

Abstract
Parkinson's disease is the second most common neurodegenerative disease, after Alzheimer's disease. Parkinson's disease is a movement disorder with characteristic motor features that arise due to the loss of dopaminergic neurons from the substantia nigra. Although symptomatic treatment by the dopamine precursor levodopa and dopamine agonists can improve motor symptoms, no disease-modifying therapy exists yet. Here, we show that Emapunil (AC-5216, XBD-173), a synthetic ligand of the translocator protein 18, ameliorates degeneration of dopaminergic neurons, preserves striatal dopamine metabolism, and prevents motor dysfunction in female mice treated with the MPTP, as a model of parkinsonism. We found that Emapunil modulates the inositol requiring kinase 1α (IRE α)/X-box binding protein 1 (XBP1) unfolded protein response pathway and induces a shift from pro-inflammatory toward anti-inflammatory microglia activation. Previously, Emapunil was shown to cross the blood–brain barrier and to be safe and well tolerated in a Phase II clinical trial. Therefore, our data suggest that Emapunil may be a promising approach in the treatment of Parkinson's disease.
SIGNIFICANCE STATEMENT Our study reveals a beneficial effect of Emapunil on dopaminergic neuron survival, dopamine metabolism, and motor phenotype in the MPTP mouse model of parkinsonism. In addition, our work uncovers molecular networks which mediate neuroprotective effects of Emapunil, including microglial activation state and unfolded protein response pathways. These findings not only contribute to our understanding of biological mechanisms of translocator protein 18 (TSPO) function but also indicate that translocator protein 18 may be a promising therapeutic target. We thus propose to further validate Emapunil in other Parkinson's disease mouse models and subsequently in clinical trials to treat Parkinson's disease.
Keywords: microglia, MPTP, neurodegeneration, Parkinson's disease, TSPO
Introduction
Parkinson's disease (PD) is a devastating age-associated movement disorder with high prevalence in the elderly population. Motor symptoms arise due to the progressive loss of dopaminergic neurons from the SNpc, but the precise cause is still unclear. Surviving neurons display intracellular inclusions of α-synuclein, known as Lewy bodies (Spillantini et al., 1998). In PD patients and animal models, neurodegeneration is accompanied by neuroinflammation (McGeer et al., 1988; Imamura et al., 2003; Miklossy et al., 2006; Brochard et al., 2009; Hirsch and Hunot, 2009; Harms et al., 2017), potentially triggered by the release of ATP (Davalos et al., 2005), neuromelanin (Wilms et al., 2003; Zhang et al., 2013), or aggregated α-synuclein (W. Zhang et al., 2005; Couch et al., 2011; Acosta et al., 2015) from decaying neurons. These pro-inflammatory stimuli may activate microglia to secrete cytokines and free radicals, which further contribute to oxidative stress, neurotoxicity, and disease progression (Block et al., 2007; Hirsch and Hunot, 2009). In PD patients, higher levels of cytokines were detected in CSF (Tufekci et al., 2011), serum (Eidson et al., 2017), substantia nigra, and striatum (Knott et al., 2000; Teismann et al., 2003; Mogi et al., 2007) compared with controls. Several genes associated with familial forms of PD, including SNCA, LRKK2, PARK2, PINK2, and DJ-1, have been linked with immune response (Waak et al., 2009; Gardet et al., 2010; Gu et al., 2010; Akundi et al., 2011; Gillardon et al., 2012; Moehle et al., 2012), and polymorphisms in genes encoding inflammatory cytokines are associated with a higher risk for sporadic PD (Wahner et al., 2007; Deleidi and Gasser, 2013). Conversely, minocycline, an inhibitor of microglial activation, can protect against dopaminergic neuron loss in the MPTP parkinsonism mouse model, and treatment with microglia-targeted glucocorticoids prevented dopaminergic neuron loss in the 6-OHDA mouse model of PD (Du et al., 2001; Wu et al., 2002; Tentillier et al., 2016).
The translocator protein TSPO is a mitochondrial cholesterol binding protein, which is upregulated in microglia during inflammatory activation. TSPO ligands have been widely used to monitor neuroinflammation by PET imaging, both in humans and in mice (DuPont et al., 2017). Several PET imaging studies demonstrated higher retention of TSPO tracers in PD patients compared with controls (Ouchi et al., 2005; Gerhard et al., 2006; Bartels et al., 2010; Edison et al., 2013; Iannaccone et al., 2013), although a more recent study failed to detect differences between both groups (Koshimori et al., 2015).
Anti-inflammatory and neuroprotective effects of TSPO ligands were observed in several clinical studies and in mouse models of neuropsychiatric diseases characterized by neurodegeneration and/or neuroinflammation, such as Alzheimer's disease (Barron et al., 2013), acute retina degeneration (Scholz et al., 2015), peripheral neuropathies (Giatti et al., 2009), traumatic brain injury (Papadopoulos and Lecanu, 2009), multiple sclerosis (Leva et al., 2017), anxiety (Rupprecht et al., 2009), and depression (Gavioli et al., 2003). There, TSPO ligands inhibit the production of ROS in activated microglia (Wang et al., 2014), although the precise mechanisms by which TPSO modulate inflammatory processes are not understood.
Currently, there are no therapies that can slow down or halt disease progression in PD. In this study, we examined the therapeutic potential of a synthetic TSPO ligand, Emapunil, in the MPTP mouse model, which recapitulates important hallmarks of PD, such as dopaminergic neuron loss, neuroinflammation, and motor deficits. Emapunil (AC-5216, XBD-173) is a selective, high-affinity phenylpurine ligand of TSPO that, in contrast to other ligands such as Ro-4864 and PK1195, does not bind and stimulate GABAA receptors. Most importantly, Emapunil was already tested in a Phase II clinical trial and found to be safe and well tolerated (Rupprecht et al., 2010). TSPO ligands can exert agonistic, antagonistic, or partial agonistic function on TSPO. Differences arise due to ligand binding to distinct subunits of TSPO, target cell type, and ligand concentration. Notably, the efficacy of TSPO ligands does not correlate well with TSPO binding affinity, which is rather determined by residence time of the ligand at its target (Costa et al., 2016). Emapunil is a TSPO agonist (Rupprecht et al., 2010) that was shown to increase neurosteroid synthesis (Ravikumar et al., 2016) and to decrease inflammation (Karlstetter et al., 2014). TSPO is part of a mitochondrial multiprotein complex together with voltage-dependent anion channels, adenine nucleotide translocators, ATPase, and steroidogenic acute regulatory protein (StAR), and TSPO effects may at least partially be mediated by TSPO interaction with these proteins (Gatliff and Campanella, 2016).
Our results show that Emapunil prevents neuronal loss, restores dopamine metabolism, and attenuates inflammation and motor symptoms in mice exposed to MPTP. In addition, we provide evidence that Emapunil mitigates the unfolded protein response (UPR) and induces a shift from pro-inflammatory to anti-inflammatory gene expression, which may explain its protective effects. Together, our data suggest that Emapunil may be a promising candidate for clinical trials to treat PD.
Materials and Methods
Reagents.
N-Benzyl-N-ethyl-2-(7-methyl-8-oxo-2-phenyl-7,-8-dihydro-9H-purin-9-yl)acetamide (Emapunil, AC-5216, XBD-173, CAS [226954-04-7]) was prepared according to Figure 1 (9) below (Zhang et al., 2007).
Figure 1.

Reagents and conditions. a, NaOEt, EtOH, 0°C-80°C, 4 h, 81%. b, POCl3, 90°C, 4 h, 99%. c, Glycine, Et3N, EtOH, 78°C, 4 h, 100%. d, N-Ethylbenzylamine, PyBOP, Et3N, DMF, 22°C, 2 h, 100%. e, NaOH, EtOH, water, 78°C, 2 h, 82%. f, Diphenyl phosphorazidate, Et3N, DMF, 100°C, 6 h, 55%. g, MeI, NaH, DMF, 22°C, 3 h, 78%.
Animals.
Eight-week-old female C57BL/6JRj mice were purchased from Janvier Labs. Mice were housed in groups of five in an air-conditioned room with a 12 h light-dark cycle and with free access to standard food and water. All procedures complied with the German law on animal protection. All protocols were reviewed and approved by the responsible animal welfare board (Landesamt für Verbraucherschutz, Braunschweig, Lower Saxony, Germany, reference number 12/2210). Mice that lost >20% of the body weight during the treatment were excluded from the experiment.
Subchronic MPTP mouse model.
Mice received five intraperitoneal injections of 30 mg/kg body weight MPTP (free base, Sigma-Aldrich) dissolved in 0.9% saline at 24 h intervals for 5 consecutive days. A control group was treated with five intraperitoneal injections of saline (see Fig. 2).
Figure 2.

Experimental design. Eight-week-old female C57BL/6JRj mice were assigned to three different experimental treatment groups: Top, NaCl group. Middle, MPTP + DMSO group. Bottom, MPTP + Emapunil group. Animals either received NaCl or 30 mg/kg body weight of MPTP by intraperitoneal injection once daily on 5 subsequent days (days 1–5). Animals in the MPTP + Emapunil group were additionally treated with intraperitoneal injections of 50 mg/kg body weight of Emapunil every second day, starting from day 1 until day 15. Five animals of each group were killed on day 3; striatum as well as substantia nigra were prepared and shock-frozen for further analysis by qPCR, HPLC, and transcriptome. The remaining animals underwent motor function tests on days 11–13 (pole test) and on day 14 (cylinder test). Animals were perfused on day 15, and brains were cryo-frozen for further stereological analysis.
Emapunil treatment.
Mice exposed to MPTP were treated with either intraperitoneal injections of Emapunil or DMSO as vehicle control. Emapunil was dissolved in DMSO and given at a dose of 50 mg/kg body weight every 48 h for 15 consecutive days, starting at the same day as the MPTP administration (see Fig. 2).
Mice were killed at two different time points after the first injection: day 3 for MPP+ measurements, transcriptome analysis, and qPCR; and at day 15 for stereological assay and quantification of brain dopamine and dopamine metabolites (see Fig. 2).
MPTP metabolism.
At day 3, 5 mice of each experimental group were killed and brains were rapidly removed 90 min after the last injection of either saline or MPTP. Striatum from the left hemisphere was dissected on ice and frozen at −80°C. MPP+ concentrations were measured by high pressure liquid chromatography (HPLC) after homogenization of tissue in 0.1 m perchloric acid (50 μl/mg wet tissue) and centrifugation (14,000 × g, 10 min, 4°C). The supernatant was injected onto a reverse-phase column (Nucleosil 100–5 C18; Macherey-Nagel) and separated on a mobile phase of 650/1000 ml acetonitrile in PB, pH 2.5. The MPP+ signal was quantified at an excitation wavelength of 295 nm and an emission wavelength of 375 nm (Fluorescence HPLC Monitor; Shimadzu) (Tönges et al., 2012).
RT-PCR.
RNA was isolated from striatum of the right hemisphere of 5 mice per experimental group. Mice were killed on day 3, 90 min after the last injection of either saline or MPTP. Tissue was dissected on ice. cDNA was reverse-transcribed from RNA with RNeasy Mini kit according to the manufacturer's instructions (QIAGEN). RT-PCR was performed using Mesa Blue qPCR MasterMix Plus for SYBR Assay low rox (Eurogentec). Fold change expressions were calculated using the 2−ΔΔCT method, with β-actin as a reference gene. The primers used are shown in Table 1.
Table 1.
List of primers for qPCR
| Gene (protein) | Forward primer | Reverse primer |
|---|---|---|
| Mrc1 (MMR) | AGTGGCAGGTGGCTTATG | GGTTCAGGAGTTGTTGTGG |
| Ym1 (YM1) | CATTCAGTCAGTTATCAGATTCC | AGTGAGTAGCAGCCTTGG |
| Fizz2 (FIZZ2) | TGGAGAATAAGGTCAAGGAAC | GTCAACGAGTAAGCACAGG |
| Il10 (IL10) | TGGCCCAGAAATCAAGGAGC | CAGCAGACTCAATACACACT |
| Il4 (IL4) | AGATGGATGTGCCAAACGTCCTCA | AATATGCGAAGCACCTTGGAAGCC |
| Arg1 (Arg1) | GGAAGACAGCAGAGGAGGTG | TATGGTTACCCTCCCGTTGA |
| Iba1 (IBA1) | GTCCTTGAAGCGAATGCTGG | CATTCTCAAGATGGCAGATC |
| Tspo (TSPO) | GGGAGCCTACTTTGTGCGTGG | CAGGTAAGGATACAGCAAGCGGG |
| Tnfa (TNF-α) | TTCCGAATTCACTGGAGCCTCGAA | TGCACCTCAGGGAAGAATCTGGAA |
| IP10 (IP10) | TGAGCAGAGATGTCTGAATCCG | TGTCCATCCATCGCAGCA |
| IL1b (IL1β) | AAGGGCTGCTTCCAAACCTTTGAC | ATACTGCCTGCCTGAAGCTCTTGT |
| Mpa2l (MPA2l) | CAAGAGGGAGAAGATTGAACATGA | CACTTGCCTTCACCCCTTTC |
| Nos2 (iNOS2) | CTGCTGGTGGTGACAAGCACATTT | ATGTCATGAGCAAAGGCGCAGAAC |
| Cox2 (COX2) | TTGCTGTACAAGCAGTGGCAAAGG | TGCAGCCATTTCCTTCTCTCCTGT |
| Il6 (IL6) | TGGCTAAGGACCAAGACCATCCAA | AACGCACTAGGTTTGCCGAGTAGA |
| Xbp1 (XBP1) mouse | CAGCACTCAGACTATGTGCA | GTCCATGGGAAGATGTTCTGG |
| Xbp1s (XBP1s) mouse | CTGAGTCCGAATCAGGTGCAG | GTCCATGGGAAGATGTTCTGG |
| XBP1 (XBP1) human | CAGCACTCAGACTACGTGCA | ATCCATGGGGAGATGTTCTGG |
| XBP1s (XBP1s) human | AACCAGGAGTTAAGACAGCGCTT | CTGCACCTGCTGCGGACT |
| Actb (β-Actin) mouse | TGTGATGGTGGGAATGGGTCAGAA | TGTGGTGCCAGATCTTCTCCATGT |
| GAPDH (GAPDH) human | GAAGGTGAAGGTCGGAGT | CATGGGTGGAATCATAATGGAA |
Neurochemical quantification of dopamine and metabolites.
