

Abstract
Objective
Amyloid deposition is linked to multiple human ailments, including neurodegenerative diseases, type 2 diabetes, and systemic amyloidosis. The assembly of misfolded proteins into amyloid fibrils involves an intermediate form, i.e., soluble amyloid precursor (AP), which exerts cytotoxic function. Insoluble amyloid also stimulates innate immune cells to elicit cytokine response and inflammation. How any of these misfolded proteins are controlled by the host remains obscure. Serum amyloid-P component (SAP) is a universal constituent of amyloid deposits. Short-chain pentraxins, which include both SAP and C-reactive protein (CRP), are pattern recognition molecules that bind to diverse ligands and promote the clearance of microbes and cell debris. Whether these pentraxins interact with AP and cofactor-containing amyloid and subsequently impact their function is not known.
Methods and Results
To detect the interaction between SAP and different types of amyloids, we performed dot blot analysis. The results showed that SAP invariably bound to protein-only, nucleic acid-containing and glycosaminoglycan-containing amyloid fibrils. This interaction required the presence of calcium. By ELISA, both SAP and CRP bound to soluble AP in the absence of divalent cations. Further characterization, by gel filtration, implied that SAP decamer may recognize AP whereas aggregated SAP preferentially associates with amyloid fibril. Although SAP binding did not affect cytotoxic function of AP, SAP potently inhibited the production of interferon-α from human plasmacytoid dendritic cells triggered by DNA-containing amyloid.
Conclusions
Our data suggest that short pentraxins differentially interact with various forms of misfolded proteins and, in particular, modulate the ability of nucleic acid-containing amyloid to stimulate aberrant type I interferon response. Hence, pentraxins may function as key players in modulating the pathogenesis of protein misfolding diseases as well as interferon-mediated autoimmune manifestation.
Keywords: Pentraxins, Serum amyloid P component, C-reactive protein, Amyloid, Protein misfolding, Interferon, Plasmacytoid dendritic cells
Introduction
The misfolding of monomeric polypeptides that assemble into insoluble amyloid fibrils is linked to protein misfolding diseases [1]. A number of proteins have been identified that exhibit amyloidogeneic potential; among the most studied are amyloid-beta (Aβ) in Alzheimer’s disease (AD) and islet amyloid polypeptide in type 2 diabetes [2–4]. Recent data suggest that the self-assembly of amyloidogenic proteins occurs through formation of misfolded intermediates that have an oligomeric structure [5,6]. These aggregate species, also known as amyloid precursors (APs), are soluble and display inherent cytotoxicity towards live cells, presumably cause neuronal damage in neurodegenerative diseases [7,8].
Increasing evidence supports the notion that many polypeptides have intrinsic properties that enable amyloid formation [3,9,10]. A recent genome-wide sequence survey identified the “amylome”, where roughly 15% of all coding segments in Escherichia coli, Saccharomyces cerevisiae and humans harbor amyloid-prone hexapeptides [9]. What is more, a list of functional amyloids have been discovered to participate in diverse cellular processes, including biofilm and spore assembly in bacteria, storage of peptide hormones within mammalian secretory granules, intracellular signaling mediated by proteins with fibril-like structures and enhanced HIV infectivity during sexual transmission [11–18].
Furthermore, it is now evident that misfolded proteins have aberrant innate immune stimulatory capability. For example, amyloid and possibly AP can activate NALP3 inflammasome and induce IL-1β secretion [19,20]. In addition, amyloid fibrils containing DNA or RNA are potent to activate plasmacytoid dendritic cells (pDCs) to produce type I interferon. This activity can be pathogenic as it induces the breakdown of humoral immune tolerance in vivo [21]. To date, the mechanism by which the host minimizes the harmful effects of misfolded proteins is not clear. Therefore, it is important to elucidate the fundamental mechanism that is protective against the pathogenicity triggered by the various forms of misfolded proteins.
Pentraxins represent an important element of the humoral innate immune system. They are characterized by a common structural organization in five or ten identical subunits arranged with pentameric radial symmetry [22,23]. These pattern-recognition molecules include the short pentraxins serum amyloid-P component (SAP) and C-reactive protein (CRP), and the long pentraxin 3. All pentraxins are able to interact with components of the complement pathway [22,24]. In addition, short pentraxins can bind to membrane phospholipids and nuclear components [25]. Their binding to different ligands is critically promoted by calcium ions, which triggers changes in the conformation of SAP [22]. The aggregated form of SAP in Ca2+-containing solutions interacts with Fcγ receptor, complement, microbes and cell debris [22,24,26,27]. The binding of pentraxins with diverse ligands is important for host defense and removal of damaged cells and nuclear components [24].
It is well known that SAP can bind to amyloid fibrils in vitro and in vivo, which renders it a universal constituent of amyloid deposits [28,29]. It has been shown that SAP binding stabilizes the amyloid fibrils, whereas antibodies against SAP can facilitate the phagocytosis and clearance of amyloids [30,31]. However, it is not known whether SAP modulates innate immune cell activation triggered by amyloids. Therefore, we investigated the biological impact of SAP through its interaction with nucleic acid-containing amyloids.
Besides SAP, CRP is often detected in amyloid plaques and protein tangles of patients, however the molecular mechanism of such association is lacking [32,33]. In addition, whether pentraxins interact with protein misfolding intermediates has not been investigated. Therefore, by studying a stabilized model of AP [34,35], we intend to determine whether SAP and CRP are capable of interacting with the precursor form of amyloid.
Methods
Materials and cells
Materials and their suppliers were as follows: purified human serum albumin (HSA), Sigma-Aldrich; Aβ(1–42) peptide, EMD Biosciences; Smith antigen/ribonucleoprotein complex (Sm/RNP), Meridian Life Sciences; epigallocatechin gallate (EGCG), Sigma-Aldrich; human SAP, Calbiochem; and human CRP, Cell Sciences. SAP and CRP were biotinylated by using EZ-link Sulfo-NHS-LC-Biotin (Invitrogen) according to the manufacturer’s instructions. Pan human interferon α (IFN-α) was detected by using Mabtech ELISA kit; interleukin-6 (IL-6) and tumour necrosis factor α (TNFα) were detected by using R&D ELISA kits. RPMI 8226 cells were grown in RPMI 1640 medium supplemented with 10% fetal bovine serum, 50 units/ml penicillin and 50 μg/ml streptomycin. Peripheral blood mononuclear cells (PBMC) were isolated from whole blood provided by the Gulf Coast Blood Center. pDCs were isolated by BDCA-4 positive selection (Miltenyi Biotech) from buffy coats provided by the Gulf Coast Blood Center.
