

Abstract
Despite a growing interest in, and commitment to, implementing pediatric clinical trials, approximately one in every five trials in children fails because of inappropriate study design, suboptimal experiment planning, or inadequate participant enrollment. This tutorial, presented from the perspectives of seasoned pediatric investigators, an experienced research coordinator, and an established pediatric clinical trials network, is designed to provide practical guidance for successfully implementing pediatric clinical trials at an academic center or another comparable institution.
The evolution in pediatric legislation (Figure 1),1 a growing commitment to pediatric studies by the pharmaceutical industry, and a renewed interest in therapeutics for rare diseases have increased the demand for timely, high‐quality, cost‐effective clinical trials in children.2, 3 Despite the urgency and the implementation of incentives to conduct these trials, approximately one in every five pediatric trials fails because of inappropriate study design, suboptimal experiment planning, or inadequate participant enrollment.4, 5 This tutorial, presented from the perspectives of seasoned pediatric investigators, an experienced research coordinator, and an established pediatric clinical trials network, is designed to provide practical guidance for successfully implementing pediatric clinical trials at your institution.
Figure 1.
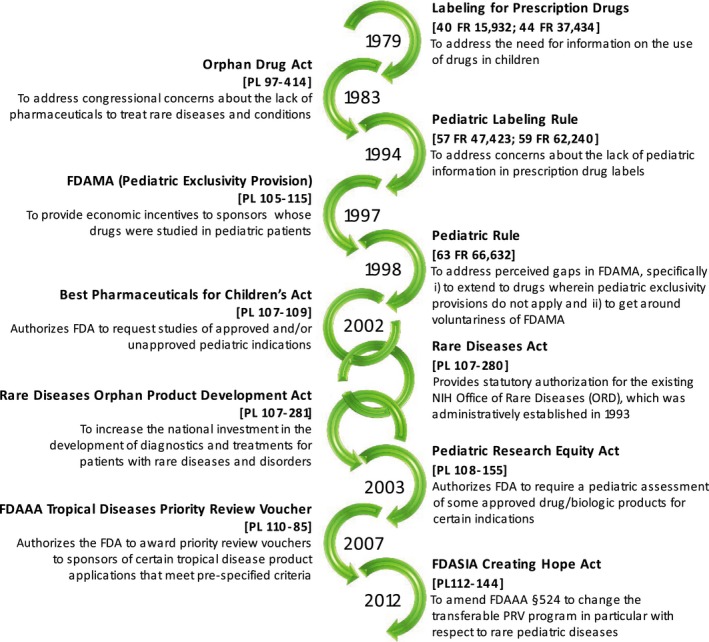
Evolution in pediatric drug legislature; image created with content from Tolbert et al.1 FDA, US Food and Drug Administration.
Planning Your Study
Easily recognizable as problematic, to an experienced pediatric clinical trialist, are sponsored protocols intended for children that are virtually indistinguishable from the antecedent adult trials. This singular protocol feature is assured to present challenges in the execution of the trial and interpretation of the study data.5 A successful pediatric trial, by contrast, takes into consideration the influence of normal growth and development on physiologic processes that are relevant to medication administration, drug disposition, the measurement of drug response, and how the intersection of development and disease impact both the logistical and scientific aspects of the study.6, 7
Medication administration
In a population where items intended for oral ingestion, including foods and medicines, are often rejected on the basis of smell, taste, and texture, the importance of drug formulation cannot be overemphasized. Consistent, reliable drug delivery during a clinical trial necessitates age‐appropriate formulations. Although stable and easy to transport, solid oral dosage forms can be difficult for young children to manage. Importantly, these formulations limit the extent to which the dose can be titrated in a population of individuals with widely disparate weights (e.g., below 500 g for some neonates8 and well in excess of 100 kg for some adolescents9). Minitablets, chewables, orodispersibles, and liquid formulations can, to varying extent, overcome the challenges of accurate weight‐based dosing; however, these formulations can be rejected as a result of their texture, taste, or smell, each of which has a unique developmental trajectory, driven by teleology and overlaid by adaptive and cultural influences.10, 11
Allowing more than one formulation within a study can broaden the range of children eligible for participation, but can introduce unexpected challenges. We recall a sponsored study incorporating both a solid and liquid formulation, where the latter was so poorly formulated that vomiting rates approached 25% (vs. 0% for the solid formulation).5 Fortunately, in this single‐dose pharmacokinetic (PK) trial, withdrawn participants could be replaced, albeit at an expense in excess of what was initially budgeted in the study. Had this study required repeated dose administration, the result could very well have been failure of the trial.