Left striatum of 5 mice per experimental group were collected at day 15 and immediately dissected on ice. Tissue samples were homogenized in 0.1 mm perchloric acid (50 μl/mg wet tissue), centrifuged at 5000 × g for 1 min, followed by centrifugation at 10,000 × g for 30 min at 4°C. The supernatant was injected onto a C18 reverse-phase HR-80 catecholamine HPLC column (ESA) with a mobile phase, pH 2.9, of 90% 75 mm sodium phosphate, 275 mg/L octane sulfonic acid and 10% methanol. Dopamine, 3,4-dihydroxyphenylacetic acid (DOPAC) and homovanillic acid (HVA) were quantified after electrochemical detection (ESA Coulochem II with a model 5010 detector) using a Chromeleon computer system (Dionex).
Behavioral analysis.
Pole and cylinder tests were performed as described previously (Ogawa et al., 1985; Tönges et al., 2012).
Pole test.
Eleven days after the first injection of either saline or MPTP, 13–15 mice per experimental group were trained for 2 d with three test trials per day. Mice were placed head-upward on top of a vertical rough-surfaced pole (diameter 12 mm; height 55 cm), with the base of pole placed in the home cage. Mice oriented themselves downward and descended along the pole to return to their home cage. The time required for orienting downward was measured at day 13. Values were averaged from five consecutive trials performed on day 13.
Cylinder test.
The same animals were subjected to the cylinder test at day 14. The cylinder test measures forelimb utilization during normal exploratory activity. Mice were placed in a transparent cylinder (diameter: 11.5 cm; height: 25 cm), and all movements were recorded during the test duration of 5 min. A mirror placed behind the cylinder allowed videotaping of forelimb movements with the mouse turned away from the camera. Forelimb use against the wall after rearing was classified into: (1) “both” forelimbs: simultaneous use of left and right forelimb to contact the cylinder wall during a full rear or alternating use of left and right forelimb during movement along the cylinder wall; (2) “free” rears: full rears of the entire body without touching the wall; (3) “left” forelimb use: first contact of the wall with the left forelimb during a full rear; and (4) “right” forelimb use: first contact of the wall with the right forelimb during a full rear. The percentage of “both,” “right,” “left,” or “free” movements of all movements within the entire observation time was calculated.
Immunohistochemistry.
At day 15, 8–10 mice per experimental group were anesthetized with 14% chloral hydrate in water and intracardially perfused with cold PBS, followed by 4% PFA in PBS. Brains were removed, postfixed in 4% PFA overnight and cryopreserved for 48 h at 4°C in 30% sucrose in PBS, snap frozen, and stored at −80°C until further processing. Free-floating coronal sections (30 μm) were prepared using a cryostat (CM1900, Leica Microsystems) and stored in TBS with 0.1% NaN3 at 4°C. Sections were quenched of endogenous peroxidase activity, blocked with 2% BSA, 0.5% Triton X-100 in TBS for 1 h at room temperature, and incubated with an anti-Tyrosine Hydroxylase (TH) antibody (rabbit polyclonal, 1:1000, AB152, Millipore) in blocking solution for 48 h at 4°C. After washing, sections were incubated with biotinylated donkey anti-rabbit IgG (1:200; RPN-1004, GE Healthcare) in blocking solution for 2 h at room temperature, followed by Extravidin-Peroxidase (1:1000, E2886, Sigma-Aldrich) in blocking solution for 1 h at room temperature. Visualization was performed using 0.03% DAB (D5637, Sigma-Aldrich) for 2 min at room temperature. All sections were counterstained in 0.5% cresyl violet for Nissl staining, dehydrated, and mounted in DPX Mountant (06522, Sigma-Aldrich).
Immunofluorescence and quantification of microglia activation and astrogliosis.
After blocking for 1 h at room temperature, every 10th section through the striatum (3 slices/brain) was stained with either antibody against ionized calcium-binding adapter molecule 1 (IBA1) (rabbit polyclonal, 1:2000, 019–19741, Wako) or antibody against Glial Fibrillary Acidic Protein (GFAP) (guinea pig polyclonal, 1:1000, 173004, Synaptic Systems) for 48 h at 4°C in blocking solution (3% BSA, 0.5% Triton X-100 in TBS). After washing, sections were incubated with corresponding secondary antibodies, AlexaFluor-488-conjugated donkey-anti-rabbit IgG antibody (1:1500, ab150129, Abcam) or AlexaFluor-647-conjugated goat anti-guinea pig IgG antibody (1:1000, Thermo Fisher Scientific) for 2 h at room temperature, followed by washing and mounting in Mowiol 4–88 (A9011, AppliChem). For coimmunofluorescence stainings of TH and TSPO, we used anti-TSPO antibody (rabbit polyclonal, 1:500, ab109497, Abcam) and anti-TH antibody (chicken, 1:1000, ab76442, Abcam) as primary antibodies and AlexaFluor-488 goat anti-rabbit IgG (1:1000, A11008, Thermo Fisher Scientific) and Cy3 goat anti-chicken IgY (1:500, ab97145, Abcam) as secondary antibodies. Fluorescent images were captured and stitched using either a Nikon Eclipse TI microscope or an Axio Observer.Z1 microscope (Carl Zeiss) connected to Apotome.2 at 10× magnification. Fluorescent images of TH and TSPO staining were captured using an LSM 700 laser scanning confocal microscope (Carl Zeiss). IBA1- and GFAP-positive cells in the striatum were counted using ImageJ software with a cell counter analysis plug-in.
Stereological quantification of substantia nigra neurons.
TH-positive, as well as Nissl-positive, neurons in the substantia nigra were quantified by stereology. Every fifth section through the substantia nigra was stained and analyzed at an Axioplan microscope (Carl Zeiss) equipped with a computer-controlled motorized stage (Ludl Electronics), using a 100× objective and Stereo Investigator software (Stereo Investigator 9.0, MicroBrightField). A 150 × 150 μm grid layout of the substantia nigra and a counting frame of 50 × 50 μm, with 15 μm dissector height and 3 μm guard were used for cell counting. All countings were performed by a blinded investigator.
Lund human mesencephalic (LUHMES) cell culture, differentiation, and toxicity assay.
LUHMES cells (Scholz et al., 2011; Szego et al., 2017) were first grown in proliferation medium: advanced DMEM/F12, 1% N2 supplement (Invitrogen), 2 mm l-glutamine (Invitrogen), and 40 ng/ml recombinant basic fibroblast growth factor (R&D Systems) in cell culture flasks (Nunc) coated with 50 ng/ml poly-L-lysine (Sigma-Aldrich). For differentiation, proliferation medium was replaced by differentiation medium: advanced DMEM/F12, 1% N2 supplement, 2 mm l-glutamine, 1 mm dibutyryl 3′,5′-cAMP (Sigma-Aldrich), 1 μg/μl tetracycline, and 2 ng/ml recombinant human glial derived neurotrophic factor (R&D Systems).
For siRNA-mediated downregulation of TSPO, cells were transfected with esiRNA EHU080541 (Sigma-Aldrich) and Lipofectamine RNAiMAX transfection reagent (13778030, Thermo Fisher Scientific) according to the manufacturer's instructions 4 d after differentiation. Control cells were transfected with control siRNA (1027310, QIAGEN). For toxicity assay, on the seventh day of differentiation, LUHMES cells were treated with either Emapunil (5 μm) or DMSO as solvent control. After 12 h, cells were treated with DMSO, MPP+ (10 μm in DMSO), or rotenone (10 μm in DMSO). Cell culture supernatants were harvested after 24 h for toxicity measurements using ToxiLight assay (ToxiLight Kit, Lonza), following the manufacturer's instruction. In brief, 50 μl of reagent was added to 50 μl of culture supernatant, and incubated at room temperature for 5 min. Luminescence was measured in a luminescence counter (Wallac).
For mRNA isolation and qPCR, LUHMES cells were randomly divided into 8 groups and treated for 12 h with DMSO, toyocamycin (Sigma-Aldrich, 1 μm in DMSO), Emapunil (5 μm), or toyocamycin plus Emapunil. Cells were subsequently incubated for 12 h with DMSO or 10 μm rotenone, and harvested for mRNA isolation (RNeasy Mini Kit, QIAGEN).
RNA sequencing.
RNA-Seq was performed in an Illumina HiSeq, 2000. Reads were aligned to mm10 using StAR (Anders et al., 2015) with the following parameters: -outFilterMismatchNmax 2, -outSAMstrandField intronMotif, and -sjdbOverhang 49. Counts were generated using htseq (Anders et al., 2015). Differential gene expression analysis was performed using DESeq2 (Love et al., 2014). Genes with an adjusted p value < 0.05 were considered significant. Gene list overlaps were calculated using Venny (2007–2015; http://bioinfogp.cnb.csic.es/tools/venny/index.html). Functional enrichment analysis was performed using Webgestalt 2.0 (B. Zhang et al., 2005; Wang et al., 2013; Kirov et al., 2014), and gene network visualization was performed using Cytoscape 3.0's plugin ClueGO (Bindea et al., 2009). Pathways with a p value < 0.1 were considered significant. Transcription factor binding site enrichment analysis was done with PSCAN (Zambelli et al., 2009) using TRANSFAC database. Heatmaps and individual gene traces were plotted in R (Team, 2008).
Experimental design and statistical analysis.
A graphical overview on the experimental design is given in Figure 2. Data were analyzed either with two-tailed unpaired t test (Welch's t test), one- or two-way ANOVA followed by Tukey's honestly significant difference test (one-way ANOVA), or Holm–Sidak test (one- or two-way ANOVA) using Prism 6.07 (GraphPad Software). Data were assessed for normality using the Shapiro–Wilk test, with F test for variances. The null hypothesis was rejected at the 0.05 p value level. Data are mean ± SEM.
Results
Emapunil prevents neuronal loss in the MPTP model of parkinsonism
First, we tested the potential of Emapunil as a therapeutic strategy by evaluating its effect on dopaminergic neuronal loss in the subchronic MPTP mouse model of parkinsonism, which recapitulates important motor features of sporadic PD. To this end, 8-week-old female mice were intraperitoneally injected with 30 mg/kg body weight MPTP on 5 consecutive days. At the same time of MPTP treatment, mice received intraperitoneal injections of either Emapunil (MPTP + Emapunil group) or DMSO (MPTP + DMSO group), as a vehicle control. WT mice treated with saline instead of MPTP served as an additional control (NaCl group) (Fig. 2). TSPO ligands mediate gonadal upregulation of testosterone production. Alterations in testosterone levels have been discussed as a potential risk factor for PD, and gonadectomy in mice was associated with loss of dopaminergic neurons and Parkinson-like symptoms (Khasnavis et al., 2013). Therefore, we decided to use female mice for our experiments. Fifteen days after the first exposure to MPTP treatment, mice were killed and stereologically analyzed for surviving dopaminergic neurons in the substantia nigra (Fig. 3A,B). While the number of TH-positive neurons was significantly lower in MPTP + DMSO-treated mice compared with NaCl control animals, neuron loss was significantly prevented in the MPTP + Emapunil group. Similar results were found for Nissl-positive neurons in the substantia nigra (Fig. 3C) (NaCl group: TH-positive neurons = 21,572 ± 417, Nissl-positive neurons = 27,125 ± 352, n = 10; MPTP + DMSO group: TH-positive neurons = 12,047 ± 246, Nissl-positive neurons = 20,258 ± 401, n = 10; MPTP + Emapunil group: TH-positive neurons = 17,547 ± 391, Nissl-positive neurons = 25,662 ± 423, n = 8; *p < 0.05; ***p < 0.001; TH-positive neurons: F(2,25) = 192.5, p < 0.0001; Nissl-positive neurons: F(2,25) = 90.46, p < 0.0001, one-way ANOVA, Tukey's post hoc test).