Preparations of AP and amyloid
HSA-derived AP (AP-HSA) was prepared as described before [34]. Briefly, HSA was cross-linked with 1-ethyl-3-[3-dimethylaminopropyl] carbodiimide hydrochloride (EDC) in MES buffer at pH 4.7. To produce DNA- and heparin-containing amyloid, AP-HSA was mixed at a 1:3 ratio with DNA (Sigma) or heparin (Lovenox, Sanofi-Aventis) and after 1 hr, precipitates were centrifuged to remove soluble components. To prepare protein-only amyloid, 10 mg/ml HSA was reconstituted in MES buffer and incubated for 4 hrs at 65°C. To crosslink HSA with dimethyl pimelimidate (DMP), further referred as HSA-DMP, a 10 fold molar excess of DMP (Thermo Scientific) was added to 5 mg/ml of HSA in 0.2M triethanolamine pH 8. After 1 hr, the reaction was stopped with glacial acetic acid. To prepare glutaraldehyde cross-linked HSA (HSA-Glut), 0.05% glutaraldehyde (Sigma Aldrich) was added to 1 mg/ml HSA for 10 min. Tris-HCl was added to terminate the reaction.
ELISA
To detect the binding of SAP and CRP to different ligands, ELISA was performed. Different concentrations of Aβ (1–42), Aβ (42-1), Sm/RNP, HSA, AP-HSA, HSA-DMP or HSA Glut in carbonate buffer (45.3 mM NaHCO3, 18.2 mM Na2CO3 pH 9.6) were coated on ELISA plates overnight at 4°C. The plates were blocked with blocking buffer (150 mM NaCl, 25 mM Tris(TBS), 1% bovine serum albumin (BSA)) for 1 hr at room temperature. Biotinylated human SAP or CRP (4 μg/ml) was added in TBS+0.01% tween 20 at different conditions, such as Ca2+, EDTA, copper (Cu2+) or magnesium (Mg2+), for 2 hrs. Then, streptavidin-horseradish peroxidase (streptavidin-HRP) was added and the 3,3′,5,5′-Tetramethylbenzidine (TMB) substrate was used to detect colour development with maximum absorbance of 450 nm.
Dot blot analysis
To identify the binding of pentraxins to cofactor-containing amyloids, 30 μg of protein-only amyloid, DNA-containing amyloid or heparin-containing amyloid was made as described above and incubated with biotinylated SAP or biotinylated BSA (5 μg/ml) in PBS or 2 mM Ca2+ in PBS overnight. After several washes, the precipitates were resuspended in 100 μl of PBS and then 4 μl of this solution was spotted onto activated Immobilon-P membrane (Millipore). The blot was then blocked in 1% BSA in TBS and streptavidin-HRP was added. After several washes, the blot was developed using SuperSignal West Pico Chemiluminescent Substrate (Pierce Biotech). To determine the effect of EGCG on pentraxin binding to AP-HSA, EGCG was incubated with 1μg of HSA or AP-HSA for 1 hr and then 4 μl of the mixture was spotted onto an activated membrane as described above. After blocking with 1% BSA in TBS for 1hr, biotinylated SAP or biotinylated CRP was incubated in blocking buffer overnight. After several washes, streptavidin-HRP was added and the blot was developed as described above.
Gel filtration
To identify the conformation of SAP at different buffer conditions, a 24 ml Superose 6 10/300 GL column was run on an AKTA (Amersham Bioscience) liquid chromatography system. After equilibration with at least 4 column volumes of buffer, 3 μg of biotinylated SAP in PBS, 2 mM Ca2+ or 2 mM Ca2+ plus 80 mg/ml HSA was loaded into the column. One ml fractions were collected and used to coat ELISA plates. Streptavidin-HRP was used to detect SAP in each fraction.
In vitro cell cultures
To determine the effect of SAP and CRP binding on the cytotoxicity of AP-HSA, RPMI 8226 cells were plated at 0.5 × 106/ml in PBS and biotinylated HSA or AP-HSA preincubated with SAP or CRP was added. One hour later, cells were washed and then FITC conjugated streptavidin and propidium iodide were added to detect binding of HSA or AP-HSA and cell death, respectively. In other experiments, human PBMC (10 × 106/ml) or isolated pDCs was cultured overnight in complete medium with 1 μg/ml HSA or AP-HSA mixed with 1 μg/ml E. coli DNA with or without SAP. Supernatants were then analyzed by ELISA for cytokine production.
Statistical analysis
Statistical analyses were performed with the two-tailed unpaired Student t test or with a paired student t test using GraphPad Prism 6.0 software. P>0.05 was considered non-significant.
Results
SAP recognizes amyloid fibrils containing cofactors
SAP can bind to different types of ligands in a calcium-dependent manner [22]. To verify that the commercially obtained SAP was functionally active, ELISA was performed to confirm that SAP bound to Aβ (1–42), an interaction that was enhanced by the presence of calcium (Figure 1B). Given that amyloid fibrils may contain various cofactors such as nucleic acids and glycosaminoglycans [36,37], we first determined whether SAP binds to in vitro generated cofactor-containing amyloids. To do that, we prepared heparin-containing amyloid, DNA-containing amyloid and protein-only amyloid as demonstrated previously [35]. The resulting insoluble fibrils did not coat ELISA plates evenly (data not shown), therefore a dot blot analysis was performed. Briefly, SAP or BSA, an irrelevant protein used as a control, was incubated with the amyloid-containing blots in the presence or absence of Ca2+. Our results showed that SAP, but not BSA, readily bound to all types of amyloid complexes examined (Figure 1B). This binding predominantly occurred in the presence of Ca2+. These data demonstrated that, in addition to protein-only amyloid, SAP readily binds to hybrid amyloid fibrils containing different cofactors.
Figure 1.
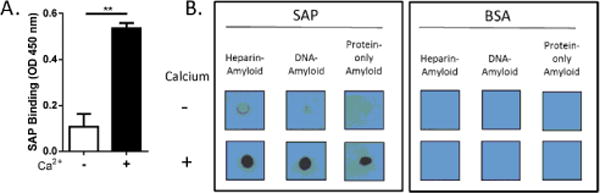
SAP binding to amyloid fibrils containing cofactors. (A) Binding of SAP to Aβ (1–42) (10 μg/ml) in the absence or presence of 2 mM Ca2+ was assessed by ELISA. Error bars are means ± SEM of duplicate wells (**p<0.005 compared with no Ca2+). Similar results were obtained from five independent experiments. (B) Heparin-containing amyloid, DNA-containing amyloid, and protein-only amyloid were mixed with biotinylated SAP or biotinylated BSA in the absence or presence of 2 mM Ca2+. After several washes, the precipitates were dotted on a membrane, and binding was detected by chemiluminescence. Similar results were obtained from three independent experiments.
