

Abstract
Developing cortical GABAergic interneurons rely on genetic programs, neuronal activity, and environmental cues to construct inhibitory circuits during early postnatal development. Disruption of these events can cause long-term changes in cortical inhibition and may be involved in neurological disorders associated with inhibitory circuit dysfunction. We hypothesized that tonic glutamate signaling in the neonatal cortex contributes to, and is necessary for, the maturation of cortical interneurons. To test this hypothesis, we used mice of both sexes to quantify extracellular glutamate concentrations in the cortex during development, measure ambient glutamate-mediated activation of developing cortical interneurons, and manipulate tonic glutamate signaling using subtype-specific NMDA receptor antagonists in vitro and in vivo. We report that ambient glutamate levels are high (≈100 nm) in the neonatal cortex and decrease (to ≈50 nm) during the first weeks of life, coincident with increases in astrocytic glutamate uptake. Consistent with elevated ambient glutamate, putative parvalbumin-positive interneurons in the cortex (identified using G42:GAD1-eGFP reporter mice) exhibit a transient, tonic NMDA current at the end of the first postnatal week. GluN2C/GluN2D-containing NMDA receptors mediate the majority of this current and contribute to the resting membrane potential and intrinsic properties of developing putative parvalbumin interneurons. Pharmacological blockade of GluN2C/GluN2D-containing NMDA receptors in vivo during the period of tonic interneuron activation, but not later, leads to lasting decreases in interneuron morphological complexity and causes deficits in cortical inhibition later in life. These results demonstrate that dynamic ambient glutamate signaling contributes to cortical interneuron maturation via tonic activation of GluN2C/GluN2D-containing NMDA receptors.
SIGNIFICANCE STATEMENT Inhibitory GABAergic interneurons make up 20% of cortical neurons and are critical to controlling cortical network activity. Dysfunction of cortical inhibition is associated with multiple neurological disorders, including epilepsy. Establishing inhibitory cortical networks requires in utero proliferation, differentiation, and migration of immature GABAergic interneurons, and subsequent postnatal morphological maturation and circuit integration. Here, we demonstrate that ambient glutamate provides tonic activation of immature, putative parvalbumin-positive GABAergic interneurons in the neonatal cortex via high-affinity NMDA receptors. When this activation is blocked, GABAergic interneuron maturation is disrupted, and cortical networks exhibit lasting abnormal hyperexcitability. We conclude that temporally precise activation of developing cortical interneurons by ambient glutamate is critically important for establishing normal cortical inhibition.
Keywords: ambient glutamate, cortex, cortical interneuron, developmental, inhibition, NMDA receptor
Introduction
Inhibitory GABAefrgic interneurons (INs) control cortical function by constraining local neuronal activity and orchestrating circuit synchronization (Hensch et al., 1998; Buzsáki and Draguhn, 2004; Markram et al., 2004; Cardin et al., 2009). During embryonic development, cells destined to become cortical INs are generated in the medial and caudal ganglionic eminences (Xu et al., 2004). These newborn INs migrate to the cortex, before radially migrating into the cortical lamina (Angevine and Sidman, 1961; Berry and Rogers, 1965). Once in the cortex, a combination of genetic programs and environmental cues drive IN localization, morphological differentiation, and synaptic connectivity (Huang et al., 1999; Levitt et al., 2004; Flames and Marín, 2005; Batista-Brito and Fishell, 2009; Bortone and Polleux, 2009). GABAergic synaptic activity is established before glutamatergic, and GABA is excitatory in early development due to developmentally specific expression of chloride transporters (Kaila et al., 2014). This engenders GABAergic INs with the ability to drive spontaneous developmental network activity (Kasyanov et al., 2004; Allène et al., 2008; Minlebaev et al., 2011), which is critical to establishing synaptic properties, local microcircuits, and long-range neuronal connections. The underlying developmental events that shape IN activity and maturation are critically important for long-term brain function but yet not fully understood.
Glutamate excitation arising from spontaneous network activity and sensory input is an important driver of cortical network maturation (Chattopadhyaya et al., 2004; Bortone and Polleux, 2009; Belforte et al., 2010; De Marco García et al., 2011; Zechel et al., 2016). IN migration, morphology, and synaptic connectivity all rely on glutamate receptor activation during development. Reducing extracellular glutamate attenuates both NMDA and GABA receptor-dependent spontaneous network activity (Allène et al., 2008). Developmental disruption of NMDA receptor (NMDAR) activation in INs has long-lasting effects on IN localization and morphology and leads to deficits in inhibitory network function and behavioral abnormalities (Fredriksson et al., 2004; Reiprich et al., 2005; Belforte et al., 2010). Indeed, dysfunctional cortical inhibition is thought to be involved in the pathophysiology of epilepsy, schizophrenia, and autism (Powell et al., 2003; Harrison and Weinberger, 2005; Liu-DeRyke et al., 2009; Belforte et al., 2010; Cohen et al., 2015). Thus, glutamate-mediated excitation of INs during development has far reaching effects, beyond the cell-specific effects on INs themselves.
In the adult cortex, astrocytes rapidly remove glutamate from the extracellular space via the excitatory amino acid transporters (EAATs), GLT-1 and GLAST (Danbolt, 2001). This ensures that extracellular glutamate levels are low and that glutamatergic neurotransmission is spatially and temporally constrained. In the first week of rodent postnatal life, however, EAAT expression and functional glutamate uptake are minimal (Ullensvang et al., 1997; Hanson et al., 2015) and astrocyte/neuron interactions are largely lacking (Bushong et al., 2004; Morel et al., 2014). During the second and third weeks of life, cortical astrocytes rapidly increase EAAT expression and expand their processes, thereby constraining synaptic glutamate signaling (Diamond and Jahr, 1997; Thomas et al., 2011). This creates a developmental window in which glutamate signaling is unconstrained by astrocytes (Hanson et al., 2015), potentially allowing the accumulation of ambient glutamate (Herman and Jahr, 2007), which is known to contribute to IN function (Cavelier et al., 2005). Thus, we suspect that, during development, elevated ambient glutamate levels contribute to IN maturation via high-affinity NMDARs.
The affinity of NMDARs for glutamate is largely determined by their subunit composition. The obligate GluN1 subunit interacts with one or more of the four unique GluN2 subunits (GluN2A-D), which are differentially expressed throughout the brain and through development (Monyer et al., 1994; Sheng et al., 1994). GluN2D expression peaks in the cortex during early postnatal development (Monyer et al., 1994). In both the developing and mature hippocampus, GluN2D is selectively expressed in GABAergic INs, not glutamatergic neurons (Monyer et al., 1994; Standaert et al., 1996; Yamasaki et al., 2014; von Engelhardt et al., 2015; Perszyk et al., 2016), suggesting a potential role in IN development. In addition to their effects on glutamate affinity, GluN2 subunit compositions affect magnesium affinities, channel kinetics, and calcium permeability of NMDARs (Paoletti et al., 2013). GluN2C and GluN2D-containing NMDARs (GluN2C/DRs) have higher affinities for glutamate, allowing them to be activated by relatively low concentrations of glutamate (Hollmann and Heinemann, 1994). Notably, GluN2C/DRs have lower affinities for magnesium, which reduces the magnesium block at resting membrane potential (Qian et al., 2005). They also a have prolonged deactivation time course following removal of glutamate, and do not show rapid desensitization (Vicini et al., 1998). These unique properties make GluN2C/DRs strong candidates for mediating tonic glutamate currents.
Here, we examined developmental regulation of ambient glutamate and how tonic activation of GluN2C/DRs during early cortical development influences the maturation of inhibitory INs. We show that ambient glutamate levels are elevated during early cortical development and act on GluN2C/D to drive a developmentally transient (P7-P9) depolarization of INs. In vivo pharmacological blockade of GluN2C/DRs during the window of transient depolarization leads to a long-term reduction in the morphological complexity of IN dendritic arbors, a decrease in the number of inhibitory synapses, and an increase in cortical network hyperexcitability. Together, these observations demonstrate that activation of GluN2C/DRs by ambient glutamate plays a critical role in cortical IN maturation and integration.
Materials and Methods
Animals
All guidelines of Tufts University's and Emory University's Institutional Animal Care and Use Committee were followed. C57Bl/6J mice, originally obtained from The Jackson Laboratory (stock #000664; RRID:IMSR_JAX:000664), were bred in house. G42 mice (CB6-Tg(Gad1-EGFP)G42Zjh/J; The Jackson Laboratory stock #007677; RRID:IMSR_JAX:007677), SST-Cre (Sst-IRES-Cre knock-in; The Jackson Laboratory stock #028864; RRID:IMSR_JAX:028864), and Ai9 (Ai9(RCL-tdT); The Jackson Laboratory stock #007909; RRID:IMSR_JAX:007909) were also obtained from The Jackson Laboratory and bred in house. Mice of both sexes were used for experiments and housed on a 12 h light/12 h dark cycle and given ad libitum access to water and standard chow.
Preparation of brain slices
Cortical brain slices containing sensorimotor cortex were prepared from mice between the ages of P2 and P21 based on previous protocols (Andresen et al., 2014). Briefly, mice were anesthetized (isoflurane), decapitated, and the brains were rapidly removed and placed in chilled (4°C) low-Ca, low-Na slicing solution consisting of (in mm as follows): 234 sucrose, 11 glucose, 24 NaHCO3, 2.5 KCl, 1.25 NaH2PO4, 10 MgSO4, and 0.5 CaCl2, equilibrated with a mixture of 95% O2:5% CO2. The brain was glued to the slicing stage of a VT1200S vibratome (Leica Microsystems), and 400-μm-thick slices were cut in a coronal orientation. Slices were then incubated in 32°C oxygenated aCSF (in mm as follows): 126 NaCl, 2.5 KCl, 1.25 NaH2PO4, 1 MgSO4, 2 CaCl2, 10 glucose, 26 NaHCO2) for 1 h, and then held at room temperature. Subsequent recordings were performed at 27°C.
Glutamate biosensor imaging
Acute cortical slices from P5-P10 C57BL/6 mice were prepared as above. Biosensor was produced and loaded into brain slices as previously described (Dulla et al., 2013). Slices were placed in a submersion chamber (Siskiyou) and perfused with aCSF containing 20 μm DNQX and 10 μm CPP at 2 ml/min. Slices were imaged using a customized Bx51 upright microscope with a 4× UPlanSApo (Olympus) objective, an Optosplit (Cairn) dual emission splitter onto a Zyla sCMOS camera (Andor). Each biosensor image had a 200 ms exposure time using 440 nm excitation. Both CFP (470 nm/30 nm bandpass) and Venus (535 nm/40 nm bandpass) fluorescence was collected. Images were acquired every 10 s. Ten frames of autofluorescence were recorded both before loading of the biosensor and in equivalent unloaded slices. After 20 frames of baseline fluorescence was recorded, 5 μm TFB-TBOA (astrocytic excitatory amino acid transporter inhibitor) and 5 μm MPEP (mGluR inhibitor) was added to assay the effects of glutamate uptake. Subsequently, 1 mm glutamate was washed in for 10–15 min to saturate the biosensor. Average fluorescence from ROIs spanning all cortical layers was quantified for autofluorescence, control, TBOA+MPEP, and high glutamate conditions. Autofluorescence was subtracted from the other conditions, and fluorescence resonance energy transfer (FRET) ratios were calculated. Saturation of the sensor with glutamate gave a consistent FRET ratio (≈1.8), ensuring consistency between slices and ages.
Electrophysiological measurement of ambient glutamate concentrations
Slices were prepared as described above from C57Bl/6J mice. DNQX (20 μm, AMPA receptor antagonist), d-serine (30 μm, NMDAR coagonist), and GABAzine (10 μm, GABAA receptor antagonist) were included in the aCSF. For nucleated patches and whole-cell voltage-clamp recording, internal solution contained the following (in mm): 100 cesium gluconate, 10 HEPES, 5 EGTA, 4 NaCl, 1 MgCl2, 10 TEACl, 10 phosphocreatine, 2 MgATP, 0.3 NaGTP, pH 7.33 (mOsm = 272).
Calculating ambient glutamate levels was based on an established protocol (Herman and Jahr, 2007). For generating concentration-response curves for NMDA and glutamate, nucleated patches were made from the soma of Layer 5 pyramidal neurons (L5Ps) by recording a cell in whole-cell configuration, voltage-clamping it at −60 mV, applying gentle negative pressure, then slowly drawing back from the cell body. Nucleated patch formation was monitored visually and with continuously repeated 1 mV, 100 ms voltage steps. Once nucleated, patches were separated from the cell body, they were brought to the surface of the slice, slowly raised to 40 mV, and placed within the flow of a rapid solution switcher (AutoMate Scientific) with a custom 6-barrel flow pipe. Solutions were pressurized to 2 PSI with 95% O2/5% CO2. For the generation of NMDA concentration-response curves solutions included bath aCSF, aCSF + 5 μm NMDA, aCSF + 10 μm NMDA, aCSF + 50 μm NMDA, aCSF + 1 mm NMDA, and aCSF + 100 μm glutamate. For the generation of glutamate concentration-response curves, solutions included aCSF, aCSF + 0.5 μm glutamate, aCSF + 1 μm glutamate, aCSF + 5 μm glutamate, aCSF + 100 μm glutamate, and aCSF + 1 mm NMDA. NMDA and glutamate dose–response curves were generated by baseline adjusting all responses and then normalizing to the maximum NMDA or glutamate responses, respectively, on a cell-by-cell basis. Estimates of the EC50 for NMDA and glutamate at P3 and P14 were obtained by fitting baseline-adjusted, normalized values with a Hill equation. Ratios of the maximum glutamate response to maximum NMDA response were obtained by dividing the baseline-adjusted 100 μm glutamate response by the baseline-adjusted 1 mm NMDA response on a cell-by-cell basis. Conversion factors were calculated for each age as follows:
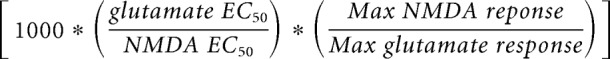 |
Multiplying a μm concentration of NMDA by this conversion factor gives an equivalent nm concentration of glutamate.