Figure 3.

Emapunil ameliorates MPTP-induced dopaminergic neuron loss. A, Representative images of the substantia nigra of mice treated with NaCl (top), MPTP (middle), or MPTP and Emapunil (bottom) (anti-TH antibody staining). Scale bar, 200 μm. B, Dopaminergic neurons were quantified as the total number of TH-immunoreactive neurons. C, The number of Nissl-stained neurons was quantified in the NaCl, MPTP, or MPTP and Emapunil treatment groups. One-way ANOVA, in between-group difference: p < 0.0001 (both in TH and Nissl stained neurons). *p < 0.05; ***p < 0.001; Tukey's post hoc test.
MPTP crosses the blood–brain barrier, where it is mainly converted by astrocytic monoamine-oxidase B (MAO B) to its toxic metabolite, 1-methyl-4-phenylpyridinium (MPP+). To exclude any interference of Emapunil itself on MPP+ production, we quantified MPP+ levels in the MPTP + DMSO and the MPTP + Emapunil groups. No significant differences were detected between the two groups (data not shown). Together, these data suggest that Emapunil protects against MPTP-induced degeneration of dopaminergic neurons.
Emapunil protects against MPTP-induced motor impairments
Next, to determine whether the protective effect of Emapunil on dopaminergic cell survival also correlated with preserved motor function in MPTP-treated animals, mice were subjected to a cylinder test (Fig. 2). We recorded limb-use asymmetry of rearing movements in mice placed into a glass cylinder, including total number of rears, percentage of free rears, rears with either the right or left forelimb, and rears using both forelimbs. MPTP + DMSO-treated mice showed less free rears compared with saline-injected control mice, whereas rears supported by both forehands were increased (Fig. 4A). In contrast, MPTP + Emapunil-treated mice scored similar to the saline-control group, with a significantly higher rate of the more difficult free rears compared with the control group (NaCl group: ratio of free rears = 22.9 ± 2.37%, ratio of left forelimb rears = 4.71 ± 1.27%, ratio of right forelimb rears = 3.48 ± 0.845%, ratio of rears with both forelimbs = 68.9 ± 3.49%, n = 15; MPTP + DMSO group: ratio of free rears = 13.5 ± 2.82%, ratio of left forelimb rears = 4.07 ± 0.947%, ratio of right forelimb rears = 5.98 ± 1.37%, ratio of rears with both forelimbs = 68.9 ± 3.49%, n = 14; MPTP + Emapunil group: ratio of free rears = 32 ± 2.36%, ratio of left forelimb rears = 6.79 ± 0.01.4%, ratio of right forelimb rears = 2.92 ± 0.99%, ratio of rears with both forelimbs = 58.3 ± 3.66%, n = 13; *p < 0.05; **p < 0.01; ***p < 0.001; free rears: F(2,39) = 12.91, p = 0.0001; both forelimbs: F(2,39) = 6.494, p = 0.00368; one-way ANOVA, Tukey's post hoc test; Fig. 4A). These data indicate that Emapunil treatment protected MPTP-treated mice against deteriorating motor function. Encouraged by these findings, we used an additional well-established motor function assay, the pole test, to assess the effects of Emapunil on coordination deficits and bradykinesia. As expected, mice treated with MPTP + DMSO turned slower downward after head-upward placement near the top of the pole compared with saline controls (Fig. 4B). However, MPTP + Emapunil mice were faster in orienting themselves downward, and scored comparable to saline controls (NaCl group: time = 1.36 ± 0.1311 s, n = 15; MPTP + DMSO group: time = 1.545 ± 0.1008 s, n = 15; MPTP + Emapunil group: time = 1.152 ± 0.0658 s, n = 13; *p < 0.05; **p < 0.01; ***p < 0.001; F(2,40) = 3.366, p = 0.04; one-way ANOVA, Tukey's post hoc test). Together, these results further confirm that Emapunil mediates neuroprotective effects in the MPTP mouse model, preserving motor coordination as well as postural control.
Figure 4.

Emapunil treatment restores motor function. A, Cylinder test of mice treated with NaCl (black bars), MPTP (red bars), or MPTP + Emapunil (green bars) 14 d after first MPTP application. Histograph represents the percentage of rears against the cylinder wall, either as free rears without paw support or rears supported with only the left, only the right or with both paws. No significant between-group difference was observed for right or left paw only assisted rears. One-way ANOVA, in between-group difference: p = 0.000108 (free rears) and p = 0.003677 (support with both paws). *p < 0.05; **p < 0.01; ***p < 0.001; Tukey's post hoc test. B, Pole test performance 11–13 d after the initial MPTP dose. Histograph represents the time taken to orient downward at the top of the pole. One-way ANOVA, in between-group difference: p = 0.04. *p < 0.05 (Tukey's post hoc test).
Emapunil attenuates loss of dopamine and dopamine metabolites in the MPTP model
Next, we asked, whether Emapunil would also preserve functional activity of nigrostriatal innervation as assessed by striatal concentrations of dopamine and its metabolites, quantified by HPLC at day 3. In MPTP-treated animals, both dopamine and its metabolites DOPAC and HVA were reduced compared with saline-treated controls, reflecting MPTP-induced toxicity toward dopaminergic neurons. Consistently with a protective effect of Emapunil, reduction of dopamine and DOPAC levels was less pronounced in the Emapunil-treated MPTP group compared with the MPTP (+ DMSO) group (Fig. 5A). No significant difference was observed for HVA levels between MPTP mice treated with Emapunil or not. Dopamine turnover rates, as defined by the ratio of ([DOPAC] + [HVA])/[dopamine], were higher in MPTP-treated mice compared with saline controls, indicating increased cellular stress. In contrast, dopamine turnover rates in MPTP-treated animals exposed to Emapunil were lower and reached a level that was comparable with saline-treated control animals. These data suggest that Emapunil decreases cellular stress and preserves nigrostriatal innervation by preventing axonal injury (NaCl group: dopamine = 8.444 ± 0.691 ng/mg, DOPAC = 7.144 ± 0.2925 ng/mg, metabolic ratio = 1.934 ± 0.1684, n = 5; MPTP + DMSO group: dopamine = 3.293 ± 0.3095 ng/mg, DOPAC = 5.534 ± 0.3939 ng/mg, metabolic ratio = 3.743 ± 0.3718, n = 5; MPTP + Emapunil group: dopamine = 5.581 ± 0.4028 ng/mg, DOPAC = 6.691 ± 0.4018 ng/mg, metabolic ratio = 2.304 ± 0.1942, n = 5; *p < 0.05; **p < 0.01; ***p < 0.001; dopamine: F(2,12) = 27.17, p < 0.0001; DOPAC: F(2,12) = 5.147, p = 0.0243; metabolic ratio: F(2,12) = 13.41, p = 0.0008; one-way ANOVA, Tukey's post hoc test).
Figure 5.

HPLC analysis of dopamine and its metabolites in the striatum. A, Dopamine. B, DOPAC. C, Metabolic ratio (HVA + DOPAC)/dopamine. One-way ANOVA, in between-group difference: p < 0.0001 (dopamine), p = 0.02 (DOPAC), and p = 0.0008 (metabolic ratio). *p < 0.05; **p < 0.01; ***p < 0.001; Tukey's post hoc test.
Emapunil mitigates ER stress
To further investigate the effects of Emapunil on cellular stress responses, we assessed the activation of X-box binding protein 1 (XBP1), a transcription factor that positively regulates the UPR. UPR serves as an adaptive cellular stress response induced by the accumulation of misfolded proteins in the endoplasmic reticulum (ER). Its downstream cascade leads to inhibition of translation, clearance of misfolded proteins through autophagy and the ER-associated degradation (ERAD) pathway, and the induction of chaperones, thereby restoring ER homeostasis. However, chronic activation of UPR can ultimately induce apoptosis (Urra et al., 2013). ER stress has been implicated in PD pathogenesis, with a protective function during initial disease stages and a deleterious, proapoptotic effect under chronically sustained stress levels (Mercado et al., 2015). The active form of XBP1 transcription factor mRNA, Xbp1s, is generated by unconventional splicing through inositol requiring kinase 1α (IRE1α) (Ron and Walter, 2007). Thus, the activity of XBP1 as a marker for ER stress can be assessed through qRT-PCR for the detection of spliced Xbp1s mRNA (van Schadewijk et al., 2012). We found that, consistently with a protective effect of Emapunil, the mRNA levels of Xbp1s, as well as the ratio of Xbp1s/Xbp1 mRNA were restored to saline control levels in mice treated with Emapunil (MPTP + Emapunil) (Fig. 6A–C). In contrast, MPTP treatment alone (MPTP + DMSO) resulted in significantly higher levels of active Xbp1s mRNA and Xbp1s/Xbp1 ratio compared with saline control and MPTP + Emapunil groups (Fig. 6A–C) (NaCl group: Xbp1 = 1 ± 0.02093, spliced Xbp1s = 1 ± 0.045, ratio = 1 ± 0.0473, n = 5; MPTP + DMSO group: Xbp1 = 0.88 ± 0.067, spliced Xbp1s = 1.35 ± 0.0527, ratio = 1.57 ± 0.155, n = 5; MPTP + Emapunil group: Xbp1 = 0.904 ± 0.05349, spliced Xbp1s = 0.896 ± 0.036, ratio = 1.01 ± 0.0834, n = 5; **p < 0.01; ***p < 0.001; Xbp1: F(2,12) = 1.543, p = 0.25329; spliced Xbp1s: F(2,12) = 27.3, p < 0.0001; ratio: F(2,12) = 9.67, p = 0.003151; one-way ANOVA, Tukey's post hoc test). To further investigate the neuroprotective effects of Emapunil, we used an immortalized human dopaminergic cell line (LUHMES) (Lotharius et al., 2002). Treatment of postmitotically differentiated LUHMES cells with either the active metabolite of MPTP (MPP+) or rotenone has been established as cellular models of human parkinsonism (Giordano et al., 2012; Krug et al., 2014). Both, MPP+ and rotenone exposure resulted in reduced dopaminergic cell survival as measured by the release of adenylate kinase relative to DMSO solvent control (ToxiLight assay, Lonza). Consistent with our in vivo findings, Emapunil treatment ameliorated toxic MPP+ and rotenone effects (Fig. 6D) (DMSO group: fold change = 1 ± 0.06748, n = 31; DMSO + 5 μm Emapunil group: fold change = 1.216 ± 0.08, n = 31; 10 μm MPP+ + DMSO group: fold change = 4.877 ± 0.4397, n = 17; 10 μm MPP+ + 5 μm Emapunil group: fold change = 4.024 ± 0.3168, n = 17; 10 μm rotenone + DMSO group: fold change = 5.624 ± 0.2506, n = 14; 10 μm rotenone + Emapunil group: fold change = 4.042 ± 0.0871, n = 14; *p < 0.05; ***p < 0.001; F(2,118) = 9.856, p < 0,0001; two-way ANOVA, Holm–Sidak post hoc test). We next assessed XBP1s/XBP1 ratios under oxidative stress induced by rotenone. Similar to our in vivo treatment with MPTP, we found a significant increase of XBP1s/XBP1 with rotenone compared with saline controls (Fig. 6E). Increased XBP1s/XBP1 ratios were restored to normal levels by additional treatment with either Emapunil or the IRE1α-XBP1 inhibitor toyocamycin (2-hydroxy-1-naphthaldehyde) (Tanabe et al., 2018) (Fig. 6E). Simultaneous treatment of rotenone challenged cells with Emapunil and toyocamycin together did not result in additive effects, further indicating that Emapunil acts on the IRE1α-XBP1 pathway (Fig. 6E). Of note, no XBP1s/XBP1 differences were detected in LUHMES cells treated with either Emapunil or toyocamycin alone (Fig. 6E) (vehicle group: XBP1 = 1 ± 0.106, spliced XBP1s = 1 ± 0.132, ratio = 1 ± 0.041, n = 9; rotenone group: XBP1 = 9.23 ± 3.41, spliced XBP1s = 67.7 ± 22.6, ratio = 8.47 ± 1, n = 9; toyocamycin group: XBP1 = 0.681 ± 0.181, spliced XBP1s = 0.607 ± 0.171, ratio = 0.896 ± 0.102, n = 9; Emapunil group: XBP1 = 0.679 ± 0.197, spliced XBP1s = 0.68 ± 0.197, ratio = 1.02 ± 0.0424, n = 9; toyocamycin + Emapunil group: XBP1 = 0.879 ± 0.212, spliced XBP1s = 0.823 ± 0.199, ratio = 0.93 ± 0.0884, n = 9; rotenone + toyocamycin group: XBP1 = 0.969 ± 0.175, spliced XBP1s = 1.23 ± 0.188, ratio = 1.46 ± 0.264, n = 9; rotenone + Emapunil group: XBP1 = 0.599 ± 0.167, spliced XBP1s = 0.756 ± 0.173, ratio = 1.58 ± 0.252, n = 9; rotenone + toyocamycin + Emapunil group: XBP1 = 1.41 ± 0.404, spliced XBP1s = 2.2 ± 0.724, ratio = 1.48 ± 0.14, n = 9; **p < 0.01; ***p < 0.001; XBP1: F(7,64) = 5.835, p < 0.0001; XBP1s: F(7,64) = 8.645, p < 0.0001; ratio: F(7,64) = 45.43, p < 0.0001; one-way ANOVA, Tukey's post hoc test). We performed coimmunofluorescence stainings with antibodies against TSPO and TH in mouse brain sections and antibodies against TSPO and β-III- tubulin in LUHMES cells to test for neuronal TSPO expression. Indeed, TSPO immunofluorescence was present in TH-positive dopaminergic neurons in mouse brains (Fig. 7A) and in differentiated LUHMES cells (Fig. 7B). TSPO expression was additionally detected by Western blot analysis of LUHMES cell lysates (Fig. 7C). The selectivity of Emapunil binding to TSPO has previously been determined by measuring binding affinities of Emapunil to a series of receptor, transporter, and ion channel proteins in vitro up to concentrations of 1 μm (Kita et al., 2004). To further validate that the effects of Emapunil are indeed mediated by TSPO, we tested Emapunil on toxicity and XBP1s/XBP1 ratio in rotenone-exposed LUHMES cells treated with either TSPO or control siRNA. TSPO siRNA treatment resulted in ∼55% reduction of TSPO protein levels as determined by Western blot analysis (TSPO siRNA group: TSPO = 0.445 ± 0.067, n = 3; control siRNA group, TSPO = 1 ± 0.071, n = 3; **p < 0.01; t = 5.693, df = 4, p = 0.0048; two-tailed unpaired t test, Welch's t test; Fig. 8A,B) without affecting TH expression (TSPO siRNA group: TH = 0.897 ± 0.096, n = 3; control siRNA group, TH = 1 ± 0.048, n = 3; t = 2.92, df = 2.92, p = 0.4121; two-tailed unpaired t test, Welch's t test; Fig. 8C). Consistent with our earlier findings, rotenone-induced toxicity in LUHMES cells was ameliorated by Emapunil in control siRNA-treated cells (DMSO group: fold change = 1 ± 0.026, n = 8; DMSO + 10 μm rotenone group: fold change = 3.682 ± 0.345, n = 8; 5 μm Emapunil + DMSO group: fold change = 1.37 ± 0.114, n = 8; 5 μm Emapunil + 10 μm rotenone group: fold change = 1.61 ± 0.152, n = 8; **p < 0.005; ***p < 0.001; two-way ANOVA, Tukey post hoc test; Fig. 9A). However, downregulation of TSPO by siRNA significantly impaired this Emapuni-mediated rescue (DMSO group: fold change = 1.165 ± 0.0768, n = 8; DMSO + 10 μm rotenone group: fold change = 4.293 ± 0.357, n = 8; 5 μm Emapunil + DMSO group: fold change = 1.385 ± 0.135, n = 8; 5 μm Emapunil + 10 μm rotenone group: fold change = 2.908 ± 0.324, n = 8; **p < 0.005; ***p < 0.001 F(3,55) = 3.365, p = 0.0251; two-way ANOVA, Tukey's post hoc test; Fig. 9A). Notably, rotenone toxicity was not further increased in TSPO siRNA-treated cells compared with control siRNA-exposed cells (Fig. 9A). This is most likely due to the fact that rotenone toxicity reached already a maximum toxicity at 10 μm and did not further increase with higher concentrations (DMSO group: fold change = 1 ± 0.00554, n = 6; 0.5 μm rotenone group: fold change = 4.18 ± 0.195, n = 6; 1 μm rotenone group: fold change = 4.55 ± 0.189, n = 6; 2.5 μm rotenone group: fold change = 6.028 ± 0.229, n = 8; 5 μm rotenone group: fold change = 7.246 ± 0.104, n = 8; 10 μm rotenone group: fold change = 8.559 ± 0.259, n = 8; 20 μm rotenone group: fold change = 8.518 ± 0.317, n = 8; **p < 0.005; ***p < 0.001; F(6,42) = 95.5, p < 0.0001; one-way ANOVA, Tukey's post hoc test; Fig. 9B). Supporting the notion that Emapunil effects require TSPO, XBP1s/XBP1 ratios did not decrease in response to Emapunil treatment in TSPO siRNA and rotenone-exposed cells, compared with rotenone and control siRNA-treated cells (DMSO with control siRNA group: fold change = 1 ± 0.029, n = 12; DMSO with TSPO siRNA group: fold change = 1.022 ± 0.069, n = 12; DMSO + 10 μm rotenone with control siRNA group: fold change = 3.66 ± 0.245, n = 12; DMSO + 10 μm rotenone with TSPO siRNA group: fold change = 3.178 ± 0.204, n = 12; 5 μm Emapunil + DMSO with control siRNA group: fold change = 0.955 ± 0.03, n = 12; 5 μm Emapunil + DMSO with TSPO group: fold change = 1.141 ± 0.153, n = 12; 5 μm Emapunil + 10 μm rotenone with control siRNA group: fold change = 1.573 ± 0.14, n = 12; 5 μm Emapunil + 10 μm rotenone with TSPO group: fold change = 2.515 ± 0.246, n = 12; **p < 0.005; ***p < 0.001; F(3,88) = 6.931, p = 0.0003; two-way ANOVA, Tukey's post hoc test; Fig. 9C).
Figure 6.

Emapunil mitigates ER stress. A, qPCR analysis of relative mRNA expression levels of unspliced Xbp1. B, qPCR analysis of relative mRNA expression levels of spliced Xbp1s. C, Ratio of spliced Xbp1s/unspliced Xbp1 normalized to NaCl control. One-way ANOVA, in between-group difference: p = 0.25329 (Xbp1), p < 0.0001 (spliced Xbp1s), and p = 0.003151 (ratio Xbp1s/Xbp1). **p < 0.01; ***p < 0.001; Tukey's post hoc test. D, Cell toxicity was measured by adenylate kinase release (relative ToxiLight luminescence) in differentiated LUHMES cells treated with DMSO, MPP+, or rotenone and with or without addition of Emapunil. Two-way ANOVA, in all interaction comparisons: p < 0.0001. *p < 0.05; ***p < 0.001; Holm–Sidak post hoc test. E, XBP1s/XBP1 ratios in LUHMES cells. One-way ANOVA, in between-group difference: p < 0.0001. **p < 0.01; ***p < 0.001; Tukey's post hoc test.
Figure 7.

TSPO expression in dopaminergic neurons. A, Immunofluorescence staining of 8-week-old WT control mice (substantia nigra). Sections were stained with antibodies against TSPO (left, green) and TH (middle, red). Right, Merged image. Scale bar, 20 μm. B, Immunofluorescence staining of LUHMES cells. Left, DAPI (blue), antibody against TSPO (green), antibody against β-III-tubulin (red). Right, Merged image. Scale bar, 50 μm. C, Western blot analysis of TSPO expression in LUHMES cells.
Figure 8.

TSPO siRNA treatment decreases TSPO expression levels. A, Western blot analysis of TSPO, β-actin, and TH levels in LUHMES cells, with and without siRNA directed against TSPO. B, Quantification of TSPO levels (Western blot analysis from A) normalized to β-actin (loading control). Two-tailed unpaired t test (Welch's t test): p = 0.0048. **p < 0.005. C, Quantification of TH levels (Western blot analysis from A) normalized to β-actin (loading control). Two-tailed unpaired t test (Welch's t test): p = 0.412. n.s. = not significant.
Figure 9.
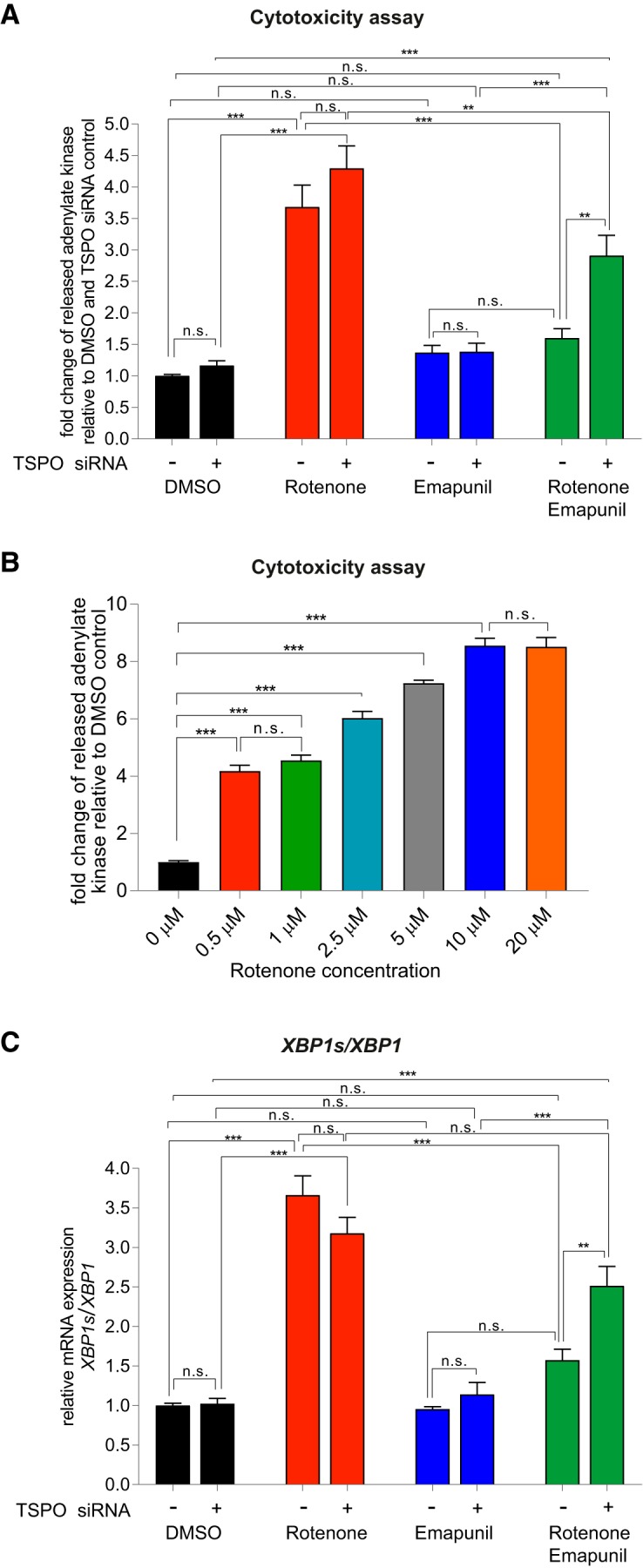
The effects of Emapunil depend on TSPO expression. A, Cytotoxicity measured by adenylate kinase release (ToxiLight luminescence) in differentiated LUHMES cells treated with either TSPO or control siRNA. Experimental groups: DMSO, 10 μm rotenone, 5 μm Emapunil, 10 μm rotenone plus 5 μm Emapunil. Two-way ANOVA: F(3,54) = 3.365, p = 0.0251. **p < 0.005; ***p < 0.001; Tukey post hoc test. B, Cytotoxicity (ToxiLight luminescence assay) in differentiated LUHMES cells with increasing rotenone concentrations. One-way ANOVA, in between-group difference: p < 0.0001. ***p < 0.001 (Tukey post hoc test). C, qPCR analysis of spliced XBP1s/unspliced XBP1 in differentiated LUHMES cells treated with TSPO or control siRNA. Experimental groups as in A. Two-way ANOVA: F(3,82) = 6.458, p = 0.0006. **p < 0.005; ***p < 0.001; Tukey post hoc test. n.s. = not significant.
Emapunil inhibits microgliosis
Because TSPO ligands have been linked to mitigated neuroinflammatory responses, we tested the effects of Emapunil on microgliosis and astrocytosis in vivo. At day 15, mice were killed and sections stained for microglia (IBA1) and astrocytes (GFAP) (Fig. 10A,B). Quantification of striatal cell counts confirmed microgliosis as well as astrocytosis in the MPTP + DMSO group compared with saline controls. Treatment with Emapunil markedly ameliorated microgliosis in MPTP-treated mice but induced only a modest, albeit significant, reduction of astrogliosis (Fig. 10C,D) (NaCl group: IBA1-positive cells = 180.4 ± 4.125/mm2, n = 9; GFAP-positive cells = 1.152 ± 0.1288/mm2, n = 6; MPTP + DMSO group: IBA1-positive cells = 234.7 ± 4.813/mm2, n = 10; GFAP-positive cells = 133.7 ± 4.283/mm2, n = 7; MPTP + Emapunil group: IBA1-positive cells = 190.8 ± 6.627/mm2, n = 8; GFAP-positive cells = 109.4 ± 3.438/mm2, n = 6; ***p < 0.001; IBA1-positive cells: F(2,158) = 33.19, p < 0.0001; GFAP-positive cells: F(2,111) = 446.5, p < 0.0001; one-way ANOVA, Tukey's post hoc test).
Figure 10.

