

Abstract
β-arrestins are classic attenuators of G-Protein coupled receptor signaling. However, they have multiple roles in cellular physiology, including carcinogenesis. The present work shows for the first time that β-arrestins have prognostic significance for predicting metastasis and response to chemotherapy in bladder cancer. β-arrestin-1 (ARRB1) and β-arrestin-2 (ARRB2) mRNA levels were measured by quantitative RT-PCR in two clinical specimen cohorts (n=63; n=43). The role of ARRBs in regulating a stem cell-like phenotype and response to chemotherapy treatments was investigated. The consequence of forced expression of ARRBs on tumor growth and response to Gemcitabine in vivo were investigated using bladder tumor xenografts in nude mice. ARRB1 levels were significantly elevated and ARRB2 levels downregulated in cancer tissues compared to normal tissues. In multivariate analysis only ARRB2 was an independent predictor of metastasis, disease-specific-mortality and failure to Gemcitabine + Cisplatin (G+C) chemotherapy; ~ 80% sensitivity and specificity to predict clinical outcome. ARRBs were found to regulate stem cell characteristics in bladder cancer cells. Depletion of ARRB2 resulted in increased cancer stem cell markers but ARRB2 overexpression reduced expression of stem cell markers (CD44, ALDH2, and BMI-1), and increased sensitivity towards Gemcitabine. Overexpression of ARRB2 resulted in reduced tumor growth and increased response to Gemcitabine in tumor xenografts. CRISPR-Cas9 mediated gene-knockout of ARRB1 resulted in the reversal of this aggressive phenotype. ARRBs regulate cancer stem cell-like properties in bladder cancer and are potential prognostic indicators for tumor progression and chemotherapy response.
Keywords: β-Arrestins, Cancer Stem Cells, Muscle Invasive Bladder Cancer, Cancer biomarkers
Introduction:
Bladder cancer is a common cancer of the urinary tract; 90% of bladder tumors are urothelial cell carcinomas (1). While low-grade bladder tumors rarely invade the bladder muscle and metastasize, high-grade tumors will become muscle invasive if not detected early (1, 2). Non-muscle invasive bladder tumors are treated with transurethral tumor resection, and with additional intravesical therapy if the tumor is high-grade; however, frequent recurrence of the tumors in the bladder require regular surveillance and is associated with morbidity and medical cost (3, 4). Patients with muscle invasive bladder cancer (MIBC) undergo cystectomy. Despite cystectomy, ≥ 50% of MIBC patients develop metastasis within 2-years. The median survival of patients with metastatic bladder cancer is 14 months despite adjuvant chemotherapy (5, 6). Gemcitabine plus cisplatin (G+C) or Methotrexate-Vinblastine-Adriamycin-Cisplatin (MVAC) are first-line chemotherapy regimens for treating patients with metastatic bladder cancer. G+C treatment is preferred in many institutions due to its favorable toxicity profile (5–8). Identification of molecular drivers that promote a muscle invasive phenotype and chemoresistance in bladder cancer, as well as an understanding of their mechanism of action should help in designing potential prognostic markers and effective targeted treatments for patients with advanced bladder cancer.
Arrestins belong to a family of proteins that consists of visual arrestin (arrestin 1), cone arrestin (arrestin 4), β-arrestin-1/ARRB1 (arrestin 2) and β-arrestin-2/ARRB2 (arrestin 3). While arrestin 1 and 4 are exclusively expressed in the retina, ARRB1 and ARRB2 are expressed ubiquitously (9). β-Arrestins act downstream of G-protein coupled receptors (GPCRs), which essentially regulate every physiological process in the human body (10). We and others have demonstrated the role of β-Arrestins in progression and metastasis in several cancers (11–13). β-arrestins are known to uncouple GPCRs from heterotrimeric G proteins and target them to clathrin coated pits for endocytosis, thus attenuating GPCR signaling. In some systems, β-arrestins can function as versatile adaptor molecules that mediate G-protein independent signaling. They can serve as scaffolds that link signaling networks (14) and regulate signaling molecules such as the mitogen activated protein kinases, Akt, and Phosphoinositide 3-kinase (PI3K) (15). There is evidence that depletion of ARRB2 promotes tumor growth in a murine model of lung cancer, as well as in prostate cancer cells, indicating that ARRB2 might function as a tumor suppressor (13, 16). In contrast, ARRB1 was reported to have pro-tumorigenic effects in several cancers (12, 17).
Recent evidence suggests a role of β-arrestins in the regulation of stem cell properties. ARRB1 promotes stemness in non-small cell lung cancers where it acts downstream of nicotinic acetylcholine receptors (18), as well as in leukemia-initiating cells where it promotes self-renewal (19). Furthermore, while ARRB1 inhibits apoptosis in intestinal stem cells induced by chemotherapy (20), ARRB2 may, under certain circumstances (e.g. ionizing radiation), promote apoptosis of intestinal crypt progenitor/stem cells (21). The role of ARRBs in bladder cancer has not yet been reported.
In the present work we investigated, for the first time, the prognostic significance of ARRBs in predicting metastasis and response to chemotherapy. Furthermore, we investigated the role of ARRB1 and ARRB2 in regulating primitive cancer stem cell (CSC)-like phenotypes, including expression of markers associated with stemness, regulation of self-renewal pathways, as well as invasive potential and resistance to chemotherapy. The self-renewal property of CSCs is the driving force behind tumor growth and therapy resistance. Thus, identifying molecules that regulate self-renewal in CSC-like cells will potentially have a significant impact on the development of novel therapeutics for MIBC.
Materials and Methods:
Clinical specimens and qPCR: Cohort 1:
Normal bladder (NBL; n = 20) and bladder tumor (TBL; n = 43) specimens were collected at the University of Miami Miller School of Medicine. The studies and the human subjects protocol were approved by the Institutional Review Board (IRB) and were conducted in accordance with the ethical guidelines of the Declaration of Helsinki. All tissues were collected after obtaining approved, signed patient consent forms. The normal tissues used in the study was isolated from the urothelial layer of the bladder, separated from muscle. Further, we ascertained the urothelial composition of the tissue by histology. De-identified, freshly-frozen specimens and associated data were transferred to Augusta University under an approved IRB protocol. Patient/specimen characteristics are shown in Supplementary Table S1. Time to metastasis or disease-specific mortality (DSM) was calculated from the date of surgery until the event. Cohort 2: Formalin-fixed paraffin-embedded cystectomy or TURBT (transurethral resection of bladder tumor) specimens (n = 43) from 31 patients were obtained from the Department of Urology, Hannover Medical School, Germany (supplementary Table S1). All patients had either synchronous metastasis or developed metastasis during follow-up. All patients with metastasis received adjuvant G+C combination as first-line (n = 278 patients) or second-line (n = 3) treatment; one patient received radiation plus G+C. Treatment failure was defined as either disease-specific mortality following first or second-line treatment or the starting of an alternate treatment due to disease progression. Total RNA isolated from tissues was subjected to reverse transcription quantitative polymerase chain reaction (q-PCR) using primers specific for ARRB1 and ARRB2 transcripts. Transcript levels were normalized to β-actin levels, as described previously (22); primer sequences are shown in supplementary Table S2.
