

 Binuclear platinum(ii) diphosphite complexes efficiently catalyzed the photo-dehydrogenation of alcohols and N-heterocycles (75 examples in total) under oxygen-free and acceptorless conditions at room temperature.
Binuclear platinum(ii) diphosphite complexes efficiently catalyzed the photo-dehydrogenation of alcohols and N-heterocycles (75 examples in total) under oxygen-free and acceptorless conditions at room temperature.
Abstract
Although photoredox catalysis employing Ru(ii) and Ir(iii) complexes as photocatalysts has emerged as a versatile tool for oxidative C–H functionalization under mild conditions, the need for additional reagents acting as electron donor/scavenger for completing the catalytic cycle undermines the practicability of this approach. Herein we demonstrate that photo-induced oxidative C–H functionalization can be catalysed with high product yields under oxygen-free and acceptorless conditions via inner-sphere atom abstraction by binuclear platinum(ii) diphosphite complexes. Both alcohols (51 examples), particularly the aliphatic ones, and saturated N-heterocycles (24 examples) can be efficiently dehydrogenated under light irradiation at room temperature. Regeneration of the photocatalyst by means of reductive elimination of dihydrogen from the in situ formed platinum(iii)-hydride species represents an alternative paradigm to the current approach in photoredox catalysis.
Introduction
Oxidative C–H functionalization reactions1 are important tools commonly used in organic synthesis but the majority of these reactions require a stoichiometric amount of oxidant and/or high temperature,2,3 thereby constraining the variety of compatible functional groups and limiting large-scale application (Scheme 1). In order to restrict the need for hazardous reagents, using air as the terminal oxidant and/or photoredox catalysis are appealing approaches for the development of environment-friendly oxidation reactions under mild conditions. In the case of photo-redox catalysis, up to now, the widely used photocatalysts include polypyridyl Ru(ii) and Ir(iii) complexes,4 which initiate the photochemical oxidation by an outer-sphere, single electron transfer process. In most scenarios, however, a sacrificial electron donor/acceptor is needed to provide/scavenge electron(s) to complete the catalytic cycle, which is undesirable from both environmental and economic points of view. To overcome this, several groups have realized oxidant-free and acceptorless dehydrogenation of alcohols and saturated N-heterocycles at room temperature (RT) by combining photo-catalysis and metal-catalysis (i.e., dual catalysis; Scheme 1c).5–9 Given the importance of sustainability in contemporary research, we conceive that a more attractive approach would be to employ a single photocatalyst capable of activating C–H bonds by hydrogen atom abstraction to form a metal-hydride intermediate that subsequently releases dihydrogen by reductive elimination or disproportionation. Herein, we show that the binuclear platinum(ii) diphosphite complexes, previously developed by Gray and coworkers,10–12 can act as versatile photocatalysts for oxidant-free and acceptorless dehydrogenation of alcohols including aliphatic alcohols, and saturated N-heterocycles, with these substrates present in limiting amounts at RT (Scheme 1d). The simple protocol we describe in this work has also been employed for the direct generation of lactones and one-pot syntheses of biologically active compounds.
Scheme 1. Strategies for the dehydrogenation of alcohols and N-heterocycles.

Results and discussion
Binuclear platinum(ii) diphosphite complexes [Pt2(P2O5H2)4]4– (abbreviated as [Pt2(pop)4]4–) and their perfluoroborated derivatives [Pt2(P2O5(BF2)2)4]4– ([Pt2(pop-BF2)4]4–) have long-lived, strongly emissive 3[5dσ* → 6pσ] excited states with emission maxima at ∼510 nm and with lifetimes of around 10 μs.13,14 Compared to [Pt2(pop)4]4–, the 3[5dσ*6pσ] excited state of [Pt2(pop-BF2)4]4– is a much stronger oxidant with an E(*3Pt2(pop-BF2)4–/Pt2(pop-BF2)5–) of +0.86 V vs. Cp2Fe+/0,14b which is advantageous for oxidative C–H functionalization. Electronically excited [Pt2(pop)4]4– has previously been known to react with a variety of substrates, including alcohols, hydrocarbons and alkylhalides, under the conditions of substrates present in large excess.13
Details of the synthesis and characterization of the platinum(ii) diphosphite complexes (Fig. 1) are given in the ESI.† (Bu4N)4[Pt2(P2O5H2)4] (Ptpop-II) and [(C16H33)2(CH3)2N]4 [Pt2(P2O5H2)4] (Ptpop-III) were obtained via cation exchange of K4[Pt2(P2O5H2)4] (Ptpop-I) with tetrabutylammonium and dihexadecyldimethylammonium salts, respectively. In accordance with Gray and Vlček's reports,14a dissolution of Ptpop-III in neat F3B·OEt2 and stirring at RT for a few days resulted in the generation of perfluoroborated [(C16H33)2(CH3)2N]4[Pt2(P2O5(BF2)2)4] (Ptpop-BF2-IV). The photophysical properties of Ptpop-III and Ptpop-BF2-IV are similar to those of their [Bu4N] counterparts reported in the literature.14a
Fig. 1. Top: structures of the binuclear platinum(ii) diphosphite complexes. Bottom: electronic absorption (solid lines) and emission (dashed lines) spectra of Ptpop-I and Ptpop-BF2-IV in degassed H2O and MeCN respectively at room temperature. The emission quantum yield (Φ) and triplet emission lifetime (τ) of Ptpop-BF2-IV are shown.