Using a single adult formulation, or permitting extemporaneous compounding of an adult formulation to accommodate children, can reveal a different set of challenges, and the impact of formulation may not become apparent until the time of data analysis. We have encountered clinical trials in which extemporaneous compounds were made to the wrong specifications, resulting in plasma concentrations that were below the lower limit of detection. We also have examples of trials in which the process of compounding interfered with delivery of the active compound, such that we were unable to disentangle formulation effects from age effects at the time of data analysis.5 Ultimately, the decision to make a provision for compounding in your protocol should not be undertaken without consideration for the frequency with which the preparation will need to be made, whether quality control measures can be put in place, and whether this approach is suitable for the study's medication administration strategy.
With respect to extemporaneous preparation, we have also observed trials in which existing dosage forms were modified, and co‐administered foods/beverages interfered with the bioavailability of the active drug compound, resulting in unexpected disposition profiles and, in some cases, failure to satisfy bioequivalence criteria.7 Through Figure 2, derived from a study in healthy volunteers, we illustrate the impact of a pediatric dietary staple (apple juice) on the systemic exposure to a medication.12 Prior to our improved understanding of the impact of drug–nutrient interactions on presystemic drug metabolism and transport, it was not uncommon to encounter pediatric trials permitting concomitant juice administration, and we still see studies that permit flavored beverages (e.g., fruit punch), despite the fact that most contain apple juice as an ingredient. Failure to recognize or account for the effect of food and formulation during data capture can introduce added levels of variability, confounding data interpretation and experimental results.
Figure 2.

Mean plasma concentrations (means ± SD) of fexofenadine (FEX) after oral administration of FEX 60 mg with water 600 mL, apple juice (AJ) 150 mL, followed by water 450 mL, AJ 300 mL, followed by water 300 mL, and AJ 600 mL. N = 10; image adapted from Luo et al.,12 https://doi.org/10.1111/cts.12400, is licensed under CC BY‐NC‐ND 4.0. ©2016 The authors.
Dosing approach
Considerations surrounding medication administration also extend to the dosing approach. Pediatric trials are frequently conducted after the exposure–response relationship for the drug of interest has been determined in adults,5 with adult data informing reference ranges for dose selection in children. To account for variability in size across the age spectrum, drug doses can be normalized to body mass (mg/kg), lean body mass (mg/kg lean body weight or fat‐free mass), or surface area (mg/m2) to approximate weight‐adjusted exposures in adults. These approaches are particularly useful when existing data offer support for a comparable pharmacodynamics (PD) profile between adults and children; however, they require an easily titratable drug formulation.
Alternatively, a fixed dose of a drug (mg) may be administered to subjects across the continuum of pediatric ages, such that drug doses span a targeted range, when corrected for body weight/size (e.g., mg/kg or mg/m2). Given the wide range in body size encountered in the pediatric population, the fixed‐dose approach may result in significant variability in weight‐adjusted or size‐adjusted drug exposures, which will need to be addressed during data analysis. Ultimately, the objective of the study, as well as the availability of an age‐appropriate drug formulation, should guide the dose selection strategy for a pediatric investigation.
Experimental Design
Knowledge of the impact of normal growth and development on drug disposition informs experimental design, as it relates to PK and PD sampling schemes and assay sensitivity requirements. Although a thorough review of these principles is outside the scope of this article, we provide several illustrative examples below.