Cell Culture and Transfection
Bladder cancer cell lines (HT1376 and 5637) were obtained from American Type Culture Collection (Rockville, MD) and were used within 10 passages of cell authenticity confirmation by genetic profiling (Genetica DNA Laboratory Inc., Cincinnati, OH). 253J cells and nonmalignant bladder epithelial cells (UROTSA) were a kind gift from Dr. Colin Dinney (MD Anderson Cancer Center, Houston, TX, USA) and Dr. Donald Sens (University of North Dakota, Grand Forks, ND, USA), respectively. Fetal bovine serum (FBS) was purchased from Atlanta Biologicals (Atlanta, GA), antibiotics (gentamycin) and cell culture media were purchased from Invitrogen (Carlsbad, CA) or Sigma-Aldrich (St. Louis, MO). All cells were grown in RPMI 1640 containing 10% FBS. For stable down regulation of ARRB2, 253J cells were transfected with HuSH/shRNA Plasmid Panels (Origene). For over expression of ARRB2 cells were transfected with an ARRB2 plasmid (RC201168, Origene). The details of transfection and other routine procedures are described in Supplementary Materials and Methods.
Colony formation and spheroid assays
Clonogenic growth was measured using colony formation assays as previously described (23). To assess anchorage-independent survival and proliferation, tumor spheroid formation was determined by culturing cells in low-adhesion 35 mm dishes in SpheroMax medium and supplement (PromoCell; Heidelberg Germany; C-28070). Spheroid viability was analyzed utilizing Celltiter-Glo 3D cell viability assay (Promega) according to the manufacturer’s instructions.
Real-time quantitative PCR (RT-q-PCR)
RT-q-PCR was performed as described previously (24). Primer sequences are listed in the supplementary Table S2.
Immunoblotting
Cell lysates were prepared using RIPA-buffer and total protein was quantified with the Pierce Micro BCA Protein Assay Kit (Thermo Scientific; 23235). Western blot analysis was performed as previously described (13, 25) (antibody details are listed in supplementary Table S3). Expression of β-actin served as a loading control (24).
Immunofluorescence
We evaluated expression of cytokeratins (CK14 and CK17), CD44, STAT3 and BMI-1 in HT1376 derived subcutaneous tumors by immunofluorescence staining. Paraffin-embedded tissue sections were boiled in 10mM sodium citrate solution+0.05% Tween-20 for 30 min (antigen retrieval), blocked in 3%BSA, 10% goat serum (1 h) and incubated with anti-CK14, anti-CK17, anti-STAT3, or anti-BMI-1 primary antibodies. Following washing with PBS, the sections were incubated with fluorophore (Alexa Fluor 488, or Alexa Fluor 546/555) labeled secondary antibodies (Thermo Fisher). Nuclei were stained with DAPI. Labeled cells were observed under a confocal microscope (BZ-X710, Keyence Corporation of America, Itasca, IL).
Transwell assays
Chemotactic motility of 253J cells and the sublines created in this study was evaluated using 8 µm-pore size Boyden chambers (Transwell plates, Corning/Costar Inc., Corning, NY) as described previously (26, 27). For invasion assays, upper well was coated with Matrigel (200 µl/cm2, Corning)
CRISPR/Cas9
CRISPR guide RNA (gRNA) sequence design was generated using CRISPR design software (Zhang Lab, MIT 2017, http://CRISPR.mit.edu) and gRNA sequences were cloned into the plentiCRISPRv2GFP backbone utilizing the Genome-Scale CRISPR Knock-Out protocols (GeCKO) (28, 29). The 20-nucleotide target sequences (Supplementary Table S2) along with the protospacer adjacent motif (PAM) sequences were confirmed to be specific to ARRB1 (Basic Local Alignment Search Tool [BLAST] search; National Center for Biotechnology Information, Bethesda MD) and further confirmed with the CasFinder algorithm31. Plasmids were transfected into HT1376-Luc cells using Lipofectamine 3000. After 48 h, GFP positive cells were sorted using the BD FACSAria II (Augusta University flow cytometry core), and plated at single cell dilutions into 96-well plates for clonal expansion. Due to random insertion or deletion mutations (InDels) resulting from CRISPR-Cas9, clonal expansion was necessary to obtain homogeneous cell populations. Colonies were tested for ARRB1 knockout via western blot.
Influx/Efflux studies of Gemcitabine
HT1376-EV and HT1376 ARRB2-OE cells were seeded in triplicate in 12-well plates and incubated with 10 nM Gem (TCI America, Cat# G0367) in presence of 10 nCi of Tritiated Gem (3H-Gem, Morvek MT 1572). Intracellular levels of 3H-Gem (influx studies) as well as 3H-Gem levels in the culture medium (efflux studies) were measured in a liquid scintillation counter at the indicated time points. Counts per minute (CPM) were normalized to plated cells/well.
Subcutaneous xenografts
All studies on mice were conducted using a protocol approved by the Augusta University Institutional Animal Care and Use Committee (IACUC). 1 × 106 HT1376-EV or HT1376 ARRB2-OE cells were subcutaneously injected into eight week old athymic nu/nu female mice (Envigo Inc, Harlan, IN). We monitored tumor growth, and response to Gem therapy weekly. Tumor volume was measured three times weekly with a handheld manual caliper (Tumorimeter and RECIST Caliper [Cancer Technologies Inc.] (30). Gem (25 mg/kg bodyweight) was administered intraperitoneally, twice per week. Treatment was initiated after tumors reached an average volume of 100 mm3. Animal weight was measured weekly.
Statistical analysis
The analysis of the ARRB expression data was conducted using the JMP-Pro 13 software program (SAS Campus Drive, Cary NC). The differences in transcript levels between various groups (e.g., normal vs. non-metastatic) from cohorts 1 and 2 were compared using the Mann-Whitney U-test, because the data showed non-normal distribution. For all analyses, P-values were two-tailed. The logistic regression single-parameter model (i.e., univariate analysis) was used to determine the association of clinical parameters, ARRB1 and ARRB2 levels with metastasis or disease-specific mortality (DSM) for cohort 1 and G+C treatment failure for cohort 2. Based on the Youden’s index (J; (31)) determined from the ROC curve, optimal cut-off values were obtained to compute sensitivity, specificity and accuracy. Boot-strap modeling with 1000 iterations was used to calculate mean, median and range for cut-off value, sensitivity and specificity. Cox-proportional hazards model (i.e., multivariate analysis) was used to determine which of the demographic and pathologic parameters and/or ARRB1/ARRB2 levels were significant in predicting metastasis and DSM (cohort 1) or treatment failure (cohort 2). Stratified Kaplan-Meier plots were prepared for ARRB1 and ARRB2 for clinical outcome parameters (i.e., metastasis and treatment failure). All quantitative data shown in Figures 2–4 and supplementary Figure S1, except the western blot quantification, were from three separate experiments; each data point represents the mean of the triplicate results. Western blots have been conducted twice. We analyzed the significance of data using the built-in statistical tools in Graph Pad Prism (Graph Pad Inc., San Diego, CA).
Figure 2: Expression of ARRBs in bladder cell lines and corresponding spheroid cultures.