The photocatalytic activity of these complexes was first examined by using Ptpop-I as a catalyst and 1-phenylethanol as a test substrate. Irradiation of a degassed aqueous solution containing 1-phenylethanol and 0.5 mol% of Ptpop-I with 410 nm LED light (12 W) under N2 afforded acetophenone (b1) in 45% yield together with the release of hydrogen gas (Table 1, entry 1). Aqueous solvent mixtures with MeCN, MeOH, DMF or acetone resulted in a high yield but with low selectivity due to the formation of a pinacol product (b0) (Table 1, entries 2–5). We found that when the reaction was conducted in a mixture of H2O and CH2Cl2, using nBu4NCl as the phase transfer reagent, acetophenone was obtained exclusively with 96% yield (Table 1, entry 6). Control experiments revealed that no reaction occurred in the absence of the photocatalyst Ptpop-I or light, indicating that both components were essential to the reaction (Table 1, entries 7–8). From these experiments, we established a simple and easily manipulated protocol: 0.5 mol% Ptpop-I, 5 mol% nBu4NCl in a mixture of H2O/CH2Cl2 (1 : 3) under N2 and 410 nm LED irradiation.
Table 1. Optimization of reaction conditions a .
| |||
| Entry | Conditions | Yield (%) |
|
| b0 | b1 | ||
| 1 | H2O | — | 45 |
| 2 | H2O/CH3CN 2 : 1 | 22 | 26 |
| 3 | H2O/CH3OH 2 : 1 | 78 | 20 |
| 4 | H2O/DMF 2 : 1 | 67 | 30 |
| 5 | H2O/CH3COCH3 2 : 1 | 76 | 21 |
| 6 | H2O/CH2Cl2 1 : 3, 5% Bu4NCl | — | 96 |
| 7 c | H2O/CH2Cl2 1 : 3, 5% Bu4NCl | — | — |
| 8 d | H2O/CH2Cl2 1 : 3, 5% Bu4NCl | — | — |
a0.4 mmol a1, 0.5 mol% Ptpop-I, 3 mL solvent, degassed for 15 min under N2, 410 nm LED irradiation, 8 hours.
bYield determined by 1H NMR spectroscopy using CH2Br2 as internal standard.
cNo light.
dNo Ptpop-I.
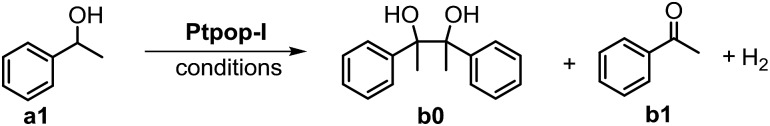
We studied various secondary benzylic alcohols to examine the substrate scope of the protocol. As shown in Table 2, a wide range of secondary benzylic alcohols with different electron-withdrawing or electron-donating substituents on the aromatic ring were oxidized to the corresponding ketones in 82–98% yields (Table 2, b1–b4, b6–b10, b21). However, the presence of an ortho-substituent on the aromatic ring led to only moderate product yields (Table 2, b5, b11), which is probably due to steric effects. This method can also be extended to the oxidation of benzhydrols with 92–99% product yields (Table 2, b12–b16). Cyclic benzylic alcohols with different substituents were also effective substrates (Table 2, b17–b20), but secondary aliphatic alcohols were ineffective substrates under the conditions examined (Table 2, b22, b23).
Table 2. Substrate scope of dehydrogenation of alcohols by Ptpop-I a .

|
|
a0.4 mmol alcohol, 5 mol% nBu4NCl, H2O/CH2Cl2 1 : 3 (3 mL), under N2, 410 nm LEDs irradiation, 8 hours; mol% loading of Ptpop-I is shown in parenthesis, isolated yields.
bIn H2O and without nBu4NCl.
cIn H2O and without nBu4NCl, yield of product was determined by GC-FID.