Emapunil prevents microgliosis and astrogliosis in MPTP animals. A, IBA1 (green) immunohistochemistry of striatal sections from NaCl, MPTP + DMSO, or MPTP + Emapunil-treated mice, killed at day 15. B, GFAP (red) immunohistochemistry of striatal sections from NaCl, MPTP + DMSO, or MPTP + Emapunil-treated mice, killed at day 15. C, Quantification of IBA1-positive cells (striatum). D, GFAP-positive cells (striatum). Data are total numbers/mm2; three sections from each animal were analyzed with n = 6 or 7 animals per group. One-way ANOVA, in between-group differences: p < 0.0001 (both in IBA1 and GFAP quantification). ***p < 0.001 (Tukey's post hoc test). Scale bars: A, 50 μm; B, 100 μm. n.s. = not significant.
Emapunil induces a shift from pro-inflammatory to anti-inflammatory microglial activation state
In contrast to the effect observed on microglia cell counts at day 15, qRT-PCR analysis of striatal tissue at day 3 revealed higher Iba1 mRNA levels in both MPTP + DMSO and MPTP + Emapunil-treated groups, compared with saline control (Fig. 11). Iba1 mRNA expression levels were comparable between MPTP + DMSO and MPTP + Emapunil groups at day 3 (NaCl group normalized to 1: 1 ± 0.1763; MPTP + DMSO group: 5.633 ± 0.6228; MPTP + Emapunil group: 6.842 ± 1.015, n = 5; **p < 0.01; ***p < 0.001; F(2,12) = 19.68, p = 0.000163; one-way ANOVA, Tukey's post hoc test). We next investigated mRNA expression levels of Tspo (TSPO) as a microglia activation marker (Fig. 11B), of the classical M1 pro-inflammatory markers: Tnfa, encoding tumor necrosis factor α (TNF-α), Cxcl10, encoding C-X-C motif chemokine 10 (CXCL10), Il1b, encoding interleukin 1-β (IL-1β), Mpa2l, encoding macrophage activating 2 like protein (Mpa2l), Il6, encoding interleukin 6 (IL6), Nos2, encoding inducible nitric oxide synthase 2 (iNOS2), Cox2, encoding cyclooxygenase 2 (COX2) as well as of the anti-inflammatory M2 marker genes Arg1, encoding arginase 1 (Arg1), Il10, encoding interleukin 10 (IL10), Il4, encoding interleukin 4 (IL4), Mrc1, encoding macrophage mannose receptor (MMR), Ym1, encoding for chitinase-3-like protein 3/YM1, and Fizz2, encoding found in inflammatory zone (FIZZ2) (Fig. 11C). Tspo and Cox2 mRNA levels were exclusively increased in the MPTP + DMSO group compared with saline controls and MPTP + Emapunil-treated animals (Fig. 11B,C). Another M1 pro-inflammatory marker, Cxcl10, was increased in MPTP + DMSO and to a lower extent in MPTP + Emapunil-treated mice (Fig. 11C). Together, these data indicate a mitigating effect of Emapunil on pro-inflammatory microglia activation (NaCl group normalized to 1: Tspo = 1 ± 0.07894, Tnfa = 1 ± 0.3178, Cxcl10 = 1 ± 0.1652, Il1b = 1 ± 0.1329, Mpa2l = 1 ± 0.07732, Il6 = 1 ± 0.1694, Nos2 = 1 ± 0.1704, Cox2 = 1 ± 0.1734, n = 5; fold change in MPTP + DMSO group: Tspo = 2.342 ± 0.379, Tnfa = 5.885 ± 0.6208, Cxcl10 = 11.6 ± 0.5612, Il1b = 1.581 ± 0.3056, Mpa2l = 1.111 ± 0.07614, Il6 = 2.227 ± 0.3152, Nos2 = 2 ± 0.1327, Cox2 = 2.746 ± 0.2961, n = 5; fold change in MPTP + Emapunil group: Tspo = 0.7979 ± 0.1375, Tnfa = 4.979 ± 0.6865, Cxcl10 = 5.365 ± 1.449, Il1b = 1.385 ± 0.1907, Mpa2l = 1.158 ± 0.279, Il6 = 3.189 ± 0.5172, Nos2 = 1.66 ± 0.305, Cox2 = 1.539 ± 0.171, n = 5; **p < 0.01; ***p < 0.001; Tspo: F(2,12) = 12.52, p = 0.001157; Tnfa: F(2,12) = 21.75, p = 0.000151; Cxcl10: F(2,12) = 34.86, p < 0.0001; Il1b: F(2,12) = 1.777, p = 0.004; Mpa2l: F(2,11) = 0.2046, p = 0.8180; Il6: F(2,12) = 9.131, p = 0.0039; Nos2: F(2,12) = 5.55, p = 0.0197; Cox2: F(2,12) = 16.32, p = 0.00038; one-way ANOVA, Tukey's post hoc test). In contrast, the anti-inflammatory M2 state markers Arg1, Il10, YM1, and FIZZ2 were exclusively increased in the MPTP + Emapunil group, suggesting an additional anti-inflammatory effect (NaCl group normalized to 1: Arg1 = 1 ± 0.06473, Il10 = 1 ± 0.04799, Il4 = 1 ± 0.08438, Mrc1 = 1 ± 0.0555, Ym1 = 1 ± 0.1139, Fizz2 = 1 ± 0.2787, n = 5; fold change in MPTP + DMSO group: Arg1 = 0.8055 ± 0.001321, Il10 = 0.9218 ± 0.02449, Il4 = 1.905 ± 0.4561, Mrc1 = 1.517 ± 0.06856, Ym1 = 1.357 ± 0.225, Fizz2 = 1.282 ± 0.2985, n = 5; fold change in MPTP + Emapunil group: Arg1 = 1.259 ± 0.1289, Il10 = 2.478 ± 0.557, IL4 = 3.082 ± 0.9407, Mrc1 = 1.955 ± 0.1961, Ym1 = 2.21 ± 0.0745, Fizz2 = 4.248 ± 0.5753, n = 5; *p < 0.05; **p < 0.01; ***p < 0.001; Arg1: F(2,12) = 6.171, p = 0.015957; Il10: F(2,12) = 7.365, p = 0.008185; Il4: F(2,12) = 2.971, p = 0.0895; Mrc1: F(2,12) = 14.84, p = 0.00057; Ym1: F(2,12) = 16.75, p = 0.000336; Fizz2: F(2,12) = 19.51, p = 0.000169; one-way ANOVA, Tukey's post hoc test). Together, these data indicate a shift from pro-inflammatory to anti-inflammatory microglial activation induced by Emapunil treatment.
Figure 11.

Emapunil mediates a shift from pro-inflammatory to anti-inflammatory gene expression. Striatal RNA was isolated on day 3 from mice treated with NaCl (black bars), MPTP + DMSO (red bars), or MPTP + Emapunil (green bars) (n = 5 per experimental group). qPCR analysis of mRNA expression: A, Iba1. B, Tspo. C, Pro-inflammatory cytokines and chemokines. D, Anti-inflammatory cytokines and chemokines. Histographs represent relative mRNA levels normalized to NaCl control condition. One-way ANOVA, in between-group difference: p = 0.000163 (Iba1), p = 0.001157 (Tspo), p = 0.000151 (Tnfa), p < 0.0001 (Cxcl10), p = 0.004103 (Il1b), p = 0.81803 (Mpa2l), p = 0.0039 (Il6), p = 0.019653 (Nos2), p = 0.000377 (Cox2), p = 0.015957 (Arg1), p = 0.008185 (Il10), p = 0.0895 (Il4), p = 0.00057 (Mrc1), p = 0.000336 (Ym1), p = 0.000169 (Fizz2). *p < 0.05; **p < 0.01; ***p < 0.001; Tukey's post hoc test. n.s. = not significant.
Effects of Emapunil on transcription
There is increasing evidence for significant changes in gene expression during PD pathogenesis (Pinho et al., 2016). Moreover, altered transcriptome plasticity is an early sign of disrupted cellular homeostasis (Fischer, 2014; Martinez Hernandez et al., 2018). When we compared striatal gene-expression levels at day 3 between the three groups (NaCl control, MPTP + DMSO, MPTP + Emapunil) by RNA sequencing, we found MPTP-associated changes in 1017 genes compared with saline controls (Fig. 12A). Administration of Emapunil to MPTP-treated animals resulted in 2464 differentially expressed genes compared with saline controls, 834 of which were also changed in MPTP + DMSO only treated animals (Fig. 12A). These data suggest that Emapunil treatment attenuated the deregulation of 183 MPTP-induced genes and elicited an Emapunil-specific gene-expression program linked to 1630 genes (Fig. 12A). This indicates that Emapunil not only partially reinstates transcriptional changes induced by MPTP but also exerts additional effects. Functional enrichment analysis revealed that genes upregulated by MPTP and normalized by additional Emapunil treatment are linked to pathways of innate immune response and cytokine production, vesicle-mediated transport, endocytosis, neurogenesis, neuron differentiation, and kinase signaling (Fig. 12B). In contrast, MPTP-specific downregulated genes, which were fully restored to normal expression levels by Emapunil, are linked to cell projection organization and assembly pathways, lipid metabolism, cell migration, and motility (Fig. 12B). Apart from restoring ∼82% of MPTP-induced gene expression changes, Emapunil treatment resulted in the 1630 differentially expressed genes that were unique to the Emapunil-treated MPTP-exposed animals (Fig. 12A). Functional network analysis of these 1630 Emapunil-related genes revealed pathways linked to immune response (e.g., regulation of inflammatory response, cytokine production, leukocyte-mediated immunity, and response to molecules of bacterial origin) (Fig. 12C). These data are in line with the reported effect of Emapunil on microglia-mediated immune function.
Figure 12.

Effects of MPTP and Emapunil on the transcriptome. A, Venn diagram showing the overlap between the transcriptional response to MPTP and Emapunil as well as genes that are only affected by MPTP treatment or only affected by additional Emapunil treatment (MPTP + Emapunil). B, Individual traces for the 183 genes that are significant in the MPTP group alone (i.e., attenuated by Emapunil treatment). Right, The most salient gene ontology (GO) categories for upregulated and downregulated genes within those 183 are represented. C, Network of GO terms associated with Emapunil-specific genes (i.e., those 1630 in the Venn diagram from A that are only changed in the MPTP + Emapunil treatment group but not in the MPTP group). LFC, Log fold change.
Discussion
Here, we investigated the effect of the TSPO ligand Emapunil in a well-established mouse model of parkinsonism, an important component of PD. We show that systemic administration of Emapunil ameliorates microgliosis, neuroinflammation, and ER stress in the MPTP-induced subchronic parkinsonism mouse model. Most importantly, we find that Emapunil treatment protects against neuron loss in the substantia nigra, striatal dopamine depletion, and motor deficits. In addition to an almost complete restoration of MPTP-associated gene expression changes, Emapunil alters the expression of genes unaffected by MPTP. We identified two potential pathways, which may explain the neuroprotective effects of Emapunil: (1) inhibition of the IREα/XBP1s related UPR; and (2) shift from pro-inflammatory to anti-inflammatory microglia polarization.
A common feature in many neurodegenerative diseases is the misfolding and accumulation of proteins. Under acute conditions, the UPR counteracts the accumulation of misfolded proteins by activation of ER-associated degradation pathways, attenuation of protein expression, and increased chaperone production. However, sustained activation of the UPR induces apoptosis, as observed in many neurodegenerative diseases, including prion (Moreno et al., 2013), Alzheimer's disease, Huntington's disease, and PD. Thus, inhibition of the UPR was suggested as a potential therapeutic approach in various neurodegenerative disorders. The UPR can be induced by three distinct pathways: PRKR-like ER kinase, IRE1, or activating transcription factor 6 (Schröder and Kaufman, 2005). In response to ER stress, activated IRE1α splices Xbp1 mRNA to the more active and stable form Xbp1s. The XBP1s protein acts as a transcription factor for UPR-related genes, which can result in protective as well as in toxic effects, depending, among others, on the pathway induced and the duration of UPR activation. Thus, overexpression of active XBP1s was previously shown to suppress neurotoxicity in MPP+-treated dopaminergic neuron cultures (Sado et al., 2009), whereas downregulation of XBP1 in dopaminergic neurons resulted in higher ER stress levels and neuronal loss (Valdés et al., 2014). Recent evidence, however, supported a protective effect of pharmacological inhibition of the IRE1/XBP1 pathway by β-asarone in the 6-OHDA rat model of parkinsonism (Ning et al., 2016). Inhibition of IRE1/XBP1 was also protective in various mouse models of other neurodegenerative diseases (Vidal et al., 2012) and activated IRE1/XBP1 signaling promoted pathology in a mouse model of Alzheimer's disease (Duran-Aniotz et al., 2017). Our findings indicate that MPTP and rotenone activate IRE1/XBP1 signaling. Emapunil restores increased XBP1s/XBP1 ratios and protects against neurotoxicity in MPP+ and rotenone-treated dopaminergic neuron cultures as well as in the MPTP mouse model. Our data suggest that Emapunil may act as an upstream inhibitor of the IRE1/XBP1 pathway, thereby attenuating cellular stress and apoptosis.