Drug disposition
For orally administered medications, physiologic changes in the gastrointestinal tract need to be considered when designing a pediatric study spanning different age groups. The relative achlorhydria during the neonatal period, for example, can protect acid labile drugs, resulting in higher systemic concentrations after oral administration relative to older children and adults.13 Conversely, weakly basic drugs may experience reduced bioavailability at these higher gastric pH values.14 Maturation of gastric emptying and intestinal motility, during the first few weeks15 to years7 of life, correspond with decreased times to reach maximum blood concentrations for many drugs (e.g., cisapride8) for the younger pediatric age range. Similarly, for drugs that require bile salts to facilitate uptake, absorption may be erratic and incomplete during the first few months of life,16, 17 secondary to the immaturity of transporters responsible for carrying them across the biliary canaliculi.18, 19, 20, 21, 22
The gastrointestinal tract has other important features that influence the bioavailability of orally administered medications, including phase I and II drug‐metabolizing enzymes and drug transporters. Although data on the intestinal expression of these proteins are sparse,23, 24, 25, 26, 27 existing data merit consideration. Reduced bioavailability, due to drug‐metabolizing enzyme ontogeny, can lead to samples that fall below the lower limit of quantification at the end of the sampling interval. Too few values on the terminal slope can compromise half‐life and area under the concentration–time curve (AUC) estimates. By extension, enhanced absorption can result in concentrations that exceed the upper limit of quantification when the working concentration range for the bioanalytical method does not span several orders of magnitude, requiring reanalysis, assuming sufficient residual sample remains. Alterations in gastric emptying and intestinal transit may require changes to sampling strategy, such that data‐rich segments of the PK profile are modified to enable adequate characterization of both the apparent time to peak concentration (Tmax) and the peak plasma concentration (Cmax).
As total and free circulating drug concentrations relate to apparent distribution volume, it is equally important to recognize that total body water stores in children are increased compared with adults, effectively lowering drug concentrations of hydrophilic drugs.10 The extent of protein binding can also be reduced, primarily in young infants, owing to higher concentrations of endogenous substances capable of binding albumin (e.g., bilirubin, fatty acids), lower α1‐acid glycoprotein concentrations, and albumin stores that are constituted, in part, by fetal albumin, which has a lower binding affinity for drugs.28, 29 The impact of these changes on moderately bound drugs are likely limited; however, for a highly protein‐bound drug, a small shift in binding may significantly increase the unbound concentration of drug available to interact with therapeutic targets, depending on the hepatic extraction ratio and apparent distribution volume of the drug.
Disposition can also be altered for drugs administered via extraoral routes. Enhanced percutaneous absorption can be expected in children, owing to a larger surface area, improved hydration status, and increased capillary perfusion of the skin.10 Because of erratic lower colonic contractions, rectal suppositories that are slower to melt may be expelled in young infants before complete release of the drug, resulting in a lower fractional absorption.30
Sampling strategy
Ontogenic changes in the rate of drug elimination influence the extent of sampling required to characterize PK and the timeframe over which to examine PD. Importantly, drug clearance pathways mature at different rates; therefore, in designing a successful pediatric trial, one must explore whether the drug under investigation is a substrate for proteins or elimination pathways that are maximally expressed perinatally (e.g., cytochrome P450 (CYP)s 2C9, 2C19, and 3A7, and sulfotransferase 1A1 and 1E1), shortly after birth (e.g., CYPs 2D6, glutathione S‐transferase (GST)A1 and GSTA2), or at varying points during infancy or childhood (e.g., CYPs 1A2, 2B6, 2C8, and 3A4/5, and UDP‐glucuronosyltransferases 1A1, 1A9, and 2B7, GST1, glomerular filtration, and active tubular secretion).31 It is also important to recognize whether redundancy exists (i.e., for polyfunctional substrates), such that ontogeny of compensatory pathways may be relevant.
Knowledge of the anticipated impact of developmental changes in clearance and distribution volume can and should be used to inform your sampling strategy. Specific attention should be paid to changes in clearance and volume that can occur in concert to intensify the developmental effect. For example, our pediatric study of linezolid revealed concentrations at the end of a 12‐hour dosing interval that were more than an order of magnitude lower than reported in adults, secondary to an expanded distribution volume and enhanced total body clearance.32
Overlaid on the developmental differences highlighted above are acute or chronic pediatric diseases that can independently interfere with drug disposition. Gastric emptying rates are influenced by pediatric comorbidities, including prematurity, gastroesophageal reflux disease, congenital heart disease, and type I diabetes.33 Intestinal dysmotility accompanies gastroschisis, Hirschsprung's disease, and infantile pyloric stenosis.34, 35, 36 Children with amino acid n‐acyltransferase deficiency, or those receiving chronic total parenteral nutrition, may experience alterations in protein binding, whereas renal filtration is expected to be compromised in children with nephropathies, such as focal segmental glomerulosclerosis.10
For anticipated scenarios, such as those described above, special consideration may need to be given to assay sensitivity and specimen volumes. However, pediatric protocols must always remain cognizant of blood volume requirements defined by institutional review boards (IRBs). A commonly overlooked flaw in sampling strategy involves study protocol requirements that call for blood volumes exceeding the maximum allowable amounts for a given age or size of a child. Although there is an inherent degree of variability among IRBs, most stipulate that no more than 3% of the estimated circulating blood volume may be removed from a child, for research and clinical purposes combined, over a specified period (typically 2–8 weeks).3, 5 This can be problematic and particularly restrictive for investigations in preterm neonates, where typical total maximum allowable volumes amount to < 5 mL (Table 1).5
Table 1.