Primary benzylic alcohols were found to be effective substrates, but generally less reactive than secondary benzylic alcohols. Various primary benzylic alcohols were oxidized to the corresponding aldehydes in 78–98% yields (Table 2, b24–b32, b36). Only 55% yield of benzaldehyde was obtained (Table 2, b33). The steric effect was also observed when the ortho-position of the aromatic ring was substituted, leading to a decrease in reaction efficiency (Table 2, b34). 4-Fluorobenzyl alcohol was found to give 4-fluorobenzaldehyde in 45% yield (Table 2, b35) together with side products including 4′-fluoroacetophenone, 2-chloro-4′-fluoroacetophenone and 4-fluorobenzoic acid. The ratio of 4-fluorobenzaldehyde : 4′-fluoroacetophenone : 2-chloro-4′-fluoroacetophenone : 4-fluorobenzoic acid was estimated to be 1 : 0.13 : 0.11 : 0.21 by 19F NMR spectroscopy. The formation of 2-chloro-4′-fluoroacetophenone suggests that a ˙CH2Cl radical may have formed (from CH2Cl2) and reacted with the α-hydroxybenzyl radical (p-F-PhCH(OH)˙) generated from hydrogen atom abstraction of 4-fluorobenzyl alcohol with triplet excited Ptpop-I in the reaction. Further chlorine atom abstraction from this acetophenone would give 4′-fluoroacetophenone. A heterocyclic primary alcohol was oxidized to give the corresponding aldehyde with 60% yield (Table 2, b37). Remarkably, vanillin, which is widely used in food, fine chemical industries and pharmaceuticals, was obtained in 90% yield by photo-dehydrogenation of 4-hydroxy-3-methoxybenzyl alcohol (Table 2, b38). Compared to the previously reported methods,15 our protocol shortened the synthetic procedures and omitted the use of oxidants and harsh conditions.
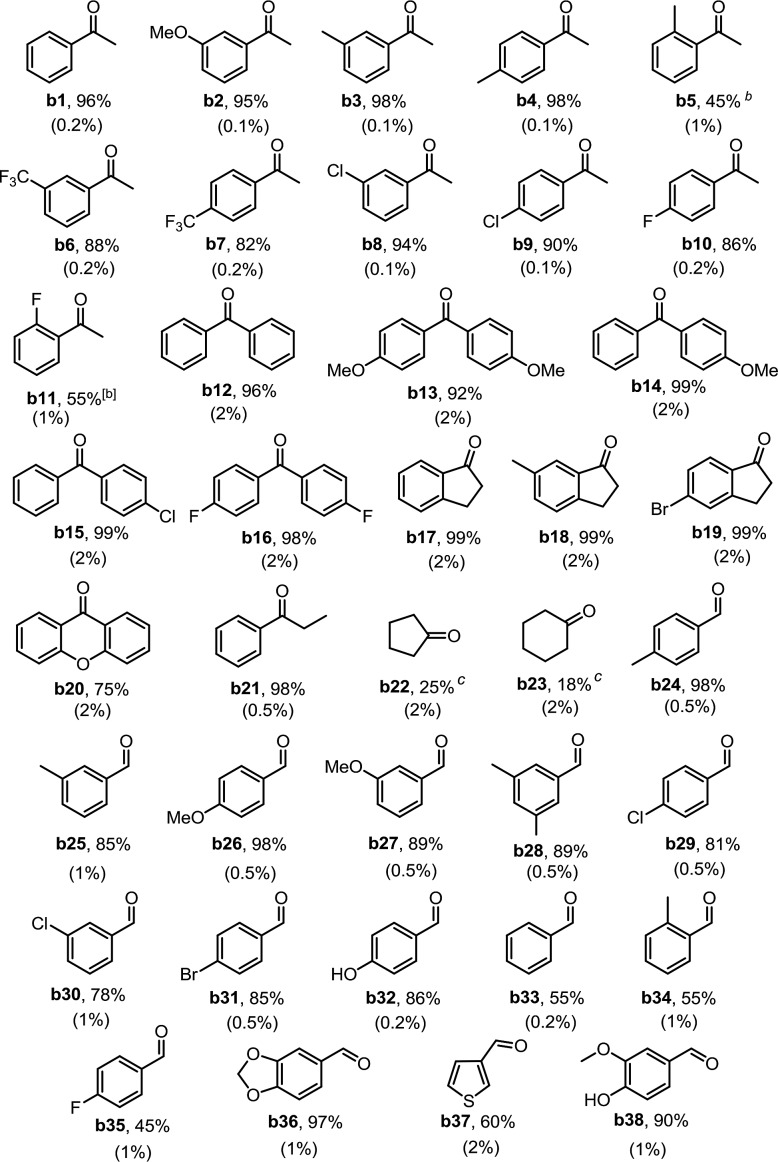
The acceptorless dehydrogenation of aliphatic alcohols is more challenging than that of benzylic alcohols due to the larger bond dissociation energies of aliphatic C–H bonds. Although Ptpop-I displayed little reactivity to aliphatic alcohols such as cyclopentanol and cyclohexanol present in limiting amounts (Table 2, b22 and b23), we found that Ptpop-BF2-IV, which is a stronger photo-oxidant than Ptpop-I,14b could efficiently catalyze aliphatic alcohols oxidation with the evolution of hydrogen gas, under N2 and 365 nm LED irradiation. (E(*3Pt2(pop-BF2)44–/Pt2(pop-BF2)45–) = +0.86 V; E(*3Pt2(pop)44–/Pt2(pop)45–) = +0.70 V. Values vs. Cp2Fe+/0). Secondary aliphatic alcohols of different chain lengths were converted into the corresponding ketones with 85–100% yields (Table 3, b39–b45). Hydrogen gas was produced at 68% yield in the dehydrogenation reaction of 2-propanol. Various cyclic alcohols were also found to be effectively oxidized to give the corresponding ketones with 90–100% yields (Table 3, b22, b23, b46, b47). Interestingly, propane-1,2-diol was converted into acetone instead of its corresponding ketone (Table 3, b39). It was proposed that the hydroxyl-alkyl radical generated after hydrogen atom abstraction of propane-1,2-diol by triplet excited Ptpop-BF2-IV underwent C–O bond cleavage, as suggested by Iglesia as a possible pathway for the formation of acetone though the mechanism of which remains elusive.16 For primary aliphatic alcohols, though no reaction was observed for methanol (Table 3, b48), a small amount of aldehyde was observed for ethanol and butan-1-ol (Table 3, b49, b50). Importantly, the lactonization of 1,4-butanediol and 1,5-pentanediol proceeded smoothly to yield γ-butyrolactone and δ-valerolactone with 80% and 83% product yields, respectively (Table 3, b51, b52). To test the lactonization selectivity of a benzylic alcohol versus an aliphatic alcohol, we subjected 2-(2-(hydroxymethyl)phenyl)ethanol to the reaction; isochroman-1-one was formed as the major product (50% yield) demonstrating the preferential oxidation of the benzylic alcohol over the aliphatic alcohol (Table 3, b53).