SAP binds to amyloid precursor protein in the absence of divalent cations
The AP of amyloidogenic proteins is only formed transiently in solution [38], which made it challenging to study their biochemical properties. However, we have recently generated a form of stabilized AP derived from HSA, referred to as AP-HSA, which displays partially misfolded structure and is capable of converting to amyloid [34]. To determine whether AP-HSA is recognized by SAP, we performed an ELISA-based assay. In contrast to what was observed regarding SAP interaction with amyloid, SAP failed to bind to native HSA or to AP-HSA in the presence of Ca2+ (Figure 2B). Instead, SAP bound considerably to AP-HSA, but not native HSA, in the absence of Ca2+ (Figure 2B). These data suggested that SAP can recognize amyloid precursor proteins under a condition distinct from its interaction with amyloid.
Figure 2.
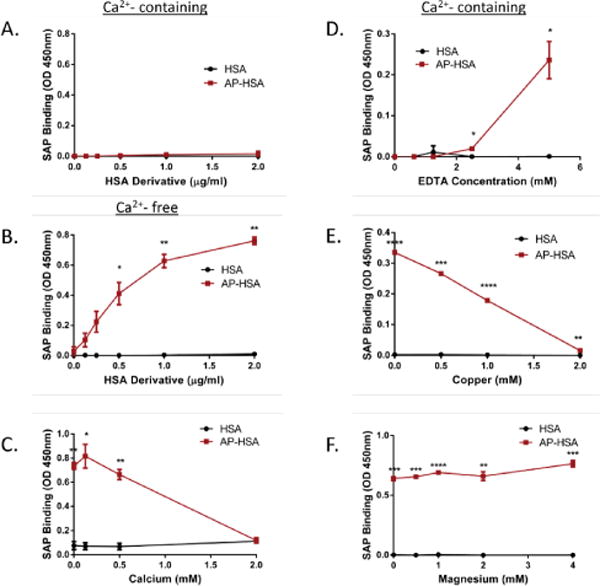
SAP binding to amyloid precursor protein. (A–F) Binding of SAP to HSA and AP-HAS in the presence of 2 mM Ca2+ (A), in PBS (B) in PBS with different concentrations of Ca2+ (C), in 2 mM Ca2+ plus different concentrations of EDTA (D), or with different concentrations of Cu2+ (E) or Mg2+ (F) was assessed by ELISA. Error bars are means ± SEM of duplicate wells. Similar results were obtained from at least 2 independent experiments (*p<0.05, **p<0.005, ***p<0.0005 and ****p<0.00005 compared with HSA).
Given that SAP bound to AP-HSA in the absence of Ca2+, we tested whether Ca2+ would influence the interaction between SAP and AP-HSA. As shown in Figure 2C, Ca2+ blocked the binding of SAP to AP-HSA in a dose-dependent manner. EDTA is a chelating agent that can sequester metal ions such as Ca2+. After the incubation of SAP with different concentrations of EDTA in the presence of Ca2+, we showed that EDTA neutralized the inhibitory effect of Ca2+ on the binding of SAP to AP-HSA (Figure 2D). These results demonstrated that Ca2+ prohibited the binding of SAP to AP.
It is established that the binding of SAP to its ligands can be affected by different divalent cations. Similar to Ca2+, Cu2+, but not Mg2+, can promote SAP binding to its ligands through the induction of conformational changes in SAP structure [39]. Therefore, we determined whether these divalent cations affected SAP-AP interaction. Expectedly, Cu2+ blocked SAP interaction with AP-HSA (Figure 2E). In contrast, Mg2+ did not affect the binding of SAP to AP-HSA (Figure 2F). These findings suggested that cations that enable conformational changes in SAP effectively inhibit its interaction with AP.
SAP decamer preferentially binds to amyloid precursor protein
Human SAP can undergo conformational changes according to the presence of calcium, pH, ligand availability and albumin concentration [39–41]. In the absence of Ca2+, SAP forms a decamer composed of two cyclic pentamers whereas in the presence of calcium, SAP aggregates into high molecular weight complexes [40]. If high concentrations of proteins are present in Ca2+-containing conditions, such as HSA in human serum, SAP reverses to a pentameric form [41]. To determine the conformation of SAP during its interaction with AP-HSA, we incubated SAP in the presence of Ca2+ and tested the effect of different concentrations of HSA (native monomeric form). As shown earlier (Figure 2), SAP, in the presence of Ca2+ but at low levels of HSA, failed to bind AP-HSA. However, it regained the ability to interact with AP-HSA in the presence of high doses of HSA (Figure 3A). These data hint to us that, when interacting with AP, SAP may adopt a pentameric conformation.
Figure 3.
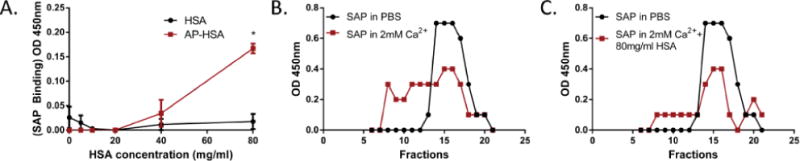
SAP conformation during amyloid precursor protein binding. (A) Effect of different doses of HSA, in the presence of 2 mM Ca2+, on SAP binding to HSA or AP-HSA was assessed by ELISA. Error bars are means ± SEM of duplicate wells (*p<0.05 compared with HSA). Similar results were obtained from at least two independent experiments. (B, C) Biotinylated SAP was incubated in the presence or absence of Ca2+ (B) or 80 mg/ml HSA plus Ca2+ (C), and the molecular weight of SAP was assessed by gel filtration. Fractions were collected and analyzed by ELISA.
To further identify the assembly of SAP, gel filtration of purified SAP in different buffer conditions was performed. SAP monomer has a molecular weight of 25 kD, therefore SAP pentamer has a molecular mass of 127 kD and SAP decamer is 254 kD [41]. In PBS, SAP eluted between fractions 13 and 17, within the expected range of decamers. When 2 mM Ca2+ was added, SAP appeared earlier, in fractions 7 to 16, suggesting the formation of heterogeneous higher molecular weight aggregates (Figure 3B). These results are consistent with our earlier assumption that, SAP exists primarily as a decamer in buffer lacking Ca2+, a conformation enabling its binding to AP-HSA (Figure 2B) [41], whereas in the presence of Ca2+, SAP autoaggregates and gained capacity to interact with amyloid fibrils (Figure 1A and 1B). Given that high concentration of HSA promoted the binding of SAP to AP-HSA in Ca2+-containing solution (Figure 3A), we further fractionated SAP under this condition. The presence of 80 mg/ml HSA in Ca2+-containing PBS solution, reverted the majority of the aggregated SAP back into the fractions 13 to 16, which contain lower molecular weight species (Figure 3C). These data supported the notion that SAP decamer predominantly interacts with AP whereas highly aggregated SAP binds to amyloids.