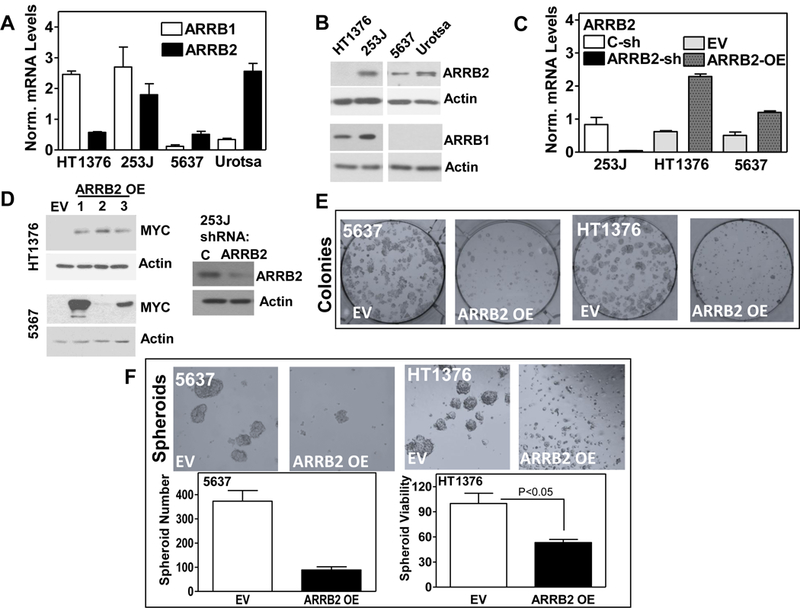
A: ARRB1 & ARRB2 mRNA levels in bladder cells measured by RT-qPCR. The levels were normalized to β-actin mRNA levels and plotted using following formula: Norm. mRNA Level= (2^(-dCT)) x 1000). Data: Mean ± SD., B: ARRB1 & ARRB2 protein levels. C: ARRB2 mRNA levels in transfectants. C-sh & ARRB2 sh: control & ARRB2 shRNA; EV: empty vector; OE: ARRB2 over-expression. D: Detection of ARRB2 protein level in stable 5637 and HT1376 clones overexpressing a Myc-DDK tagged ARRB2 construct and in 253J cells transfected with an ARRB2 shRNA plasmid E: Effect of ARRB2 over expression on colony formation. F: Effect of ARRB2 over expression on spheroid formation. Viability of HT1376 spheroids measured in Cell Titer-Glo 3D Cell Viability assay. Data: triplicate; Mean ± SD.
Figure 4: Response to Gemcitabine in ARRB2 overexpressing and ARRB2 depleted transfectants.
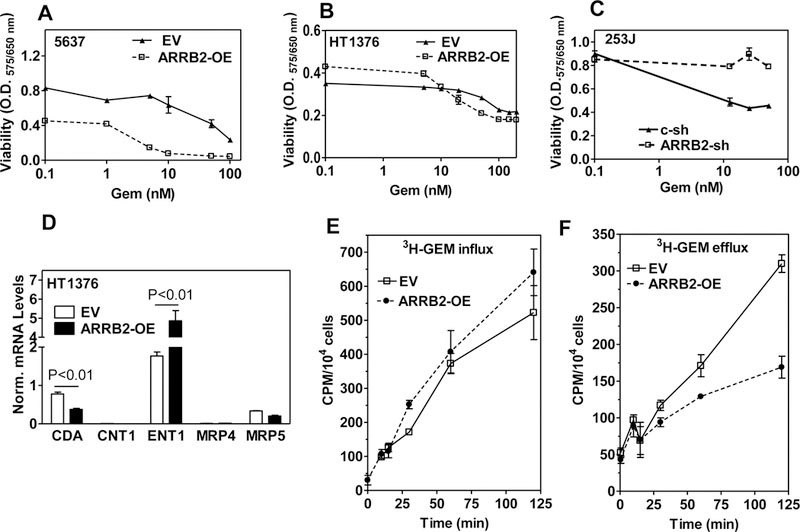
A, B, C: 5637, HT1376 (EV, ARRB2 OE) and 253J (C-sh, ARRB2-sh) transfectants were treated with GEM (0 – 100 nM) in growth medium in a 72 h assay. Cytotoxicity was determined by MTT assay. MTT assay was performed after 72 h. Data: Mean ± SD; triplicate. D: Detection of genes associated with GEM resistance by qPCR analysis. E: 2 × 104 cells/well were seeded in 12-well plates and incubated with 3H-Gem (10 nCi ) plus 10 nM unlabeled Gem up to 2 h. 3H-Gem uptake was measured at indicated time intervals by liquid scintillation counting. F: Cells were cultured as in E and incubated with 3H-Gem for 2 hours and efflux into the medium was measured at indicated time intervals.
Results:
Association of ARRB1 and ARRB2 expression with bladder cancer metastasis
We measured ARRB1 and ARRB2 transcript expression by RT-qPCR in normal bladder and bladder cancer specimens to determine whether they were differentially expressed. ARRB1 transcript levels in tumor specimens from patients who developed metastasis were 7.7-fold elevated compared to normal bladder and 5.2-fold elevated compared to bladder cancer specimens from patients who did not develop metastasis (Fig. 1A). In contrast, ARRB2 levels were significantly (2.1- and 4.7-fold) elevated in bladder cancer specimens from patients who did not develop metastasis, and in normal bladder tissues, respectively (Fig. 1B). ARRB1 levels were 2.7-fold higher and ARRB2 levels were 2-fold lower in bladder cancer specimens from patients who experienced disease specific mortality (DSM) during follow-up, when compared to those who did not (Fig. 1 A, B). In univariate analysis, lymph node invasion, ARRB1 and ARRB2 transcript levels significantly correlated with metastasis (Table 1). In multivariate analysis, only ARRB1 and ARRB2 levels were independent predictors of metastasis (Table 1). At cut-off values generated from the respective ROC curves, ARRB1 and ARRB2 markers had higher specificity (87% - 91%) than sensitivity (~ 70%) to predict metastasis. Based on bootstrap modeling, the median sensitivity and specificity values for both ARRB1 and ARRB2 were ~ 80% (Table 1). Both ARRB1 and ARRB2 levels also predicted DSM in univariate analysis, however, only ARRB2 was an independent predictor of DSM in multivariate analysis. At cut-off values generated from the ROC curves, ARRB2 showed higher specificity (84%) but slightly lower sensitivity (73.3%) than ARRB1 to predict DSM; although based on bootstrap modeling, both markers showed ~ 80% median sensitivity and specificity (Table 1).
Figure 1: Association of ARRB1 and ARRB2 levels with clinical outcome.
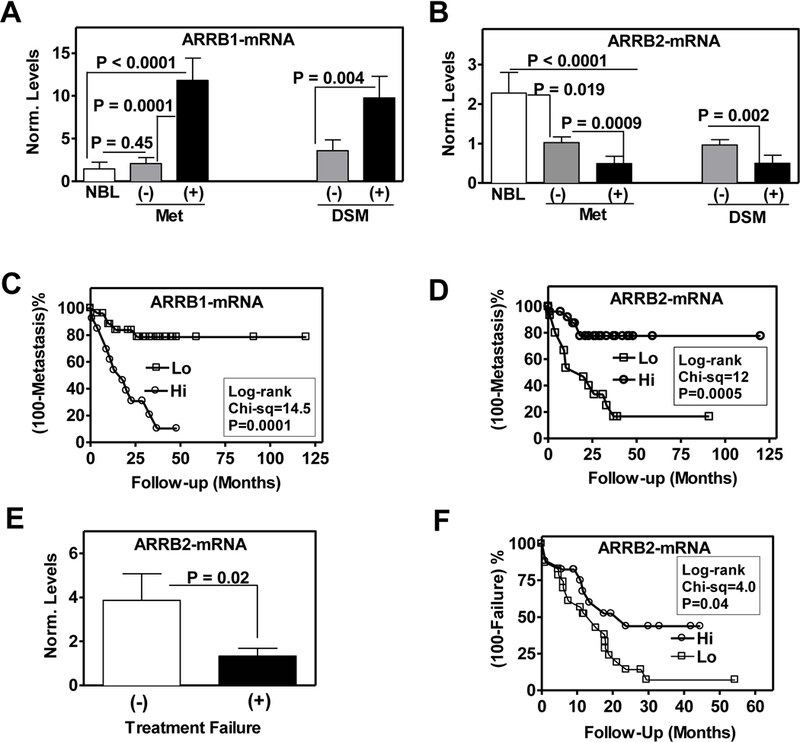
A and B: ARRB1and ARRB2 transcript levels in specimens from cohort 1 i.e., in normal bladder (NBL) tissues and in BC tissues from patients who either did not (−) or did (+) develop metastasis (Met) or died from the disease (DSM). P value: Mann-Whitney U test. C and D: Data on ARRB1 and ARRB2 transcript levels were stratified as high (Hi) and low (Lo) based on the Youden’s index from the ROC curves generated from the logistic regression (univariate) analysis. Stratified data were used to generate Kaplan-Meier plots with respect to metastasis. E: ARRB2 transcript levels in specimens from cohort 2; i.e., from patients who either failed (+) or did not fail (−) G+C treament for metastatic disease. F: BARR2 levels were stratified as Hi or Lo based on the Youden index from the ROC curve. Stratified data were used to generate Kaplan-Meier plots with respect to treatment failure. Norm. levels: target mRNA levels normalized to β-Actin mRNA.