Until recently, it was assumed that TSPO is required for the transport of cholesterol from the outer to the inner mitochondrial membrane, as the rate-limiting step in steroid synthesis (Mukhin et al., 1989; Krueger and Papadopoulos, 1990; Rupprecht et al., 2010). This was challenged by findings in TSPO KO mice, which failed to show impaired steroidogenesis (Banati et al., 2014; Tu et al., 2014; Batoko et al., 2015), although conflicting results were obtained by other groups (Fan et al., 2015; Owen et al., 2017; Barron et al., 2018). Emapunil effects may thus at least partially be caused by alterations in neurosteroid levels. This notion is also supported by our transcriptome network analysis, which indicates Emapunil-mediated changes in steroid hormone response networks (Fig. 12C). Neurosteroids allosterically bind GABA receptors and may enhance GABA signaling, in line with the anxiolytic effects of Emapunil (Rupprecht et al., 2009). Neurosteroids can additionally regulate gene expression, thus potentially ameliorating inflammatory activation. Consistently, a TSPO ligand of the N,N-dialkyl-2-phenylindol-3-ylglyoxylamide class (PIGAs) was shown to attenuate LPS/interferon-γ-induced upregulation of iNOS and COX2 in an astroglioma cell line (Santoro et al., 2016). Our functional network analysis of gene expression changes, induced by MPTP and reinstated by Emapunil, revealed that Emapunil counteracts the activation of innate immune response and cytokine production pathways upregulated by MPTP. Unexpectedly, the majority of Emapunil-associated transcriptome alterations do not involve genes affected by MPTP. Network analysis of these Emapunil-specific genes again included networks linked to the regulation of cytokine production and other, immune-related pathways. This is in line with previous reports of anti-inflammatory effects observed with different synthetic TPSO ligands (Choi et al., 2011), for example, in experimental autoimmune encephalomyelitis (Daugherty et al., 2013), Alzheimer's disease (Barron et al., 2013), or retina degeneration (Scholz et al., 2015; Barichello et al., 2017).
Similar to macrophages, microglia can adopt different activation states, formerly distinguished into a pro-inflammatory M1, and an alternative, anti-inflammatory activation state M2. Recently, instead of this oversimplistic model, a concept of multiple or mixed microglia activation states was proposed. We found several of the M1 associated pro-inflammatory marker transcripts upregulated under MPTP treatment, including Tnfa, Cxcl10, Nos2, and Cox2. Enhanced mRNA levels of Cxcl10 and Cox2 were reversed under Emapunil administration, whereas a trend toward a reduction of Nos2 and Tnfa was observed but did not reach significance. In parallel, we found upregulation of various M2 linked phenotypic marker genes in MPTP + Emapunil-treated animals: Arg1, Il10, Il4 (trend), Mrc1, Ym1, and Fizz2. M2 activation states can further be divided into M2a, M2b, and M2c polarization states, which are associated with different functions (e.g., the M2a state with tissue repair). M2a-like activation is induced by Il4 and is characterized by increased Arg1, Ym1, and Fizz2 expression as well as Il10 secretion. Il10 in turn can stimulate M2c polarization, which suppresses M1-associated pro-inflammatory activity. Thus, Emapunil treatment appears to activate a microglial M2a and M2c anti-inflammatory response. Microglia can shift from M1 to M2 activation state, which may lead to mixed M1/M2 profiles as we observed. Thus, manipulating microglial activation to enhance anti-inflammatory phenotypes has been suggested as a possible therapeutic strategy in PD (Subramaniam and Federoff, 2017). Our findings suggest that Emapunil could constitute a promising molecule for therapeutic intervention in PD, especially given that Emapunil is orally available and readily crosses the blood–brain barrier (Owen et al., 2014). Emapunil was previously tested in 70 healthy volunteers in a panic disorder paradigm and in a clinical Phase II trial in patients with generalized anxiety disorder (Rupprecht et al., 2009; Owen et al., 2012). Importantly, the side effects of Emapunil were comparable with those of placebo. Thus, the safety profile and blood–brain barrier permeability make Emapunil ideally suited for clinical applications in CNS disorders. We have tested Emapunil effects only in female mice as upregulation of gonadal testosterone production by TSPO ligands had been reported previously (Chung et al., 2013). Therefore, it is possible that the effects of Emapunil differ between sexes. In addition, TSPO expression levels may differ between male and female mice. For example, significantly higher myocardial TSPO levels were reported in male compared with female mice during myocarditis (Fairweather et al., 2014). In brain, TSPO mRNA levels were significantly higher in male Drosophila compared with females (Lin et al., 2015), further supporting the notion that treatment response to TSPO ligands may differ in a sex-dependent manner. Another limitation of our study is the comparatively high dose of 50 mg Emapunil/kg body weight. It cannot be excluded that this may lead to off-target effects, although the effects of Emapunil on the UPR IRE1α-XBP1 pathway depended on the presence of TSPO. This supports the notion that the effects of Emapunil are mediated by TSPO rather than by off-target effects. TSPO bears a single nucleotide polymorphism rs6971, which results in the substitution of alanine 147 to threonine (A147T). Homozygous A147T carriers are characterized by a 15-fold lower binding affinity of TSPO to most of its synthetic ligands, including Emapunil (Owen et al., 2011a, b). The A147T polymorphism is frequent in the Caucasian population with ∼9% homozygous carriers and <1% in the Asian population (Kurumaji et al., 2000; Owen et al., 2011b). In future clinical studies, this limitation could be overcome by increasing the dose of Emapunil in low-affinity binders. In conclusion, we propose that Emapunil should be further validated in clinical trials to treat PD.
Footnotes
J.G. was supported by a China Scholarship Council scholarship. E.M.S. was supported by the Dorothea Schlözer fellowship. A.F. was supported by core funds from the German Center for Neurodegenerative Diseases, Deutsche Forschungsgemeinschaft Projects FI981/9-1 and SPP1738 (FI981-11-1), and the European Union ERC Consolidator Grant DEPICODE. M.Z. was supported by Deutsche Forschungsgemeinschaft Collaborative Research Center 803 (Project A11). T.O. was supported by Deutsche Forschungsgemeinschaft Center for Nanoscale Microscopy and Molecular Physiology of the Brain and by SFB1286 (Project B6). A.S. was supported by Deutsche Forschungsgemeinschaft Center for Nanoscale Microscopy and Molecular Physiology of the Brain, German Center for Neurodegenerative Diseases core funds, Deutsche Forschungsgemeinschaft Project SCHN1265/1-1, and Michael J. Fox Foundation Grant.
The authors declare no competing financial interests.
References
- Acosta SA, Tajiri N, de la Pena I, Bastawrous M, Sanberg PR, Kaneko Y, Borlongan CV (2015) Alpha-synuclein as a pathological link between chronic traumatic brain injury and Parkinson's disease. J Cell Physiol 230:1024–1032. 10.1002/jcp.24830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akundi RS, Huang Z, Eason J, Pandya JD, Zhi L, Cass WA, Sullivan PG, Büeler H (2011) Increased mitochondrial calcium sensitivity and abnormal expression of innate immunity genes precede dopaminergic defects in Pink1-deficient mice. PLoS One 6:e16038. 10.1371/journal.pone.0016038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, Huber W (2015) HTSeq: a python framework to work with high-throughput sequencing data. Bioinformatics 31:166–169. 10.1093/bioinformatics/btu638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banati RB, Middleton RJ, Chan R, Hatty CR, Kam WW, Quin C, Graeber MB, Parmar A, Zahra D, Callaghan P, Fok S, Howell NR, Gregoire M, Szabo A, Pham T, Davis E, Liu GJ (2014) Positron emission tomography and functional characterization of a complete PBR/TSPO knockout. Nat Commun 5:5452. 10.1038/ncomms6452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barichello T, Simões LR, Collodel A, Giridharan VV, Dal-Pizzol F, Macedo D, Quevedo J (2017) The translocator protein (18kDa) and its role in neuropsychiatric disorders. Neurosci Biobehav Rev 83:183–199. 10.1016/j.neubiorev.2017.10.010 [DOI] [PubMed] [Google Scholar]
- Barron AM, Garcia-Segura LM, Caruso D, Jayaraman A, Lee JW, Melcangi RC, Pike CJ (2013) Ligand for translocator protein reverses pathology in a mouse model of Alzheimer's disease. J Neurosci 33:8891–8897. 10.1523/JNEUROSCI.1350-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barron AM, Ji B, Kito S, Suhara T, Higuchi M (2018) Steroidogenic abnormalities in translocator protein knockout mice and significance in the aging male. Biochem J 475:75–85. 10.1042/BCJ20170645 [DOI] [PubMed] [Google Scholar]
- Bartels AL, Willemsen AT, Doorduin J, de Vries EF, Dierckx RA, Leenders KL (2010) [11C]-PK11195 PET: quantification of neuroinflammation and a monitor of anti-inflammatory treatment in Parkinson's disease? Parkinsonism Relat Disord 16:57–59. 10.1016/j.parkreldis.2009.05.005 [DOI] [PubMed] [Google Scholar]
- Batoko H, Veljanovski V, Jurkiewicz P (2015) Enigmatic translocator protein (TSPO) and cellular stress regulation. Trends Biochem Sci 40:497–503. 10.1016/j.tibs.2015.07.001 [DOI] [PubMed] [Google Scholar]
- Bindea G, Mlecnik B, Hackl H, Charoentong P, Tosolini M, Kirilovsky A, Fridman WH, Pagès F, Trajanoski Z, Galon J (2009) ClueGO: a cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 25:1091–1093. 10.1093/bioinformatics/btp101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Block ML, Zecca L, Hong JS (2007) Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci 8:57–69. 10.1038/nrn2038 [DOI] [PubMed] [Google Scholar]
- Brochard V, Combadière B, Prigent A, Laouar Y, Perrin A, Beray-Berthat V, Bonduelle O, Alvarez-Fischer D, Callebert J, Launay JM, Duyckaerts C, Flavell RA, Hirsch EC, Hunot S (2009) Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J Clin Invest 119:182–192. 10.1172/JCI36470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J, Ifuku M, Noda M, Guilarte TR (2011) Translocator protein (18 kDa)/peripheral benzodiazepine receptor specific ligands induce microglia functions consistent with an activated state. Glia 59:219–230. 10.1002/glia.21091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung JY, Chen H, Midzak A, Burnett AL, Papadopoulos V, Zirkin BR (2013) Drug ligand-induced activation of translocator protein (TSPO) stimulates steroid production by aged brown Norway rat Leydig cells. Endocrinology 154:2156–2165. 10.1210/en.2012-2226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa B, Da Pozzo E, Giacomelli C, Barresi E, Taliani S, Da Settimo F, Martini C (2016) TSPO ligand residence time: a new parameter to predict compound neurosteroidogenic efficacy. Sci Rep 6:18164. 10.1038/srep18164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couch Y, Alvarez-Erviti L, Sibson NR, Wood MJ, Anthony DC (2011) The acute inflammatory response to intranigral alpha-synuclein differs significantly from intranigral lipopolysaccharide and is exacerbated by peripheral inflammation. J Neuroinflammation 8:166. 10.1186/1742-2094-8-166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daugherty DJ, Selvaraj V, Chechneva OV, Liu XB, Pleasure DE, Deng W (2013) A TSPO ligand is protective in a mouse model of multiple sclerosis. EMBO Mol Med 5:891–903. 10.1002/emmm.201202124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, Littman DR, Dustin ML, Gan WB (2005) ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci 8:752–758. 10.1038/nn1472 [DOI] [PubMed] [Google Scholar]
