGA and qPCR (Figure 5A and B). Six (9%) had oropharyngeal samples positive for redondovirus (3 male, 3 female), indicating relevance to both genders (male 7% vs female 12%; p=0.66, Fisher’s Exact test). Post-SWGA quantities were, on average, 104 fold greater than in healthy subjects, although the use of SWGA complicates quantitative comparisons between groups. Four of these six critically ill subjects also had lung secretions (endotracheal aspirates) available for testing; three were positive for redondovirus. In subjects with serial samples, redondovirus was generally detectable over a period of 2–3 weeks, suggesting persistent colonization or infection. We conclude that redondoviruses are found in both healthy and critically ill individuals, but levels are elevated in illness. Furthermore, the upper and lower respiratory tracts appear to represent common niches with stable redondovirus detection over time.
Redondovirus Sequence Reads are Associated with Periodontitis
The set of 97 metagenomic studies assessed for redondovirus sequences (Table S3) contained samples from several disease states, allowing us to assess possible associations of redondoviruses with human disorders. In addition to our initial detections in BAL from organ donors and lung transplant recipients (Abbas et al., 2017, Abbas et al., 2018), redondoviruses were found in 1) BAL from subjects with sarcoidosis and healthy adults (Clarke et al., 2017a), 3) gingival samples from subjects with periodontitis (Wang et al., 2013, Shi et al., 2015, Califf et al., 2017), 4) oropharyngeal and nasopharyngeal samples from febrile subjects (Mokili et al., 2013, Wang et al., 2016), 5) oral samples from subjects with rheumatoid arthritis (Zhang et al., 2015), 6) stool samples from healthy individuals, 7) stool samples from subjects with inflammatory bowel disease (Norman et al., 2015) and 8) stool samples from subjects with HIV-associated immunodeficiency (Monaco et al., 2016) (Figure 4B).
A considerable proportion of redondovirus-positive samples were from studies of periodontal disease (Figure 4B), so we analyzed these further. Three studies queried gingival or oral samples from subjects suffering or recovered from periodontitis (detailed metadata in Table S4). One study queried samples before and after corrective treatment by scaling and root planing together with improved oral hygiene (Shi et al., 2015). Redondovirus representation was high prior to treatment, and then fell substantially after treatment, as measured by the number of reads aligning to the most broadly covered redondovirus genome in each sample (Figure 5C). We averaged redondovirus reads across all individual tooth sites sampled for each subject and found lower redondovirus prevalence after recovery (Figure 5C, p=0.014, Wilcoxon signed-rank test). The second study compared disease severity in two groups with chronic periodontitis; one group received treatment with 0.25% sodium hypochlorite rinse, while the other received a water rinse (Califf et al., 2017). We compared redondovirus representation in sub and supra-gingival sites from subjects whose periodontitis did or did not improve, and found that subjects that did not show improvement had greater numbers of reads mapping to redondovirus genomes (Figure 5D, p=0.028, Wilcoxon rank-sum test). A third study analyzed two patients with severe periodontal disease, before and after treatment; both subjects were positive for redondovirus prior to treatment but no detections were found in samples taken after successful treatment (Kumar et al., 2018). Thus we conclude that redondoviruses are associated with periodontitis in multiple studies, and that levels are reduced with effective treatment.
Discussion
Here we introduce Redondoviridae, a family of small, circular DNA viruses discovered in metagenomic sequence data that is found selectively in human lung and oro-pharyngeal samples. We first identified redondovirus genomes by aligning metagenomic sequences from lungs of two organ donors to a viral genome database, resulting in weak hits to PoSCV-5. Assembly of shotgun metagenomic reads yielded complete circular genomes, which were then used to interrogate our collection of lung virome samples, allowing us to identify seven genomes. We then used these genomes to interrogate 7,581 metagenomic samples from diverse environmental sites, hosts, body sites, and disease states, detecting redondoviruses in 67 human samples and building 12 additional genomes. Independently, another group reported a single genome (Cui et al., 2017) in a sample from the throat of a febrile patient that we find is most closely related to Human oral-associated brisavirus YH (accession MK059758). Of the DNA viruses we surveyed in 20 human virome datasets, redondoviruses were the second most abundant, exceeded only by anelloviruses. The prevalence of redondoviruses was similar in cohorts of healthy subjects and critically ill subjects, although higher genome quantities suggested higher absolute levels in the ill subjects. Analysis of metagenomic samples revealed an association of redondoviruses with periodontal disease.
It is possible that redondovirus infection and replication may help maintain the inflammatory state associated with periodontitis and contribute to disease progression. A role in disease initiation seems less likely given the established roles of bacteria and oral hygiene (Edlund et al., 2015, Costalonga and Herzberg, 2014). Previous studies have tentatively implicated viruses in periodontitis based on alterations of subgingival bacteriophage communities (Ly et al., 2014) and increased representation of some eukaryotic viruses including HIV, HCMV and HSV-1 (Cappuyns et al., 2005, Li et al., 2017). The role of redondoviruses in periodontitis warrants further study. Similarly, the role redondoviruses play in diseases of the respiratory tract can now be investigated.
Do redondoviruses require helper viruses to replicate? The Dependoparvoviruses, which include AAV, are small, linear ssDNA viruses that require co-infection with larger DNA viruses to condition cells for efficient replication. Samples containing redondoviruses were scanned for other DNA viruses, but no large double stranded DNA viruses were consistently identified. Anelloviruses, small, circular ssDNA viruses, did co-occur. While we do not rule out that anelloviruses support redondovirus replication, it seems more likely that the inflammatory states known to promote anellovirus replication may do the same for redondoviruses, or alternatively, that the methods for virome sampling preferentially recover both redondoviruses and anelloviruses.