Table 1. Determination of the association between clinical outcome and demographic and clinical parameters or marker levels.
Logistic regression (univariate analysis) was used to evaluate the association between metastasis and DSM and demographic/clinical parameters or marker levels for the patient dataset in cohort 1. OR: odds ratio; p-vales are two-tailed. For multivariate analysis, Cox proportional hazards analysis was performed. For the multivariate analysis data, Chi-square and P-values for each significant parameter are based on Wald’s test. For the ROC curve, area under the curve for Metastasis: BARR1: 0.855; BARR2: 0.844; for DSM: BARR1: 0.786; BARR2: 0.827. Median and range (10th – 90th percentile) cut-off values, sensitivity and specificity were determined by bootstrap modeling.
| Metastasis | DSM | |||||
|---|---|---|---|---|---|---|
| Univariate analysis | ||||||
| Parameter | Chi-sq | P-value | OR; 95% CI | Chi-sq | P-value | OR; 95% CI |
| Age | 0.41 | 0.521 | NS | 1.18 | 0.276 | NS |
| Gender | 1.19 | 0.275 | NS | 0.45 | 0.504 | NS |
| Grade (Hi/Lo) | 0.01 | 0.998 | NS | 0.0 | 0.999 | NS |
| Stage (< T2; ≥ T2) | 5.6 | 0.018 | 13.8; 1.6 – 122 | 0.00 | 0.996 | NS |
| Lymph node | 7.51 | 0.006 | 11.3; 2 – 63.6 | 8.86 | 0.0029 | 15; 2.5 – 89.2 |
| CIS | 1.52 | 0.218 | NS | 2.2 | 0.138 | NS |
| ARRB1 | 6.21 | 0.0137 | 1.35; 1.06–1.71 | 4.48 | 0.0342 | 1.12; 1.01 – 1.25 |
| ARRB2 | 7.94 | 0.005 | 0.03; 0.35 – 0.003 | 7.21 | 0.007 | 0.029; 0.38 – 0.002 |
| Multivariate analysis | ||||||
| Parameter | Chi-sq | P-value | RR; 95% CI | Chi-sq | P-value | RR; 95% CI |
| ARRB1 | 5.91 | 0.015 | 1.07; 1.01 – 1.13 | |||
| ARRB2 | 7.65 | 0.006 | 0.13; 0.86– 0.02 | 5.16 | 0.0231 | 0.17; 0.75 – 0.031 |
| Efficacy analysis | ||||||
| Marker | Cut-off | Sensitivity | Specificity | Cut-off | Sensitivity | specificity |
| ARRB1 |
ROC: 5.3 Mean: 4.3 Median: 4.0 Range: 3.9 – 5.3 |
68.8% 82% 83.3% 75% - 88.9% |
91.2% 85.6% 87.5% 80% - 91.7% |
ROC: 4.0 Mean: 4.0 Median: 4.0 Range: 2.3 – 5.1 |
78.6% 83.1% 83.3% 71.8% - 100% |
72% 75.8% 76% 67.4% - 84.6% |
| ARRB2 | ROC: 0.31 Mean: 0.5 Median: 0.31 Range: 0.3– 0.61 |
70.6% 75.6% 77.8% 66.7% – 87.5% |
87% 82.3% 84.7% 75% - 91.7% |
ROC: 0.31 Mean: 0.68 Median: 0.44 Range: 0.3 – 0.61 |
73.3% 74.7% 78.9% 66.7% - 85.7% |
84% 81.9% 83.3% 75% - 91.7% |
Kaplan-Meier plots showed that higher levels of ARRB1 significantly stratified the cohort into higher risk for metastasis (Fig 1C). Contrarily, lower levels of ARRB2 significantly stratified the cohort into higher risk for metastasis (Fig. 1D). Lower ARRB2 levels also significantly stratified patients into higher risk for DSM (P=0.039; supplementary Fig. S1).
Association of ARRB2 expression with G+C adjuvant treatment failure
G+C is the preferred first-line chemotherapy for treating metastatic bladder cancer. We measured ARRB1 and/or ARRB2 transcript levels in formalin fixed paraffin embedded (FFPE) cystectomy specimens from bladder cancer patients who later received adjuvant G+C chemotherapy for the treatment of metastatic disease. Although different primer sets were tried, ARRB1 transcript levels could not be measured in FFPE specimens. ARRB2 levels were significantly (3-fold) downregulated in cystectomy specimens from patients who failed G+C treatment when compared to those who did not fail treatment (Fig. 1E). In univariate analysis, pathologic stage and ARRB2 transcript levels significantly correlated with treatment failure (supplementary Table S4). In multivariate analysis, only ARRB2 was an independent predictor of treatment failure (supplementary Table S4). At cut-off values generated from the ROC curve, ARRB2 had 71% sensitivity and 75% specificity to predict treatment failure. Based on bootstrap modeling, the median sensitivity and specificity values for ARRB2 were 73% and 83.3%, respectively (supplementary Table S4). Kaplan-Meier plots showed that lower ARRB2 levels stratified patients into higher risk of G+C treatment failure (Fig. 1F).
Expression of beta-arrestins in bladder cancer cell lines
We evaluated expression of ARRBs in established bladder cancer cell lines using qPCR and western blot analysis. The immortalized normal bladder cell line UROTSA expressed ARRB2, however, it did not express ARRB1 at the protein level (Fig. 2A, B). The stage 2 bladder cancer cell line 5637 expressed low levels of ARRB2, while ARRB1 levels were not detectable at the protein level. 253J cells exhibited high expression of both beta-arrestins, whereas highly tumorigenic HT1376 cells only expressed ARRB1 (Fig. 2A, B). This heterogeneity of ARRB expression in normal vs cancer cell lines prompted us to investigate their role in bladder cancer cell proliferation. We generated stable 253J cells with depleted ARRB2 using shRNA, and 5637 and HT1376 clones overexpressing ARRB2. ARRB2 levels in the transfectants were monitored by qPCR and immunoblotting (Figure 2C, D).
ARRB2 overexpression abrogates colony- and spheroid-formation potential
The stable ARRB2 over-expressing HT1376 and 5637 cells formed significantly less and smaller colonies compared to corresponding controls (Fig. 2E). Since MIBC harbors CSCs (32), we inquired whether ARRBs regulate the CSC population which spheroids when seeded in low-adhesion plates with stem-cell medium (33). Spheroid formation was decreased by 4.6-fold in ARRB2-OE 5637 transfectants compared to corresponding controls (Fig 2F i, ii). Consistently, ARRB2-OE transfectants of HT1376 showed decreased spheroid viability (Fig. 2F iii, iv).
ARRB2 expression inversely correlates with CSC-like phenotypes
We investigated the expression of cytoskeletal markers that are associated with basal urothelial cells (34, 35) which are the origin of MIBC carcinomas (36). Depletion of ARRB2 in 253J - a cell line expressing high levels of ARRB2- resulted in a remarkable increase of both CK14 and CK17 (Fig. 3A). Consistently, both CK14 and CK17 mRNA levels were 4.4–7-fold reduced in ARRB2-OE transfectants of HT1376 cells (Fig. 3B); similar results were obtained in 5637 transfectants (supplementary Figure S2). The luminal marker Uroplakin was not detectable by qPCR in HT1376 parental as well as ARRB2-OE cells.