- Deleidi M, Gasser T (2013) The role of inflammation in sporadic and familial Parkinson's disease. Cell Mol Life Sci 70:4259–4273. 10.1007/s00018-013-1352-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y, Ma Z, Lin S, Dodel RC, Gao F, Bales KR, Triarhou LC, Chernet E, Perry KW, Nelson DL, Luecke S, Phebus LA, Bymaster FP, Paul SM (2001) Minocycline prevents nigrostriatal dopaminergic neurodegeneration in the MPTP model of Parkinson's disease. Proc Natl Acad Sci U S A 98:14669–14674. 10.1073/pnas.251341998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont AC, Largeau B, Santiago Ribeiro MJ, Guilloteau D, Tronel C, Arlicot N (2017) Translocator Protein-18 kDa (TSPO) positron emission tomography (PET) imaging and its clinical impact in neurodegenerative diseases. Int J Mol Sci 18:E785. 10.3390/ijms18040785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duran-Aniotz C, Cornejo VH, Espinoza S, Ardiles ÁO, Medinas DB, Salazar C, Foley A, Gajardo I, Thielen P, Iwawaki T, Scheper W, Soto C, Palacios AG, Hoozemans JJ, Hetz C (2017) IRE1 signaling exacerbates Alzheimer's disease pathogenesis. Acta Neuropathol 134:489–506. 10.1007/s00401-017-1694-x [DOI] [PubMed] [Google Scholar]
- Edison P, Ahmed I, Fan Z, Hinz R, Gelosa G, Ray Chaudhuri K, Walker Z, Turkheimer FE, Brooks DJ (2013) Microglia, amyloid, and glucose metabolism in Parkinson's disease with and without dementia. Neuropsychopharmacology 38:938–949. 10.1038/npp.2012.255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eidson LN, Kannarkat GT, Barnum CJ, Chang J, Chung J, Caspell-Garcia C, Taylor P, Mollenhauer B, Schlossmacher MG, Ereshefsky L, Yen M, Kopil C, Frasier M, Marek K, Hertzberg VS, Tansey MG (2017) Candidate inflammatory biomarkers display unique relationships with alpha-synuclein and correlate with measures of disease severity in subjects with Parkinson's disease. J Neuroinflammation 14:164. 10.1186/s12974-017-0935-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairweather D, Coronado MJ, Garton AE, Dziedzic JL, Bucek A, Cooper LT Jr, Brandt JE, Alikhan FS, Wang H, Endres CJ, Choi J, Pomper MG, Guilarte TR (2014) Sex differences in translocator protein 18 kDa (TSPO) in the heart: implications for imaging myocardial inflammation. J Cardiovasc Transl Res 7:192–202. 10.1007/s12265-013-9538-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J, Campioli E, Midzak A, Culty M, Papadopoulos V (2015) Conditional steroidogenic cell-targeted deletion of TSPO unveils a crucial role in viability and hormone-dependent steroid formation. Proc Natl Acad Sci U S A 112:7261–7266. 10.1073/pnas.1502670112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer A. (2014) Epigenetic memory: the Lamarckian brain. EMBO J 33:945–967. 10.1002/embj.201387637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardet A, Benita Y, Li C, Sands BE, Ballester I, Stevens C, Korzenik JR, Rioux JD, Daly MJ, Xavier RJ, Podolsky DK (2010) LRRK2 is involved in the IFN-gamma response and host response to pathogens. J Immunol 185:5577–5585. 10.4049/jimmunol.1000548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatliff J, Campanella M (2016) TSPO: kaleidoscopic 18-kDa amid biochemical pharmacology, control and targeting of mitochondria. Biochem J 473:107–121. 10.1042/BJ20150899 [DOI] [PubMed] [Google Scholar]
- Gavioli EC, Duarte FS, Bressan E, Ferrara P, Farges RC, De Lima TC (2003) Antidepressant-like effect of Ro5–4864, a peripheral-type benzodiazepine receptor ligand, in forced swimming test. Eur J Pharmacol 471:21–26. 10.1016/S0014-2999(03)01789-8 [DOI] [PubMed] [Google Scholar]
- Gerhard A, Pavese N, Hotton G, Turkheimer F, Es M, Hammers A, Eggert K, Oertel W, Banati RB, Brooks DJ (2006) In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson's disease. Neurobiol Dis 21:404–412. 10.1016/j.nbd.2005.08.002 [DOI] [PubMed] [Google Scholar]
- Giatti S, Pesaresi M, Cavaletti G, Bianchi R, Carozzi V, Lombardi R, Maschi O, Lauria G, Garcia-Segura LM, Caruso D, Melcangi RC (2009) Neuroprotective effects of a ligand of translocator protein-18 kDa (Ro5–4864) in experimental diabetic neuropathy. Neuroscience 164:520–529. 10.1016/j.neuroscience.2009.08.005 [DOI] [PubMed] [Google Scholar]
- Gillardon F, Schmid R, Draheim H (2012) Parkinson's disease-linked leucine-rich repeat kinase 2(R1441G) mutation increases proinflammatory cytokine release from activated primary microglial cells and resultant neurotoxicity. Neuroscience 208:41–48. 10.1016/j.neuroscience.2012.02.001 [DOI] [PubMed] [Google Scholar]
- Giordano S, Lee J, Darley-Usmar VM, Zhang J (2012) Distinct effects of rotenone, 1-methyl-4-phenylpyridinium and 6-hydroxydopamine on cellular bioenergetics and cell death. PLoS One 7:e44610. 10.1371/journal.pone.0044610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu XL, Long CX, Sun L, Xie C, Lin X, Cai H (2010) Astrocytic expression of Parkinson's disease-related A53T alpha-synuclein causes neurodegeneration in mice. Mol Brain 3:12. 10.1186/1756-6606-3-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harms AS, Delic V, Thome AD, Bryant N, Liu Z, Chandra S, Jurkuvenaite A, West AB (2017) α-Synuclein fibrils recruit peripheral immune cells in the rat brain prior to neurodegeneration. Acta Neuropathol Commun 5:85. 10.1186/s40478-017-0494-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch EC, Hunot S (2009) Neuroinflammation in Parkinson's disease: a target for neuroprotection? Lancet Neurol 8:382–397. 10.1016/S1474-4422(09)70062-6 [DOI] [PubMed] [Google Scholar]
- Iannaccone S, Cerami C, Alessio M, Garibotto V, Panzacchi A, Olivieri S, Gelsomino G, Moresco RM, Perani D (2013) In vivo microglia activation in very early dementia with Lewy bodies, comparison with Parkinson's disease. Parkinsonism Relat Disord 19:47–52. 10.1016/j.parkreldis.2012.07.002 [DOI] [PubMed] [Google Scholar]
- Imamura K, Hishikawa N, Sawada M, Nagatsu T, Yoshida M, Hashizume Y (2003) Distribution of major histocompatibility complex class II-positive microglia and cytokine profile of Parkinson's disease brains. Acta Neuropathol 106:518–526. 10.1007/s00401-003-0766-2 [DOI] [PubMed] [Google Scholar]
- Karlstetter M, Nothdurfter C, Aslanidis A, Moeller K, Horn F, Scholz R, Neumann H, Weber BH, Rupprecht R, Langmann T (2014) Translocator protein (18 kDa) (TSPO) is expressed in reactive retinal microglia and modulates microglial inflammation and phagocytosis. J Neuroinflammation 11:3. 10.1186/1742-2094-11-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khasnavis S, Ghosh A, Roy A, Pahan K (2013) Castration induces Parkinson disease pathologies in young male mice via inducible nitric-oxide synthase. J Biol Chem 288:20843–20855. 10.1074/jbc.M112.443556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirov S, Ji R, Wang J, Zhang B (2014) Functional annotation of differentially regulated gene set using WebGestalt: a gene set predictive of response to ipilimumab in tumor biopsies. Methods Mol Biol 1101:31–42. 10.1007/978-1-62703-721-1_3 [DOI] [PubMed] [Google Scholar]
- Kita A, Kohayakawa H, Kinoshita T, Ochi Y, Nakamichi K, Kurumiya S, Furukawa K, Oka M (2004) Antianxiety and antidepressant-like effects of AC-5216, a novel mitochondrial benzodiazepine receptor ligand. Br J Pharmacol 142:1059–1072. 10.1038/sj.bjp.0705681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knott C, Stern G, Wilkin GP (2000) Inflammatory regulators in Parkinson's disease: iNOS, lipocortin-1, and cyclooxygenases-1 and -2. Mol Cell Neurosci 16:724–739. 10.1006/mcne.2000.0914 [DOI] [PubMed] [Google Scholar]
- Koshimori Y, Ko JH, Mizrahi R, Rusjan P, Mabrouk R, Jacobs MF, Christopher L, Hamani C, Lang AE, Wilson AA, Houle S, Strafella AP (2015) Imaging striatal microglial activation in patients with Parkinson's disease. PLoS One 10:e0138721. 10.1371/journal.pone.0138721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger KE, Papadopoulos V (1990) Peripheral-type benzodiazepine receptors mediate translocation of cholesterol from outer to inner mitochondrial membranes in adrenocortical cells. J Biol Chem 265:15015–15022. [PubMed] [Google Scholar]
- Krug AK, Gutbier S, Zhao L, Pöltl D, Kullmann C, Ivanova V, Förster S, Jagtap S, Meiser J, Leparc G, Schildknecht S, Adam M, Hiller K, Farhan H, Brunner T, Hartung T, Sachinidis A, Leist M (2014) Transcriptional and metabolic adaptation of human neurons to the mitochondrial toxicant MPP(+). Cell Death Dis 5:e1222. 10.1038/cddis.2014.166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurumaji A, Nomoto H, Yoshikawa T, Okubo Y, Toru M (2000) An association study between two missense variations of the benzodiazepine receptor (peripheral) gene and schizophrenia in a Japanese sample. J Neural Transm (Vienna) 107:491–500. 10.1007/s007020070090 [DOI] [PubMed] [Google Scholar]
- Leva G, Klein C, Benyounes J, Halle F, Bihel F, Collongues N, De Seze J, Mensah-Nyagan AG, Patte-Mensah C (2017) The translocator protein ligand XBD173 improves clinical symptoms and neuropathological markers in the SJL/J mouse model of multiple sclerosis. Biochim Biophys Acta 1863:3016–3027. 10.1016/j.bbadis.2017.09.007 [DOI] [PubMed] [Google Scholar]
- Lin R, Rittenhouse D, Sweeney K, Potluri P, Wallace DC (2015) TSPO, a mitochondrial outer membrane protein, controls ethanol-related behaviors in Drosophila. PLoS Genet 11:e1005366. 10.1371/journal.pgen.1005366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotharius J, Barg S, Wiekop P, Lundberg C, Raymon HK, Brundin P (2002) Effect of mutant alpha-synuclein on dopamine homeostasis in a new human mesencephalic cell line. J Biol Chem 277:38884–38894. 10.1074/jbc.M205518200 [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550. 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez Hernandez A, Urbanke H, Gillman AL, Lee J, Ryazanov S, Agbemenyah HY, Benito E, Jain G, Kaurani L, Grigorian G, Leonov A, Rezaei-Ghaleh N, Wilken P, Arce FT, Wagner J, Fuhrmann M, Caruana M, Camilleri A, Vassallo N, Zweckstetter M, et al. (2018) The diphenylpyrazole compound anle138b blocks Aβ channels and rescues disease phenotypes in a mouse model for amyloid pathology. EMBO Mol Med 10:32–47. 10.15252/emmm.201707825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeer PL, Itagaki S, Boyes BE, McGeer EG (1988) Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson's and Alzheimer's disease brains. Neurology 38:1285–1291. 10.1212/WNL.38.8.1285 [DOI] [PubMed] [Google Scholar]
- Mercado G, Castillo V, Vidal R, Hetz C (2015) ER proteostasis disturbances in Parkinson's disease: novel insights. Front Aging Neurosci 7:39. 10.3389/fnagi.2015.00039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miklossy J, Doudet DD, Schwab C, Yu S, McGeer EG, McGeer PL (2006) Role of ICAM-1 in persisting inflammation in Parkinson disease and MPTP monkeys. Exp Neurol 197:275–283. 10.1016/j.expneurol.2005.10.034 [DOI] [PubMed] [Google Scholar]
- Moehle MS, Webber PJ, Tse T, Sukar N, Standaert DG, DeSilva TM, Cowell RM, West AB (2012) LRRK2 inhibition attenuates microglial inflammatory responses. J Neurosci 32:1602–1611. 10.1523/JNEUROSCI.5601-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogi M, Kondo T, Mizuno Y, Nagatsu T (2007) p53 protein, interferon-gamma, and NF-kappaB levels are elevated in the parkinsonian brain. Neurosci Lett 414:94–97. 10.1016/j.neulet.2006.12.003 [DOI] [PubMed] [Google Scholar]
- Moreno JA, Halliday M, Molloy C, Radford H, Verity N, Axten JM, Ortori CA, Willis AE, Fischer PM, Barrett DA, Mallucci GR (2013) Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice. Sci Transl Med 5:206ra138. 10.1126/scitranslmed.3006767 [DOI] [PubMed] [Google Scholar]
- Mukhin AG, Papadopoulos V, Costa E, Krueger KE (1989) Mitochondrial benzodiazepine receptors regulate steroid biosynthesis. Proc Natl Acad Sci U S A 86:9813–9816. 10.1073/pnas.86.24.9813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ning B, Deng M, Zhang Q, Wang N, Fang Y (2016) β-Asarone inhibits IRE1/XBP1 endoplasmic reticulum stress pathway in 6-OHDA-induced parkinsonian rats. Neurochem Res 41:2097–2101. 10.1007/s11064-016-1922-0 [DOI] [PubMed] [Google Scholar]
- Ogawa N, Hirose Y, Ohara S, Ono T, Watanabe Y (1985) A simple quantitative bradykinesia test in MPTP-treated mice. Res Commun Chem Pathol Pharmacol 50:435–441. [PubMed] [Google Scholar]