Average reference weights and volumes for children representing various age groups
| Age | Weight (kg) | Normal daily fluid requirement (mL) | Circulating blood volume (mL) | Max allowable sample volume over 2 weeks (mL) |
|---|---|---|---|---|
| Preterm neonate | 1.5 | 144 | 120 | 3.6 |
| Full‐term neonate | 4 | 384 | 144 | 9.6 |
| Infant/toddler (3 years) | 15 | 1,250 | 320 | 36 |
| Child (12 years) | 40 | 1,920 | 1,200 | 96 |
Knowing the stipulations of your IRB with respect to allowable blood volume is essential to successful protocol design. For example, some sampling constraints can be overcome with sparse sampling schemes, scavenged samples (i.e., those left over after medical procedures), or population PK approaches.3, 5 Similarly, careful evaluation of the minimum amount of biological sample necessary for analytical integrity and close communication with the sample processing laboratory can be incredibly valuable for reducing unnecessary sample collection and achieving a protocol design that is acceptable to the investigator, the participant, their parent/legal guardian, and the IRB.
Drug response
Physiologic alterations unique to the growing and developing child, as described above, may also influence PDs and the selection of appropriate PD markers (usually, drug safety and efficacy) for your investigation. Some PD markers, like minimum inhibitory concentration for antimicrobial agents, can be extrapolated from adult studies. However, other “well‐accepted” PD end points (e.g., exercise capacity measured by a 6‐minute walk test) are simply inappropriate for younger children, and pediatric investigators have to rely on alternate markers of PD. Some general guidance regarding appropriate biomarker selection and PD extrapolation can be gleaned from the US Food and Drug Administration (FDA)37 and the European Medicines Agency (EMA);38, 39 however, many knowledge gaps remain for children,40 and it is the responsibility of the pediatric investigator to consider natural variability in PD end points across the pediatric age range. Cutoffs for the upper limit of normal for commonly used screening laboratories (e.g., hemoglobin and hematocrit), hepatic safety parameters (e.g., aminotransferases and alkaline phosphatase), and biomarkers of inflammation (e.g., erythrocyte sedimentation rate and C‐reactive protein), for example, all vary with the age of the child; therefore, age‐corrected ranges should be used in the context of a pediatric trial.
A popular strategy for measuring drug response that facilitates PD comparison and extrapolation across studies is study end‐point parameterization, in which markers of response (e.g., disease severity) are turned into numeric scores; however, few existing scores are validated for children.40 Investigators must remain cognizant of this shortcoming and, at times, be prepared to take on investigation to validate a particular assessment tool to demonstrate its utility for pediatric research. In order to be useful, assessment tools, especially ones that rely on self‐report, need to be age appropriate for nonverbal and preschool children if these younger children are included in the targeted study population. Similarly, monitoring devices for assessing PD (e.g., blood pressure cuffs for an antihypertensive trial, otoscope tips for an otitis media trial) must be age appropriate and size appropriate for the pediatric population under investigation.