For quantifying ambient glutamate in cortical slices, L5Ps were patched in whole-cell configuration at P3 and P14. Cells were voltage-clamped at 40 mV, and then NMDA (5 μm) was bath-applied. After reaching steady state, NMDA was washed out, and APV (50 μm) was bath-applied. APV-sensitive holding currents were normalized to NMDA-induced holding currents on a cell-by-cell basis. Normalized currents at each age were converted to equivalent concentrations of nm glutamate by multiplying them by the conversion factors for each age as follows:
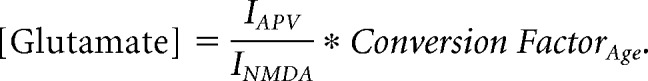 |
SEM was calculated for ambient glutamate estimates by propagating proportional errors from the EC50 values, maximum response ratios, and the NMDA-normalized APV-sensitive holding currents to the final estimated [Glutamate] (Young, 1962).
 |
Whole-cell electrophysiology
Slices were prepared as described above. DNQX (20 μm), d-serine (30 μm), and GABAzine (10 μm) were included in the aCSF. When applicable, APV (50 μm), DQP-1105 (20 μm, GluN2C/D-containing NMDAR antagonist), TBF-TBOA (50 nm), and/or TBOA (100 μm) were applied directly to the recirculating solution. Slices were visualized with a 4× objective on an BX51 inverted microscope (Olympus). L5Ps were identified based on location and soma morphology using a 60× water-immersion objective (Olympus) and DIC optics. Neurons were patched with borosilicate glass electrodes (4–6 mΩ). Cell type was confirmed based on electrophysiological properties, including membrane resistance and action potential (AP) firing frequency, as well as reporter expression.
For voltage-clamp recordings, internal solution contained the following (in mm): 100 cesium gluconate, 10 HEPES, 5 EGTA, 4 NaCl, 1 MgCl2, 10 TEACl, 10 phosphocreatine, 2 MgATP, 0.3 NaGTP, pH 7.33 (mOsm = 272). L5Ps were voltage-clamped at −60 mV, slowly increased to 40 mV, and allowed to equilibrate until a stable baseline holding current was achieved. Baseline holding currents were recorded for at least 90 s before addition of APV, DQP-1105, or TBOA. The change in holding current was recorded and access resistance was monitored throughout drug applications. For TBOA experiments, applications of TBOA were followed after 7 min by applications of APV. APV-sensitive holding currents were calculated as the average of 30 s of stable holding current after APV/DQP-1105 application subtracted from the average of 90 s of stable baseline before APV/DQP-1105. TBOA-induced holding currents were calculated as the average of 90 s of stable baseline before TBOA application subtracted from the average of 30 s of stable holding current in TBOA. Holding current values from voltage-clamp recordings were normalized to the capacitance of the recorded neuron to calculate current density. Capacitance was calculated as where τ is the decay of the current response to a 5 mV depolarizing step, Ra is access resistance, and Rm is membrane resistance. For TFB-TBOA current-clamp experiments, ΔVm was calculated by subtracting the average of 7 min of Vm after 50 nm TFB-TBOA exposure from the average of 30 s of Vm before the application of TFB-TBOA.
For current-clamp recordings, internal solution contained the following (in mm): 130 potassium gluconate, 10 HEPES, 5 KCl, 5 EGTA, 2 NaCl, 1 MgCl2, 2 MgATP, 0.3 NaGTP, pH 7.33 (mOsm = 290). INs or pyramidal cells were identified and whole-cell recordings were established as described above. Neurons were then current-clamped at I = 0 to record the resting potential over time. After achieving a stable baseline, APV or DQP-1105 was added to the recirculating aCSF. Resting membrane potentials were recorded for > 6 min after drug applications. Drug-induced hyperpolarizations were calculated by subtracting the baseline resting potential (average of 90 s) from the resting potential after drug wash-on (average of 30 s). Additionally, current injections were performed before and after drug applications in current-clamped cells. Current injections consisted of 250 ms current steps from −20 pA to 170 pA in ascending 10 pA intervals, returning to 0 pA for 750 ms between each step.
For recording mIPSCs, 300-μm-thick slices were prepared; 300-μm-thick sections were used in this experiment to allow for better visualization and patch-clamp recording in more mature brain tissue. Internal solution contained the following (in mm): 140 CsCl, 10 HEPES, 4 NaCl, 0.5 CaCl, 5 EGTA, 2 MgATP, 0.3 NaGTP, pH 7.25 (mOsm = 290). Bath aCSF included DNQX (10 μm), CPP (20 μm), and TTX (1 μm). L5Ps were held at −70 mV for >5 min. In some cases, GABAzine (20 μm) was then applied to verify that miniature currents were mediated by GABAA receptors. mIPSCs were quantified from 2 min of stable baseline using MiniAnalysis software (Synaptosoft). The amplitude threshold was set to 15 pA, and all events were confirmed manually. For all electrophysiological experiments, access resistance was monitored before and after experiments and cells >25% change in access were excluded from analysis.
FACS and qPCR
FACS was used to isolate eGFP-expressing neurons in P7-P9 G42 pups. Mice were killed, cortical tissue extracted, minced, and incubated at 37°C in a digest solution. The digest contained Collagenase D (2 mg/ml, Roche Diagnostics, #11088866001), DNase (2 mg/ml, Sigma-Aldrich, #DN25-1G), and Dispase II (5 mg/ml, Sigma-Aldrich, #D4693), dissolved in Ca2+/Mg2+ HBSS. After 30 min, 10% heat-activated FBS was added to double the final volume, and the solution was strained through a 100 μm filter. After centrifuging remaining tissue at 1400 RPM for 10 min, the cells were resuspended in L15 media and added to a Percoll gradient (upper concentration 12.5%, a lower concentration 28%). Tissue was centrifuged for another 10 min at 1300 RPM, and the upper half of the interface containing myelin and debris was removed. The lower gradient was diluted with L15 and centrifuged at 1300 RPM for 6 min, after which the supernatant was discarded. The remaining cells were taken to the Flow Cytometry Core at Tufts University, and a FACSARIA cell sorter (BD Biosciences) was used to isolate neurons that expressed eGFP based on fluorescence at 488 nm.
RNA extraction and cDNA synthesis were then performed on ∼1000 FACS-sorted GFP+ cells and GFP− cells using the REPLI-g WTA Single Cell Kit (QIAGEN). The cDNA was diluted 1:100. For qPCR, 2 μl of cDNA was amplified using 600 nm primers and 2× SYBR Green master mix in a volume of 25 μl in the Mx3000P system (Agilent Technologies) in quadruplets using the following primers: GAD1 forward, 5′-TCGATTTTTCAACCAGCTCTCTACT, reverse, 5′-GTGCAATTTCATATGTGAACATATT; GFP forward, 5′-AAGCTGACCCTGAAGTTCATCTGC, reverse, 5′-CTTGTAGTTGCCGTCGTCCTTGAA; and ACTB forward, 5′-GTGACGTTGACATCCGTAAAGA, reverse, 5′-GCCGGACTCATCGTACTCC.
The gene dose was calculated relative to ACTB using 2ΔΔCt method.
FISH
C57Bl/6 mice age P3-P4 and P7-P8 were transcardially perfused with 0.9% saline followed by 4% PFA in PB, pH 7.4. Brains were removed and placed in 4% PFA at 4°C for 16 h and cryoprotected in 30% sucrose at 4°C for 48 h. Frozen brain sections of 20 μm thickness were cut with a cryostat, mounted on glass slides, and dried overnight. Riboprobes were in vitro transcribed from plasmids containing cDNA for GAD65, GAD67, and GluN2D. GAD65 and GAD67 plasmids were generously provided by Dr. Shane Hentges (Colorado State Unviersity), and GAD65/67 riboprobes were made as previously described (Jarvie and Hentges, 2012). The GluN2D plasmid was made by amplifying a portion of rat GluN2D cDNA (2029–3167 bp) and inserting the PCR product into an EcoR1 cut pCI-Neo vector using InFusion (Clontech). GluN2D cDNA was amplified with the following primers: CTAGCCTCGAGAATTCTTCTTGTCATACATCGAGGTGC and TACCACGCGTGAATTCGACACAGTGCCATTGCTGCGTG. GluN2D sense and antisense riboprobes were in vitro transcribed from linearized plasmids with T7 and T3 polymerases, respectively (Roche Diagnostics). Digoxigenin-labeled dNTPs were used for GAD65 and GAD67, and fluorescein-labeled dNTPs were used for GluN2D. Riboprobes were precipitated, subjected to alkaline hydrolysis for 3.5 min (GAD65/67) or 4 min (GluN2D), diluted in hybridization buffer, and used at a final concentration of 1 ng/μl. The prehybridization treatments, riboprobe hybridization, and posthybridization washes were performed on brain sections as previously described (Swanger et al., 2011). For riboprobe detection, the sections were incubated with peroxidase-conjugated anti-fluorescein antibodies (1:1000, Roche Diagnostics) at room temperature for 1 h, washed with TBS with 0.05% Tween 5 times for 10 min each, and incubated with fluorescein tyramide signal amplification reagent and washed as per the manufacturer's instructions (PerkinElmer). The sections were then incubated with alkaline phosphatase-conjugated anti-digoxigenin antibodies (1:500, Roche Diagnostics) followed by five 10 min washes with TBS with 0.05% Tween buffer. HNPP (2-hydroxy-3-naphtoic acid-2′-phenylanilide phosphate) and Fast Red TR (4-chloro-2-methylbenzenediazonium hemi-zinc chloride salt) were used to detect digoxigenin-labeled GAD65/67 mRNA as per the manufacturer's instructions (Roche Diagnostics). Slides were stained with DAPI, rinsed briefly with H2O, and coverslipped with polyvinyl chloride mounting media containing propyl gallate (Swanger et al., 2011). Brain sections were imaged with an inverted IX51 microscope (Olympus) equipped with a disk-spinning unit, an ORCA-ER CCD camera (Hamamatsu), and MicroManager software (Edelstein et al., 2010). Fluorophores were imaged with the following emission, excitation, and dichroic filter sets: fluorescein (470/40, 525/50, and 495 nm), Fast Red TR (545/25, 605/70, and 565 nm), and DAPI (350/50, 460/50, and 400 nm). Sense and antisense probes were hybridized on separate slides in parallel, detected with the same detection solutions, and imaged with the same acquisition settings. ImageJ was used to prepare the images for presentation, and the intensity settings for each channel are the same across all images at a given magnification.
Neonatal DQP-1105 treatment
Gad-1:GFP or C57B6/J mice were treated daily from either P7-P9 or from P11-P13 with DQP-1105 (Tocris Bioscience) at 28 mg/kg intraperitoneally (Lozovaya et al., 2014). DQP-1105 was first dissolved to 100 mm stock solution in DMSO and then diluted, before injections, to a final concentration of 5 mm in vehicle. Vehicle formulation was 4% ethanol, 5% Tween 80, 5% PEG 400, in sterile 0.9% saline (DMSO final concentration was <5%). DQP-1105 solution was warmed and sonicated and then immediately injected intraperitoneally at 10 μl/g.
Immunohistochemistry
Immunofluorescence quantification of GABAergic synapses.
For immunofluorescence-based quantification of GABAergic synapse density, DQP- and vehicle-treated littermates were processed, stained, and imaged in parallel. DQP- and vehicle-treated C57B6/J mice were anesthetized with isoflurane and perfused with PBS. Following perfusion, brains were dissected out and placed in 4% PFA for 12–18 h. Fixed brains were then cryoprotected in a high sucrose solution (in mm 818 sucrose, 77 mm sodium phosphate, 23 mm sodium phosphate), and later sectioned into 14 μm slices using a Microm HM 525 cryostat (Thermo Fisher Scientific). Brain sections were washed in PBS and placed in blocking buffer (20% normal goat serum in PBS) for 1 h at room temperature. VGAT (1:500, Sigma-Aldrich, AMAb91043) and Gephyrin (1:500, Invitrogen, PA5–19589) antibodies were diluted in antibody solution (PBS containing 0.3% Triton X-100, 10% goat serum). Brain sections were incubated in diluted primary antibody solutions for 48 h at 4°C and then rinsed three times in PBS. Secondary antibodies (goat anti-rabbit 488 and goat anti-mouse Cy3.5, The Jackson Laboratory; diluted 1:500 in antibody solution) were added to the sections for 2 h at room temperature. Following incubation in the secondary antibodies, sections were washed three times in PBS, with DAPI included in the final wash. Sections were mounted using Fluoromount-G (Southern Biotechnology). Mounted sections were then used for confocal imaging. Sections were imaged on an A1R confocal microscope (Nikon) with a 60× oil objective (Olympus) and 10× optical zoom. Images were taken from L4–5 of somatosensory cortex. Laser and imaging settings were identical from section to section and across imaging days. Z stacks consisting of 3 confocal images taken at 1 μm intervals were collected from each section. Stacks were then maximum intensity projected in ImageJ to condense 2 μm of Z space into a single image. Synapses were counted in the projected images using ImageJ. Briefly, a mask was generated for both VGAT and Gephyrin images using a background subtraction and rolling ball radius of 50. Colocalized puncta were then quantified using the “Puncta Analyzer” ImageJ plugin as previously reported (Ippolito and Eroglu, 2010).