Table 3. Substrate scope of dehydrogenation of aliphatic alcohols a .

|
|
a0.2 mmol alcohol, 1 mol% Ptpop-BF2-IV, under N2, CD3CN (1 mL) in an NMR tube, 365 nm LED irradiation, 8 hours. Yield determined by 1H NMR spectroscopy. For dehydrogenation of benzyl alcohols, the reactions were stopped after complete consumption of substrates.
Although Ptpop-BF2-IV exhibited superior catalytic activity with aliphatic alcohols and appeared to be a better catalyst than Ptpop-I, dehydrogenation of benzyl alcohols with Ptpop-BF2-IV as the catalyst was found to give hydrobenzoins as major products (74–85%; pinacol coupling product) and relatively low yields of benzaldehydes (0–17%; cf. 45–98% with Ptpop-I as photocatalyst, Table 2, b24–b35). Further light irradiation of the reaction mixture could convert hydrobenzoins to benzoins. The possibility that the hydrobenzoins were formed by reductive coupling of aldehydes was excluded since no reaction was observed when 4-chlorobenzaldehyde and 4-methoxybenzaldehyde were subjected to light irradiation with Ptpop-BF2-IV under the same reaction conditions for 3 hours. However, when the solvent was changed to CD2Cl2, aldehydes were obtained as the major products in good yields (b29: 81%; b26: 73%) when 4-chlorobenzyl alcohol and 4-methoxybenzyl alcohol were subjected to steady state photolysis with Ptpop-BF2-IV. Only very low yields (5–12%) of hydrobenzoins were obtained. We also examined the solvent effect on product distribution in the dehydrogenation of 1-phenylethanol by Ptpop-I (Table 1).
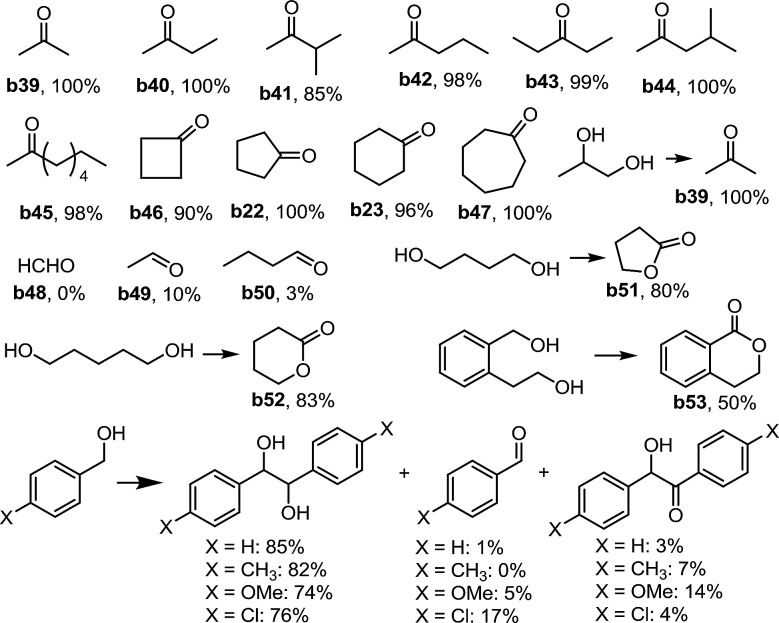
Next, we examined the acceptorless dehydrogenation of N-heterocycles with [Pt2(pop)4]4– as the photocatalyst. Both Ptpop-II and Ptpop-III were found to be efficient catalysts for this transformation under N2 and 410 nm LED irradiation. Various NH-free indolines containing electron-donating or electron-withdrawing groups were converted to the corresponding indoles with 78–99% yield (Table 4, d1–d11). Dehydrogenation of indoline with Ptpop-III also furnished hydrogen with 76% yield. When 2-carboxyl indoline was subjected to the reaction, the decarboxylated indole product d1 was formed. Dehydrogenation of N-methylindoline proceeded smoothly to afford N-methylindole with 98% yield (Table 4, d12). In the work by Li et al.,8 phenyl or acetyl groups on NH completely inhibited the transformation, while with the present protocol, various N-acetyl or phenyl indolines could be transformed into the corresponding indoles with 95–99% yields (Table 4, d13–d17). Remarkably, for N-Boc indoline containing a benzylic alcohol at the 2-position, selective oxidation of the N-heterocycle rather than benzylic alcohol was observed (Table 4, d18). 1,2,3,4-Tetrahydroquinoline, 1,2,3,4-tetrahydroisoquinoline and 6-methoxy-1,2,3,4-tetrahydroisoquinoline were also found to give the desired products in moderate to good product yields (Table 4, d19–d21). Furthermore, 65% yield of pyrrole was obtained by dehydrogenation of 3-pyrroline (Table 4, d22). Also, a quantitative yield of benzothiazole was afforded via dehydrogenation of 2,3-dihydrobenzothiazole (Table 4, d23).