Figure 3: Expression of stem cell markers and analysis of motility and invasive potential following modulation of ARRB2 levels.
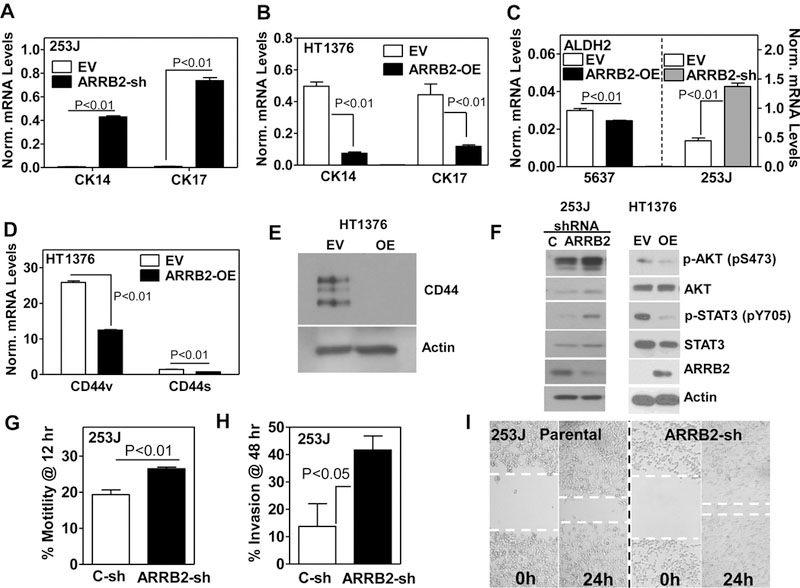
A, B & C: qPCR analysis for CK14, CK17 ALDH2 in bladder cancer cell transfectants. Data: Mean ± SD. D: qPCR analysis for CD44s and CD44v in HT1376 transfectants, Norm. levels: target mRNA levels normalized to β-Actin mRNA. E: Immunoblot for detection of CD44 in HT1376 transfectants. OE: ARRB2 over expression. F: Detection of AKT and STAT3 activation by Western Blot analysis following shRNA-mediated depletion of ARRB2 in 253J cells or ARRB2 over expression in HT1376 cells. β-Actin served as a loading control. OE: ARRB2 over expression. G, H: Chemotactic motility and invasion of 253J cells and ARRB2-sh transfectants in a Transwell assay. In the invasion assay, the top well was coated with Matrigel. % motility and invasion were calculated as [O.D. Bottom/ (O.D. top + bottom chamber)] x100. I: Migration of 253J c-sh and ARRB2-sh transfectants was examined in a wound healing assay. Dotted lines show the width of the scratch area that is not covered. EV: Empty Vector, OE: ARRB2 over expression.
Since ARRB2 overexpression reduced anchorage-independent growth (spheroid formation), we tested whether ARRB2 regulates the CSC-like phenotype. A typical stem cell marker ALDH2 decreased in 5637 ARRB2-OE cells, but showed an increase in 253J ARRB2-sh transfectants compared to corresponding controls (3-fold; P<0.001; Fig. 3C). Modulation of the ARRB2 levels did not affect the expression of another stem-cell marker, SOX2 (supplementary Figure S3). Moreover, overexpression of ARRB2 resulted in a significant decrease of the CD44 standard form (CD44s) and variants (CD44v) in HT1376 cells (Fig. 3D, E). Furthermore, we looked at STAT3 signaling, which is involved in CSC maintenance and self-renewal (37). Depletion of ARRB2 resulted in activation of STAT3 in 253J cells, whereas over expression of ARRB2 in HT1376 cells reduced the level of activated STAT3 (Fig. 3F). In addition, the levels of B-cell-specific Moloney murine leukemia virus insertion site 1 (BMI-1), a polycomb protein associated with CSC stemness (38), was significantly decreased in ARRB2-OE transfectants of 5637 cells compared to corresponding controls (supplementary Figure S2). In conclusion, depletion of ARRB2 in bladder cancer cells resulted in an increase of markers associated with low differentiation and stem cell properties, whereas opposing results were observed when ARRB2 was over expressed.
Depletion of ARRB2 promotes migration and invasion
Since the CSC phenotype associates with increased invasiveness, we examined the chemotactic motility and invasive potential of ARRB2 shRNA transfectants compared to controls in wound healing/scratch and transwell assays (24). Depletion of ARRB2 increased motility and invasion of 253J cells through basement membrane (Matrigel) (> 3.5-fold) (Fig. 3G, H) and enhanced migration in wound healing assays (>2, Fig. 3I). These results confirm our observations in tumor specimens from patients (Fig. 1).
ARRB2 regulates CDA-mediated Gem resistance
One of the hallmarks of CSCs is their resistance to chemotherapy. Therefore, we investigated whether over expression of ARRB2 affects sensitivity towards Gemcitabine (Gem) and Cisplatin (CIS), since Gemcitabine + Cisplatin (G + C) combination is the standard first-line treatment for MIBC (5–8). ARRB2 overexpression sensitized 5637 cells to Gem (IC50: EV: > 100 nM; OE: 16.5 nM, Fig. 4A). Similar results were observed in HT1376 cells (Fig. 4B). In 253J cells (Fig. 4C), ARRB2 depletion induced resistance to Gem induced cytotoxicity. Modulation of ARRB2 levels in bladder cancer cells did not affect sensitivity to CIS (supplementary Figure S4), suggesting, that ARRB2 overexpression specifically sensitizes bladder cells towards Gem.
Further, we determined the effect of over expression of ARRB2 on ENT1 –a protein involved in Gem influx- and CDA, an enzyme that converts Gem into inactive metabolites that are pumped out of the cell (39, 40). ARRB2 overexpression induced expression of ENT1, whereas it inhibited expression of CDA (Fig. 4D). Therefore, we further investigated whether ARRB2 regulates Gem influx into the cells as well as conversion of Gem to inactive metabolites. We measured the rate of 3H-Gem uptake and efflux in HT1376-EV and HT1376 ARRB2-OE cells. The cellular accumulation of 3H-Gem was significantly higher in HT1376 ARRB2-OE cells compared to HT1376-EV cells (Fig. 4 E). The efflux of 3H-Gem was slower in HT1376 ARRB2-OE cells (Fig. 4 F). Furthermore, qPCR analysis showed no significant change in the expression ABC transporters [(ABCC1 (MRP1), ABCC3 (MRP3), ABCC5 (MRP5), ABCC10 (MRP7), ABCB1 (MDR1), and ABCG2 (CD338)] in ARRB2-shRNA or ARRB2-OE transfectants compared to control cells (supplementary Figure S5 A, B). ABC transport inhibitors such as Verapamil (non-selective ABC transporter inhibitor) and Probenecid (inhibitor of MRP4 and MRP5) also did not sensitize cells to Gem (supplementary Figure S5 C, D). These results suggest that resistance to Gem following ARRB2 depletion was not attributed to elevated levels of ABC transporters.
In summary, over expression of ARRB2 sensitized bladder cancer cells towards Gem, whereas depletion of ARRB2 conferred resistance towards Gem in a mechanism that involved ENT1 and CDA, which play a crucial role in Gem uptake and its metabolic conversion to inactive intermediates, respectively.