The high level of sequence variation in redondovirus Rep proteins is intriguing. Viruses encoding Reps are ubiquitous in both prokaryotes and eukaryotes. There are even transposon families that mobilize via ssDNA intermediates using Rep-like enzymes (Grabundzija et al., 2016). Cells have likely been opposing parasitism by Rep-encoding elements since the origins of cellular life. We conjecture that Rep amino acid variation reflects an ongoing Red Queen’s Race between host intrinsic immunity and Rep enzymes. If so, there should be active host cell mechanisms targeting and inhibiting Rep proteins. The redondovirus Rep enzymes reported here provide an entry point to investigating this possibility.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Frederic Bushman (bushman@pennmedicine.upenn.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human Studies:
Samples analyzed here obtained in the context of studies previously reported were obtained with informed consent and under protocols approved by the Institutional Review Board at their respective institutes as detailed in (Abbas et al., 2017, Clarke et al., 2017a, Abbas et al., 2018, Charlson et al., 2011, Charlson et al., 2010). Human subjects >18 years of age with varying critical illnesses were enrolled within 24 hours of admission at the Hospital of the University of Pennsylvania medical Intensive Care Unit (ICU). Individuals with an anticipated ICU length of stay <48 hours were excluded. Informed consent was obtained under IRB protocol 823392. Subjects were not involved in any other experimental procedures. Oropharyngeal swabs (n = 198), endotracheal aspirates (n = 87), and stool (n = 16) from 69 (44 males and 25 females) subjects were available to be queried by qPCR. Redondovirus positivity was similar in males and females, suggesting no obvious influence of gender. DNA from oropharyngeal swabs was extracted in single-tube DNeasy PowerSoil Kit (Qiagen; Hilden, Germany) and followed manufacturer’s protocol except for two 50 L elutions with buffer C6. Endotracheal aspirate was extracted with a 96-well format of the same kit. Due to sample availability limitations, a single biological replicate was used for each sample. No estimation for optimal sample size to detect statistical significance was performed for this initial survey. Additional metagenomic sequence data (n = 7,581 human, animal, and environmental samples) were derived from publically available data repositories or unpublished studies performed in our lab (n = 173 individual studies or datasets) (Table S3).
METHOD DETAILS
Discovery and Detection in Clinical Samples
Acellular bronchoalveolar lavage (BAL) samples were obtained from prior studies of organ donors and lung transplant recipients (Abbas et al., 2017, Abbas et al., 2018), subjects with sarcoidosis (Clarke et al., 2017a) and healthy adults (Charlson et al., 2011). Virus-like particle purification, preparation of shotgun DNA libraries, metagenomic sequencing, within-sample contig assembly and annotation based on alignment to the NCBI viral database has been previously described (Abbas et al., 2017, Clarke et al., 2017a, Abbas et al., 2018). Contigs found to have homology to redondovirus genomes were amplified and cloned from 7 samples.
Primers (Table S1) were designed to amplify redondovirus genome sequences from DNA extracted from BAL that underwent whole genome amplification with Illustra GenomiPhi V2 DNA (GE Healthcare; Little Chalfont, UK). PCR was performed with AccuPrime™ Taq DNA Polymerase System (ThermoFisher; Waltham, MA, USA) using 1 μL of whole-genome-amplified product, 20 μM of forward and reverse primers and 0.2 μL Taq polymerase in a total volume of 50 μL. Products were visualized on 1–1.5% ethidium bromide agarose gels (Figure S1). Amplicons were cloned and validated by using the Sanger sequence method on an ABI 3730XL (Applied BioSystems; Waltham, MA, USA) instrument. Full redondovirus genomes were either de-novo synthesized (BioBasic; Markham, ON, CA) or cloned by Gibson assembly (NEB; Ipswitch, MA, USA) and also verified by Sanger sequencing.
To detect redondovirus sequences in BAL samples, a TaqMan-based qPCR assay (Table S1) was designed targeting the genomic region encoding the capsid gene. For each sample, triplicate 20 μL reactions containing 4 μL of template DNA (depending on sample availability), 0.33 μL forward and reverse primers (18 μM), 0.33 μL probe (5 μM), 10 μL TaqMan Fast Universal PCR Master Mix (Applied Biosystems; Waltham, MA, USA) and 5 μL water were analyzed on a QuantStudio 5 Real Time PCR System (Applied Biosystems; Waltham, MA, USA) with the following cycling profile: 20 sec at 95°C for 1 cycle, and 40 cycles of 95°C for 3 sec and 60°C for 30 sec (signal collection). A linearized plasmid containing the complete Human lung-associated brisavirus RC genome in a pUC57 vector was used as a 7 point standard curve ranging from 75 to 30,000,000 copies/reaction. Amplification signal was required in 2 out of 3 wells to be scored as positive.
Endotracheal aspirates (lung secretions), oropharyngeal swabs and stool were collected from critically ill subjects and oropharyngeal swabs were collected from healthy volunteers, following written informed consent, under protocols approved by the University of Pennsylvania Institutional Review Board (protocols 823392 and 810987, respectively).
Extracted DNA was first subjected to SWGA using primers designed with the software described in Clarke et al., 2017b. Each reaction contained 2 μL Phi29 10x Buffer (NEB; Ipswich, MA), 1 μL Phi29 polymerase, 0.2 μL bovine serum albumin (10 mg/mL), 100 μM total of 20 primers (final concentration of each primer was 2 M), 2 μL of 10mM dNTPs, and 1 μL of template DNA in a total volume of 20 μL. Reactions underwent a step-down amplification process by incubating at 35°C for 5 min, 34°C for 10 min, 33°C for 15 min, 32°C for 20 min, 31°C for 30 min and th en 30°C for 16 hours, followed by a heat inactivation step (65°C for 15 min) as previou sly described (Clarke et al., 2017b). After SWGA, 4 μL of product was queried in duplicate using a more sensitive TaqMan-based qPCR assay (Table S1) that was designed targeting a conserved region of the Cp gene. A linearized plasmid containing the complete genome of human lung-associated brisavirus RC was used a 9-point standard curve ranging from 10 to 106 copies per reaction. Negative and positive controls were included in each run to evaluate inter-assay variability. The positive control was 104 copies of the standard curve plasmid containing the viral genome spiked into DNA extracted from a redondovirus-negative endotracheal aspirate sample and also subjected to SWGA.