Table 4. Substrate scope of dehydrogenation of saturated N-heterocycles a .
|
a0.3 mmol saturated N-heterocycle, 3 mL solvent, condition A or condition B, under N2, 410 nm LED irradiation, 4–12 hours; isolated yield.
bDetermined by GC with FID.
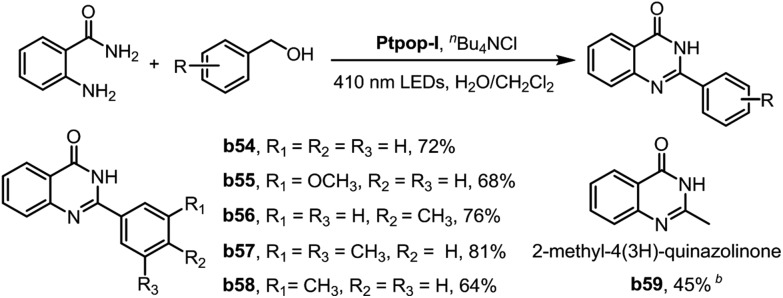
The cascade reaction of aldehydes with o-aminobenzamides is a well-known method to construct a quinazolin-4(3H)-one skeleton which is prevalent in natural and biological active molecules. The traditional methods for the synthesis of quinazolin-4(3H)-ones usually require stoichiometric oxidants or high temperature.17 In this work, we have realized a one-pot acceptorless dehydrogenation coupling of primary alcohols with o-aminobenzamides to furnish quinazolin-4(3H)-ones. The photo-reaction of primary benzylic alcohols with o-aminobenzamides proceeded smoothly under N2 to form quinazolin-4(3H)-ones with 64–81% yield using Ptpop-I as the photo-catalyst (Table 5, b54–b58). It is noteworthy that the biologically active compound 2-methyl-4(3H)-quinazolinone was synthesized in one-step with 45% yield using ethanol as the C2 source and Ptpop-BF2-IV as the photocatalyst (Table 5, b59).
Table 5. One-pot cascade synthesis of quinazolin-4(3H)-ones a .
|
a0.4 mmol alcohol, 0.2 mmol anthranilamide, 2 mol% Ptpop-I, 5 mol% nBu4NCl, H2O/CH2Cl2 1 : 3 (3 mL), under N2, 410 nm LEDs irradiation, 8 hours; isolated yields.
bThe ethanol solution containing 0.2 mmol anthranilamide and 2 mol% Ptpop-BF2-IV was under N2 and 365 nm LEDs irradiation; isolated yield.
The photocatalytic reactions proceeded smoothly on a gram scale with 92% yield of acetophenone, 85% yield of vanillin, 90% yield of indole and 95% yield of acetone (Fig. 2). These results highlight the promise of this simple and easily manipulated protocol for gram-scale application.
Fig. 2. Gram-scale reactions.