Immunofluorescence characterization of GFP+ INs in G42 mice.
To quantify colocalization of eGFP with other molecular markers, immunohistochemical approaches were used to label neuronal tissue from G42 animals. The 40 μm brain slices were stained, as described above, with primary antibodies against GFP (1:1000, chicken, Abcam) and one of the following markers: GABA (1:5000, rabbit, Sigma-Aldrich), parvalbumin (PV, 1:2000, mouse, Swant), glutamate decarboxylase 67 (GAD67, 1:10,000, mouse, Millipore), somatostatin (SST, 1:100, rat, Millipore), and calretinin (CalR, 1:5000, mouse, Swant). Secondary antibodies against mouse, rat, chicken, or rabbit, tagged with Alexa-488, -Cy3, or -647, were applied at a concentration of 1:500. For cell counting, layers 5–6 of somatosensory cortex were imaged on either a Keyence microscope with a 10× or 20× optical zoom or A1R confocal microscope (Nikon) with a 40× oil objective (Olympus), with identical laser and imaging settings across sections. Images were analyzed in ImageJ, GFP+ cells were identified manually, and then examined for their colabeling with other immunolabels.
IN filling and reconstruction
For filling INs with biocytin, 4% biocytin was added to the current-clamp internal solution described above. After in vivo treatment with DQP-1105 from P7-P9 or P11-P13, 300-μm-thick slices were prepared from G42 animals at P19-P25. Bath aCSF contained DNQX, CPP, and GABAzine. GFP+ INs were patched in deep cortical layers, and current steps from −20 to 180 pA were injected in 10 pA intervals to measure firing properties. All INs showed characteristic fast-spiking properties. Cells were then held for 5–15 min at resting potential, and current steps were repeated. The patch pipette was then withdrawn slowly to form an outside-out patch and allow the cell to reseal. Cells sat for at least 10 min after removal of the patch pipette. Slices were then immersed in 4% PFA overnight at 4°C. Following fixation, slices were washed 4× in PBS and then incubated overnight at 4°C in PBS containing streptavidin-conjugated Cy3.5 (1:50), 2% BSA, 10% normal goat serum, and 0.1% Triton X-100. Slices were then washed 3× in PBS followed by a 30 min incubation in 50% glycerol at room temperature. Slices were whole-mounted in Fluoromount-G (Southern Biotechnology). Mounted sections were imaged on an A1R confocal microscope (Nikon), with a 20× objective (Olympus). Z stacks were taken, spanning the visible range of the filled cell, at 1 μm intervals. Two-dimensional reconstructions were made in ImageJ by SUM-projecting Z stacks of filled neurons and then tracing with the simple neurite tracer ImageJ plugin. Sholl analysis was done on the two-dimensional reconstructions in ImageJ with the Sholl analysis plugin. Additionally, three-dimensional reconstructions were made with Imaris (Bitplane) and branch points were quantified in Imaris, using the three-dimensional reconstructions.
Cortical field potential recording
Slices were placed in an interface chamber maintained at 34°C, superfused with oxygenated aCSF at 2 ml/min, and cortical projections were stimulated with a tungsten concentric bipolar electrode at the layer 6-white matter boundary. Electrical stimulation consisted of 10–50 μA, 100 μs pulses at 30 s intervals delivered by a stimulus isolator (World Precision Instruments). Glass micropipettes (resistance ≅ 1 mΩ) were filled with aCSF and placed in layer 5 of the cortex directly above the stimulation electrode. Electrophysiological data were recorded with an Axon Multiclamp 700A amplifier and Digidata 1322A digitizer (sampling rate = 20 kHz) with Lab Chart software (AD Instruments). Threshold stimulation intensity was identified as the minimum amount of current required to elicit a detectable cortical field potential response (≥0.05 mV).
Chemical sources
All drugs were obtained from commercial sources. All chemicals for aCSF, drug treatments, and buffers were obtained from Sigma-Aldrich, including d-serine, DNQX, MPEP, and biocytin, unless noted below. DQP-1105, TFB-TBOA, GABAzine, NMDA, APV, and TBOA were obtained from Tocris Bioscience. CPP and TTX citrate were obtained from Abcam.
Experimental design and statistical analysis
Power analyses were performed to determine the sample size for each experiment, and the sample populations used for each experiment are detailed below. Mice of either sex were used for all experiments, with an approximate 1:1 ratio of female to male mice. Data were not analyzed by gender, but no abnormal distributions of responses were seen. For all electrophysiology analyses, data analysis was automated with MATLAB scripts and applied in the same fashion to datasets across all experimental groups. For all experiments, analysis was done using between-subject comparisons; no within-subject comparisons were made. Electrophysiology: Data collected from at least 3 animals/group, 3–5 slices per animal. Field EPSP recordings: Data collected from at least 3 animals/group, 3–5 slices per animal. Biocytin cell filling: Data collected from at least 3 animals/group, 3–5 slices per animal, 1–3 cells per slice. Immunofluorescence experiments: Data collected from at least 3 animals/group, 3–5 slices per animal. Glutamate imaging: Data collected from 2 or 3 animals per group, 3 slices per animal. For comparisons of one or two experimental groups, a Student's t test was used. For three or more experimental groups, a one-way ANOVA with the Tukey post hoc test was used. The Kolmogorov–Smirnov test was used to compare distributions of synaptic current properties. The Wilcoxon rank-sum test was used to compare two groups when data were not normally distributed. Values of p < 0.05 were considered statistically significant. In addition, a linear mixed model (LMM) was used to perform more rigorous statistical analysis of GABAergic synapse staining and IPSC recordings. In LMM analysis, data from the same animal are fitted with random intercepts to account for the correlation between repeated measurements on the same mouse. The fixed effects in these analyses were DQP treatment versus vehicle treatment; 95% CIs that exclude 0 and t values >1.96 and <−1.96 were considered to be statistically significant. Data are reported as mean ± SEM unless otherwise noted. Bar graphs depicting mean ± SEM values were used to represent the data unless a scatter plot could better depict variability within an experimental group.
Results
Ambient glutamate is elevated during early cortical development
Extracellular glutamate levels are controlled by astrocytic EAATs (Danbolt, 2001; Herman and Jahr, 2007; Le Meur et al., 2007). Because EAAT expression and function are dynamic during cortical development (Ullensvang et al., 1997; Hanson et al., 2015), we hypothesized that extracellular glutamate levels may vary as the cortex matures. To monitor extracellular glutamate levels, we first used fluorescent glutamate biosensor imaging in acute cortical slices (Dulla et al., 2013). This approach allows semiquantitative evaluation of glutamate levels across broad areas of the cortex in acute cortical slices. The glutamate biosensor is a FRET-based reporter in which FRET efficacy decreases in response to glutamate (Fig. 1a). Therefore, the ratio of CFP to YFP fluorescence reports the relative amount of glutamate present. We found that the glutamate biosensor FRET ratio was significantly lower at P5-P6 (2.89 ± 0.15) compared with P9-P10 (3.85 ± 0.09, p = 0.008), indicative of increased glutamate levels at younger ages. Intermediate glutamate biosensor FRET ratios were seen at P7-P8 (3.52 ± 0.26), consistent with dynamic extracellular glutamate levels in the developing cortex (Fig. 1b).
Figure 1.

Ambient glutamate is elevated in the neonatal cortex and decreases with development. a, Heat maps of the glutamate biosensor FRET ratio in acute cortical slices in aCSF, after the wash on of TBOA, and after the addition of saturating glutamate. FRET ratio is inversely proportional to glutamate concentration. b, Box-and-whisker plot showing baseline glutamate biosensor FRET ratios (in aCSF) in acute cortical slices at ages P5–P6 (n = 6 slices/2 animals), P7–P8 (n = 9 slices/3 animals), and P9–P10 (n = 9 slices/3 animals). **p = 0.008 (one-way ANOVA with Tukey post hoc test). Box boundaries represent quartiles. Circles represent individual slices. c, Example recording of a P3 L5P-nucleated patch voltage-clamped at 40 mV as NMDA (10, 20, 50, 1000 μm) and glutamate (100 μm) are locally perfused. Calibration: 30 s. d, Concentration-response curves generated for NMDA (dashed line, open circles) and glutamate (solid line, filled circles) at P3 (red) and P14 (black). P3 NMDA, n = 9 cells/4 animals; P14 NMDA, n = 10 cells/3 animals; P3 glutamate, n = 8 cells/4 animals; P14 glutamate, n = 7 cells/3 animals. Glutamate concentrations tested were 0.5, 1, 5, and 100 μm. Inset, Ratio of maximum glutamate response to maximum NMDA response amplitudes in L5P-nucleated patches at P3 and P14. e, Example traces represent whole-cell recordings of P3 (red) and P14 (black) L5Ps during bath application of 5 μm NMDA and, separately, 50 μm APV. Calibration: x = 60 s, y = 50 pA. f, Box-and-whisker plot of ambient glutamate values calculated from individual cells. Box boundaries represent quartiles. Box whiskers represent range. **p = 0.009 (two-sample t test). Error bars indicate SEM.
Next, to quantify the concentration of ambient glutamate in the cortex at different ages, we used an electrophysiological method (Herman and Jahr, 2007). This approach allows more precise quantification of extracellular glutamate levels and enables evaluation of glutamate levels at the neuronal membrane, as NMDARs are used as the glutamate detector. Briefly, the NMDAR concentration-response curves to glutamate and NMDA are generated using nucleated patches. Then, the neuronal response to APV, an NMDAR antagonist, and NMDA application are sequentially quantified. These values, combined with the concentration response curves, are used to extrapolate the micromolar concentration of glutamate. First, we established the concentration-response relationships for NMDA and glutamate in nucleated patches from layer 5 pyramidal neuron (L5P) somas at P3 and P14 (Fig. 1c,d) to calculate the apparent affinity of the NMDA receptors in situ. We calculated the EC50 values for NMDA and glutamate at P3 and P14 and used these values to generate conversion factors that allow us to convert concentrations of NMDA into functionally equivalent concentrations of glutamate at each age. Additionally, because glutamate is a more potent agonist of NMDARs than NMDA, we adjusted the conversion factor by the ratio of the maximum response in saturating NMDA to the maximum response in saturating glutamate at P3 (1.52 ± 0.12) and P14 (1.53 ± 0.16) (Fig. 1d, inset). Finally, the conversion factor was multiplied by 1000 to convert μm NMDA into nm glutamate. To determine ambient glutamate concentrations in cortical slices, whole-cell recordings were established in L5Ps in voltage-clamp mode. Neurons were voltage-clamped at 40 mV. NMDA (5 μm) was bath-applied, followed by wash out of NMDA, and bath application of APV (50 μm), a wide spectrum NMDA antagonist (Fig. 1e). For each cell, the APV-sensitive holding current was normalized to the NMDA-induced holding current, converting the APV effect into an equivalent concentration of NMDA (P3, 1.24 ± 0.03 μm; P14, 0.60 ± 0.05 μm). Using the conversion factors calculated in the first set of experiments, we determined the approximate concentrations of ambient glutamate to be 102 ± 13.8 nm at P3 and 56.9 ± 8.14 nm at P14. These results indicate that ambient glutamate is significantly elevated in the neonatal cortex compared with later time points (p = 0.009) (Fig. 1f).
The change from high to low extracellular glutamate coincided with developmental upregulation of EAATs in the cortex (Hanson et al., 2015). To test whether reductions in ambient glutamate levels after P7 are due to increased astrocytic EAAT function, we recorded tonic NMDAR-mediated currents from L5Ps held at 40 mV. Holding L5Ps at 40 mV relieved the magnesium block of NMDARs and allowed us to use NMDARs as detectors of extracellular glutamate, regardless of subunit composition. Once a stable baseline holding current was achieved, we bath-applied 100 μm TBOA (EAAT antagonist). Changes in the holding current upon transport blockade were normalized to capacitance (Cm) on a cell-by-cell basis to account for developmental changes in cell surface area (Fig. 2a). EAAT blockade induced a significantly larger change in capacitance-normalized holding current at P14 and older compared with P7-P8 and younger than P7 (Fig. 2b; p < 7 vs P14+: p = 0.00082; P7-P8 vs P14+: p = 0.0080; p < 7 vs P7-P8: p = 0.49; one-way ANOVA with Tukey post hoc test). EAAT blockade induced an increase in the holding current at each time point, consistent with increased EAAT function with postnatal age (p < 7: 1.82 ± 0.19 pA/pF, p = 0.00019; P7-P8: 2.20 ± 0.18 pA/pF, p = 0.000021; p > 14: 3.30 ± 0.39 pA/pF, p = 0.0000042; one-sample t test). Combined, these data suggest that, after P7-P8, increased astrocytic EAAT function reduces ambient glutamate levels and suppresses tonic NMDAR activation in pyramidal neurons.