CRP binds to amyloid precursor protein
CRP shares properties with SAP, including calcium-dependent ligand binding [22]. To determine whether CRP could also interact with AP, an ELISA-based assay was performed. As previously described, CRP bound to Sm/RNP in the presence and absence of Ca2+, but did not bind to Aβ peptide (Figure 4A) [25]. The failure of CRP to recognize amyloid is in direct contrast with the binding of SAP to amyloid under the same condition (Figure 1A). Surprisingly, CRP bound to AP-HSA but not native HSA, in the absence (Figure 4B) and, to a lesser extent, in the presence of Ca2+ (Figure 4C). These findings therefore indicated that, both SAP and CRP, two short pentraxins, have the ability to bind AP in vitro.
Figure 4.
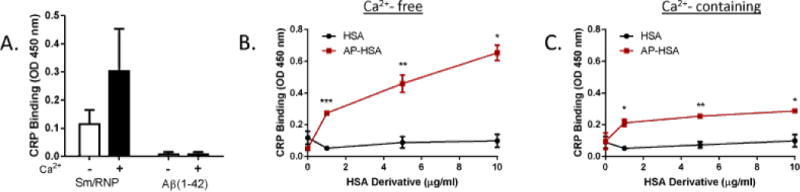
CRP binding to amyloid precursor protein. (A) Binding of CRP to Aβ or Sm/RNP in the absence or presence of 2 mM Ca2+ was assessed by ELISA. (B, C) Binding of biotinylated CRP to HSA or AP-HSA in the absence (B) or presence (C) of 2 mM Ca2+ was determined by ELISA. Error bars are means ± SEM of duplicate wells. Similar results were obtained from at least three independent experiments (*p<0.05, **p<0.005 and ***p<0.0005 compared with HSA).
Misfolded structure in amyloid precursor protein is crucial for pentraxin binding
EGCG is a natural compound that interferes the formation of β-sheet structure by complexing with AP [42]. To determine whether the misfolded structure of AP-HSA is critical for its interaction with short pentraxins, different amounts of EGCG were pre-incubated with HSA or AP-HSA. By dot blot analysis, we found that high doses of EGCG prohibited the binding of SAP to AP-HSA (Figure 5A). A similar effect of EGCG was seen on CRP binding to AP-HSA (Figure 5B). These data suggested that a misfolded structure of APs might be crucial for their recognition by pentraxins.
Figure 5.
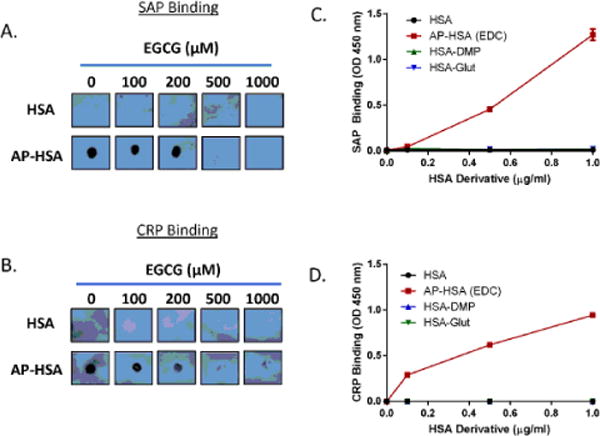
Misfolded structure in amyloid precursor protein is crucial for pentraxin binding. (A, B) EGCG was incubated with HSA or AP-HSA for 1 hr and binding of SAP (A) or CRP (B) to HSA or AP-HSA was assessed by dot blot. Similar results were obtained from at least two independent experiments. (C, D) Binding of SAP (C) or CRP (D) to HSA or HSA crosslinked with EDC (AP-HSA), DMP (HSA-DMP) or glutaraldehyde (HSA-Glut) was assessed by ELISA. Error bars are means ± SEM of duplicate wells. Similar results were obtained from at least two independent experiments.
Acidic conditions can promote structural misfolding of proteins; therefore, acidic conditions can facilitate the formation of APs in vitro [43,44]. Consistently, the generation of stabilized AP-HSA by crosslinking requires an acidic pH. Conversely, other crosslinkers, such as glutaraldehyde and DMP, that only react at basic pH, failed to produce AP [35]. Therefore, we crosslinked HSA with glutaraldehyde (referred as HSA-Glut) and DMP (referred as HAS-DMP) to determine the specific determinant enabling pentraxin binding. Even though all three cross-linked HSA products had similar HSA oligomerization (Supplementary Information 1), SAP and CRP selectively recognized EDC-stabilized AP-HSA but not glutaraldehyde or DMP cross-linked proteins (Figure 5C and 5D). Therefore, instead of indiscriminately interacting with any form of aggregated proteins, short pentraxins likely recognize specific misfolded structure present within AP.
Short pentraxins do not affect amyloid precursor protein mediated cytotoxicity
In light of our findings on pentraxin binding to APs, we wanted to determine whether such interactions have functional consequence on the cytotoxicity of APs. As previously shown, AP-HSA can bind to the cell membrane and exert cytotoxic function [35]. First, to determine whether SAP and CRP interfere with the binding of AP-HSA to the cell membrane, we incubated SAP or CRP with biotinylated HSA or AP-HSA before adding it to RPMI 8226 cells, a human plasmacytoma cell line. After 1hr on ice, the cells were washed to remove unbound HSA or AP-HSA followed by staining with Alexa Fluor 488-labeled neutravidin. The binding of HSA or AP-HSA to RPMI 8226 cells was then examined by flow cytometry. At a concentration up to 50 μg/ml, SAP or CRP had no significant effect on the surface attachment of AP-HSA (Figure 6A and 6B). Second, to determine whether pentraxin affects cell death induced by AP, we stained RPMI 8226 with propidium iodide to quantify dead cell population by flow cytometry. Neither SAP (Figure 6C) nor CRP (Figure 6D) affected the cytotoxicity of AP-HSA at the dose tested (up to 50 μg/ml). These data suggested that, at the doses tested, pentraxin binding to APs does not affect AP-induced cytotoxicity.
Figure 6.
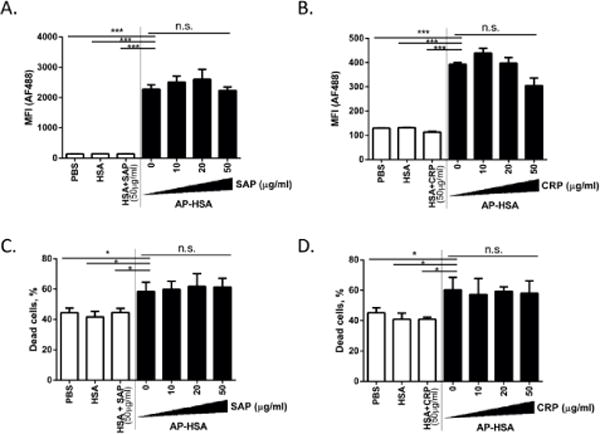
Effect of pentraxin binding to amyloid precursor protein in cellular cytotoxicity. (A–D) Biotinylated HSA or AP-HSA (1 μg/ml) preincubated with different concentrations of SAP or CRP was added to RPMI 8226 cells. After 1 hr on ice, streptavidin-AF488 and propidium iodide were added to analyze binding of HSA or AP-HSA (A and B) and cell death (C and D) by flow cytometry. Error bars are means ± SEM of 3 pooled experiments (*p<0.05 and ***p<0.0005).