To gain more insight into the reaction mechanism, the emission quenching rate constants (kq) of Ptpop-BF2-IV by alcohols were measured and are shown in Table 6. The kH/kD of 2-propanol and p-methoxybenzyl alcohol, 6.33 and 2.37 respectively, reveal that C–H bond cleavage is the rate determining step. The smaller kH/kD ratio for p-methoxy-benzyl alcohol may be due to a competitive oxidative quenching pathway which generates the cation radical of the electron-rich benzyl alcohol. We investigated the kinetic isotope effect (KIE) of p-methoxybenzyl alcohol and 2-propanol (see ESI† for details). As shown in Fig. 3, the KIE for intramolecular and intermolecular competition of photo-dehydrogenation of p-methoxybenzyl alcohol by Ptpop-I are 3.35 and 3.35, respectively, and that of 2-propanol by Ptpop-BF2-IV is 4.2, suggesting that the rate-determining step in these reactions involves C–H bond cleavage by the triplet excited [Pt2(pop)4]4– (*Pt2). Based on these results, a plausible reaction pathway is proposed (Fig. 3b). Upon light irradiation, α-hydrogen atom abstraction from the alcohol by *Pt2 generates an α-hydroxybenzyl/α-hydroxyalkyl radical and H–Pt2; further hydrogen atom abstraction from the radical leads to the formation of carbonyl compounds and H2–Pt2. Reductive elimination of H2 from H2–Pt2 completes the catalytic cycle.11,18 For the dehydrogenation of indoline, it was found that in the presence of 1.1 equivalents of styrene, the Markovnikov hydroamination product, 1-(1-phenylethyl)-1H-indole, was obtained with 46% yield. The formation of 1-(1-phenylethyl)-1H-indole might give support to the involvement of Pt2-hydride. With styrene as a trap reagent, Pt2-hydride would be captured to form Pt2–CH(CH3)Ph which further reacted with indoline/indole to give the hydroamination product 1-(1-phenylethyl)-1H-indole.19
Table 6. Rate constants for the quenching of Ptpop-BF2-IV by alcohols.
| Substrates | BDE a (kcal mol–1) | k q c (M–1 s–1) |
| 2-Butanol | 93.1 | 4.1 × 104 |
| 2-Pentanol | — b | 6.0 × 104 |
| Cyclopentanol | — b | 6.0 × 104 |
| 2-Propanol | 91 | 9.5 × 104, kH/kD = 6.33 d |
| p-Methoxybenzyl alcohol | — b | 2.2 × 106, kH/kD = 2.37 d |
aC–H bond dissociation energy of the carbinol carbon.
bNot known.
cRate constants for the quenching of phosphorescence of Ptpop-BF2-IV by alcohol.
d k H refers to kq of non-deuterated alcohols whereas kD refers to kq of deuterated alcohols. For 2-propanol, kD was obtained with CD3CD(OD)CD3; for p-methoxybenzyl alcohol, kD was obtained with p-(CH3O)C6H4CD2OH.
Fig. 3. Mechanistic study. (a) Investigation of kinetic isotopic effect on photo-dehydrogenation of alcohols. (b) Proposed reaction pathway for photo-dehydrogenation of alcohols by [Pt2(pop)4]4–.

We conducted nanosecond time-resolved absorption (TA) measurements to gain further insight into the reaction mechanism. Ptpop-BF2-IV exhibits a TA spectrum with maxima at 327 and 430 nm, and a bleaching signal at 367 nm in degassed CH3CN with a decay lifetime of 9.8 μs (Fig. 4a). In the presence of p-methoxybenzyl alcohol, the absorption evolved to give a long-lived (>100 μs; Fig. 4b) spectral profile with maxima at 337 and 408 nm. This can be ascribed to the direct reaction of the substrate and the triplet excited [Pt2(pop-BF2)4]4–, likely via hydrogen atom abstraction to give a mixed-valence [Pt2(pop-BF2)4(H)]4–. For Ptpop-III, the addition of indoline (c1) resulted in emission quenching with a kq of 4.7 × 108 dm3 mol–1 s–1. The intensity of the TA maximum at ∼470 nm decayed quickly with a time constant of 0.86 μs, and a concomitant growth of the TA signal at 390 nm, with a time constant of 0.83 μs (Fig. 4c and d). The latter TA spectral profile is long-lived (Fig. S1;† τ = 72 μs), and resembles that obtained in the presence of H-atom donors such as 2-propanol and nBuSnH.20 This suggests that the triplet excited Ptpop-III reacts with indoline via hydrogen atom abstraction to give a [Pt2(pop)4(H)]4– species during the photocatalysis.
Fig. 4. (a) Nanosecond time-resolved absorption spectra of Ptpop-BF2-IV (3 × 10–5 M) and (b) that with 4-methoxybenzyl alcohol (0.1 M) in degassed CH3CN at room temperature. (c) Nanosecond time-resolved absorption spectra of Ptpop-III (3 × 10–5 M) and (d) that in the presence of indoline (2 mM) in degassed MeOH at room temperature.

Steady state photolysis of Ptpop-BF2-IV in the presence of p-methoxybenzyl alcohol (0.1 M) in degassed CH3CN gave a new, broad absorption band centered at 350 nm within 20 seconds as shown in Fig. 5. This absorption band is stable without apparent decay in 1 hour at room temperature. In addition, light irradiation of Ptpop-BF2-IV in the presence of p-methoxybenzyl alcohol (30 equiv., 0.15 M) in CD3CN generated new 1H NMR signals from –7.0 to –9.4 ppm (Fig. S2†), which could be assigned to the formation of platinum hydride species in the photochemical reactions. Therefore, the formation of broad absorption band centered at 350 nm during steady state photolysis as shown in Fig. 5 may be ascribed to the conversion of Ptpop-BF2-IV to platinum hydride species via hydrogen atom abstraction.