ARRB2 over-expressing HT1376 tumors are more responsive to Gem treatment
To analyze the role of ARRB2 in growth and chemo-resistance of subcutaneous tumor xenografts, we injected HT1376 EV or HT1376 ARRB2-OE cells subcutaneously into mice and monitored tumor growth, and response to Gem therapy. Tumor take rates were >95% in both groups. We observed a decreased tumor growth (tumor volume and weight) in tumors overexpressing ARRB2 (HT1376 ARRB2-OE) compared to EV tumors (Fig. 5A, B). Importantly, ARRB2-OE xenografts responded better than EV tumors to Gem.
Figure 5: Tumor growth of ARRB2-OE and ARRB2-sh transfectants.
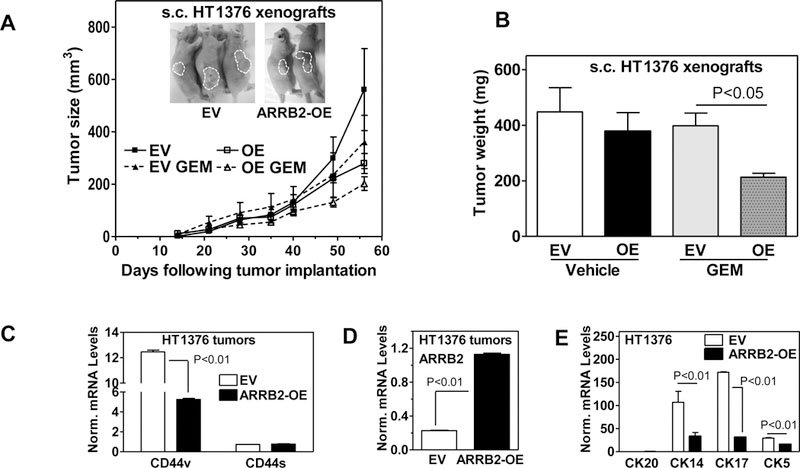
A, B: Xenograft tumors were generated by injecting 1 × 106 HT1376-EV or HT1376 ARRB2-OE cells subcutaneously into nude mice. Xenograft tumor growth, and response to Gemcitabine treatment was monitored weekly. Gem (25 mg/kg bodyweight) was administered intraperitoneally, twice per week. C, D, E: Analysis of CD44s, CD44v, ARRB2, and cytokeratin mRNA levels in HT1376 subcutaneous tumor xenografts by qPCR. Norm. levels: target mRNA levels normalized to β-Actin mRNA.
We examined expression of CD44 and ARRB2 in HT1376 EV vs HT1376 ARRB2-OE tumors by qPCR analysis (Fig. 5C, G). CD44v mRNA (qPCR) was significantly reduced in HT1376-ARRB2-OE subcutaneous tumor xenografts compared to HT1376-EV tumors (Fig. 5C). We did not detect CK20 in HT1376 cells (Fig. 5E). However, basal cell markers (CK14, CK17, CK5) were decreased in tumor cells over expressing ARRB2 compared to tumors derived from corresponding controls, confirming that ARRB2 expression inversely correlates with expression of basal markers in vivo (Fig. 5E). Consistently, we observed a dramatic reduction of CK17, CK14 expression, as well as expression of CSC markers BMI-1, STAT3 and CD44 using immunofluorescence (supplementary Fig. 6A-F). While ARRB2 overexpression in tumors did not completely abrogate expression of CD44, we did not observe any co-localization of ARRB2 and CD44 (supplementary Fig. 6E, F). Taken together, HT1376 ARRB2-OE tumors exhibited remarkably reduced levels of CSC markers.
ARRB1 positively regulates the CSC phenotype
Next, we examined the role of ARRB1 in regulating CSC properties. shRNA mediated ARRB1 depletion inhibited clonogenic growth and anchorage-independent spheroid formation (supplementary Fig. S7A). We utilized the CRISPR/Cas9 technology to completely abrogate ARRB1 expression in HT1376luc cells (stably expressing luciferase). The clones were screened for ARRB1 deletion using western blot analysis (supplementary Fig. S7B). Importantly, deletion of ARRB1 resulted in abrogation of stem cell marker CD44 and reduced expression of BMI-1 (supplementary Fig. S7B, C). These results suggest that ARRB1 and ARRB2 have opposing functions in regulating the CSC markers BMI-1 and CD44.
Discussion:
This is the first study demonstrating an inverse pattern of expression for ARRB1 and ARRB2 in the same set of bladder cancer specimens. Further, this expression pattern correlates with prognostic significance for predicting metastasis and failure of G+C adjuvant chemotherapy. The finding that ARRBs correlate with clinical outcome is significant for several reasons. First, we observed a reciprocal expression of ARRB1 and ARRB2 in localized and metastatic tumors. The up-regulation of ARRB1 or down-regulation of ARRB2 both positively correlated with metastasis. This clinical observation strongly supports our mechanistic studies, which show that while ARRB1 is a positive regulator, ARRB2 is a negative regulator of invasive and stem cell phenotype in preclinical models of bladder cancer. Second, in cohort 1, the majority of the clinical specimens (74.4%) were from patients with MIBC. This is a high-risk population for developing metastasis and even in this patient cohort ARRB1 and ARRB2 predicted metastasis. Moreover, ARRB2 and ARRB1 levels were independent predictors of metastasis and ARRB2 was an independent predictor for DSM. Third, our mechanistic data establish the basis for ARRB2 (and ARRB1) as a functional biomarker because ARRB2 expression induces Gem resistance by increasing CDA expression. Fourth, among patients with metastatic bladder cancer, ARRB2 downregulation correlated with response to G+C treatment; low ARRB2 expression was an independent predictor of G+C treatment failure. Fifth, since the expression of ARRB2 can be measured both in fresh-frozen and in archival formalin-fixed specimens, it should allow for the flexibility of measuring ARRB2 expression in archived cystectomy specimens later, i.e. when a patient develops metastasis and is a candidate for adjuvant therapy. G+C is the preferred first-line adjuvant chemotherapy regimen for patients with metastatic bladder cancer. Therefore, measurement of ARRB2 levels in cystectomy specimens may not only allow identification of patients who have a high risk for metastasis, but may also further stratify these patients as either responders or non-responders before starting G+C treatment. Patients stratified as non-responders may then be offered another treatment such as, MVAC or PD-1/PD-L1 immunotherapy (41, 42).
Increasing evidence suggests that the high recurrence rate of bladder cancer is attributed to the presence of a small population of cells within a tumor that are characterized by stem cell properties. We observed that expression of ARRB2 inversely correlated with CSC properties such as expression of CSC markers and self-renewal potential in bladder cancer cell lines. A recent study demonstrated that ALDH2 and SOX2 provide a 2-gene stem-like signature that discriminates muscle-invasive from non-muscle invasive tumors with an accuracy of 93% (43). Interestingly, we did not observe any significant effect on expression of SOX2 upon overexpression of ARRB2 in HT1376 and 5637 cells (Supplementary Figure S3). SOX2 expression was not detectable in 253J cells. However, we detected a significant increase of ALDH2 in ARRB2 depleted cells (Fig 3C). Consistently, over expression of ARRB2 resulted in a decrease of ALDH2 as well as other CSC markers such as CD44- a CSC marker that is associated with metastasis and cisplatin resistance (27, 32, 38, 44). There is compelling evidence that MIBCs exclusively derive from stem cells in the basal urothelium (36). Therefore, we also examined how ARRB2 regulates markers associated with the basal phenotype, such as CK14 and CK17. De novo expression of CK14 is indicative of squamous differentiation that is associated with an unfavorable prognosis, whereas CK17, a basal type cytokeratin, is associated with poor prognosis (34, 45, 46). We demonstrated that ARRB2 negatively regulated both CK14 and CK17. These data suggest that ARRB2 is absent in tumor initiating cells. It is noteworthy, that while HT1376 cells -which lack expression of ARRB2- formed fast growing tumors in vivo, cell lines that endogenously expressed ARRB2 (e.g. 253J) failed to form tumors in nude mice.