SAP inhibits DNA-containing amyloid-stimulated IFN-α production
Earlier observation that SAP binds to diverse types of amyloids (Figure 1B) prompted us to investigate whether the association of SAP might affect the innate immune property of amyloid fibrils. Given that DNA-containing amyloid can potently activate pDCs [21], we investigated whether SAP binding could impact the ability of pDCs to produce IFN-α triggered by DNA-containing amyloid. First, we pre-incubated SAP with comparable amounts of DNA-containing amyloid or native HSA plus DNA (referred to as control) then added it to the culture of PBMC. DNA-containing amyloid induced significant amounts of secreted IFN-α; in contrast, SAP at 50 μg/ml decreased the levels of IFN-α considerably (Figure 7A). No IFN-α was detected in cultures incubated with control. SAP had no effect on the production of IL-6 and TNFα, two proinflammatory cytokines, in PBMC culture (Figure 7B and 7C). To further confirm the inhibitory effect of SAP, we isolated primary human pDCs from peripheral blood and cultured with DNA-containing amyloid together with SAP. Consistently, pDCs produced less amounts of IFN-α when exposed to SAP-amyloid complexes (Figure 7D). These data revealed that SAP binding can limit the activation of pDCs and inhibit the production of type I interferon stimulated by nucleic acid-containing amyloids.
Figure 7.
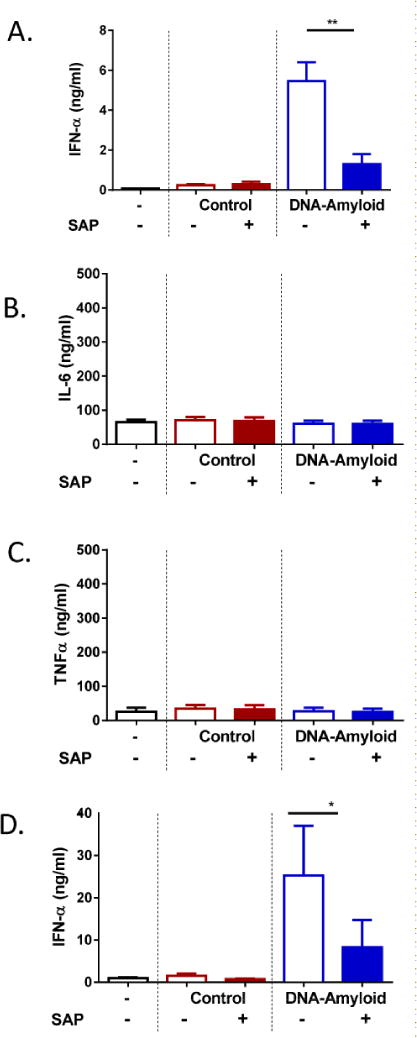
SAP inhibits IFN-α production triggered by DNA-containing amyloids. (A–C) PBMC were incubated with control (HSA+DNA) or DNA-amyloid with or without SAP (50 μg/ml) for 24 hrs. Supernatants were analyzed by ELISA for IFN-α (A), IL-6 (B) and TNFα (C) secretion. Error bars are means ± SEM of six donors. (D) Purified pDCs were incubated with control or DNA-amyloid, and IFN-α was quantified by ELISA after 24 hrs of culture. Error bars are means ± SEM of six donors (*p<0.05 and **p<0.005 compared with DNA-amyloid without SAP).
Discussion
In this study, we discovered a previously unrecognized interaction between short-chain pentraxins and amyloid precursor protein. Our investigation further reveals a molecular mechanism by which SAP differentially recognizes amyloid fibrils and AP by adopting different conformations. Additionally, we demonstrated that SAP binding can inhibit the innate immune function of amyloid, particularly IFN-α production triggered by nucleic acid-containing amyloid.
Proteins lose structural integrity due to genetic mutations or when subjected to assorted stress, which may lead to cellular damage and eventually diseases if not controlled [1]. Therefore, mechanisms must exist to prevent the accumulation of extracellular misfolded proteins. Here, we report that SAP and CRP can interact with terminal amyloid fibrils and/or their soluble precursor species, suggesting a fundamental role of short pentraxins in protein homeostasis. Interestingly, we observed that the interaction between SAP and amyloid required Ca2+, whereas binding of pentraxins to AP was inhibited by divalent cations. Human SAP and CRP share 51% amino acid sequence homology; however, only SAP can bind amyloid fibrils. Even though the motif enabling the binding of SAP to amyloid fibrils is still unknown, it has been speculated that the aggregated structure of SAP creates a pocket formed by calcium-ligating loops located on each SAP subunit that allows recognition of a specific motif shared by all amyloids [45].
It is known that inflammation may increase the extracellular levels of calcium ion in the tissue [46]. Therefore, an inflammatory condition may favour Ca2+-mediated SAP aggregation and complex formation between SAP and amyloid. Since neuroinflammation is exceedingly associated with AD [47], deposition of SAP with amyloid is therefore promoted in the brain of AD patients. On the contrary, human peripheral blood contains high levels of HSA and Ca2+, in which SAP predominantly exist as pentamers [41]. Suggested by our study, AP may be recognized by pentameric SAP; thus it is likely that SAP complexes with AP primarily in the blood. Given that pentraxins readily activate complement to facilitate cargo clearance, it is reasonable to hypothesize that circulating pentraxins mediate the removal of misfolded proteins. Indeed, we have observed that AP-HSA was rapidly cleared from circulation after intravenous injection in mice (data not shown). Therefore, the interaction between pentraxins and misfolded proteins may serve as an important protective mechanism to eliminate pathogenic misfolded proteins.
Although it was unexpected initially to observe that both SAP and CRP bound to AP, several published reports have suggested a unique affinity between these short pentraxins and proteins with altered structures. For example, SAP binds to denatured lactate dehydrogenase independently of calcium ion [48]. Moreover, CRP could interact with diverse proteins that are aggregated and adopt altered conformations at acidic pH [49,50]. We present experimental evidence to suggest that SAP and CRP specifically recognize the misfolded structure within AP, a novel finding worthy of further investigation. It is likely that the exposed surface of SAP pentamers contains binding sites for the misfolded structures on APs. Less is known about functional disparity associated with the conformational changes of CRP. Here we demonstrate that CRP binds to AP more efficiently in low Ca2+ condition (Figure 4B and 4C). Although circulating human CRP is in pentameric form, monomeric CRP has been recently shown to selectively deposit in AD amyloid plaques with co-localized complement proteins [51]. Further study is necessary to delineate the specific conformation that enables the binding between CRP and AP.