Fig. 5. UV-vis absorption spectral change during steady state photolysis of Ptpop-BF2-IV (3 × 10–5 M) in the presence of p-methoxybenzyl alcohol (0.1 M) in degassed CH3CN. The arrow indicates the direction of the change in absorbance upon light irradiation.

Conclusion
In summary, here we have described binuclear platinum(ii) diphosphite complexes as practical and efficient photocatalysts for oxidant-free and acceptorless dehydrogenation of alcohols, including aliphatic alcohols, and N-heterocycles in high yields even under substrate-limiting conditions. The reaction can be used for constructing lactones, quinazolin-4(3H)-ones and C–N bonds. The unique reactivity of binuclear platinum(ii) diphosphite complexes, as well as the wide substrate scope, mild reaction conditions and scalability of the protocol, underscore the utility and versatility of these photocatalysts for organic transformations with practical relevance.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
This work was supported by the National Key Basic Research Program of China (No. 2013CB834802), the National Natural Science Foundation of China (21801163), STU Scientific Research Foundation for Talents (NTF18003) and Basic Research Program of Shenzhen (JCYJ20160229123546997, JCYJ20160530184056496). We thank Dr Xiao-Yong Chang for solving the X-ray crystal structure of (Bu4N)4[Pt2(P2O5(BF2)2)4].
Footnotes
†Electronic supplementary information (ESI) available: Experimental details and NMR spectral data. See DOI: 10.1039/c8sc05600e
References
- (a) Larock R. C., Comprehensive Organic Transformations, Wiley-VCH, New York, 2nd edn, 1999, p. 1234. [Google Scholar]; (b) Arends I. W. C. E. and Sheldon R. A. in Modern Oxidation Methods, ed. J.-E. Bäckvall, Wiley-VCH, Weinheim, 2nd edn, 2010, p. 147. [Google Scholar]; (c) Dobereiner G. E., Crabtree R. H. Chem. Rev. 2010;110:681. doi: 10.1021/cr900202j. [DOI] [PubMed] [Google Scholar]
- (a) Dess D. B., Martin J. C. J. Am. Chem. Soc. 1991;113:7277. [Google Scholar]; (b) Markó I. E., Giles P. R., Tsukazaki M., Brown S. M., Urch C. J. Science. 1996;274:2044. doi: 10.1126/science.274.5295.2044. [DOI] [PubMed] [Google Scholar]; (c) Bolm C., Magnus A. S., Hildebrand J. P. Org. Lett. 2000;2:1173. doi: 10.1021/ol005792g. [DOI] [PubMed] [Google Scholar]; (d) Gonsalvi L., Arends I. W. C. E., Sheldon R. A. Org. Lett. 2002;4:1659. doi: 10.1021/ol025705f. [DOI] [PubMed] [Google Scholar]; (e) Guan B., Xing D., Cai G., Wan X., Yu N., Fang Z., Yang L., Shi Z. J. Am. Chem. Soc. 2005;127:18004. doi: 10.1021/ja055398j. [DOI] [PubMed] [Google Scholar]; (f) Ni J., Yu W.-J., He L., Sun H., Cao Y., He H.-Y., Fan K.-N. Green Chem. 2009;11:756. [Google Scholar]; (g) Xie X., Stahl S. S. J. Am. Chem. Soc. 2015;137:3767. doi: 10.1021/jacs.5b01036. [DOI] [PubMed] [Google Scholar]
- (a) Yamaguchi R., Ikeda C., Takahashi Y., Fujita K.-i. J. Am. Chem. Soc. 2009;131:8410. doi: 10.1021/ja9022623. [DOI] [PubMed] [Google Scholar]; (b) Fujita K.-i., Yoshida T., Imori Y., Yamaguchi R. Org. Lett. 2011;13:2278. doi: 10.1021/ol2005424. [DOI] [PubMed] [Google Scholar]; (c) Wu J., Talwar D., Johnston S., Yan M., Xiao J. Angew. Chem., Int. Ed. 2013;52:6983. doi: 10.1002/anie.201300292. [DOI] [PubMed] [Google Scholar]; (d) Chakraborty S., Brennessel W. W., Jones W. D. J. Am. Chem. Soc. 2014;136:8564. doi: 10.1021/ja504523b. [DOI] [PubMed] [Google Scholar]; (e) Maier A. F. G., Tussing S., Schneider T., Flörke U., Qu Z.-W., Grimme S., Paradies J. Angew. Chem., Int. Ed. 2016;55:12219. doi: 10.1002/anie.201606426. [DOI] [PubMed] [Google Scholar]; (f) Kojima M., Kanai M. Angew. Chem., Int. Ed. 2016;55:12224. doi: 10.1002/anie.201606177. [DOI] [PubMed] [Google Scholar]; (g) Crabtree R. H. Chem. Rev. 2017;117:9228. doi: 10.1021/acs.chemrev.6b00556. [DOI] [PubMed] [Google Scholar]