Figure 2.
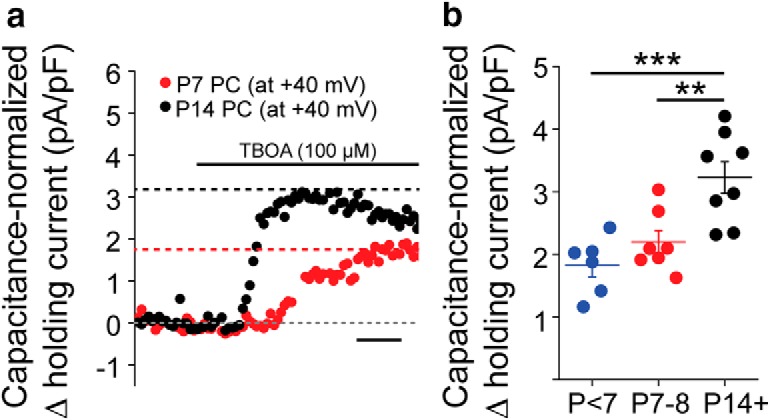
Maturation of glutamate uptake limits ambient glutamate after P7. a, Example recording, binned in 5 s increments, from a P7 L5P voltage-clamped at 40 mV during bath application of 100 μm TBOA. Gray dashed line indicates baseline. Black dashed line indicates holding current in TBOA at P14. Red dashed line indicates holding current in TBOA at P7. Calibration, 60 s. b, Capacitance-normalized, TBOA-induced holding currents binned by age (p < 7, n = 6 cells/3 animals; P7–P8, n = 7 cells/3 animals; P14+, n = 8 cells/4 animals). Circles represent individual cells. Lines indicate mean ± SEM. **p = 0.0080 (one-way ANOVA with Tukey post hoc test). ***p = 0.00082 (one-way ANOVA with Tukey post hoc test).
Neurons in the neonatal cortex are transiently sensitive to elevated ambient glutamate
To determine whether neurons in the developing cortex are sensitive to developmental changes in ambient glutamate, we recorded tonic NMDAR-mediated currents from L5Ps during cortical development. We then bath-applied APV (50 μm) and monitored the change in holding current required to maintain L5Ps at 40 mVs (Fig. 3a). APV-sensitive holding currents were normalized to Cm to account for developmental changes in cell size. The APV-sensitive holding current, normalized to Cm, reflects the density of NMDARs activated by ambient glutamate on the recorded neuron. Using this approach, we found that, before P7, L5Ps had consistently small APV-sensitive holding currents, even in the environment of elevated ambient glutamate (0.20 ± 0.01 pA/pF). This is likely due to low NMDAR expression (see Fig. 5f), as assessed by GluN2D FISH (Wenzel et al., 1997). At P7-P8, the APV-sensitive holding current was significantly increased compared with earlier time points (0.35 ± 0.04 pA/pF, p = 0.0010, one-way ANOVA with Tukey post hoc test), consistent with increased NMDAR expression during a period of elevated ambient glutamate. After P8, APV-sensitive holding currents were significantly decreased compared with P7-P8 (Fig. 3b,c; 0.22 ± 0.02 pA/pF, p = 0.014, one-way ANOVA with Tukey post hoc test), consistent with increased developmental expression of astrocytic EAATs and decreased ambient glutamate levels.
Figure 3.

Neurons in the neonatal cortex are transiently sensitive to elevated ambient glutamate. a, Example traces from voltage-clamped, cortical L5Ps held at 40 mV during application of APV (50 μm). Dashed lines indicate baseline and final holding currents. Calibration: x = 60 s, y = 25 pA. b, APV-sensitive holding currents (baseline − APV) in individual cells, normalized to capacitance on a cell-by-cell basis. Line indicates spline fit. c, Data in b, binned by age at P3–P6 (n = 11 cells/4 animals), P7–P8 (n = 9 cells/3 animals), and P10+ (n = 7 cells/5 animals). *p = 0.014 (one-way ANOVA with Tukey post hoc test). **p = 0.0010 (one-way ANOVA with Tukey post hoc test). Data are mean ± SEM.
Figure 5.

INs are tonically depolarized by ambient glutamate acting at GluN2C/DRs in the neonatal cortex. a, Averaged, baseline-adjusted, current-clamp traces, binned in 5 s increments, from L5Ps (n = 12 cells/4 animals) and INs (n = 9 cells/3 animals) during application of APV (50 μm). Traces represent the change in resting membrane potential upon APV bath application (ΔVm). b, Total change in resting potential (ΔVm) upon APV application in INs ages P3–P4 (n = 9 cells/ 3 animals), P7–P8 (n = 9 cells/ 4 animals), and P10+ (n = 10 cells/3 animals), as well as for L5Ps at P7 (n = 12 cells/5 animals). ##p = 0.0025 (two-tailed t test). ###p < 0.001 (two-tailed t test). **p < 0.01 (one-way ANOVA with Tukey post hoc test). ***p < 0.001 (one-way ANOVA with Tukey post hoc test). Lines indicate mean ± SEM. c, Total change in resting potential (ΔVm) upon 50 nm TFB-TBOA application in INs (n = 17 cells/7 animals) and L5Ps (14 cells/6 animals) at P7. **p < 0.01 (two-sample t test). d, Averaged, baseline-adjusted, current-clamp traces, binned in 5 s increments, from L5Ps (n = 8 cells/3 animals) and INs (n = 8 cells/4 animals) during bath application of DQP (20 μm). Traces represent ΔVm upon DQP application. e, Change in resting membrane potential after application DQP-1105 to P7 INs (n = 8 cells/4 animals) and L5Ps (n = 8 cells/3 animals). ##p = 0.0053 (two-tailed t test). *p = 0.012 (two-sample t test). Lines indicate mean ± SEM. f, Images represent absence of GluN2D mRNA signal at P3 and presence at P7, as detected by FISH using tyramide signal amplification with fluorescein (green). g, Images represent GluN2D and GAD56/67 mRNA signals detected by FISH using tyramide signal amplification with fluorescein (green) and HNPP/Fast Red TR (magenta), respectively. Nuclei were stained with DAPI (blue). Scale bars, 40 μm. h, Representative traces from a P7 L5P and a P7 IN demonstrating responses to positive and negative current injections before (top) and after (bottom) application of APV. Calibration: x = 50 ms, y = 20 mV. i, For each current injection: #APs before APV; #APs after APV = ΔAP. *p < 0.05 (one-sample t test comparing mean to zero). **p = 0.0043 (one-sample t test comparing mean to zero). Data are mean ± SEM.
Characterization of GFP+ neurons in neonatal G42 mice
To determine the effects of elevated ambient glutamate on inhibitory cortical development, we used the G42 mouse line to identify INs. In this mouse line, eGFP+ cells are ≈90% PV+ INs in the mature cortex (Chattopadhyaya et al., 2004). We found a similar colabeling of GFP+ cells with PV immunolabeling in cortical layers 5/6 in P28 G42 mice (data not shown). In the neonatal cortex, eGFP+ cells are present in the somatosensory cortex as early as P3, allowing us to identify INs very early in development, before the expression of PV. This approach is advantageous because, as PV is not expressed in the cortex until approximately P12-P14, meaning that PVCRE-based approaches are not feasible for identifying immature PV INs. To characterize the identity of GFP+ cells in the neonatal cortex, we examined their expression of markers of inhibitory neurons, characterized their electrophysiological properties, and examined GAD1 mRNA expression (Fig. 4). In neonatal animals (P9), >80% of eGFP+ cells colabeled for GABA, suggesting that they are immature inhibitory INs (Fig. 4a,b). Additionally, we observed robust SST expression in the P9 cortex but found little to no colocalization with eGFP (1 of 262 eGFP+ cells expressed SST). Calretinin was also not expressed at P9, so no colocalization of GFP+ cells with calretinin was seen (Fig. 4a,b).
Figure 4.
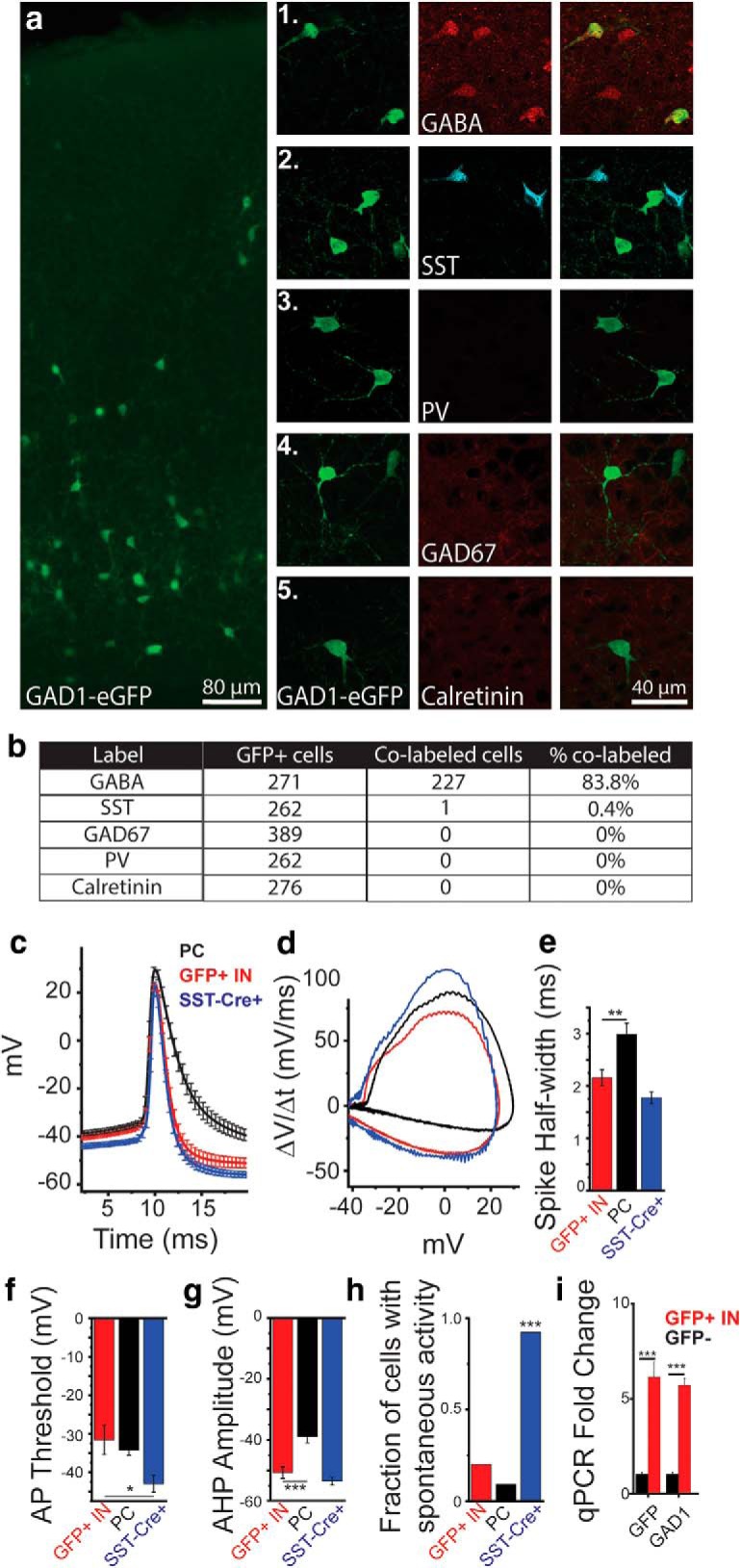
G42 mice identify GABAergic non-SST INs at P9. a, Immunohistochemical labeling of GFP in the somatosensory cortex (SSC) of G42 (GAD1-eGFP) mice at P9. Coimmunolabeling of GFP in G42 mice with markers on inhibitory INs. a1–a5, GFP (left). a1, GABA. a2, Somatostatin. a3, PV. a4, GAD67. a5, Calretinin (middle), colabel (right) in layer V of the SSC. b, Table showing abundant colabeling of GFP+ cells with GABA immunolabeling at P9. Sections from 4 mice were used to quantify colocalization. c, Average evoked APs from whole-cell recordings from GFP+ cells in G42 mice (red), L5Ps (black), and Td-tomato+ neurons in SST-Cre X Ai9 mice (blue). d, dV/dt analysis of APs shown in c. e, AP half-width. f, AP threshold. g, Afterhyperpolarization amplitude. h, Fraction of cells showing spontaneous AP firing in GFP+ cells in G42 mice (red), L5P neurons (black), and Td-Tomato+ neurons in SST-Cre X Ai9 mice (blue). *p < 0.05 (one-way ANOVA). **p < 0.01 (one-way ANOVA). ***p < 0.001 (one-way ANOVA). i, qPCR analysis of GFP+ and GFP− cells at P9 in G42 mice separated via FACS. ***p < 0.001 (paired t test).