Querying Viral Metagenomic Datasets
Reads from viral and other shotgun metagenomic projects available in the Sequence Read Archive (SRA) (n = 146) or MG-RAST (n = 23) or the Human Oral Microbiome Database (n = 1) and from 3 unpublished datasets from the University of Pennsylvania (Table S4) were processed in the following steps: 1) adaptor-trimmed single or paired-end reads were downloaded using fastq-dump (Leinonen et al., 2011), 2) a sensitive local alignment of either single reads or read pairs to redondovirus genomes was performed using Bowtie2 (Langmead and Salzberg, 2012); 3) alignments were processed and genome coverage was calculated with SAMtools (Li et al., 2009) and BEDtools (Quinlan and Hall, 2010) and 4) alignments were visualized with a custom R (version 3.2.3) script (Ihaka and Gentleman, 1996) (R packages used: magrittr, ggplot2, reshape2). Based on the sigmoidal relationship between aligning reads and genome coverage (Figure S2), a minimum threshold of 25% genome coverage was used to determine detection of a redondovirus.
Genome Assembly
Samples in which 25% of any redondovirus genome was covered were further analyzed using the Sunbeam pipeline (Clarke et al., 2018) to process reads and to build and annotate contigs using MEGAhit (Li et al., 2015) and BLASTn (Altschul et al., 1990). Contigs were further refined by overlap consensus assembly using Cap3 (Huang and Madan, 1999) and manually checked for circularity and presence of key genomic features with CloneManager 9 (Scientific & Educational Software; Denver, CO).
DNA and Amino Acid Sequence Analysis
The EMBOSS einverted utility (Rice et al., 2000), Mfold (Zuker, 2003) or CloneManager Professional 9 (Scientific & Educational Software; Denver, CO) was used to predict and visualize energetically favorable DNA structural features potentially important for replication. Forna was used to plot stem loop structures (Kerpedjiev et al., 2015). Nucleotide and protein alignments were performed using MUSCLE (version 3.8.31) (Edgar, 2004). Phylogenetic trees were built using PhyML (Guindon et al., 2010) using the LG amino acid substitution model (Le and Gascuel, 2008) with sequences from 2–3 representative species of established viral families and all full-length protein sequences of novel redondoviruses. Branch support was quantified by the approximate likelihood-ratio test (Anisimova and Gascuel, 2006) and visualized using FigTree v1.4.3 (http://tree.bio..ac.uk/software/figtree/). Consensus motif logos were generated using WebLogo (Crooks et al., 2004). Conserved domains within the Rep protein were detected using NCBI’s CD-search against the Pfam database (v30.0, E-value < 10−2). Protein folding predictions were done using PHYRE2 (Kelley et al., 2015) using default parameters.
To predict prokaryotic ribosomal binding sites (RBS), we implemented the algorithm described in Krishnamurthy and Wang, 2018 in Python (version 3.6). Briefly, we extracted 18 nucleotides in the untranslated region immediately upstream of start codons and searched for prokaryotic ribosomal binding sites (full: AGGAGG; partial: AGGAG, GGAGG, AGGA, GGAG, GAGG).
We performed an exploratory analysis of synonymous and non-synonymous substitution rates, as dN/dS as a marker of selective pressure is untested in CRESS viruses and may be confounded by overlap of unidentified coding sequence and/or functionally important DNA secondary structure elements (Zanini and Neher, 2013, Muhire et al., 2014). First, we aligned protein sequences using MUSCLE (Edgar, 2004), and built phylogenetic trees with PhyML (Guindon et al., 2010). We then generated codon alignments using PAL2NAL (Suyama et al., 2006) and used HyPhy (Pond et al., 2005) to perform dN/dS analysis. We used FUBAR (Murrell et al., 2013) to predict sites under positive selection. As dN/dS analysis in overlapping genes is overwhelmed by pressure to maintain the amino acid sequence of two genes, we only analyzed the portion of the Cp coding sequence that did not overlap with the ORF3 protein— overlapping coding regions were identified and excluded using pyviko (Taylor and Strebel, 2017).
QUANTIFICATION AND STATISTICAL ANALYSIS
Co-occurrence of Redondoviridae and Animal-cell Viruses
Twenty datasets in which redondovirus genomes were found or were comprehensive studies of the human DNA virome were chosen for a targeted analysis of human viruses. Specifically, reads from these datasets (n = 2,675 samples) were aligned to 133 vertebrate viruses from the Adenoviridae, Anelloviridae, Herpesviridae, Papillomaviridae, Parvoviridae and Polyomaviridae families (downloaded from NCBI RefSeq on 20 August 2018). Alignments were done using the hisss pipeline as described above and analyzed in R (R packages used: tidyverse, reshape2, Biostrings, taxonomizr, UpSetR). Samples were considered positive for small (<10 kb) DNA viruses if greater than 25% of the target genome was covered. Samples were considered positive for large DNA viruses (>10 kb genomes), if greater than 10% of the target genome was covered (see Figure S3). The distribution of the frequency of Redondoviridae and other viral family detection was tested using the Fisher’s exact test with Bonferroni correction for multiple testing.
Association with Human Clinical Disease States
In studies of periodontitis, the difference in number of redondovirus reads in disease versus non-disease states were tested using non-parametric Wilcoxon signed-rank or rank-sum tests, depending on whether samples were paired or not.