- Ouchi Y, Yoshikawa E, Sekine Y, Futatsubashi M, Kanno T, Ogusu T, Torizuka T (2005) Microglial activation and dopamine terminal loss in early Parkinson's disease. Ann Neurol 57:168–175. 10.1002/ana.20338 [DOI] [PubMed] [Google Scholar]
- Owen DR, Lewis AJ, Reynolds R, Rupprecht R, Eser D, Wilkins MR, Bennacef I, Nutt DJ, Parker CA (2011a) Variation in binding affinity of the novel anxiolytic XBD173 for the 18 kDa translocator protein in human brain. Synapse 65:257–259. 10.1002/syn.20884 [DOI] [PubMed] [Google Scholar]
- Owen DR, Gunn RN, Rabiner EA, Bennacef I, Fujita M, Kreisl WC, Innis RB, Pike VW, Reynolds R, Matthews PM, Parker CA (2011b) Mixed-affinity binding in humans with 18-kDa translocator protein ligands. J Nucl Med 52:24–32. 10.2967/jnumed.110.079459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen DR, Yeo AJ, Gunn RN, Song K, Wadsworth G, Lewis A, Rhodes C, Pulford DJ, Bennacef I, Parker CA, StJean PL, Cardon LR, Mooser VE, Matthews PM, Rabiner EA, Rubio JP (2012) An 18-kDa translocator protein (TSPO) polymorphism explains differences in binding affinity of the PET radioligand PBR28. J Cereb Blood Flow Metab 32:1–5. 10.1038/jcbfm.2011.147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen DR, Guo Q, Kalk NJ, Colasanti A, Kalogiannopoulou D, Dimber R, Lewis YL, Libri V, Barletta J, Ramada-Magalhaes J, Kamalakaran A, Nutt DJ, Passchier J, Matthews PM, Gunn RN, Rabiner EA (2014) Determination of [(11)C]PBR28 binding potential in vivo: a first human TSPO blocking study. J Cereb Blood Flow Metab 34:989–994. 10.1038/jcbfm.2014.46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen DR, Fan J, Campioli E, Venugopal S, Midzak A, Daly E, Harlay A, Issop L, Libri V, Kalogiannopoulou D, Oliver E, Gallego-Colon E, Colasanti A, Huson L, Rabiner EA, Suppiah P, Essagian C, Matthews PM, Papadopoulos V (2017) TSPO mutations in rats and a human polymorphism impair the rate of steroid synthesis. Biochem J 474:3985–3999. 10.1042/BCJ20170648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos V, Lecanu L (2009) Translocator protein (18 kDa) TSPO: an emerging therapeutic target in neurotrauma. Exp Neurol 219:53–57. 10.1016/j.expneurol.2009.04.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinho R, Guedes LC, Soreq L, Lobo PP, Mestre T, Coelho M, Rosa MM, Gonçalves N, Wales P, Mendes T, Gerhardt E, Fahlbusch C, Bonifati V, Bonin M, Miltenberger-Miltényi G, Borovecki F, Soreq H, Ferreira JJ, Outeiro TF (2016) Gene expression differences in peripheral blood of Parkinson's disease patients with distinct progression profiles. PLoS One 11:e0157852. 10.1371/journal.pone.0157852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravikumar B, Crawford D, Dellovade T, Savinainen A, Graham D, Liere P, Oudinet JP, Webb M, Hering H (2016) Differential efficacy of the TSPO ligands etifoxine and XBD-173 in two rodent models of multiple sclerosis. Neuropharmacology 108:229–237. 10.1016/j.neuropharm.2016.03.053 [DOI] [PubMed] [Google Scholar]
- Ron D, Walter P (2007) Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 8:519–529. 10.1038/nrm2199 [DOI] [PubMed] [Google Scholar]
- Rupprecht R, Rammes G, Eser D, Baghai TC, Schüle C, Nothdurfter C, Troxler T, Gentsch C, Kalkman HO, Chaperon F, Uzunov V, McAllister KH, Bertaina-Anglade V, La Rochelle CD, Tuerck D, Floesser A, Kiese B, Schumacher M, Landgraf R, Holsboer F, et al. (2009) Translocator protein (18 kD) as target for anxiolytics without benzodiazepine-like side effects. Science 325:490–493. 10.1126/science.1175055 [DOI] [PubMed] [Google Scholar]
- Rupprecht R, Papadopoulos V, Rammes G, Baghai TC, Fan J, Akula N, Groyer G, Adams D, Schumacher M (2010) Translocator protein (18 kDa) (TSPO) as a therapeutic target for neurological and psychiatric disorders. Nat Rev Drug Discov 9:971–988. 10.1038/nrd3295 [DOI] [PubMed] [Google Scholar]
- Sado M, Yamasaki Y, Iwanaga T, Onaka Y, Ibuki T, Nishihara S, Mizuguchi H, Momota H, Kishibuchi R, Hashimoto T, Wada D, Kitagawa H, Watanabe TK (2009) Protective effect against Parkinson's disease-related insults through the activation of XBP1. Brain Res 1257:16–24. 10.1016/j.brainres.2008.11.104 [DOI] [PubMed] [Google Scholar]
- Santoro A, Mattace Raso G, Taliani S, Da Pozzo E, Simorini F, Costa B, Martini C, Laneri S, Sacchi A, Cosimelli B, Calignano A, Da Settimo F, Meli R (2016) TSPO-ligands prevent oxidative damage and inflammatory response in C6 glioma cells by neurosteroid synthesis. Eur J Pharm Sci 88:124–131. 10.1016/j.ejps.2016.04.006 [DOI] [PubMed] [Google Scholar]
- Scholz D, Pöltl D, Genewsky A, Weng M, Waldmann T, Schildknecht S, Leist M (2011) Rapid, complete and large-scale generation of post-mitotic neurons from the human LUHMES cell line. J Neurochem 119:957–971. 10.1111/j.1471-4159.2011.07255.x [DOI] [PubMed] [Google Scholar]
- Scholz R, Caramoy A, Bhuckory MB, Rashid K, Chen M, Xu H, Grimm C, Langmann T (2015) Targeting translocator protein (18 kDa) (TSPO) dampens pro-inflammatory microglia reactivity in the retina and protects from degeneration. J Neuroinflammation 12:201. 10.1186/s12974-015-0422-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schröder M, Kaufman RJ (2005) ER stress and the unfolded protein response. Mutat Res 569:29–63. 10.1016/j.mrfmmm.2004.06.056 [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M (1998) α-Synuclein in filamentous inclusions of Lewy bodies from Parkinson's disease and dementia with Lewy bodies. Proc Natl Acad Sci U S A 95:6469–6473. 10.1073/pnas.95.11.6469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramaniam SR, Federoff HJ (2017) Targeting microglial activation states as a therapeutic avenue in Parkinson's disease. Front Aging Neurosci 9:176. 10.3389/fnagi.2017.00176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szego EM, Gerhardt E, Outeiro TF (2017) Sirtuin 2 enhances dopaminergic differentiation via the AKT/GSK-3beta/beta-catenin pathway. Neurobiol Aging 56:7–16. 10.1016/j.neurobiolaging.2017.04.001 [DOI] [PubMed] [Google Scholar]
- Tanabe Y, Suehara Y, Kohsaka S, Hayashi T, Akaike K, Mukaihara K, Kurihara T, Kim Y, Okubo T, Ishii M, Kazuno S, Kaneko K, Saito T (2018) IRE1α-XBP1 inhibitors exerted anti-tumor activities in Ewing's sarcoma. Oncotarget 9:14428–14443. 10.18632/oncotarget.24467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Team R. (2008) R: A language and environment for statistical computing. Vienna: R Foundation for Statistical Computing. [Google Scholar]
- Teismann P, Tieu K, Choi DK, Wu DC, Naini A, Hunot S, Vila M, Jackson-Lewis V, Przedborski S (2003) Cyclooxygenase-2 is instrumental in Parkinson's disease neurodegeneration. Proc Natl Acad Sci U S A 100:5473–5478. 10.1073/pnas.0837397100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tentillier N, Etzerodt A, Olesen MN, Rizalar FS, Jacobsen J, Bender D, Moestrup SK, Romero-Ramos M (2016) Anti-inflammatory modulation of microglia via CD163-targeted glucocorticoids protects dopaminergic neurons in the 6-OHDA Parkinson's disease model. J Neurosci 36:9375–9390. 10.1523/JNEUROSCI.1636-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tönges L, Frank T, Tatenhorst L, Saal KA, Koch JC, Szego EM, Bähr M, Weishaupt JH, Lingor P (2012) Inhibition of rho kinase enhances survival of dopaminergic neurons and attenuates axonal loss in a mouse model of Parkinson's disease. Brain 135:3355–3370. 10.1093/brain/aws254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu LN, Morohaku K, Manna PR, Pelton SH, Butler WR, Stocco DM, Selvaraj V (2014) Peripheral benzodiazepine receptor/translocator protein global knock-out mice are viable with no effects on steroid hormone biosynthesis. J Biol Chem 289:27444–27454. 10.1074/jbc.M114.578286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tufekci KU, Genc S, Genc K (2011) The endotoxin-induced neuroinflammation model of Parkinson's disease. Parkinsons Dis 2011:487450. 10.4061/2011/487450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urra H, Dufey E, Lisbona F, Rojas-Rivera D, Hetz C (2013) When ER stress reaches a dead end. Biochim Biophys Acta 1833:3507–3517. 10.1016/j.bbamcr.2013.07.024 [DOI] [PubMed] [Google Scholar]
- Valdés P, Mercado G, Vidal RL, Molina C, Parsons G, Court FA, Martinez A, Galleguillos D, Armentano D, Schneider BL, Hetz C (2014) Control of dopaminergic neuron survival by the unfolded protein response transcription factor XBP1. Proc Natl Acad Sci U S A 111:6804–6809. 10.1073/pnas.1321845111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Schadewijk A, van't Wout EF, Stolk J, Hiemstra PS (2012) A quantitative method for detection of spliced X-box binding protein-1 (XBP1) mRNA as a measure of endoplasmic reticulum (ER) stress. Cell Stress Chaperones 17:275–279. 10.1007/s12192-011-0306-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal RL, Figueroa A, Court FA, Thielen P, Molina C, Wirth C, Caballero B, Kiffin R, Segura-Aguilar J, Cuervo AM, Glimcher LH, Hetz C (2012) Targeting the UPR transcription factor XBP1 protects against Huntington's disease through the regulation of FoxO1 and autophagy. Hum Mol Genet 21:2245–2262. 10.1093/hmg/dds040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waak J, Weber SS, Waldenmaier A, Görner K, Alunni-Fabbroni M, Schell H, Vogt-Weisenhorn D, Pham TT, Reumers V, Baekelandt V, Wurst W, Kahle PJ (2009) Regulation of astrocyte inflammatory responses by the Parkinson's disease-associated gene DJ-1. FASEB J 23:2478–2489. 10.1096/fj.08-125153 [DOI] [PubMed] [Google Scholar]
- Wahner AD, Sinsheimer JS, Bronstein JM, Ritz B (2007) Inflammatory cytokine gene polymorphisms and increased risk of Parkinson disease. Arch Neurol 64:836–840. 10.1001/archneur.64.6.836 [DOI] [PubMed] [Google Scholar]
- Wang J, Duncan D, Shi Z, Zhang B (2013) WEB-based GEne SeT AnaLysis toolkit (WebGestalt): update 2013. Nucleic Acids Res 41:W77–W83. 10.1093/nar/gkt439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Wang X, Zhao L, Ma W, Rodriguez IR, Fariss RN, Wong WT (2014) Macroglia-microglia interactions via TSPO signaling regulates microglial activation in the mouse retina. J Neurosci 34:3793–3806. 10.1523/JNEUROSCI.3153-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilms H, Rosenstiel P, Sievers J, Deuschl G, Zecca L, Lucius R (2003) Activation of microglia by human neuromelanin is NF-kappaB dependent and involves p38 mitogen-activated protein kinase: implications for Parkinson's disease. FASEB J 17:500–502. 10.1096/fj.02-0314fje [DOI] [PubMed] [Google Scholar]
- Wu DC, Jackson-Lewis V, Vila M, Tieu K, Teismann P, Vadseth C, Choi DK, Ischiropoulos H, Przedborski S (2002) Blockade of microglial activation is neuroprotective in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson disease. J Neurosci 22:1763–1771. 10.1523/JNEUROSCI.22-05-01763.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zambelli F, Pesole G, Pavesi G (2009) Pscan: finding over-represented transcription factor binding site motifs in sequences from co-regulated or co-expressed genes. Nucleic Acids Res 37:W247–W252. 10.1093/nar/gkp464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Kirov S, Snoddy J (2005) WebGestalt: an integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res 33:W741–W748. 10.1093/nar/gki475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang MR, Kumata K, Maeda J, Yanamoto K, Hatori A, Okada M, Higuchi M, Obayashi S, Suhara T, Suzuki K (2007) 11C-AC-5216: a novel PET ligand for peripheral benzodiazepine receptors in the primate brain. J Nucl Med 48:1853–1861. 10.2967/jnumed.107.043505 [DOI] [PubMed] [Google Scholar]
- Zhang W, Wang T, Pei Z, Miller DS, Wu X, Block ML, Wilson B, Zhang W, Zhou Y, Hong JS, Zhang J (2005) Aggregated alpha-synuclein activates microglia: a process leading to disease progression in Parkinson's disease. FASEB J 19:533–542. 10.1096/fj.04-2751com [DOI] [PubMed] [Google Scholar]
- Zhang W, Zecca L, Wilson B, Ren HW, Wang YJ, Wang XM, Hong JS, Shahsavar S (2013) Human neuromelanin: an endogenous microglial activator for dopaminergic neuron death. Front Biosci 5:1–11. 10.2741/E591 [DOI] [PMC free article] [PubMed] [Google Scholar]