We have shown that SAP binding significantly inhibited the magnitude of IFN-α production induced by DNA-containing amyloid, despite the fact that, at the same dose, SAP had no effect on the cytotoxicity of AP. DNA has been detected within the amyloid plaques in AD brain; however, its biological relevance is vague and only under speculation [36]. Several recent studies have established a remarkable link between IFN-α in the brain and the pathogenesis of cognitive decline and AD [52–54]. However, the molecular entity that triggers IFN-α production in CNS has not been identified. We would speculate that DNA-containing amyloid fibrils likely serve as a self-derived ligand to induce IFN-α in AD brain, a process that can be modulated by SAP.
Both SAP and CRP have been implicated in the process of autoimmune pathogenesis, playing largely regulatory roles. In systemic autoimmune conditions such as systemic lupus erythematosus (SLE), dead cell material and nuclear antigens may accumulate and stimulate autoimmune reactions [55]. Shown in studies where SAP and CRP were transferred or overexpressed in autoimmune prone mice, these pentraxins potently ameliorate the disease progression presumably by promoting debris clearance [56–59]. However, the relevance of these results in human disease has been difficult due to differences between mouse and human pentraxins. For example, SAP is an acute phase protein in mouse; whereas in humans, SAP is constitutively expressed [22]. Moreover, human SAP has higher affinity than mouse SAP at binding to amyloids [29].
Here we provide another potential mechanism by which short pentraxins dampen the innate immune activation pathway critical for autoimmune response. Type I interferon (IFN-α, β, ω and τ) is a family of pluripotent cytokines that are centrally associated with SLE and other autoimmune diseases [60–64]. We have shown that amyloid fibrils containing DNA induced the development of anti-nuclear antibody and a lupus-like syndrome, mimicking SLE, after inoculated into non-autoimmune mice [21]. Interestingly, not only SAP inhibits IFN-α production by pDCs as we have shown here, but also human CRP limits the pDCs’ interferon response to autoimmune complexes [65]. Hence, by regulating type I interferon response and debris removal, short pentraxins may play an important role in guarding against the development of autoimmunity at multiple stages.
Conclusions
Pentraxins are known to critically mediate the clearance of dead cells and invading pathogens. Yet, their presence in amyloid deposits of human pathologies suggests additional functions. Our findings that both SAP and CRP can bind to the soluble precursor form of amyloid implicate them as participants in the homeostatic regulation of misfolded proteins. On the other hand, short pentraxins are involved in modulating autoimmunity. In this study, we have shown that SAP binding inhibits the ability of DNA-containing amyloid to trigger type I interferon production, providing a mechanism by which SAP regulates autoimmune development. Therefore, we conclude that short-chain pentraxins interact with misfolded protein species and diminish the innate immune activity of amyloid. The novel interaction and biological effect we have observed suggest that pentraxins may function as key players in controlling the pathogenesis of protein misfolding diseases as well as interferon-mediated autoimmune manifestation.
Supplementary Material
Acknowledgments
This research was supported by an Institutional Research Grant from The University of Texas MD Anderson Cancer Center, National Institutes of Health (NIH) grant AI074809 (to W.C), and NIH Cancer Center Support Grant CA016672. S.D-E. was supported by NIH Training Program Grant T32 CA009598.
References
- 1.Dobson CM. Getting out of shape. Nature. 2002;418:729–730. doi: 10.1038/418729a. [DOI] [PubMed] [Google Scholar]
- 2.Westermark P, Wernstedt C, Wilander E, Hayden DW, O’Brien TD, et al. Amyloid fibrils in human insulinoma and islets of Langerhans of the diabetic cat are derived from a neuropeptide-like protein also present in normal islet cells. Proc Natl Acad Sci U S A. 1987;84:3881–3885. doi: 10.1073/pnas.84.11.3881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Selkoe DJ. Folding proteins in fatal ways. Nature. 2003;426:900–904. doi: 10.1038/nature02264. [DOI] [PubMed] [Google Scholar]
- 4.Savelieff MG, Lee S, Liu Y, Lim MH. Untangling amyloid-β, tau, and metals in Alzheimer’s disease. ACS Chem Biol. 2013;8:856–865. doi: 10.1021/cb400080f. [DOI] [PubMed] [Google Scholar]
- 5.Cheng B, Gong H, Xiao H, Petersen RB, Zheng L, et al. Inhibiting toxic aggregation of amyloidogenic proteins: a therapeutic strategy for protein misfolding diseases. Biochim Biophys Acta. 2013;1830:4860–4871. doi: 10.1016/j.bbagen.2013.06.029. [DOI] [PubMed] [Google Scholar]
- 6.Cheng B, Gong H, Xiao H, Petersen RB, Zheng L, et al. Inhibiting toxic aggregation of amyloidogenic proteins: a therapeutic strategy for protein misfolding diseases. Biochim Biophys Acta. 2013;1830:4860–4871. doi: 10.1016/j.bbagen.2013.06.029. [DOI] [PubMed] [Google Scholar]
- 7.Kelly JW. The alternative conformations of amyloidogenic proteins and their multi-step assembly pathways. Curr Opin Struct Biol. 1998;8:101–106. doi: 10.1016/s0959-440x(98)80016-x. [DOI] [PubMed] [Google Scholar]
- 8.Bucciantini M, Giannoni E, Chiti F, Baroni F, Formigli L, et al. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature. 2002;416:507–511. doi: 10.1038/416507a. [DOI] [PubMed] [Google Scholar]
- 9.Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, et al. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- 10.Goldschmidt L, Teng PK, Riek R, Eisenberg D. Identifying the amylome, proteins capable of forming amyloid-like fibrils. Proc Natl Acad Sci U S A. 2010;107:3487–3492. doi: 10.1073/pnas.0915166107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schnabel J. Protein folding: The dark side of proteins. Nature. 2010;464:828–829. doi: 10.1038/464828a. [DOI] [PubMed] [Google Scholar]
- 12.Münch J, Rücker E, Ständker L, Adermann K, Goffinet C, et al. Semen-derived amyloid fibrils drastically enhance HIV infection. Cell. 2007;131:1059–1071. doi: 10.1016/j.cell.2007.10.014. [DOI] [PubMed] [Google Scholar]