DATA AND SOFTWARE AVAILABILITY
The accession numbers for the viruses sequenced and reported in this paper are GenBank: MK059754-MK059772. Full details of each step of the Snakemake pipeline used in this report are available at https://github.com/louiejtaylor/hisss. The script used for RBS analysis is available at https://github.com/louiejtaylor/find-prok-rbs.
Supplementary Material
Table S1: List of Primers Used, Related to Figure 1, S1 and 5
Table S2: Summary of Taxa within Redondoviridae, Related to Figure 2 Based on definitions in (Varsani and Krupovic, 2018) and the analysis of the diversity of viral Rep proteins, Redondoviridae genomes can be grouped into two genera, demarcated by 50% Rep protein identity, which we propose be called Vientovirus and Brisavirus, from the Spanish words for “wind” and “breeze”, alluding to their discovery in the respiratory tract.
Table S3- List of Studies Queried, Related to Figure 1 and Figure 4 The number of samples in each study represents the total samples uploaded to the central database. However, only samples that were determined not to be 16S, ITS or other targeted amplicons were analyzed by the pipeline described here. SRA; Sequence Read Archive, MG-RAST; Metagenomic Rapid Annotations using Subsystems Technology, HOMD, Human Oral Microbiome Database
Table S4: Periodontitis Dataset Metadata, Related to Figure 5 Metadata from Shi et al. 2015, Califf et al. 2017, and Kumar et al. 2018 related to periodontitis disease and treatment status. The “Mapped Reads” and “Fraction Cov.” columns correspond to the number of reads mapping to and fraction coverage of the best (highest-coverage) redondovirus hit, respectively. The “Total Reads” and “Total Bases” columns refer to the total number of reads and bases in the metagenomic data. Periodontal Disease (PD) status is enumerated in the “Disease Status” column. In studies that tracked the change in periodontal status over time, the change in disease status is listed in the “Disease Change” column (samples from Shi et al. were listed as Not Applicable (NA) as recovery from periodontal disease was an inclusion criterion for the study, rather than a measured variable). The Treatment/Control column lists the participants’ study group for Califf et al. (T =Treatment, C = Control). Inflammation score is included from Shi et al. IL1-B levels (pg/ml), rating, and treatment time point are included from Kumar et al.
Highlights.
A family of human DNA viruses was identified and named Redondoviridae.
Redondoviruses were the second most common virus in human respiratory virome samples.
Some subjects with periodontitis and critical illness had higher redondovirus levels.
Acknowledgements
This work was supported by NIH grants R61-HL137063 (Lung Virome in Health and Disease) and R01-HL113252 (Lung Transplant Microbiome and Chronic Allograft Dysfunction). A.A.A was supported by NSF grant DGE-1321851 and L.J.T by T32-AI-007324. We thank J. Christie and members of the lung transplant program for access to transplant specimens and ICU staff for assistance with specimen collection. We are grateful to members of the Bushman and Collman laboratories for suggestions, Arvind Varsani for help with virus taxonomy, and for the laboratories of X.J. Meng and Matthew Weitzman for assistance and support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
ADDITIONAL RESOURCES
Supplemental Information
Supplemental Information includes 3 figures and 4 tables and can be found online.
References
- ABBAS AA, DIAMOND JM, CHEHOUD C, CHANG B, KOTZIN JJ, YOUNG JC, IMAI I, HAAS AR, CANTU E, LEDERER DJ, MEYER KC, MILEWSKI RK, OLTHOFF KM, SHAKED A, CHRISTIE JD, BUSHMAN FD & COLLMAN RG 2017. The Perioperative Lung Transplant Virome: Torque Teno Viruses Are Elevated in Donor Lungs and Show Divergent Dynamics in Primary Graft Dysfunction. Am J Transplant, 17, 1313–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ABBAS AA, YOUNG JC, CLARKE EL, DIAMOND JM, IMAI I, HAAS AR, CANTU E, LEDERER DJ, MEYER K, MILEWSKI RK, OLTHOFF KM, SHAKED A, CHRISTIE JD, BUSHMAN FD & COLLMAN RG 2018. Bidirectional transfer of anelloviridae lineages between graft and host during lung transplantation. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ABELES SR, LY M, SANTIAGO-RODRIGUEZ TM & PRIDE DT 2015. Effects of Long Term Antibiotic Therapy on Human Oral and Fecal Viromes. PloS one, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AGGARWALA V, LIANG G & BUSHMAN FD 2017. Viral communities of the human gut: metagenomic analysis of composition and dynamics. Mobile DNA, 8, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ALTSCHUL SF, GISH W, MILLER W, MYERS EW & LIPMAN DJ 1990. Basic local alignment search tool. Journal of molecular biology, 215, 403–410. [DOI] [PubMed] [Google Scholar]
- ANISIMOVA M & GASCUEL O 2006. Approximate likelihood-ratio test for branches: A fast, accurate, and powerful alternative. Systematic biology, 55, 539–552. [DOI] [PubMed] [Google Scholar]
- BREITBART M, DELWART E, ROSARIO K, SEGALÉS J, VARSANI A & ICTV REPORT C. 2017. ICTV Virus Taxonomy Profile: Circoviridae. The Journal of general virology, 98, 1997–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CALIFF KJ, SCHWARZBERG-LIPSON K, GARG N, GIBBONS SM, CAPORASO JG, SLOTS J, COHEN C, DORRESTEIN PC & KELLEY ST 2017. Multi-omics Analysis of Periodontal Pocket Microbial Communities Pre- and Posttreatment. mSystems, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CAPPUYNS I, GUGERLI P & MOMBELLI A 2005. Viruses in periodontal disease - a review. Oral diseases, 11, 219–229. [DOI] [PubMed] [Google Scholar]