Next, we characterized the AP properties in GFP+ cells and compared them with L5Ps and SST+ INs (identified in SST-Cre X Ai9 mice; Fig. 4c–h). eGFP+ cells had AP properties consistent with immature INs, compared with L5Ps, including short AP half-width and more hyperpolarized afterhyperpolarizations (Fig. 4c–g). The electrophysiological properties of eGFP+ cells were also distinct from SST+ INs at P9 based on AP threshold and spontaneous AP firing (Fig. 4g,h). This is consistent with SST+ IN′s propensity to have low thresholds for AP initiation (Urban-Ciecko and Barth, 2016) and their characteristic exhibition of frequent spontaneous activity that is often independent of GABAergic or glutamatergic synaptic activity (Fanselow et al., 2008). Finally, we isolated GFP+ cells at P9 via FACS and performed qPCR on mRNA isolated from GFP+ and GFP− cells. mRNA for GFP was significantly increased in the GFP sorted cells, confirming robust sorting. GAD1 (GAD65/67) levels were also increased in GFP+ cells, confirming that these cells are GABAergic (Fig. 4i). Together, these studies show that, at P9, eGFP+ cells in G42 mice are immature non-SST GABAergic INs. It is highly likely that these GFP+ GABAergic INs are putative immature PV+ INs that will express PV later in life.
INs are tonically depolarized by ambient glutamate in the neonatal cortex
Recording tonic NMDA currents in L5Ps held at 40 mV, as above, is an assay of ambient glutamate sensitivity, but it does not address whether NMDA receptors are activated by ambient glutamate at the normal neuronal resting membrane potential (≈−60 mV). To determine whether cortical INs and L5Ps are tonically depolarized by NMDAR activation, we made current-clamp recordings from L5Ps and cortical INs, as identified by GFP+ labeling in G42 mice (Fig. 5a). First, we recorded resting membrane potential (Vm) before and during bath application of APV (50 μm). Baseline resting potentials were significantly depolarized in P7 INs (Vm −55.51 ± 2.80 mV) compared with P7 L5Ps (Fig. 5b; Vm −63.30 ± 2.10 mV, p = 0.037, two-sample t test). Upon bath application of APV at P7, we observed that INs, but not PCs, were significantly hyperpolarized (Fig. 5b; IN ΔVm = −3.77 ± 0.44 mV, p = 0.000027; PC ΔVm = 0.83 ± 0.58 mV, p = 0.18; two-tailed t test). This suggests that tonic NMDAR activation depolarizes INs, but not PCs, at resting Vm. We next examined whether NMDAR-mediated tonic depolarization of INs was uniform across cortical development. Compared with P7, INs were significantly less hyperpolarized by APV at P3-P5 (ΔVm = −1.46 ± 0.22 mV, p = 0.0071) and P10-P12 (ΔVm = −0.98 ± 0.57 mV, p = 0.0034, one-way ANOVA with Tukey post hoc test). If tonic activation of INs is constrained by astrocytic control of ambient glutamate, then blocking EAAT function should have differential effects on IN versus PC Vm at P7. To test this, we partially blocked EAATs using 50 nm TFB-TBOA at P7, the age of peak ambient glutamate-mediated depolarization of INs. Consistent with our hypothesis, TFB-TBOA caused small, but significantly different, changes in IN (0.41 ± 0.45 mV) versus PC (−1.21 ± 0.27 mV) Vm (Fig. 5c; p = 0.005; two-sample t test). These findings confirm that tonic NMDAR-mediated depolarization of INs is transient in the developing cortex and is largest at P7.
To determine whether GluN2C/D-containing NMDARs mediate this tonic depolarization, we repeated these experiments using DQP-1105 (20 μm), a GluN2C/D-selective antagonist (Acker et al., 2011) (Fig. 5d). Upon bath application of DQP-1105, we found that pharmacological blockade of GluN2C/Ds also hyperpolarized INs, but not L5Ps, at P7 (Fig. 5d,e; IN ΔVm = −3.37 ± 0.85 mV, p = 0.0053; PC ΔVm = −0.37 ± 0.58 mV, p = 0.30; two-tailed t test). As GluN2C mRNA is not present in the cortex during early development (Akazawa et al., 1994; Monyer et al., 1994; Karavanova et al., 2007), we examined when GluN2D was detectable in the neonatal cortex. Using ISH, we found that GluN2D was absent from the cortex at P3 but was present at P7 (Fig. 5f). This is consistent with multiple previous studies (Akazawa et al., 1994; Monyer et al., 1994; von Engelhardt et al., 2015; Loo et al., 2019). To confirm that the GluN2D receptor is present in developing cortical INs, we performed further in situ hybridization studies. We found that GAD (GAD65/GAD67) and the GluN2D (Grin2D) subunit of the NMDA receptor showed colocalization in P9 somatosensory cortex (Fig. 5f), confirming the presence of high-affinity NMDA receptors in GABAergic cortical INs. Together, these data are consistent with our electrophysiological studies showing tonic activation of developing cortical INs at P7, but not before.
Finally, we examined whether tonic NMDAR activation affected IN and L5P excitability. Current steps were injected into INs and L5Ps, and the number of APs generated was quantified before and after NMDAR blockade (Fig. 5f). We found that, following blockade of all NMDARs with APV, INs fired fewer APs in response to current injection (Fig. 5g; ΔAP# at 90 pA = −1.8 ± 0.58, p = 0.018, one-tailed t test). Interestingly, PCs fired more APs in response to positive current injection after APV application (ΔAP# at 90 pA = 1.6 ± 0.68, p = 0.039, one-tailed t test), perhaps due to a net disinhibitory effect of APV acting to reduce IN activity.
Transient blockade of GluN2C/DRs from P7-P9 decreases inhibitory synaptic activity and GABAergic synapse density in the mature cortex
We next tested whether GluN2C/D-mediated tonic depolarization of developing cortical INs contributes to their functional and morphological maturation. G42 mice were treated in vivo with DQP-1105 (28 mg/kg, i.p.) (Lozovaya et al., 2014) or vehicle, once daily. Two treatment windows were examined, from P7-P9, during the peak of tonic GluN2C/D-mediated depolarization of INs, and from P11-P13, after the peak of GluN2C/D-mediated depolarization of INs. At P21-P27, animals were killed, and brains were collected for electrophysiology or immunohistochemistry. First, we measured inhibitory synaptic activity by monitoring mIPSCs in L5Ps. Voltage-clamp recordings were made from L5Ps using a high-chloride internal and held at −70 mV. Recordings were made in the presence of TTX and glutamate receptor blockers. Under these conditions, mIPSCs are detected as fast inward currents, which are blocked by the application of GABAzine (Fig. 6a). Treatment with DQP-1105 from P7-P9 significantly reduced mIPSC frequency at P21-P28, as demonstrated by longer interevent intervals (p = 5.0e-6, two sample Kolmogorov–Smirnov test) and a reduced average frequency (Fig. 6b,c; mIPSCDQP = 3.43 ± 0.37 Hz, mIPSCVEH = 4.60 ± 0.37 Hz, p = 0.028, two-sample t test). mIPSC amplitude was unaffected by DQP treatment (Fig. 6d,e; DQP = 28.5 ± 2.3 pA, Veh = 31.9 ± 1.7 pA, p = 0.25, two-sample t test). When animals were treated with DQP during a later window, from P11-P13, neither mIPSC amplitudes (DQP = 34.0 ± 2.8 pA, Veh = 39.7 ± 3.9 pA, p = 0.25, two-sample t test) nor frequencies (p = 0.12, two-sample Kolmogorov–Smirnov test) were different between DQP and vehicle-treated groups. These data suggest that specific activation of GluN2C/D-containing NMDARs during P7-P9 contributes significantly to the maturation of inhibitory synaptic activity in the cortex.
Figure 6.

In vivo blockade of GluN2C/D-containing NMDARs from P7–P9, but not P11–P13, causes long-term reductions in inhibitory synaptic activity and GABAergic synapse density. a, Representative mIPSC traces from animals treated with either DQP-1105 or vehicle at P7. mIPSCs are abolished by the application of GABAzine. Calibration: x = 10 s, y = 25 pA. b, Cumulative probability distribution (CPD) of mIPSC interevent intervals (IEIs) at P21 after treatment from P7–P9 with DQP-1105 (n = 27 cells/4 animals) or vehicle (n = 26 cells/4 animals). Inset, Average frequency. *p = 0.028 (two-sample t test). ###p = 5.0e-6 (two-sample Kolmogorov–Smirnov test). c, CPD of mIPSC IEIs at P21 after treatment from P11–P13 with DQP-1105 (n = 12 cells/2 animals) or vehicle (n = 11 cells/2 animals; p = 0.12, two-sample Kolmogorov–Smirnov test). Inset, Average frequency (p = 0.59, two-sample t test). d, CPD of mIPSC amplitudes at P21 after treatment from P7–P9 with DQP or vehicle (p = 0.48, two-sample Kolmogorov–Smirnov test). Inset, Average mIPSC amplitudes (p = 0.25, two-sample t test). e, CPD of mIPSC amplitudes at P21 after treatment from P11–P13 with DQP or vehicle (p = 0.089, two-sample Kolmogorov–Smirnov test). Inset, Average mIPSC amplitude (p = 0.25, two-sample t test). f, Enhanced-contrast z stack projection images showing colocalizaion of VGAT and Gephyrin staining at P21. Animals were treated with vehicle (n = 17 sections/7 animals) or DQP (n = 18 sections/7 animals) from P7–P9. Arrows indicate colocalization as identified in unprocessed z stack projection images. Scale bars, 2 μm. g, Quantification of the density of synapses. Circles represent synapse density in individual sections. Lines indicate mean ± SEM. *p = 0.046 (two-sample t test).
Next, we used immunofluorescence to examine whether the decrease in mIPSC frequency was a consequence of decreased GABAergic synapse density. C57Bl/6J mice were treated with DQP-1105 or vehicle in vivo from P7-P9. On P21, mice were killed, and the brains were fixed for sectioning and immunolabeled for the vesicular GABA transporter (VGAT) and the GABA receptor binding protein gephyrin (Geph). Colocalization of VGAT and Geph was used as a marker of GABAergic putative inhibitory synapses (Fig. 6f). We observed that the density of colocalized VGAT and Geph puncta was slightly, but significantly, reduced in P7-P9 DQP-treated animals (Veh, 50.1 ± 2.6 synapses/100 μm2; DQP, 43.4 ± 1.9 synapses/100 μm2; p = 0.046, two-sample t test), indicating a reduction in GABAergic synapse density (Fig. 6g).
To increase the statistical rigor of these experiments, we also performed an LMM analysis of DQP-induced effects on GABAergic staining and IPSC frequency. Briefly, LMM allows for statistical analysis of samples taken from the same experimental animal, which are not truly independent of each other, by including a random effect to account for animal-to-animal variability. Using LMM, we first reanalyzed the effects of neonatal DQP treatment on the number of putative GABAergic synapses. Using this approach, we found that DQP treatment, versus vehicle, had a statistically significant effect size of −6.63 (t value = −2.077; 95% CI = −12.87 to −0.39). This confirms our finding and demonstrates that neonatal DQP treatment has a significant effect on GABAergic synapse density, even when interanimal variability is taken into account. We next used LMM analysis to reanalyze the effects of neonatal DQP treatment on mean IPSC frequency. Here we found that neonatal DQP treatment had an effect size of −1.06, although this effect did not quite reach statistical significance (t value = −1.214; 95% CI = −2.71 to 0.63). Therefore, there is a chance that DQP's effects on mean IPSC frequency may be due to animal-to-animal variability.
Transient blockade of GluN2C/DRs from P7-P9 reduces the complexity of IN dendritic arbors in the adult cortex
The effect of in vivo GluN2C/DR blockade on IN morphology was examined by biocytin-filling and reconstructing eGFP+ neurons in DQP and vehicle-treated G42 mice (Fig. 7a). Only dendritic arbors, and not axonal arbors, were filled, imaged, and analyzed. In agreement with the effects on inhibition, DQP-1105 treatment from P7-P9 strongly affected IN morphological maturation. When assayed at P21, we found that filled and reconstructed eGFP+ INs had significantly reduced dendritic arbor complexity. Sholl analysis revealed that DQP-1105 treatment at P7-P9 significantly reduced the complexity of IN dendritic arbors (Fig. 7b; intersections at 65 μm: DQP, 9.0 ± 0.87; Veh, 11.27 ± 0.49; p = 0.025). Additionally, DQP treatment significantly reduced the number of dendritic branch points (Fig. 7c; DQP, 30.25 ± 4.9; Veh, 54.27 ± 7.37; p = 0.015). The reductions in morphological complexity also corresponded to reduced capacitance of INs treated with DQP from P7-P9 compared with vehicle-treated INs (Fig. 6d; DQP, 77.1 ± 4.7 pF; Veh, 105.0 ± 7.3 pF; p = 0.0019). DQP-1105 treatment did not change the resting potential (DQP, −65.0 ± 1.1 mV; Veh, −61.8 ± 1.8 mV; p = 0.12) or membrane resistance (DQP, 201.4 ± 18.8 mΩ; Veh, 202.8 ± 20.5 mΩ; p = 0.96) of cortical INs. Importantly, DQP-1105 treatment did not change the density of eGFP+ neurons in the cortex (DQP, 16.25 ± 0.78 eGFP+ cells/mm2; Veh, 15.25 ± 1.57 eGFP+ cells/mm2; p = 0.5, data not shown). We did not examine DQP-1105's effects on IN axonal projections, and thus cannot exclude that important changes in axonal structure and output may also occur. These results demonstrate that in vivo pharmacological attenuation of GluN2C/Ds from P7-P9 significantly alters the morphological development of cortical IN dendrites without affecting IN number.