- 13.Roan NR, Müller JA, Liu H, Chu S, Arnold F, et al. Peptides released by physiological cleavage of semen coagulum proteins form amyloids that enhance HIV infection. Cell Host Microbe. 2011;10:541–550. doi: 10.1016/j.chom.2011.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barnhart MM, Chapman MR. Curli biogenesis and function. Annu Rev Microbiol. 2006;60:131–147. doi: 10.1146/annurev.micro.60.080805.142106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Claessen D, Rink R, de Jong W, Siebring J, de Vreugd P, et al. A novel class of secreted hydrophobic proteins is involved in aerial hyphae formation in Streptomyces coelicolor by forming amyloid-like fibrils. Genes Dev. 2003;17:1714–1726. doi: 10.1101/gad.264303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maji SK, Perrin MH, Sawaya MR, Jessberger S, Vadodaria K, et al. Functional amyloids as natural storage of peptide hormones in pituitary secretory granules. Science. 2009;325:328–332. doi: 10.1126/science.1173155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Badtke MP, Hammer ND, Chapman MR. Functional amyloids signal their arrival. Sci Signal. 2009;2:pe43. doi: 10.1126/scisignal.280pe43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li J, McQuade T, Siemer AB, Napetschnig J, Moriwaki K, et al. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell. 2012;150:339–350. doi: 10.1016/j.cell.2012.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hou F, Sun L, Zheng H, Skaug B, Jiang QX, et al. MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell. 2011;146:448–461. doi: 10.1016/j.cell.2011.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Masters SL, Dunne A, Subramanian SL, Hull RL, Tannahill GM, et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1β in type 2 diabetes. Nat Immunol. 2010;11:897–904. doi: 10.1038/ni.1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol. 2008;9:857–865. doi: 10.1038/ni.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Di Domizio J, Dorta-Estremera S, Gagea M, Ganguly D, Meller S, et al. Nucleic acid-containing amyloid fibrils potently induce type I interferon and stimulate systemic autoimmunity. Proc Natl Acad Sci U S A. 2012;109:14550–14555. doi: 10.1073/pnas.1206923109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Clos TWD. Pentraxins: structure, function, and role in inflammation. ISRN Inflamm. 2013;2013:379040. doi: 10.1155/2013/379040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lu J, Marjon KD, Mold C, Du Clos TW, Sun PD. Pentraxins and Fc receptors. Immunol Rev. 2012;250:230–238. doi: 10.1111/j.1600-065X.2012.01162.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Clos TWD, Mold C. Pentraxins (CRP, SAP) in the process of complement activation and clearance of apoptotic bodies through Fcγ receptors. Curr Opin Organ Transplant. 2011;16:15–20. doi: 10.1097/MOT.0b013e32834253c7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Du Clos TW. The interaction of C-reactive protein and serum amyloid P component with nuclear antigens. Mol Biol Rep. 1996;23:253–260. doi: 10.1007/BF00351177. [DOI] [PubMed] [Google Scholar]
- 27.Lu J, Marnell LL, Marjon KD, Mold C, Du Clos TW, et al. Structural recognition and functional activation of FcgammaR by innate pentraxins. Nature. 2008;456:989–992. doi: 10.1038/nature07468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.de Beer FC, Baltz ML, Holford S, Feinstein A, Pepys MB. Fibronectin and C4-binding protein are selectively bound by aggregated amyloid P component. J Exp Med. 1981;154:1134–1139. doi: 10.1084/jem.154.4.1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pepys MB, Dyck RF, de Beer FC, Skinner M, Cohen AS. Binding of serum amyloid P-component (SAP) by amyloid fibrils. Clin Exp Immunol. 1979;38:284–293. [PMC free article] [PubMed] [Google Scholar]
- 30.Yamada T. Studies on extracorporeal circulation with large volume hemodilution using lactate ringer’s solution and low molecular weight dextran: alterations of acid-base balance associated with intentional hemodilution (author’s transl) Hokkaido Igaku Zasshi. 1975;50:169–196. [PubMed] [Google Scholar]
- 31.Hawkins PN, Lavender JP, Pepys MB. Evaluation of systemic amyloidosis by scintigraphy with 123I-labeled serum amyloid P component. N Engl J Med. 1990;323:508–513. doi: 10.1056/NEJM199008233230803. [DOI] [PubMed] [Google Scholar]
- 32.Tennent GA, Lovat LB, Pepys MB. Serum amyloid P component prevents proteolysis of the amyloid fibrils of Alzheimer disease and systemic amyloidosis. Proc Natl Acad Sci U S A. 1995;92:4299–4303. doi: 10.1073/pnas.92.10.4299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bodin K, Ellmerich S, Kahan MC, Tennent GA, Loesch A, et al. Antibodies to human serum amyloid P component eliminate visceral amyloid deposits. Nature. 2010;468:93–97. doi: 10.1038/nature09494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Duong T, Nikolaeva M, Acton PJ. C-reactive protein-like immunoreactivity in the neurofibrillary tangles of Alzheimer’s disease. Brain Res. 1997;749:152–156. doi: 10.1016/s0006-8993(96)01359-5. [DOI] [PubMed] [Google Scholar]
- 35.Iwamoto N, Nishiyama E, Ohwada J, Arai H. Demonstration of CRP immunoreactivity in brains of Alzheimer’s disease: immunohistochemical study using formic acid pretreatment of tissue sections. Neurosci Lett. 1994;177:23–26. doi: 10.1016/0304-3940(94)90035-3. [DOI] [PubMed] [Google Scholar]
- 36.Dorta-Estremera SM, Li J, Cao W. Rapid generation of amyloid from native proteins in vitro. J Vis Exp. 2013:50869. doi: 10.3791/50869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Di Domizio J, Zhang R, Stagg LJ, Gagea M, Zhuo M, et al. Binding with nucleic acids or glycosaminoglycans converts soluble protein oligomers to amyloid. J Biol Chem. 2012;287:736–747. doi: 10.1074/jbc.M111.238477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jiménez JS. Protein-DNA interaction at the origin of neurological diseases: a hypothesis. J Alzheimers Dis. 2010;22:375–391. doi: 10.3233/JAD-2010-100189. [DOI] [PubMed] [Google Scholar]
- 39.Snow AD, Mar H, Nochlin D, Kimata K, Kato M, et al. The presence of heparan sulfate proteoglycans in the neuritic plaques and congophilic angiopathy in Alzheimer’s disease. Am J Pathol. 1988;133:456–463. [PMC free article] [PubMed] [Google Scholar]