- CHARLSON ES, BITTINGER K, HAAS AR, FITZGERALD AS, FRANK I, YADAV A, BUSHMAN FD & COLLMAN RG 2011. Topographical Continuity of Bacterial Populations in the Healthy Human Respiratory Tract. American Journal of Respiratory and Critical Care Medicine, 184, 957–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHARLSON ES, CHEN J, CUSTERS-ALLEN R, BITTINGER K, LI H, SINHA R, HWANG J, BUSHMAN FD & COLLMAN RG 2010. Disordered microbial communities in the upper respiratory tract of cigarette smokers. PloS one, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEUNG AK, NG TFF, LAGER KM, ALT DP, DELWART EL & POGRANICHNIY RM 2014. Identification of a novel single-stranded circular DNA virus in pig feces. Genome announcements, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CLARKE EL, LAUDER AP, HOFSTAEDTER CE, HWANG Y, FITZGERALD AS, IMAI I, BIERNAT W, RĘKAWIECKI B, MAJEWSKA H, DUBANIEWICZ A, LITZKY LA, FELDMAN MD, BITTINGER K, ROSSMAN MD, PATTERSON KC, BUSHMAN FD & COLLMAN RG 2017a. Microbial Lineages in Sarcoidosis: A Metagenomic Analysis Tailored for Low Microbial Content Samples. American journal of respiratory and critical care medicine. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CLARKE EL, SUNDARARAMAN SA, SEIFERT SN, BUSHMAN FD, HAHN BH & BRISSON D 2017b. swga: a primer design toolkit for selective whole genome amplification. Bioinformatics (Oxford, England), 33, 2071–2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CLARKE EL, TAYLOR LJ, ZHAO C, CONNELL J, BUSHMAN FD & BITTINGER K 2018. Sunbeam: an extensible pipeline for analyzing metagenomic sequencing experiments. bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COSTALONGA M & HERZBERG MC 2014. The oral microbiome and the immunobiology of periodontal disease and caries. Immunology letters, 162, 22–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CROOKS GE, HON G, CHANDONIA J-MM & BRENNER SE 2004. WebLogo: a sequence logo generator. Genome research, 14, 1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CUI L, WU B, ZHU X, GUO X, GE Y, ZHAO K, QI X, SHI Z, ZHU F, SUN L & ZHOU M 2017. Identification and genetic characterization of a novel circular single-stranded DNA virus in a human upper respiratory tract sample. Archives of virology, 162, 3305–3312. [DOI] [PubMed] [Google Scholar]
- EDGAR RC 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic acids research, 32, 1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EDLUND A, SANTIAGO-RODRIGUEZ TM, BOEHM TK & PRIDE DT 2015. Bacteriophage and their potential roles in the human oral cavity. Journal of oral microbiology, 7, 27423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ELLIS J 2014. Porcine circovirus: a historical perspective. Veterinary pathology, 51, 315–327. [DOI] [PubMed] [Google Scholar]
- FAHSBENDER E, BURNS JM, KIM S, KRABERGER S, FRANKFURTER G, EILERS AA, SHERO MR, BELTRAN R, KIRKHAM A, MCCORKELL R, BERNGARTT RK, MALE MF, BALLARD G, AINLEY DG, BREITBART M & VARSANI A 2017. Diverse and highly recombinant anelloviruses associated with Weddell seals in Antarctica. Virus evolution, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FAUQUET CM, MAYO MA, MANILOFF J, DESSELBERGER U & BALL L 2005. Virus taxonomy: VIIIth report of the International Committee on Taxonomy of Viruses, Academic Press. [Google Scholar]
- GORBALENYA AE, KOONIN EV & WOLF YI 1990. A new superfamily of putative NTP-binding domains encoded by genomes of small DNA and RNA viruses. FEBS letters, 262, 145–148. [DOI] [PubMed] [Google Scholar]
- GRABUNDZIJA I, MESSING SA, THOMAS J, COSBY RL, BILIC I, MISKEY C, GOGOL-DÖRING A, KAPITONOV V, DIEM T, DALDA A, JURKA J, PRITHAM EJ, DYDA F, IZSVÁK Z & IVICS Z 2016. A Helitron transposon reconstructed from bats reveals a novel mechanism of genome shuffling in eukaryotes. Nature communications, 7, 10716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GUINDON S, DUFAYARD J-FF, LEFORT V, ANISIMOVA M, HORDIJK W & GASCUEL O 2010. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Systematic biology, 59, 307–321. [DOI] [PubMed] [Google Scholar]
- HARRISON BD, RKER H, BOCK KR, GUTHRIE EJ, MEREDITH G & ATKINSON M 1977. Plant viruses with circular single-stranded DNA. Nature, 270. [Google Scholar]
- HUANG X & MADAN A 1999. CAP3: A DNA sequence assembly program. Genome research, 9, 868–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IHAKA R & GENTLEMAN R 1996. R: A Language for Data Analysis and Graphics. Journal of Computational and Graphical Statistics, 5, 299–314. [Google Scholar]