Study Population
To some extent, the study population you choose will be guided by the nature of the drug being studied and the target medical condition to which it applies. However, there are nuanced considerations that need to be kept in mind when designing a pediatric trial. For example, will study cohort selection be guided by well‐accepted, albeit arbitrary, age ranges (e.g., neonate: 0–1 month; infant: 1 month–2 years; child: 2–12 years; adolescent: 12–18 years) that may have little bearing on the developmental trajectory of relevant disposition/response pathways? When relying on these categories, trialists should consider that physiologic differences within individual age categories can still contribute substantially to variability in study results. Neonates, for example, demonstrate striking differences in organ maturation depending on their gestational age.6 Organ function in this population is also affected by prenatal/perinatal events (e.g., prenatal drug exposure, asphyxia at birth), pathophysiologic conditions, and postnatal events (e.g., concomitant treatments). Similarly, variability in exposure–response relationships within the adolescent population is observed as a consequence of body composition, pubertal status, and circulating hormone profiles.6 Failure to account for the extent of intersubject variability, contributed by these factors, may result in study sample size that compromises the statistical power of the investigation, and some investigators may choose to base study cohorts on maturity ratings, like Tanner Staging, rather than age for this reason.
Apart from the variability contributed by growth and development to individual age groups, consideration should also be given to differences in the prevalence and/or etiology of disease across the range of cohorts intended for the protocol. For example, a study designed to assess the influence of age on the disposition of an oral antihypertensive agent may be difficult to perform, given that essential hypertension rarely exists below the period of adolescence and the incidence of secondary hypertension, without significant comorbidity, is extremely low. Failure to consider these age‐related differences can negatively impact overall enrollment and the balance of enrollment across the various age cohorts. Worse, the study may be so slow to enroll that it exceeds planned time frames, adds costs, or terminates prior to completion. Familiarity with the population you are attempting to engage in a critical trial, including their usual patterns of care, is essential to the success of the trial.
Recruitment and enrollment
In a recent review of pediatric clinical trials registered on ClinicalTrials.gov, 19% were terminated early, largely because of difficulty with patient accrual.4 The timing of your study is extremely relevant to its success, with respect to enrollment and retention of pediatric participants. For example, a PK study that requires more than a 4‐hour sampling period will be very slow to enroll during the academic year, as parents/children are unwilling to miss school. Conducting such studies during the summer months or during school breaks, when children are already out of school, is incredibly helpful, in our experience. Similarly, being prepared to schedule study visits on holidays and weekends greatly improves pediatric enrollment, as parents are, generally, not challenged by the demands of work or providing child care.
Thoughtful evaluation of research protocol fasting and resting requirements is equally important to optimizing study enrollment by minimizing the disruption to a child's/parent's daily schedule. In our experience, studies that require extensive fasting or resting (e.g., > 2–4 hours) are best conducted in the morning, or after nap time, depending on the child's age, when children are naturally fasted and have not yet had a chance to exert themselves with play or other physical activity. Restricting intake of certain foods (e.g., caffeine‐containing soft drinks, chocolate, milk, and fruit juices) for prolonged periods of time (e.g., days) prior to the study visit can present significant challenges during the school year, when children are receiving school meals, and it may not be feasible, or practical, for parents to modify their child's diet to accommodate a research requirement.
We experience these “expected” pediatric challenges when engaging healthy children; however, studies that target enrollment of otherwise healthy children are generally limited to medications from which the child may benefit, or be reasonably exposed to, during the course of his/her normal “healthy” childhood (e.g., antibiotics, cough/cold products, etc.). In reality, most investigations are looking to identify and enroll children with specific disease processes. For children with diabetes, attention deficit/hyperactivity disorder (ADHD), or inflammatory bowel disease, investigators need to consider the impact of disease on the participants' and the family's daily living and design a protocol that is minimally disruptive to their quality of life.
When these same patients are participating in your study at a clinical research unit, investigators must consider whether the facility is equipped with the necessary supplies to make the study visit a successful and enjoyable experience for the child. A diabetes trial that requires excessively frequent finger sticks (we have seen > 20 over an 8‐hour period proposed in one investigation) will not be pleasant for the participant. Similarly, confinement of a patient with ADHD to a study unit that does not have a sufficient array of activities to occupy the child will have even the most experienced research team frazzled by day's end. In the example of ADHD, investigators should have a plan in place with the research team, the child life department, volunteer services, or the family to ensure the availability of age‐specific and gender‐specific opportunities that the child will find engaging. Providing this extra service, and addressing it at the time of recruitment, may help patients and families decide whether or not they would like to participate in the trial. A brief tour of the research unit at the time of study recruitment can be very reassuring for a child/family, as is an introduction to the key members of the study team who will be conducting the study visit.