There are several known CSC markers, nevertheless, bladder cancer is a highly heterogeneous disease and it is likely that this heterogeneity exists at the stem cell level (43). Similar to other cancers, there is no universal marker to identify CSC in bladder cancer. However, CSCs are universally characterized by the ability to self-renew (47). Thus, unveiling the molecular pathways that regulate self-renewal of CSCs will open new therapeutic avenues for targeting CSCs resulting in a decrease in the tumor relapse rates. At first, we analyzed the effect of ARRB2 modulation on STAT3 -a transcription factor regulating the self-renewal and invasive potential of bladder CSC-like cells (37, 48). We demonstrated that ARRB2 negatively regulated STAT3 activation. Similarly, ARRB2 negatively regulated expression of the transcription factor and CSC marker BMI-1. BMI-1 is an essential component of the polycomb regulatory complex 1 (PRC1) which plays a key role in chromatin organization and regulation of CSC marker expression and self-renewal (49). High expression of BMI-1 is associated with poor prognosis in bladder cancer (50). Importantly, CRISPR/Cas9 deletion of ARRB1 dramatically reduced BMI-1 levels indicating that, in contrast to ARRB2, ARRB1 positively regulated BMI-1. Furthermore, our results from functional self-renewal assays (spheroid formation assays) confirmed that ARRB1 positively regulates self-renewal, whereas ARRB2 suppressed this CSC property.
In summary, the current work demonstrates that ARRBs regulate the CSC-like phenotype in bladder cancer cell lines. We showed that ARRB1 and ARRB2 have opposing functions, with ARRB1 inducing and ARRB2 negatively regulating the CSC-like phenotype and self-renewal potential- the real driving force behind tumor growth. Importantly, the present work demonstrates the prognostic significance of ARRBs for predicting metastasis and response to chemotherapy in bladder cancer.
Supplementary Material
Acknowledgements:
We thank Mr. Luis Lopez for help with some of the animal experiments and western blotting of CD44 antigen, members of Dr. Ali S. Arbab laboratory, and various facilities managers at the Georgia Cancer Center, Augusta University.
Financial support: This work was supported in parts by: Biomedical Laboratory Research and Development, VA Office of Research and Development: BX 001517–01 and BX003862–01A2 (B. L. Lokeshwar); the United States Health and Human Services Awards: NIH-NCI-1R01CA156776–01 (B. L. Lokeshwar); NIH-NCI-R01 CA 227277–01A1 (V. B. Lokeshwar) and the Department of Defense Congressionally Mandated Medical Research Award: W81XWH1810277 (V. B. Lokeshwar).
Footnotes
Conflict of interest: The authors declare that they have no conflict of interest.
REFERENCES
- 1.Amin MB, McKenney JK, Paner GP, Hansel DE, Grignon DJ, Montironi R, et al. ICUD-EAU International Consultation on Bladder Cancer 2012: Pathology. Eur Urol 2013;63(1):16–35. [DOI] [PubMed] [Google Scholar]
- 2.Amin MB, Smith SC, Reuter VE, Epstein JI, Grignon DJ, Hansel DE, et al. Update for the practicing pathologist: The International Consultation On Urologic Disease-European association of urology consultation on bladder cancer. Mod Pathol 2015;28(5):612–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chang SS, Boorjian SA, Chou R, Clark PE, Daneshmand S, Konety BR, et al. Diagnosis and Treatment of Non-Muscle Invasive Bladder Cancer: AUA/SUO Guideline. J Urol 2016;196(4):1021–9. [DOI] [PubMed] [Google Scholar]
- 4.Woldu SL, Bagrodia A, Lotan Y. Guideline of guidelines: non-muscle-invasive bladder cancer. BJU Int 2017;119(3):371–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Funt SA, Rosenberg JE. Systemic, perioperative management of muscle-invasive bladder cancer and future horizons. Nat Rev Clin Oncol 2017;14(4):221–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gupta S, Mahipal A. Role of systemic chemotherapy in urothelial urinary bladder cancer. Cancer Control 2013;20(3):200–10. [DOI] [PubMed] [Google Scholar]
- 7.Galsky MD, Pal SK, Chowdhury S, Harshman LC, Crabb SJ, Wong YN, et al. Comparative effectiveness of gemcitabine plus cisplatin versus methotrexate, vinblastine, doxorubicin, plus cisplatin as neoadjuvant therapy for muscle-invasive bladder cancer. Cancer 2015;121(15):2586–93. [DOI] [PubMed] [Google Scholar]
- 8.Gandhi NM, Baras A, Munari E, Faraj S, Reis LO, Liu JJ, et al. Gemcitabine and cisplatin neoadjuvant chemotherapy for muscle-invasive urothelial carcinoma: Predicting response and assessing outcomes. Urol Oncol 2015;33(5):204 e1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ma L, Pei G. Beta-arrestin signaling and regulation of transcription. J Cell Sci 2007;120(Pt 2):213–8. [DOI] [PubMed] [Google Scholar]
- 10.Eichel K, Jullie D, Barsi-Rhyne B, Latorraca NR, Masureel M, Sibarita JB, et al. Catalytic activation of beta-arrestin by GPCRs. Nature 2018;557(7705):381–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lan T, Wang HR, Zhang ZH, Zhang MS, Qu YM, Zhao ZT, et al. Downregulation of beta-arrestin 1 suppresses glioblastoma cell malignant progression vis inhibition of Src signaling. Experimental Cell Research 2017;357(1):51–8. [DOI] [PubMed] [Google Scholar]
- 12.Buchanan FG, Gorden DL, Matta P, Shi Q, Matrisian LM, DuBois RN. Role of beta-arrestin 1 in the metastatic progression of colorectal cancer. Proceedings of the National Academy of Sciences of the United States of America 2006;103(5):1492–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kallifatidis G, Munoz D, Singh RK, Salazar N, Hoy JJ, Lokeshwar BL. Beta-Arrestin-2 Counters CXCR7-mediated EGFR Transactivation and Proliferation. Mol Cancer Res 2016. [DOI] [PMC free article] [PubMed]
- 14.DeWire SM, Ahn S, Lefkowitz RJ, Shenoy SK. Beta-arrestins and cell signaling. Annu Rev Physiol 2007;69:483–510. [DOI] [PubMed] [Google Scholar]
- 15.Shenoy SK, Han S, Zhao YL, Hara MR, Oliver T, Cao Y, et al. beta-arrestin1 mediates metastatic growth of breast cancer cells by facilitating HIF-1-dependent VEGF expression. Oncogene 2012;31(3):282–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Raghuwanshi SK, Nasser MW, Chen X, Strieter RM, Richardson RM. Depletion of beta-arrestin-2 promotes tumor growth and angiogenesis in a murine model of lung cancer. J Immunol 2008;180(8):5699–706. [DOI] [PubMed] [Google Scholar]
- 17.Yang Y, Guo Y, Tan S, Ke B, Tao J, Liu H, et al. beta-Arrestin1 enhances hepatocellular carcinogenesis through inflammation-mediated Akt signalling. Nat Commun 2015;6:7369. [DOI] [PubMed] [Google Scholar]
- 18.Perumal D, Pillai S, Nguyen J, Schaal C, Coppola D, Chellappan SP. Nicotinic acetylcholine receptors induce c-Kit ligand/Stem Cell Factor and promote stemness in an ARRB1/ beta-arrestin-1 dependent manner in NSCLC. Oncotarget 2014;5(21):10486–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shu Y, Zhou X, Qi X, Liu S, Li K, Tan J, et al. beta-Arrestin1 promotes the self-renewal of the leukemia-initiating cell-enriched subpopulation in B-lineage acute lymphoblastic leukemia related to DNMT1 activity. Cancer Lett 2015;357(1):170–8. [DOI] [PubMed] [Google Scholar]