- ILYINA TV & KOONIN EV 1992. Conserved sequence motifs in the initiator proteins for rolling circle DNA replication encoded by diverse replicons from eubacteria, eucaryotes and archaebacteria. Nucleic acids research, 20, 3279–3285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KELLEY LA, MEZULIS S, YATES CM, WASS MN & STERNBERG MJ 2015. The Phyre2 web portal for protein modeling, prediction and analysis. Nature protocols, 10, 845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KERPEDJIEV P, HAMMER S & HOFACKER IL 2015. Forna (force-directed RNA): Simple and effective online RNA secondary structure diagrams. Bioinformatics (Oxford, England), 31, 3377–3379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KIM K-H & BAE J-W 2011. Amplification Methods Bias Metagenomic Libraries of Uncultured Single-Stranded and Double-Stranded DNA Viruses. Applied and Environmental Microbiology, 77, 7663–7668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KIM K-HH, CHANG H-WW, NAM Y-DD, ROH SW, KIM M-SS, SUNG Y, JEON CO, OH H-MM & BAE J-WW 2008. Amplification of uncultured single-stranded DNA viruses from rice paddy soil. Applied and environmental microbiology, 74, 5975–5985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KRISHNAMURTHY SR & WANG D 2017. Origins and challenges of viral dark matter. Virus research. [DOI] [PubMed] [Google Scholar]
- KRISHNAMURTHY SR & WANG D 2018. Extensive conservation of prokaryotic ribosomal binding sites in known and novel picobirnaviruses. Virology, 516, 108–114. [DOI] [PubMed] [Google Scholar]
- KRUPOVIC M, GHABRIAL SA, JIANG D & VARSANI A 2016. Genomoviridae: a new family of widespread single-stranded DNA viruses. Archives of virology, 161, 2633–2643. [DOI] [PubMed] [Google Scholar]
- Kumar PK, GOTTLIEB RA, LINDSAY S, DELANGE N, PENN TE, CALAC D & Kelley ST 2018. Metagenomic analysis uncovers strong relationship between periodontal pathogens and vascular dysfunction in American Indian population. bioRxiv, 250324. [Google Scholar]
- LABONTÉ JM & SUTTLE CA 2013. Previously unknown and highly divergent ssDNA viruses populate the oceans. The ISME journal, 7, 2169–2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LANGMEAD B & SALZBERG SL 2012. Fast gapped-read alignment with Bowtie 2. Nature methods, 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LE SQ & GASCUEL O 2008. An improved general amino acid replacement matrix. Molecular biology and evolution, 25, 1307–1320. [DOI] [PubMed] [Google Scholar]
- LEFEUVRE P, LETT JMM, VARSANI A & MARTIN DP 2009. Widely conserved recombination patterns among single-stranded DNA viruses. Journal of virology, 83, 2697–2707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEINONEN R, SUGAWARA H, SHUMWAY M & COLLABORATION I 2011. The sequence read archive. Nucleic acids research, 39, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEPPIK L, GUNST K, LEHTINEN M, DILLNER J, STREKER K & DE VILLIERS E-MM 2007. In vivo and in vitro intragenomic rearrangement of TT viruses. Journal of virology, 81, 9346–9356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LI D, LIU C-MM, LUO R, SADAKANE K & LAM T-WW 2015. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics (Oxford, England), 31, 1674–1676. [DOI] [PubMed] [Google Scholar]
- LI F, ZHU C, DENG F-YY, WONG MCMCM, LU H-XX & FENG X-PP 2017. Herpesviruses in etiopathogenesis of aggressive periodontitis: A meta-analysis based on case-control studies. PloS one, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LI H, HANDSAKER B, WYSOKER A, FENNELL T, RUAN J, HOMER N, MARTH G, ABECASIS G, DURBIN R & SUBGROUP 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics (Oxford, England), 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LI L, KAPOOR A, SLIKAS B, BAMIDELE OS, WANG C, SHAUKAT S, MASROOR MA, WILSON ML, NDJANGO J-BNB, PEETERS M, GROSS-CAMP ND, MULLER MN, HAHN BH, WOLFE ND, TRIKI H, BARTKUS J, ZAIDI SZ & DELWART E 2010. Multiple diverse circoviruses infect farm animals and are commonly found in human and chimpanzee feces. Journal of virology, 84, 1674–1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LY M, ABELES SR, BOEHM TK, ROBLES-SIKISAKA R, NAIDU M, SANTIAGO-RODRIGUEZ T & PRIDE DT 2014. Altered oral viral ecology in association with periodontal disease. mBio, 5, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MA C-MM, HON C-CC, LAM T-YY, LI VY, WONG CK, DE OLIVEIRA T & LEUNG FC 2007. Evidence for recombination in natural populations of porcine circovirus type 2 in Hong Kong and mainland China. The Journal of general virology, 88, 1733–1737. [DOI] [PubMed] [Google Scholar]
- MAGGI F, FORNAI C, ZACCARO L, MORRICA A, VATTERONI ML, ISOLA P, MARCHI S, RICCHIUTI A, PISTELLO M & BENDINELLI M 2001. TT virus (TTV) loads associated with different peripheral blood cell types and evidence for TTV replication in activated mononuclear cells. Journal of medical virology, 64, 190–194. [DOI] [PubMed] [Google Scholar]
- MANKERTZ A, PERSSON F, MANKERTZ J, BLAESS G & BUHK HJ 1997. Mapping and characterization of the origin of DNA replication of porcine circovirus. Journal of virology, 71, 2562–2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MARISCAL LF, LÓPEZ-ALCOROCHO JM, RODRÍGUEZ-IÑIGO E, ORTIZ-MOVILLA N, DE LUCAS S, BARTOLOMÉ J & CARREÑO V 2002. TT virus replicates in stimulated but not in nonstimulated peripheral blood mononuclear cells. Virology, 301, 121–129. [DOI] [PubMed] [Google Scholar]