- Prier C. K., Rankic D. A., MacMillan D. W. C. Chem. Rev. 2013;113:5322. doi: 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Q., Oestreich M. Angew. Chem., Int. Ed. 2017;56:7716. doi: 10.1002/anie.201703536. [DOI] [PubMed] [Google Scholar]
- Chai Z., Zeng T.-T., Li Q., Lu L.-Q., Xiao W.-J., Xu D. J. Am. Chem. Soc. 2016;138:10128. doi: 10.1021/jacs.6b06860. [DOI] [PubMed] [Google Scholar]
- Zhao L.-M., Meng Q.-Y., Fan X.-B., Ye C., Li X.-B., Chen B., Ramamurthy V., Tung C.-H., Wu L.-Z. Angew. Chem., Int. Ed. 2017;56:3020. doi: 10.1002/anie.201700243. [DOI] [PubMed] [Google Scholar]
- He K.-H., Tan F.-F., Zhou C.-Z., Zhou G.-J., Yang X.-L., Li Y. Angew. Chem., Int. Ed. 2017;56:3080. doi: 10.1002/anie.201612486. [DOI] [PubMed] [Google Scholar]
- Kato S., Saga Y., Kojima M., Fuse H., Matsunaga S., Fukatsu A., Kondo M., Masaoka S., Kanai M. J. Am. Chem. Soc. 2017;139:2204. doi: 10.1021/jacs.7b00253. [DOI] [PubMed] [Google Scholar]
- (a) Che C.-M., Butler L. G., Gray H. B. J. Am. Chem. Soc. 1981;103:7796. [Google Scholar]; (b) Che C.-M., Herbstein F. H., Schaefer W. P., Marsh R. E., Gray H. B. J. Am. Chem. Soc. 1983;105:4604. [Google Scholar]
- Roundhill D. M. J. Am. Chem. Soc. 1985;107:4354. [Google Scholar]
- Záliš S., Lam Y.-C., Gray H. B., Vlček A. Inorg. Chem. 2015;54:3491. doi: 10.1021/acs.inorgchem.5b00063. [DOI] [PubMed] [Google Scholar]
- Roundhill D. M., Gray H. B., Che C.-M. Acc. Chem. Res. 1989;22:55. [Google Scholar]
- (a) Durrell A. C., Keller G. E., Lam Y.-C., Sýkora J., Vlček A., Gray H. B. J. Am. Chem. Soc. 2012;134:14201. doi: 10.1021/ja305666b. [DOI] [PubMed] [Google Scholar]; (b) Darnton T. V., Hunter B. M., Hill M. G., Záliš S., Vlček A., Gray H. B. J. Am. Chem. Soc. 2016;138:5699. doi: 10.1021/jacs.6b02559. [DOI] [PubMed] [Google Scholar]
- Fu W., Yue L., Duan X., Li J., Lu G. Green Chem. 2016;18:6136. [Google Scholar]
- Díaz E., Sad M. E., Iglesia E. ChemSusChem. 2010;3:1063. doi: 10.1002/cssc.201000142. [DOI] [PubMed] [Google Scholar]
- Parua S., Das S., Sikari R., Sinha S., Paul N. D. J. Org. Chem. 2017;82:7165. doi: 10.1021/acs.joc.7b00643. [DOI] [PubMed] [Google Scholar]
- (a) Marshall J. L., Stiegman A. E., Gray H. B. ACS Symp. Ser. 1986;307:166. [Google Scholar]; (b) Smith D. C., Gray H. B. ACS Symp. Ser. 1989;394:356. [Google Scholar]; (c) Kalsbeck W. A., Gingell D. M., Malinsky J. E., Thorp H. H. Inorg. Chem. 1994;33:3313. [Google Scholar]; (d) Esswein A. J., Nocera D. G. Chem. Rev. 2007;107:4022. doi: 10.1021/cr050193e. [DOI] [PubMed] [Google Scholar]
- (a) Vo L. K., Singleton D. A. Org. Lett. 2004;6:2469. doi: 10.1021/ol049137a. [DOI] [PubMed] [Google Scholar]; (b) Hartwig J. F. Pure Appl. Chem. 2004;76:507. [Google Scholar]; (c) Pirnot M. T., Wang Y.-M., Buchwald S. L. Angew. Chem., Int. Ed. 2016;55:48. doi: 10.1002/anie.201507594. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Kang O.-Y., Kim B. E., Park S. J., Ryu D. H., Lim H. J. Asian J. Org. Chem. 2018;7:451. [Google Scholar]
- (a) Roundhill D. M., Atherton S. J., Shen Z.-P. J. Am. Chem. Soc. 1987;109:6076. [Google Scholar]; (b) Che C.-M., Lee W. M., Cho K. C., Harvey P. D., Gray H. B. J. Phys. Chem. 1989;93:3095. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.