- 40.Stefani M. Structural features and cytotoxicity of amyloid oligomers: implications in Alzheimer’s disease and other diseases with amyloid deposits. Prog Neurobiol. 2012;99:226–245. doi: 10.1016/j.pneurobio.2012.03.002. [DOI] [PubMed] [Google Scholar]
- 41.Isaac O, Thiemer K. [Biochemical studies on camomile components/III. In vitro studies about the antipeptic activity of (−)-alpha-bisabolol (author’s transl)] Arzneimittelforschung. 1975;25:1352–1354. [PubMed] [Google Scholar]
- 42.Potempa LA, Kubak BM, Gewurz H. Effect of divalent metal ions and pH upon the binding reactivity of human serum amyloid P component, a C-reactive protein homologue, for zymosan. Preferential reactivity in the presence of copper and acidic pH. J Biol Chem. 1985;260:12142–12147. [PubMed] [Google Scholar]
- 43.Baltz ML, De Beer FC, Feinstein A, Pepys MB. Calcium-dependent aggregation of human serum amyloid P component. Biochim Biophys Acta. 1982;701:229–236. doi: 10.1016/0167-4838(82)90118-2. [DOI] [PubMed] [Google Scholar]
- 44.Hutchinson WL, Hohenester E, Pepys MB. Human serum amyloid P component is a single uncomplexed pentamer in whole serum. Mol Med. 2000;6:482–493. [PMC free article] [PubMed] [Google Scholar]
- 45.Ehrnhoefer DE, Bieschke J, Boeddrich A, Herbst M, Masino L, et al. EGCG redirects amyloidogenic polypeptides into unstructured, off-pathway oligomers. Nat Struct Mol Biol. 2008;15:558–566. doi: 10.1038/nsmb.1437. [DOI] [PubMed] [Google Scholar]
- 46.McParland VJ, Kalverda AP, Homans SW, Radford SE. Structural properties of an amyloid precursor of beta(2)-microglobulin. Nat Struct Biol. 2002;9:326–331. doi: 10.1038/nsb791. [DOI] [PubMed] [Google Scholar]
- 47.Liu K, Cho HS, Lashuel HA, Kelly JW, Wemmer DE. A glimpse of a possible amyloidogenic intermediate of transthyretin. Nat Struct Biol. 2000;7:754–757. doi: 10.1038/78980. [DOI] [PubMed] [Google Scholar]
- 48.Pepys MB, Herbert J, Hutchinson WL, Tennent GA, Lachmann HJ, et al. Targeted pharmacological depletion of serum amyloid P component for treatment of human amyloidosis. Nature. 2002;417:254–259. doi: 10.1038/417254a. [DOI] [PubMed] [Google Scholar]
- 49.Olszak IT, Poznansky MC, Evans RH, Olson D, Kos C, et al. Extracellular calcium elicits a chemokinetic response from monocytes in vitro and in vivo. J Clin Invest. 2000;105:1299–1305. doi: 10.1172/JCI9799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chewick S. Infection control demand administrative support. Dimens Health Serv. 1975;52:40–41. [PubMed] [Google Scholar]
- 51.Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015;14:388–405. doi: 10.1016/S1474-4422(15)70016-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Coker AR, Purvis A, Baker D, Pepys MB, Wood SP. Molecular chaperone properties of serum amyloid P component. FEBS Lett. 2000;473:199–202. doi: 10.1016/s0014-5793(00)01530-1. [DOI] [PubMed] [Google Scholar]
- 53.Hammond DJ, Jr, Singh SK, Thompson JA, Beeler BW, Rusiñol AE, et al. Identification of acidic pH-dependent ligands of pentameric C-reactive protein. J Biol Chem. 2010;285:36235–36244. doi: 10.1074/jbc.M110.142026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Singh SK, Hammond DJ, Jr, Beeler BW, Agrawal A. The binding of C-reactive protein, in the presence of phosphoethanolamine, to low-density lipoproteins is due to phosphoethanolamine-generated acidic pH. Clin Chim Acta. 2009;409:143–144. doi: 10.1016/j.cca.2009.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Strang F, Scheichl A, Chen YC, Wang X, Htun NM, et al. Amyloid plaques dissociate pentameric to monomeric C-reactive protein: a novel pathomechanism driving cortical inflammation in Alzheimer’s disease? Brain Pathol. 2012;22:337–346. doi: 10.1111/j.1750-3639.2011.00539.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Taylor JM, Minter MR, Newman AG, Zhang M, Adlard PA, et al. Type-1 interferon signaling mediates neuro-inflammatory events in models of Alzheimer’s disease. Neurobiol Aging. 2014;35:1012–1023. doi: 10.1016/j.neurobiolaging.2013.10.089. [DOI] [PubMed] [Google Scholar]
- 57.Baruch K, Deczkowska A, David E, Castellano JM, Miller O, et al. Aging. Aging-induced type I interferon response at the choroid plexus negatively affects brain function. Science. 2014;346:89–93. doi: 10.1126/science.1252945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hofer MJ, Campbell IL. Type I interferon in neurological disease-the devil from within. Cytokine Growth Factor Rev. 2013;24:257–267. doi: 10.1016/j.cytogfr.2013.03.006. [DOI] [PubMed] [Google Scholar]
- 59.Muñoz LE, Lauber K, Schiller M, Manfredi AA, Herrmann M. The role of defective clearance of apoptotic cells in systemic autoimmunity. Nat Rev Rheumatol. 2010;6:280–289. doi: 10.1038/nrrheum.2010.46. [DOI] [PubMed] [Google Scholar]
- 60.Zhang W, Wu J, Qiao B, Xu W, Xiong S. Amelioration of lupus nephritis by serum amyloid P component gene therapy with distinct mechanisms varied from different stage of the disease. PLoS One. 2011;6:e22659. doi: 10.1371/journal.pone.0022659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ardenne M, Reitnauer PG. Demonstration of tumor inhibiting properties of a strongly immunostimulating low-molecular weight substance. Comparative studies with ifosfamide on the immuno-labile DS carcinosarcoma. Stimulation of the autoimmune activity for approx. 20 days by BA 1, a N-(2-cyanoethylene)-urea. Novel prophylactic possibilities. Arzneimittelforschung. 1975;25:1369–1379. [PubMed] [Google Scholar]
- 62.Clos TWD, Zlock LT, Hicks PS, Mold C. Decreased autoantibody levels and enhanced survival of (NZB × NZW) F1 mice treated with C-reactive protein. Clin Immunol Immunopathol. 1994;70:22–27. doi: 10.1006/clin.1994.1005. [DOI] [PubMed] [Google Scholar]
- 63.Szalai AJ, Weaver CT, McCrory MA, van Ginkel FW, Reiman RM, et al. Delayed lupus onset in (NZB × NZW)F1 mice expressing a human C-reactive protein transgene. Arthritis Rheum. 2003;48:1602–1611. doi: 10.1002/art.11026. [DOI] [PubMed] [Google Scholar]
- 64.Rodriguez W, Mold C, Kataranovski M, Hutt J, Marnell LL, et al. Reversal of ongoing proteinuria in autoimmune mice by treatment with C-reactive protein. Arthritis Rheum. 2005;52:642–650. doi: 10.1002/art.20846. [DOI] [PubMed] [Google Scholar]
- 65.Rönnblom L, Pascual V. The innate immune system in SLE: type I interferons and dendritic cells. Lupus. 2008;17:394–399. doi: 10.1177/0961203308090020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.