- MINOT S, BRYSON A, CHEHOUD C, WU GD, LEWIS JD & BUSHMAN FD 2013. Rapid evolution of the human gut virome. Proceedings of the National Academy of Sciences of the United States of America, 110, 12450–12455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MINOT S, SINHA R, CHEN J, LI H, KEILBAUGH SA, WU GD, LEWIS JD & BUSHMAN FD 2011. The human gut virome: inter-individual variation and dynamic response to diet. Genome research, 21, 1616–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MOKILI JL, DUTILH BE, LIM YW, SCHNEIDER BS, TAYLOR T, HAYNES MR, METZGAR D, MYERS CA, BLAIR PJ, NOSRAT B, WOLFE ND & ROHWER F 2013. Identification of a novel human papillomavirus by metagenomic analysis of samples from patients with febrile respiratory illness. PloS one, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MONACO CL, GOOTENBERG DB, ZHAO G, HANDLEY SA, GHEBREMICHAEL MS, LIM ES, LANKOWSKI A, BALDRIDGE MT, WILEN CB, FLAGG M, NORMAN JM, KELLER BC, LUÉVANO JMM, WANG D, BOUM Y, MARTIN JN, HUNT PW, BANGSBERG DR, SIEDNER MJ, KWON DS & VIRGIN HW 2016. Altered Virome and Bacterial Microbiome in Human Immunodeficiency Virus-Associated Acquired Immunodeficiency Syndrome. Cell host & microbe, 19, 311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MUHIRE BM, GOLDEN M, MURRELL B, LEFEUVRE P, LETT J-MM, GRAY A, POON AY, NGANDU NK, SEMEGNI Y, TANOV EP, MONJANE ALL, HARKINS GW, VARSANI A, SHEPHERD DN & MARTIN DP 2014. Evidence of pervasive biologically functional secondary structures within the genomes of eukaryotic single-stranded DNA viruses. Journal of virology, 88, 1972–1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MURRELL B, MOOLA S, MABONA A, WEIGHILL T, SHEWARD D, KOSAKOVSKY POND SL & SCHEFFLER K 2013. FUBAR: a fast, unconstrained bayesian approximation for inferring selection. Molecular biology and evolution, 30, 1196–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NACCACHE SN, GRENINGER AL, LEE D, COFFEY LL, PHAN T, REIN-WESTON A, ARONSOHN A, HACKETT J, DELWART EL & CHIU CY 2013. The perils of pathogen discovery: origin of a novel parvovirus-like hybrid genome traced to nucleic acid extraction spin columns. Journal of virology, 87, 11966–11977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NORMAN JM, HANDLEY SA, BALDRIDGE MT, DROIT L, LIU CY, KELLER BC, KAMBAL A, MONACO CL, ZHAO G, FLESHNER P, STAPPENBECK TS, MCGOVERN DP, KESHAVARZIAN A, MUTLU EA, SAUK J, GEVERS D, XAVIER RJ, WANG D, PARKES M & VIRGIN HW 2015. Disease-specific alterations in the enteric virome in inflammatory bowel disease. Cell, 160, 447–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PAEZ-ESPINO D, ELOE-FADROSH EA, PAVLOPOULOS GA, THOMAS AD, HUNTEMANN M, MIKHAILOVA N, RUBIN E, IVANOVA NN & KYRPIDES NC 2016. Uncovering Earth’s virome. Nature, 536, 425–430. [DOI] [PubMed] [Google Scholar]
- PÉREZ-BROCAL V & MOYA A 2018. The analysis of the oral DNA virome reveals which viruses are widespread and rare among healthy young adults in Valencia (Spain). PloS one, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PHAN TG, LUCHSINGER V, AVENDAÑO LF, DENG X & DELWART E 2014. Cyclovirus in nasopharyngeal aspirates of Chilean children with respiratory infections. The Journal of general virology, 95, 922–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- POND SL, FROST SD & MUSE SV 2005. HyPhy: hypothesis testing using phylogenies. Bioinformatics (Oxford, England), 21, 676–679. [DOI] [PubMed] [Google Scholar]
- QUINLAN AR & HALL IM 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics (Oxford, England), 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RICE P, LONGDEN I & BLEASBY A 2000. EMBOSS: the European Molecular Biology Open Software Suite. Trends in genetics : TIG, 16, 276–277. [DOI] [PubMed] [Google Scholar]
- ROSARIO K & BREITBART M 2011. Exploring the viral world through metagenomics. Current opinion in virology, 1, 289–297. [DOI] [PubMed] [Google Scholar]
- ROSARIO K, DUFFY S & BREITBART M 2012. A field guide to eukaryotic circular single-stranded DNA viruses: insights gained from metagenomics. Archives of virology, 157, 1851–1871. [DOI] [PubMed] [Google Scholar]
- SALTER SJ, COX MJ, TUREK EM, CALUS ST, COOKSON WO, MOFFATT MF, TURNER P, PARKHILL J, LOMAN NJ & WALKER AW 2014. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biology, 12, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHI B, CHANG M, MARTIN J, MITREVA M, LUX R, KLOKKEVOLD P, SODERGREN E, WEINSTOCK GM, HAAKE SK & LI H 2015. Dynamic changes in the subgingival microbiome and their potential for diagnosis and prognosis of periodontitis. mBio, 6, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SIMMONDS P, ADAMS MJ, BENKŐ M, BREITBART M, BRISTER JR, CARSTENS EB, DAVISON AJ, DELWART E, GORBALENYA AE, HARRACH B, HULL R, KING AM, KOONIN EV, KRUPOVIC M, KUHN JH, LEFKOWITZ EJ, NIBERT ML, ORTON R, ROOSSINCK MJ, SABANADZOVIC S, SULLIVAN MB, SUTTLE CA, TESH RB, VAN DER VLUGT RAA, VARSANI A & ZERBINI FM 2017. Consensus statement: Virus taxonomy in the age of metagenomics. Nature reviews. Microbiology, 15, 161–168. [DOI] [PubMed] [Google Scholar]
- SMITS SL, SCHAPENDONK CM, VAN BEEK J, VENNEMA H, SCHÜRCH AC, SCHIPPER D, BODEWES R, HAAGMANS BL, OSTERHAUS AD & KOOPMANS MP 2014. New viruses in idiopathic human diarrhea cases, the Netherlands. Emerging infectious diseases, 20, 1218–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SPANDOLE S, CIMPONERIU D, BERCA LM & MIHĂESCU G 2015. Human anelloviruses: an update of molecular, epidemiological and clinical aspects. Archives of virology, 160, 893–908. [DOI] [PubMed] [Google Scholar]
- SUYAMA M, TORRENTS D & BORK P 2006. PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic acids research, 34, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TAYLOR LJ & STREBEL K 2017. Pyviko: an automated Python tool to design gene knockouts in complex viruses with overlapping genes. BMC microbiology, 17, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TODD D 2000. Circoviruses: immunosuppressive threats to avian species: a review. Avian pathology : journal of the W.V.P.A, 29, 373–394. [DOI] [PubMed] [Google Scholar]