Figure 7.

In vivo blockade of GluN2C/D-containing NMDARs from P7–P9 leads to long-term reductions in IN dendritic arbor complexity. a, Representative projections of reconstructed INs in the cortex at P21 after treatment from P7–P9 with DQP (red, n = 8 cells/4 animals) or vehicle (black, n = 11 cells/3 animals). b, Sholl analysis using 2 d projections of reconstructed INs to quantify the number of intersections at increasing distances from the soma from 10 to 580 μm in 30 μm intervals. *p < 0.05 (two-sample t test). Data are mean ± SEM. c, Box-and-whisker plot showing quantification of total branch point number made on 3 d reconstructed neurons. Box boundaries indicate quartiles. Scatterplot of solid circles represents individual data points. Solid squares represent group means. **p = 0.015 (two-sample t test). d, Box-and-whisker plot showing quantification of capacitance from INs at P21 that were treated with DQP-1105 (n = 21 cells/7 animals) or vehicle (n = 13 cells/4 animals) from P7–P9. Box boundaries indicate quartiles. Scatter plot of solid circles represents individual data points. Solid squares represent group means. **p = 0.0019 (two-sample t test).
Attenuation of GluN2C/DRs from P7-P9 leads to hyperexcitability in the mature cortex
To examine cortical network activity, C57Bl/6J mice were treated with DQP-1105, or vehicle, in vivo from P7-P9, as described above. On P21, acute cortical brain slices were prepared and cortical fEPSPs were evoked by electrical stimulation of the white matter beneath the cortex while recording in layer 5 (Fig. 8a). Cortical fEPSPs are typically brief and monophasic due to powerful IN-mediated feedforward inhibition. However, in models of cortical hyperexcitability, fEPSPs are often prolonged and polyphasic (Jacobs et al., 1996). As expected, fEPSPs evoked in vehicle-treated animals were uniformly monophasic. In line with previous results indicating that DQP-1105 treatment from P7-P9 decreases cortical inhibition, the incidence of hyperexcitable network activity (polyphasic, high-frequency activity) was increased in P7-P9 DQP-1105-treated animals compared with P7-P9 vehicle-treated animals (Fig. 8b; 28.4 ± 9.2% epileptiform traces/slice, p = 0.0021, Wilcoxon rank-sum test). This type of activity is commonly seen in models of cortical hyperexcitability (Jacobs et al., 1996; Hunt et al., 2009), and is consistent with compromised cortical inhibition. Importantly, animals treated from P11-P13 with either DQP or vehicle showed uniformly monophasic fEPSPs, suggesting that GluN2C/D-mediated signaling specifically from P7-P9, but not from P11-P13, is necessary for the development of normal cortical inhibition and excitability.
Figure 8.
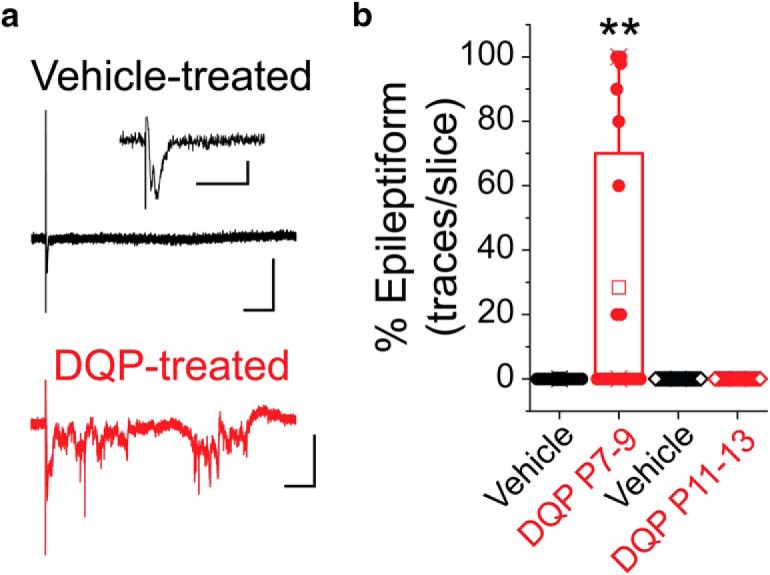
In vivo blockade of GluN2C/D-containing NMDARs from P7–P9 leads to network hyperexcitability in the adult cortex. a, Representative cortical field potential traces from animals treated with DQP or vehicle from P7–P9. Calibration: x = 100 ms, y = 0.2 mV. Recordings were made at P21. The DQP-treated example trace represents epileptiform activity, which was only observed in animals treated with DQP from P7–P9. Inset (vehicle-treated panel), Magnified field potential. Calibration: Inset, x = 20 ms, y = 0.05 mV. b, Box-and-whisker plot showing the distribution of the percentage epileptiform traces in slices from each group at 2× threshold stimulation (P7–P9 DQP: n = 20 slices/3 animals; P7–P9 Veh: n = 17 slices/3 animals; P11–P13 DQP: n = 20 slices/3 animals; P11–P13 Veh: n = 20 slices/3 animals). Box boundaries mark quartiles. Filled circles represent data from individual slices. Open squares represent the mean for each group. **p = 0.0021 (Wilcoxon rank-sum test).
Discussion
In these studies, we demonstrate a novel mechanism by which ambient glutamate contributes to the development of cortical inhibitory networks. First, we show that ambient glutamate is elevated in the neonatal cortex, which leads to tonic depolarization of cortical INs, but not pyramidal cells. This tonic depolarization is mediated by GluN2C/DRs and is greatest at the end of the first postnatal week. We go on to demonstrate that reducing GluN2C/D-mediated signaling from P7-P9 is sufficient to disrupt the development of IN dendritic arbors, decrease inhibitory synapse number, and cause lasting cortical network hyperexcitability. These studies indicate that tonic activation of GluN2C/DRs at the end of the first postnatal week plays an important role in the maturation of cortical INs and inhibition. Together, the results suggest that low glutamate transporter expression in the neonatal cortex is an important feature of cortical development, creating a permissive environment for tonic glutamate signaling and facilitating the maturation of inhibitory systems.
Ambient extracellular glutamate is elevated in the neonatal cortex
It is well established that the expression of the glutamate transporters GLT-1 and GLAST is low in the neonatal brain, and that expression increases over the first few weeks of postnatal development (Furuta et al., 1997; Ullensvang et al., 1997; Danbolt, 2001; Hanson et al., 2015). More recently, it has been shown that reduced transporter expression results in prolonged extracellular glutamate transients (Thomas et al., 2011). Together, these findings imply that glutamate may remain in the extracellular space for prolonged periods in the neonatal cortex, when transporter expression is low (Hanson et al., 2015). However, synaptic glutamatergic signaling is minimal in the neonatal cortex. Indeed, in the developing cortex, astrocytic expression of glutamate transporters is triggered by the onset of neuronal activity and synaptic glutamate release (Morel et al., 2014). This would seem to suggest that transporter expression is primarily required to manage glutamate clearance after synaptic release. There is ample evidence, however, supporting a role for nonsynaptic, tonic glutamate signaling during early cortical development. For example, functional NMDARs are present on cortical neurons long before the formation of functional synapses, and even before the completion of neuronal migration (LoTurco et al., 1991; Rossi and Slater, 1993). Indeed, the migration of cortical INs relies, in part, on the activation of nonsynaptic NMDARs (Bortone and Polleux, 2009). Additionally, glutamate is known to mediate early forms of spontaneous network activity in the cortex, which occur before the formation of glutamatergic synapses (Garaschuk et al., 2000). Enzymatically scavenging glutamate, which preferentially reduces nonsynaptic glutamate, abolishes spontaneous glutamatergic activity and alters concurrent GABAergic activity (Allène et al., 2008). Here, we demonstrate that low glutamate transporter expression in the neonatal cortex results in higher ambient glutamate concentrations, supporting the idea that slow glutamate uptake is permissive for tonic glutamate signaling. It is important to recognize that estimates of glutamate levels vary significantly depending on the methodology used; electrophysiological measurements tend to report much lower concentrations than microdialysis. This is thought to be due to regional variation in local glutamate concentration combined with region-selective assays (Moussawi et al., 2011). Therefore, our findings may have more relevance to extrasynaptic glutamate levels experienced by NMDARs, as measured using electrophysiology, rather than bulk extracellular glutamate concentration, as measured using microdialysis. Also, important to note, our electrophysiological assay of ambient glutamate (Fig. 1) reflects the concentration at the surface of L5Ps. The concentration of ambient glutamate at the surface of GABAergic INs may be unique from what is measures using L5Ps.
IN sensitivity to ambient glutamate
In the adult brain, the concentration of ambient glutamate is thought to be too low to significantly activate most NMDARs (Herman and Jahr, 2007). Furthermore, the strong voltage-dependent magnesium block of most NMDARs limits their ability to pass a physiologically relevant tonic current (Vicini et al., 1998). Despite this, tonic activation of NMDARs has been shown to influence excitability in a variety of neuronal subtypes across multiple brain regions (Sah et al., 1989; Chergui et al., 1993; Moos et al., 1997; Dalby and Mody, 2003; Le Meur et al., 2007; Povysheva and Johnson, 2012), including at times when the target neurons are not artificially depolarized (Yang and Xu-Friedman, 2015; Riebe et al., 2016). Based on their unique properties, including high glutamate affinity and low-magnesium affinity, GluN2C/DRs have been hypothesized to be the primary mediators of a glutamatergic tonic current (Le Meur et al., 2007; Riebe et al., 2016). Expression of these subunits in the cortex and hippocampus is largely restricted to GABAergic INs (Monyer et al., 1994; Xi et al., 2009). Accordingly, use-dependent NMDAR antagonists (blocking mostly extrasynaptic and nonsynaptic NMDARs) (Wu and Johnson, 2015; Sapkota et al., 2016) strongly affect cortical and hippocampal INs, leading to net disinhibition (Homayoun and Moghaddam, 2007; Riebe et al., 2016). In the current study, we demonstrate that cortical INs, but not L5Ps, are tonically depolarized by ambient glutamate acting at GluN2C/DRs. These results confirm that INs are uniquely sensitive to ambient glutamate and show that tonic depolarization depends on IN GluN2C and/or GluN2D expression.
Importantly, GluN2C and GluN2D expression is developmentally regulated in the cortex, with minimal GluN2C in the cortex even at its peak at P21 (Akazawa et al., 1994) and GluN2D expression absent at P3 and peaking at ∼P7 (Monyer et al., 1994) (Fig. 5). Together with the early elevation in ambient glutamate, the developmental expression of GluN2C/DRs suggests that tonic depolarization of INs would be greatest during early cortical development. The results of the current study support this hypothesis by demonstrating that the GluN2C/DR-mediated tonic depolarization of cortical INs is greatest at P7. Furthermore, the developmental expression profiles of GluN2C and GluN2D led us to expect that tonic depolarization at P7 is mediated mostly by GluN2D (Monyer et al., 1994; Loo et al., 2019).
Influence of tonic glutamate signaling on IN development
The maturation of cortical INs and GABAergic synaptogenesis is ongoing at P7 (Le Magueresse and Monyer, 2013). It has been established that IN migration, maturation, and inhibitory synaptogenesis rely, in part, on neuronal activity and changes in intrinsic excitability (Meier et al., 2003; Bortone and Polleux, 2009; De Marco García et al., 2011). Along these lines, our study demonstrates that blockade of GluN2C/DRs from P7-P9 led to long-term decreases in the number of GABAergic synapses, cortical network hyperexcitability, and suggest deficits in synaptic inhibition. Based on the timing of the treatment, these reductions are likely due to a failure of synaptogenesis (Le Magueresse and Monyer, 2013); however, deficits in GABAergic synapse maintenance have not been ruled out. Intriguingly, global GluN2C KO mice show an increase in mIPSC frequency, the opposite of the effects we report here (Gupta et al., 2016). Additionally, we find that treatment from P11-P13, at the peak of GluN2C expression, does not alter mIPSC frequency. These results suggest that treating mice with DQP-1105 from P7-P9 specifically disrupts aspects of IN maturation dependent on GluN2D-containing NMDARs. Inhibitory and excitatory synaptogenesis has not been investigated in GluN2D KO mice, although these mice exhibit changes in cortical gamma oscillations, which imply broad changes in inhibitory circuitry (Yamamoto et al., 2017).
Mechanisms driving the maturation of IN dendritic arbors are poorly understood. While it has been shown that specific subtypes of caudal ganglionic eminence-derived INs rely on cell-intrinsic activity to drive their overall morphological maturation (De Marco García et al., 2011), it is unclear to what extent this is generalizable to medial ganglionic eminence-derived and PV-expressing INs. Additionally, mechanisms driving axonal and dendritic arborization are likely distinct and, therefore, should be considered separately. In general, dendritic arborization depends on a variety of intrinsic and environmental signals, including neuronal activity and NMDAR activation (Cline, 2001). In the current study, we find that the complexity of IN dendritic arbors is substantially reduced by neonatal GluN2C/DR blockade. An interesting possibility is that the maturation of IN dendritic arbors relies on Ca2+ entry through the GluN2C/DRs, as has been suggested in other systems (Konur and Ghosh, 2005). Furthermore, nonsynaptic and extrasynaptic GluN2C/D-containing NMDARs may act synergistically with synaptic NMDARs by helping to depolarize the neuronal membrane, which may augment their influence on dendritic Ca2+ signaling. In the current study, we do not differentiate between the Ca2+ signaling and depolarization-mediated effects of GluN2C/DRs, which remains an interesting unanswered question.