- 20.Zhan Y, Xu C, Liu Z, Yang Y, Tan S, Yang Y, et al. beta-Arrestin1 inhibits chemotherapy-induced intestinal stem cell apoptosis and mucositis. Cell Death Dis 2016;7:e2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu Z, Tian H, Jiang J, Yang Y, Tan S, Lin X, et al. beta-Arrestin-2 modulates radiation-induced intestinal crypt progenitor/stem cell injury. Cell Death Differ 2016;23(9):1529–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kramer MW, Escudero DO, Lokeshwar SD, Golshani R, Ekwenna OO, Acosta K, et al. Association of hyaluronic acid family members (HAS1, HAS2, and HYAL-1) with bladder cancer diagnosis and prognosis. Cancer 2011;117(6):1197–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rafehi H, Orlowski C, Georgiadis GT, Ververis K, El-Osta A, Karagiannis TC. Clonogenic assay: adherent cells. J Vis Exp 2011(49). [DOI] [PMC free article] [PubMed]
- 24.Hoy JJ, Kallifatidis G, Smith DK, Lokeshwar BL. Inhibition of androgen receptor promotes CXC-chemokine receptor 7-mediated prostate cancer cell survival. Scientific Reports 2017;7(1):3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Singh RK, Lokeshwar BL. The IL-8-regulated chemokine receptor CXCR7 stimulates EGFR signaling to promote prostate cancer growth. Cancer Res 2011;71(9):3268–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lokeshwar BL, Selzer MG, Zhu BQ, Block NL, Golub LM. Inhibition of cell proliferation, invasion, tumor growth and metastasis by an oral non-antimicrobial tetracycline analog (COL-3) in a metastatic prostate cancer model. Int J Cancer 2002;98(2):297–309. [DOI] [PubMed] [Google Scholar]
- 27.Lokeshwar BL, Lokeshwar VB, Block NL. Expression of CD44 in prostate cancer cells: association with cell proliferation and invasive potential. Anticancer Res 1995;15(4):1191–8. [PubMed] [Google Scholar]
- 28.Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 2014;343(6166):84–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 2014;11(8):783–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tomayko MM, Reynolds CP. Determination of subcutaneous tumor size in athymic (nude) mice. Cancer Chemother Pharmacol 1989;24(3):148–54. [DOI] [PubMed] [Google Scholar]
- 31.Youden WJ. Index for rating diagnostic tests. Cancer 1950;3(1):32–5. [DOI] [PubMed] [Google Scholar]
- 32.Goodwin Jinesh G, Willis DL, Kamat AM. Bladder cancer stem cells: biological and therapeutic perspectives. Curr Stem Cell Res Ther 2014;9(2):89–101. [DOI] [PubMed] [Google Scholar]
- 33.Kallifatidis G, Rausch V, Baumann B, Apel A, Beckermann BM, Groth A, et al. Sulforaphane targets pancreatic tumour-initiating cells by NF-kappaB-induced antiapoptotic signalling. Gut 2009;58(7):949–63. [DOI] [PubMed] [Google Scholar]
- 34.He X, Marchionni L, Hansel DE, Yu W, Sood A, Yang J, et al. Differentiation of a highly tumorigenic basal cell compartment in urothelial carcinoma. Stem Cells 2009;27(7):1487–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Volkmer JP, Sahoo D, Chin RK, Ho PL, Tang C, Kurtova AV, et al. Three differentiation states risk-stratify bladder cancer into distinct subtypes. Proc Natl Acad Sci U S A 2012;109(6):2078–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shin K, Lim A, Odegaard JI, Honeycutt JD, Kawano S, Hsieh MH, et al. Cellular origin of bladder neoplasia and tissue dynamics of its progression to invasive carcinoma. Nat Cell Biol 2014;16(5):469–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang Z, He L, Lin K, Zhang Y, Deng A, Liang Y, et al. The KMT1A-GATA3-STAT3 Circuit Is a Novel Self-Renewal Signaling of Human Bladder Cancer Stem Cells. Clin Cancer Res 2017;23(21):6673–85. [DOI] [PubMed] [Google Scholar]
- 38.Zhu D, Wan X, Huang H, Chen X, Liang W, Zhao F, et al. Knockdown of Bmi1 inhibits the stemness properties and tumorigenicity of human bladder cancer stem cell-like side population cells. Oncol Rep 2014;31(2):727–36. [DOI] [PubMed] [Google Scholar]
- 39.Neff T, Blau CA. Forced expression of cytidine deaminase confers resistance to cytosine arabinoside and gemcitabine. Exp Hematol 1996;24(11):1340–6. [PubMed] [Google Scholar]
- 40.Zhang J, Visser F, King KM, Baldwin SA, Young JD, Cass CE. The role of nucleoside transporters in cancer chemotherapy with nucleoside drugs. Cancer Metastasis Rev 2007;26(1):85–110. [DOI] [PubMed] [Google Scholar]
- 41.Rotte A, Jin JY, Lemaire V. Mechanistic overview of immune checkpoints to support the rational design of their combinations in cancer immunotherapy. Ann Oncol 2018;29(1):71–83. [DOI] [PubMed] [Google Scholar]
- 42.Szabados B, van Dijk N, Tang YZ, van der Heijden MS, Wimalasingham A, Gomez de Liano A, et al. Response Rate to Chemotherapy After Immune Checkpoint Inhibition in Metastatic Urothelial Cancer. Eur Urol 2018;73(2):149–52. [DOI] [PubMed] [Google Scholar]
- 43.Ferreira-Teixeira M, Parada B, Rodrigues-Santos P, Alves V, Ramalho JS, Caramelo F, et al. Functional and molecular characterization of cancer stem-like cells in bladder cancer: a potential signature for muscle-invasive tumors. Oncotarget 2015;6(34):36185–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chan KS, Espinosa I, Chao M, Wong D, Ailles L, Diehn M, et al. Identification, molecular characterization, clinical prognosis, and therapeutic targeting of human bladder tumor-initiating cells. Proc Natl Acad Sci U S A 2009;106(33):14016–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Choi W, Porten S, Kim S, Willis D, Plimack ER, Hoffman-Censits J, et al. Identification of distinct basal and luminal subtypes of muscle-invasive bladder cancer with different sensitivities to frontline chemotherapy. Cancer Cell 2014;25(2):152–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gruver AM, Amin MB, Luthringer DJ, Westfall D, Arora K, Farver CF, et al. Selective immunohistochemical markers to distinguish between metastatic high-grade urothelial carcinoma and primary poorly differentiated invasive squamous cell carcinoma of the lung. Arch Pathol Lab Med 2012;136(11):1339–46. [DOI] [PubMed] [Google Scholar]
- 47.Ohishi T, Koga F, Migita T. Bladder Cancer Stem-Like Cells: Their Origin and Therapeutic Perspectives. Int J Mol Sci 2016;17(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ho PL, Lay EJ, Jian W, Parra D, Chan KS. Stat3 activation in urothelial stem cells leads to direct progression to invasive bladder cancer. Cancer Res 2012;72(13):3135–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wicha MS. Targeting self-renewal, an Achilles’ heel of cancer stem cells. Nat Med 2014;20(1):14–5. [DOI] [PubMed] [Google Scholar]
- 50.Qin ZK, Yang JA, Ye YL, Zhang X, Xu LH, Zhou FJ, et al. Expression of Bmi-1 is a prognostic marker in bladder cancer. BMC Cancer 2009;9:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.