- VARSANI A & KRUPOVIC M 2017. Sequence-based taxonomic framework for the classification of uncultured single-stranded DNA viruses of the family Genomoviridae. Virus evolution, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VARSANI A & KRUPOVIC M 2018. Smacoviridae: a new family of animal-associated single-stranded DNA viruses. Archives of virology, 163, 2005–2015. [DOI] [PubMed] [Google Scholar]
- WANG J, QI J, ZHAO H, HE S, ZHANG Y, WEI S & ZHAO F 2013. Metagenomic sequencing reveals microbiota and its functional potential associated with periodontal disease. Scientific reports, 3, 1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WANG Y, ZHU N, LI Y, LU R, WANG H, LIU G, ZOU X, XIE Z & TAN W 2016. Metagenomic analysis of viral genetic diversity in respiratory samples from children with severe acute respiratory infection in China. Clinical microbiology and infection : the official publication of the European Society of Clinical Microbiology and Infectious Diseases, 22, 4580–4589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WILLNER D, FURLAN M, HAYNES M, SCHMIEDER R, ANGLY FE, SILVA J, TAMMADONI S, NOSRAT B, CONRAD D & ROHWER F 2009. Metagenomic analysis of respiratory tract DNA viral communities in cystic fibrosis and non-cystic fibrosis individuals. PloS one, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WYLIE KM, MIHINDUKULASURIYA KA, SODERGREN E, WEINSTOCK GM & STORCH GA 2012. Sequence analysis of the human virome in febrile and afebrile children. PloS one, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YOUNG JC, CHEHOUD C, BITTINGER K, BAILEY A, DIAMOND JM, CANTU E, HAAS AR, ABBAS A, FRYE L, CHRISTIE JD, BUSHMAN FD & COLLMAN RG 2015. Viral metagenomics reveal blooms of anelloviruses in the respiratory tract of lung transplant recipients. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons, 15, 200–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZANINI F & NEHER RA 2013. Quantifying selection against synonymous mutations in HIV-1 env evolution. Journal of virology, 87, 11843–11850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHANG X, ZHANG D, JIA H, FENG Q, WANG D, LIANG D, WU X, LI J, TANG L, LI Y, LAN Z, CHEN B, LI Y, ZHONG H, XIE H, JIE Z, CHEN W, TANG S, XU X, WANG X, CAI X, LIU S, XIA Y, LI J, QIAO X, AL-AAMA JY, CHEN H, WANG L, WU Q-JJ, ZHANG F, ZHENG W, LI Y, ZHANG M, LUO G, XUE W, XIAO L, LI J, CHEN W, XU X, YIN Y, YANG H, WANG J, KRISTIANSEN K, LIU L, LI T, HUANG Q, LI Y & WANG J 2015. The oral and gut microbiomes are perturbed in rheumatoid arthritis and partly normalized after treatment. Nature medicine, 21, 895–905. [DOI] [PubMed] [Google Scholar]
- ZUKER M 2003. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic acids research, 31, 3406–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: List of Primers Used, Related to Figure 1, S1 and 5
Table S2: Summary of Taxa within Redondoviridae, Related to Figure 2 Based on definitions in (Varsani and Krupovic, 2018) and the analysis of the diversity of viral Rep proteins, Redondoviridae genomes can be grouped into two genera, demarcated by 50% Rep protein identity, which we propose be called Vientovirus and Brisavirus, from the Spanish words for “wind” and “breeze”, alluding to their discovery in the respiratory tract.
Table S3- List of Studies Queried, Related to Figure 1 and Figure 4 The number of samples in each study represents the total samples uploaded to the central database. However, only samples that were determined not to be 16S, ITS or other targeted amplicons were analyzed by the pipeline described here. SRA; Sequence Read Archive, MG-RAST; Metagenomic Rapid Annotations using Subsystems Technology, HOMD, Human Oral Microbiome Database
Table S4: Periodontitis Dataset Metadata, Related to Figure 5 Metadata from Shi et al. 2015, Califf et al. 2017, and Kumar et al. 2018 related to periodontitis disease and treatment status. The “Mapped Reads” and “Fraction Cov.” columns correspond to the number of reads mapping to and fraction coverage of the best (highest-coverage) redondovirus hit, respectively. The “Total Reads” and “Total Bases” columns refer to the total number of reads and bases in the metagenomic data. Periodontal Disease (PD) status is enumerated in the “Disease Status” column. In studies that tracked the change in periodontal status over time, the change in disease status is listed in the “Disease Change” column (samples from Shi et al. were listed as Not Applicable (NA) as recovery from periodontal disease was an inclusion criterion for the study, rather than a measured variable). The Treatment/Control column lists the participants’ study group for Califf et al. (T =Treatment, C = Control). Inflammation score is included from Shi et al. IL1-B levels (pg/ml), rating, and treatment time point are included from Kumar et al.
Data Availability Statement
The accession numbers for the viruses sequenced and reported in this paper are GenBank: MK059754-MK059772. Full details of each step of the Snakemake pipeline used in this report are available at https://github.com/louiejtaylor/hisss. The script used for RBS analysis is available at https://github.com/louiejtaylor/find-prok-rbs.