In conclusion, imbalances in excitatory and inhibitory neurotransmission in the cortex are part of the pathophysiology underlying a variety of neurodevelopmental disorders, including autism, schizophrenia, and epilepsy (McCormick and Contreras, 2001; Gogolla et al., 2009; Gandal et al., 2012; Uhlhaas and Singer, 2012). These imbalances can arise from changes in synapse density or synaptic activity, they can be acute or developmental, and they drive aberrant network activity which disrupts the function of the cortex (Paz and Huguenard, 2015). After neonatal GluN2C/DR blockade, we observed a lasting reduction in both structural inhibitory synapses and inhibitory synaptic activity, suggesting that the excitatory-inhibitory balance is shifted away from inhibition. Accordingly, we find that neonatal GluN2C/DR blockade induces lasting cortical hyperexcitability. Together, the current results demonstrate that GluN2C/DRs mediate a transient tonic signal onto INs in the neonatal cortex, which is necessary for establishing inhibitory cortical networks. We have not yet examined how interrupting neonatal GluN2C/DR affects excitatory systems. This is an important limitation of our study as there may be compensatory changes in excitation that occur in parallel with the changes in inhibition we demonstrate here. Importantly, these results imply that briefly modulating GluN2C/DRs on INs during a critical developmental window is sufficient to cause lasting cortical network deficits. It is therefore possible that factors either acutely affecting IN activity or disturbing the maturation of astrocytic glutamate uptake may drive pathological cortical development by altering the development of cortical inhibition. These findings may have important implications for the pathogenic mechanisms of disorders, such as schizophrenia, where cortical inhibitory networks are disrupted, and epilepsy, where the excitatory-inhibitory balance is altered leading to hyperexcitable cortical networks.
Footnotes
This work was supported by the Epilepsy Foundation to C.G.D., National Institute of Neurological Disease and Stroke R01-NS076885 to C.G.D., R56-NS076885 to C.G.D., NS036554 to S.F.T., Synapse Neurobiology Training Grant NINDS T32-NS061764 to E.H., and Tufts Center for Neuroscience Research P30 S047243. We thank Jennifer Shih, Thomas Papouin, Paul Rosenberg, Chinfei Chen, Christian Bjorbaek, Dan Cox, and John Lisman for helpful comments; and Pedram Hamrah, Arisa Jamali, and Allen Parmelee for help in establishing a FACS protocol.
S.F.T. is on the Scientific Advisory Board for Sage Therapeutics, and a cofounder of NeurOp Inc. The remaining authors declare no competing financial interests.
References
- Acker TM, Yuan H, Hansen KB, Vance KM, Ogden KK, Jensen HS, Burger PB, Mullasseril P, Snyder JP, Liotta DC, Traynelis SF (2011) Mechanism for noncompetitive inhibition by novel GluN2C/D N-methyl-D-aspartate receptor subunit-selective modulators. Mol Pharmacol 80:782–795. 10.1124/mol.111.073239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akazawa C, Shigemoto R, Bessho Y, Nakanishi S, Mizuno N (1994) Differential expression of five N-methyl-D-aspartate receptor subunit mRNAs in the cerebellum of developing and adult rats. J Comp Neurol 347:150–160. 10.1002/cne.903470112 [DOI] [PubMed] [Google Scholar]
- Allène C, Cattani A, Ackman JB, Bonifazi P, Aniksztejn L, Ben-Ari Y, Cossart R (2008) Sequential generation of two distinct synapse-driven network patterns in developing neocortex. J Neurosci 28:12851–12863. 10.1523/JNEUROSCI.3733-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andresen L, Hampton D, Taylor-Weiner A, Morel L, Yang Y, Maguire J, Dulla CG (2014) Gabapentin attenuates hyperexcitability in the freeze-lesion model of developmental cortical malformation. Neurobiol Dis 71:305–316. 10.1016/j.nbd.2014.08.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angevine JB Jr, Sidman RL (1961) Autoradiographic study of cell migration during histogenesis of cerebral cortex in the mouse. Nature 192:766–768. 10.1038/192766b0 [DOI] [PubMed] [Google Scholar]
- Batista-Brito R, Fishell G (2009) The developmental integration of cortical interneurons into a functional network. Curr Top Dev Biol 87:81–118. 10.1016/S0070-2153(09)01203-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belforte JE, Zsiros V, Sklar ER, Jiang Z, Yu G, Li Y, Quinlan EM, Nakazawa K (2010) Postnatal NMDA receptor ablation in corticolimbic interneurons confers schizophrenia-like phenotypes. Nat Neurosci 13:76–83. 10.1038/nn.2447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry M, Rogers AW (1965) The migration of neuroblasts in the developing cerebral cortex. J Anat 99:691–709. [PMC free article] [PubMed] [Google Scholar]
- Bortone D, Polleux F (2009) KCC2 expression promotes the termination of cortical interneuron migration in a voltage-sensitive calcium-dependent manner. Neuron 62:53–71. 10.1016/j.neuron.2009.01.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushong EA, Martone ME, Ellisman MH (2004) Maturation of astrocyte morphology and the establishment of astrocyte domains during postnatal hippocampal development. Int J Dev Neurosci 22:73–86. 10.1016/j.ijdevneu.2003.12.008 [DOI] [PubMed] [Google Scholar]
- Buzsáki G, Draguhn A (2004) Neuronal oscillations in cortical networks. Science 304:1926–1929. 10.1126/science.1099745 [DOI] [PubMed] [Google Scholar]
- Cardin JA, Carlén M, Meletis K, Knoblich U, Zhang F, Deisseroth K, Tsai LH, Moore CI (2009) Driving fast-spiking cells induces gamma rhythm and controls sensory responses. Nature 459:663–667. 10.1038/nature08002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavelier P, Hamann M, Rossi D, Mobbs P, Attwell D (2005) Tonic excitation and inhibition of neurons: ambient transmitter sources and computational consequences. Prog Biophys Mol Biol 87:3–16. 10.1016/j.pbiomolbio.2004.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyaya B, Di Cristo G, Higashiyama H, Knott GW, Kuhlman SJ, Welker E, Huang ZJ (2004) Experience and activity-dependent maturation of perisomatic GABAergic innervation in primary visual cortex during a postnatal critical period. J Neurosci 24:9598–9611. 10.1523/JNEUROSCI.1851-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chergui K, Charléty PJ, Akaoka H, Saunier CF, Brunet JL, Buda M, Svensson TH, Chouvet G (1993) Tonic activation of NMDA receptors causes spontaneous burst discharge of rat midbrain dopamine neurons in vivo. Eur J Neurosci 5:137–144. 10.1111/j.1460-9568.1993.tb00479.x [DOI] [PubMed] [Google Scholar]
- Cline HT. (2001) Dendritic arbor development and synaptogenesis. Curr Opin Neurobiol 11:118–126. 10.1016/S0959-4388(00)00182-3 [DOI] [PubMed] [Google Scholar]
- Cohen SM, Tsien RW, Goff DC, Halassa MM (2015) The impact of NMDA receptor hypofunction on GABAergic neurons in the pathophysiology of schizophrenia. Schizophr Res 167:98–107. 10.1016/j.schres.2014.12.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalby NO, Mody I (2003) Activation of NMDA receptors in rat dentate gyrus granule cells by spontaneous and evoked transmitter release. J Neurophysiol 90:786–797. 10.1152/jn.00118.2003 [DOI] [PubMed] [Google Scholar]
- Danbolt NC. (2001) Glutamate uptake. Prog Neurobiol 65:1–105. 10.1016/S0301-0082(00)00067-8 [DOI] [PubMed] [Google Scholar]
- De Marco García NV, Karayannis T, Fishell G (2011) Neuronal activity is required for the development of specific cortical interneuron subtypes. Nature 472:351–355. 10.1038/nature09865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond JS, Jahr CE (1997) Transporters buffer synaptically released glutamate on a submillisecond time scale. J Neurosci 17:4672–4687. 10.1523/JNEUROSCI.17-12-04672.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulla CG, Tani H, Brill J, Reimer RJ, Huguenard JR (2013) Glutamate biosensor imaging reveals dysregulation of glutamatergic pathways in a model of developmental cortical malformation. Neurobiol Dis 49:232–246. 10.1016/j.nbd.2012.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelstein A, Amodaj N, Hoover K, Vale R, Stuurman N (2010) Computer control of microscopes using microManager. Curr Protoc Mol Biol Chapter 14:Unit14.20. 10.1002/0471142727.mb1420s92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanselow EE, Richardson KA, Connors BW (2008) Selective, state-dependent activation of somatostatin-expressing inhibitory interneurons in mouse neocortex. J Neurophysiol 100:2640–2652. 10.1152/jn.90691.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flames N, Marín O (2005) Developmental mechanisms underlying the generation of cortical interneuron diversity. Neuron 46:377–381. 10.1016/j.neuron.2005.04.020 [DOI] [PubMed] [Google Scholar]
- Fredriksson A, Archer T, Alm H, Gordh T, Eriksson P (2004) Neurofunctional deficits and potentiated apoptosis by neonatal NMDA antagonist administration. Behav Brain Res 153:367–376. 10.1016/j.bbr.2003.12.026 [DOI] [PubMed] [Google Scholar]
- Furuta A, Rothstein JD, Martin LJ (1997) Glutamate transporter protein subtypes are expressed differentially during rat CNS development. J Neurosci 17:8363–8375. 10.1523/JNEUROSCI.17-21-08363.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandal MJ, Anderson RL, Billingslea EN, Carlson GC, Roberts TP, Siegel SJ (2012) Mice with reduced NMDA receptor expression: more consistent with autism than schizophrenia? Genes Brain Behav 11:740–750. 10.1111/j.1601-183X.2012.00816.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garaschuk O, Linn J, Eilers J, Konnerth A (2000) Large-scale oscillatory calcium waves in the immature cortex. Nat Neurosci 3:452–459. 10.1038/74823 [DOI] [PubMed] [Google Scholar]
- Gogolla N, Leblanc JJ, Quast KB, Südhof TC, Fagiolini M, Hensch TK (2009) Common circuit defect of excitatory-inhibitory balance in mouse models of autism. J Neurodev Disord 1:172–181. 10.1007/s11689-009-9023-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta SC, Ravikrishnan A, Liu J, Mao Z, Pavuluri R, Hillman BG, Gandhi PJ, Stairs DJ, Li M, Ugale RR, Monaghan DT, Dravid SM (2016) The NMDA receptor GluN2C subunit controls cortical excitatory-inhibitory balance, neuronal oscillations and cognitive function. Sci Rep 6:38321. 10.1038/srep38321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson E, Armbruster M, Cantu D, Andresen L, Taylor A, Danbolt NC, Dulla CG (2015) Astrocytic glutamate uptake is slow and does not limit neuronal NMDA receptor activation in the neonatal neocortex. Glia 63:1784–1796. 10.1002/glia.22844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison PJ, Weinberger DR (2005) Schizophrenia genes, gene expression, and neuropathology: on the matter of their convergence. Mol Psychiatry 10:40–68. 10.1038/sj.mp.4001558 [DOI] [PubMed] [Google Scholar]
- Hensch TK, Fagiolini M, Mataga N, Stryker MP, Baekkeskov S, Kash SF (1998) Local GABA circuit control of experience-dependent plasticity in developing visual cortex. Science 282:1504–1508. 10.1126/science.282.5393.1504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman MA, Jahr CE (2007) Extracellular glutamate concentration in hippocampal slice. J Neurosci 27:9736–9741. 10.1523/JNEUROSCI.3009-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollmann M, Heinemann S (1994) Cloned glutamate receptors. Annu Rev Neurosci 17:31–108. 10.1146/annurev.ne.17.030194.000335 [DOI] [PubMed] [Google Scholar]
- Homayoun H, Moghaddam B (2007) NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. J Neurosci 27:11496–11500. 10.1523/JNEUROSCI.2213-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang ZJ, Kirkwood A, Pizzorusso T, Porciatti V, Morales B, Bear MF, Maffei L, Tonegawa S (1999) BDNF regulates the maturation of inhibition and the critical period of plasticity in mouse visual cortex. Cell 98:739–755. 10.1016/S0092-8674(00)81509-3 [DOI] [PubMed] [Google Scholar]
- Hunt RF, Scheff SW, Smith BN (2009) Posttraumatic epilepsy after controlled cortical impact injury in mice. Exp Neurol 215:243–252. 10.1016/j.expneurol.2008.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ippolito DM, Eroglu C (2010) Quantifying synapses: an immunocytochemistry-based assay to quantify synapse number. J Vis Exp 45:2270. 10.3791/2270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs KM, Gutnick MJ, Prince DA (1996) Hyperexcitability in a model of cortical maldevelopment. Cereb Cortex 6:514–523. 10.1093/cercor/6.3.514 [DOI] [PubMed] [Google Scholar]
- Jarvie BC, Hentges ST (2012) Expression of GABAergic and glutamatergic phenotypic markers in hypothalamic proopiomelanocortin neurons. J Comp Neurol 520:3863–3876. 10.1002/cne.23127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaila K, Price TJ, Payne JA, Puskarjov M, Voipio J (2014) Cation-chloride cotransporters in neuronal development, plasticity and disease. Nat Rev Neurosci 15:637–654. 10.1038/nrn3819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karavanova I, Vasudevan K, Cheng J, Buonanno A (2007) Novel regional and developmental NMDA receptor expression patterns uncovered in NR2C subunit-beta-galactosidase knock-in mice. Mol Cell Neurosci 34:468–480. 10.1016/j.mcn.2006.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasyanov AM, Safiulina VF, Voronin LL, Cherubini E (2004) GABA-mediated giant depolarizing potentials as coincidence detectors for enhancing synaptic efficacy in the developing hippocampus. Proc Natl Acad Sci U S A 101:3967–3972. 10.1073/pnas.0305974101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konur S, Ghosh A (2005) Calcium signaling and the control of dendritic development. Neuron 46:401–405. 10.1016/j.neuron.2005.04.022 [DOI] [PubMed] [Google Scholar]
- Le Magueresse C, Monyer H (2013) GABAergic interneurons shape the functional maturation of the cortex. Neuron 77:388–405. 10.1016/j.neuron.2013.01.011 [DOI] [PubMed] [Google Scholar]
- Le Meur K, Galante M, Angulo MC, Audinat E (2007) Tonic activation of NMDA receptors by ambient glutamate of non-synaptic origin in the rat hippocampus. J Physiol 580:373–383. 10.1113/jphysiol.2006.123570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitt P, Eagleson KL, Powell EM (2004) Regulation of neocortical interneuron development and the implications for neurodevelopmental disorders. Trends Neurosci 27:400–406. 10.1016/j.tins.2004.05.008 [DOI] [PubMed] [Google Scholar]
- Liu-DeRyke X, Collingridge DS, Orme J, Roller D, Zurasky J, Rhoney DH (2009) Clinical impact of early hyperglycemia during acute phase of traumatic brain injury. Neurocrit Care 11:151–157. 10.1007/s12028-009-9228-6 [DOI] [PubMed] [Google Scholar]
- Loo L, Simon JM, Xing L, McCoy ES, Niehaus JK, Guo J, Anton ES, Zylka MJ (2019) Single-cell transcriptomic analysis of mouse neocortical development. Nat Commun 10:134. 10.1038/s41467-018-08079-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LoTurco JJ, Blanton MG, Kriegstein AR (1991) Initial expression and endogenous activation of NMDA channels in early neocortical development. J Neurosci 11:792–799. 10.1523/JNEUROSCI.11-03-00792.1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozovaya N, Gataullina S, Tsintsadze T, Tsintsadze V, Pallesi-Pocachard E, Minlebaev M, Goriounova NA, Buhler E, Watrin F, Shityakov S, Becker AJ, Bordey A, Milh M, Scavarda D, Bulteau C, Dorfmuller G, Delalande O, Represa A, Cardoso C, Dulac O, et al. (2014) Selective suppression of excessive GluN2C expression rescues early epilepsy in a tuberous sclerosis murine model. Nat Commun 5:4563. 10.1038/ncomms5563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markram H, Toledo-Rodriguez M, Wang Y, Gupta A, Silberberg G, Wu C (2004) Interneurons of the neocortical inhibitory system. Nat Rev Neurosci 5:793–807. 10.1038/nrn1519 [DOI] [PubMed] [Google Scholar]
- McCormick DA, Contreras D (2001) On the cellular and network bases of epileptic seizures. Annu Rev Physiol 63:815–846. 10.1146/annurev.physiol.63.1.815 [DOI] [PubMed] [Google Scholar]
- Meier J, Akyeli J, Kirischuk S, Grantyn R (2003) GABA(A) receptor activity and PKC control inhibitory synaptogenesis in CNS tissue slices. Mol Cell Neurosci 23:600–613. 10.1016/S1044-7431(03)00079-4 [DOI] [PubMed] [Google Scholar]
- Minlebaev M, Colonnese M, Tsintsadze T, Sirota A, Khazipov R (2011) Early gamma oscillations synchronize developing thalamus and cortex. Science 334:226–229. 10.1126/science.1210574 [DOI] [PubMed] [Google Scholar]
- Monyer H, Burnashev N, Laurie DJ, Sakmann B, Seeburg PH (1994) Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron 12:529–540. 10.1016/0896-6273(94)90210-0 [DOI] [PubMed] [Google Scholar]
- Moos FC, Rossi K, Richard P (1997) Activation of N-methyl-D-aspartate receptors regulates basal electrical activity of oxytocin and vasopressin neurons in lactating rats. Neuroscience 77:993–1002. 10.1016/S0306-4522(96)00536-2 [DOI] [PubMed] [Google Scholar]
- Morel L, Higashimori H, Tolman M, Yang Y (2014) VGluT1+ neuronal glutamatergic signaling regulates postnatal developmental maturation of cortical protoplasmic astroglia. J Neurosci 34:10950–10962. 10.1523/JNEUROSCI.1167-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moussawi K, Riegel A, Nair S, Kalivas PW (2011) Extracellular glutamate: functional compartments operate in different concentration ranges. Front Syst Neurosci 5:94. 10.3389/fnsys.2011.00094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoletti P, Bellone C, Zhou Q (2013) NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat Rev Neurosci 14:383–400. 10.1038/nrn3504 [DOI] [PubMed] [Google Scholar]
- Paz JT, Huguenard JR (2015) Microcircuits and their interactions in epilepsy: is the focus out of focus? Nat Neurosci 18:351–359. 10.1038/nn.3950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perszyk RE, DiRaddo JO, Strong KL, Low CM, Ogden KK, Khatri A, Vargish GA, Pelkey KA, Tricoire L, Liotta DC, Smith Y, McBain CJ, Traynelis SF (2016) GluN2D-containing NMDA receptors mediate synaptic transmission in hippocampal interneurons and regulate interneuron activity. Mol Pharmacol 90:689–702. 10.1124/mol.116.105130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Povysheva NV, Johnson JW (2012) Tonic NMDA receptor-mediated current in prefrontal cortical pyramidal cells and fast-spiking interneurons. J Neurophysiol 107:2232–2243. 10.1152/jn.01017.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell EM, Campbell DB, Stanwood GD, Davis C, Noebels JL, Levitt P (2003) Genetic disruption of cortical interneuron development causes region- and GABA cell type-specific deficits, epilepsy, and behavioral dysfunction. J Neurosci 23:622–631. 10.1523/JNEUROSCI.23-02-00622.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian A, Buller AL, Johnson JW (2005) NR2 subunit dependence of NMDA receptor channel block by external Mg2+. J Physiol 562:319–331. 10.1113/jphysiol.2004.076737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiprich P, Kilb W, Luhmann HJ (2005) Neonatal NMDA receptor blockade disturbs neuronal migration in rat somatosensory cortex in vivo. Cereb Cortex 15:349–358. 10.1093/cercor/bhh137 [DOI] [PubMed] [Google Scholar]
- Riebe I, Seth H, Culley G, Dósa Z, Radi S, Strand K, Fröjd V, Hanse E (2016) Tonically active NMDA receptors: a signalling mechanism critical for interneuronal excitability in the CA1 stratum radiatum. Eur J Neurosci 43:169–178. 10.1111/ejn.13128 [DOI] [PubMed] [Google Scholar]
- Rossi DJ, Slater NT (1993) The developmental onset of NMDA receptor-channel activity during neuronal migration. Neuropharmacology 32:1239–1248. 10.1016/0028-3908(93)90018-X [DOI] [PubMed] [Google Scholar]
- Sah P, Hestrin S, Nicoll RA (1989) Tonic activation of NMDA receptors by ambient glutamate enhances excitability of neurons. Science 246:815–818. 10.1126/science.2573153 [DOI] [PubMed] [Google Scholar]
- Sapkota K, Mao Z, Synowicki P, Lieber D, Liu M, Ikezu T, Gautam V, Monaghan DT (2016) GluN2D N-methyl-D-aspartate receptor subunit contribution to the stimulation of brain activity and gamma oscillations by ketamine: implications for schizophrenia. J Pharmacol Exp Ther 356:702–711. 10.1124/jpet.115.230391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng M, Cummings J, Roldan LA, Jan YN, Jan LY (1994) Changing subunit composition of heteromeric NMDA receptors during development of rat cortex. Nature 368:144–147. 10.1038/368144a0 [DOI] [PubMed] [Google Scholar]
- Standaert DG, Landwehrmeyer GB, Kerner JA, Penney JB Jr, Young AB (1996) Expression of NMDAR2D glutamate receptor subunit mRNA in neurochemically identified interneurons in the rat neostriatum, neocortex and hippocampus. Brain Res Mol Brain Res 42:89–102. 10.1016/S0169-328X(96)00117-9 [DOI] [PubMed] [Google Scholar]
- Swanger SA, Bassell GJ, Gross C (2011) High-resolution fluorescence in situ hybridization to detect mRNAs in neuronal compartments in vitro and in vivo. Methods Mol Biol 714:103–123. 10.1007/978-1-61779-005-8_7 [DOI] [PubMed] [Google Scholar]
- Thomas CG, Tian H, Diamond JS (2011) The relative roles of diffusion and uptake in clearing synaptically released glutamate change during early postnatal development. J Neurosci 31:4743–4754. 10.1523/JNEUROSCI.5953-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlhaas PJ, Singer W (2012) Neuronal dynamics and neuropsychiatric disorders: toward a translational paradigm for dysfunctional large-scale networks. Neuron 75:963–980. 10.1016/j.neuron.2012.09.004 [DOI] [PubMed] [Google Scholar]
- Ullensvang K, Lehre KP, Storm-Mathisen J, Danbolt NC (1997) Differential developmental expression of the two rat brain glutamate transporter proteins GLAST and GLT. Eur J Neurosci 9:1646–1655. 10.1111/j.1460-9568.1997.tb01522.x [DOI] [PubMed] [Google Scholar]
- Urban-Ciecko J, Barth AL (2016) Somatostatin-expressing neurons in cortical networks. Nat Rev Neurosci 17:401–409. 10.1038/nrn.2016.53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicini S, Wang JF, Li JH, Zhu WJ, Wang YH, Luo JH, Wolfe BB, Grayson DR (1998) Functional and pharmacological differences between recombinant N-methyl-D-aspartate receptors. J Neurophysiol 79:555–566. 10.1152/jn.1998.79.2.555 [DOI] [PubMed] [Google Scholar]
- von Engelhardt J, Bocklisch C, Tönges L, Herb A, Mishina M, Monyer H (2015) GluN2D-containing NMDA receptors-mediate synaptic currents in hippocampal interneurons and pyramidal cells in juvenile mice. Front Cell Neurosci 9:95. 10.3389/fncel.2015.00095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzel A, Fritschy JM, Mohler H, Benke D (1997) NMDA receptor heterogeneity during postnatal development of the rat brain: differential expression of the NR2A, NR2B, and NR2C subunit proteins. J Neurochem 68:469–478. 10.1046/j.1471-4159.1997.68020469.x [DOI] [PubMed] [Google Scholar]
- Wu YN, Johnson SW (2015) Memantine selectively blocks extrasynaptic NMDA receptors in rat substantia nigra dopamine neurons. Brain Res 1603:1–7. 10.1016/j.brainres.2015.01.041 [DOI] [PubMed] [Google Scholar]
- Xi D, Keeler B, Zhang W, Houle JD, Gao WJ (2009) NMDA receptor subunit expression in GABAergic interneurons in the prefrontal cortex: application of laser microdissection technique. J Neurosci Methods 176:172–181. 10.1016/j.jneumeth.2008.09.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, Cobos I, De La Cruz E, Rubenstein JL, Anderson SA (2004) Origins of cortical interneuron subtypes. J Neurosci 24:2612–2622. 10.1523/JNEUROSCI.5667-03.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto H, Kamegaya E, Hagino Y, Takamatsu Y, Sawada W, Matsuzawa M, Ide S, Yamamoto T, Mishina M, Ikeda K (2017) Loss of GluN2D subunit results in social recognition deficit, social stress, 5-HT2C receptor dysfunction, and anhedonia in mice. Neuropharmacology 112:188–197. 10.1016/j.neuropharm.2016.07.036 [DOI] [PubMed] [Google Scholar]
- Yamasaki M, Okada R, Takasaki C, Toki S, Fukaya M, Natsume R, Sakimura K, Mishina M, Shirakawa T, Watanabe M (2014) Opposing role of NMDA receptor GluN2B and GluN2D in somatosensory development and maturation. J Neurosci 34:11534–11548. 10.1523/JNEUROSCI.1811-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Xu-Friedman MA (2015) Different pools of glutamate receptors mediate sensitivity to ambient glutamate in the cochlear nucleus. J Neurophysiol 113:3634–3645. 10.1152/jn.00693.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young HD. (1962) Statistical treatment of experimental data. New York: McGraw-Hill. [Google Scholar]
- Zechel S, Nakagawa Y, Ibáñez CF (2016) Thalamo-cortical axons regulate the radial dispersion of neocortical GABAergic interneurons. eLife 5:e20770. 10.7554/eLife.20770 [DOI] [PMC free article] [PubMed] [Google Scholar]