Approaching potential study participants as a team with both the principal investigator and the research coordinator present can be to the study team's advantage. Generally, the principal investigator can better address the scientific focus of the investigation, whereas the coordinator can speak best to the actual breakdown of the study visit. Explaining study procedures in terms age appropriate and understandable for the child as well as the parent is essential for their engagement in research. We find the use of props for demonstration of common research procedures, such as i.v. catheter starts, tremendously helpful. We also always remind participants that once the i.v. goes in, the needle does not stay in them (a common pediatric fear); rather, what remains is “a bendy, plastic straw,” which we always show to them and let them examine. The number of fears we have alleviated with this simple technique is innumerable.
Research Ethics
Historically, there has been some reluctance to involve children in clinical trials due to fear of exposing children to harm from previously untested, equivocal treatments;3 however, tragic examples of what can happen when treatments not investigated in children are prescribed to children (e.g., gray baby syndrome from chloramphenicol41) highlight the importance of including children in clinical trials, especially when children comprise a portion of the targeted treatment population. In 1989, the right of children to enjoy the highest attainable standard of health, including the right to have research evidence for treatments commonly used in children, was formally recognized by the United Nations Convention on the Right of the Child.3
Informed consent and assent
Similar to other types of human subject research, pediatric research is protected by The Nuremberg Code and requires informed consent for research participation.3, 42, 43 However, informed consent for participation in pediatric trials is more complex than adult trials because informed consent is granted by proxy, from a parent or legal guardian responsible for protecting the welfare of the child, in accordance with the Declaration of Helsinki.3, 42, 43 In the United States, pediatric research involving minimal risk or direct potential benefit to the child requires signature from one parent/guardian, whereas all other risk categories of pediatric research, as determined by the IRB, require signatures from both parents/guardians.42 Specifically, Subpart D of the Common Rule (Protection of Human Subjects, 45 Code of Federal Regulations (CFR) 46) stipulates that studies involving greater than minimal risk and no direct benefit to the child are generally not allowed, unless they (i) are likely to yield generalizable knowledge about the participant's condition or (ii) present an opportunity to understand, prevent, or alleviate a serious problem affecting the health/welfare of children;44 however, interpretation of these stipulations resides with the IRB.
In addition to parental/guardian permission (i.e., research consent), informed assent (or dissent) to participate in research also needs to be obtained from the child.42 Barring any special circumstances regarding the child's cognitive abilities, 7 years of age is generally considered the age of assent by most institutions.42
As with any human subject research, all efforts should be made to minimize the risks to the pediatric participant. When obtaining informed consent/assent for pediatric research, it is important to remember that research risks constitute any and all harms, discomforts, indignities, embarrassments, and potential breaches of privacy/confidentiality associated with research, and this must be explained not only to the parents/guardians but also to children of assent age, using age‐appropriate terminology that they can understand. In our experience, illustrated research consent/assent forms are very useful for engaging participants and can be very helpful to facilitate age‐appropriate dialogue. Notably, parents/guardians also see the value of creatively modified consent forms, and, when thoughtfully constructed, IRBs are very willing to consider abbreviated‐illustrated documents in lieu of longer text‐based forms.45
Payment for participation
Payment for participation in pediatric research is allowed in the United States and parts of Europe, although different IRBs may have different stipulations with respect to payment type (i.e., reimbursement, compensation, incentive, or appreciation46) and payment value. In our experience, to avoid undue influence while still demonstrating appreciation for study engagement, the payment type and value ought to reflect the effort and commitment required on the part of the study participant first and the parent/guardian second.
Monitoring and safety
When it comes to safety and data monitoring, studies conducted in children are subject to the same regulatory requirements as adult investigations.5, 42, 47 Drug safety assessments are paramount to pediatric trials, as even the complete efficacy extrapolation approach endorsed by the FDA (option C in the FDA Pediatric Study Decision Tree from the Office of Pediatric Therapeutics; Figure 3) requires the collection of safety data in children.48 At a minimum, all trials should have a clear data safety and monitoring plan, which can be augmented by a formal data safety and monitoring board that convenes at regular intervals to assess data integrity, protocol deviations/violations, subject withdrawals, study hold or stopping rules, and any other applicable issues.
Figure 3.
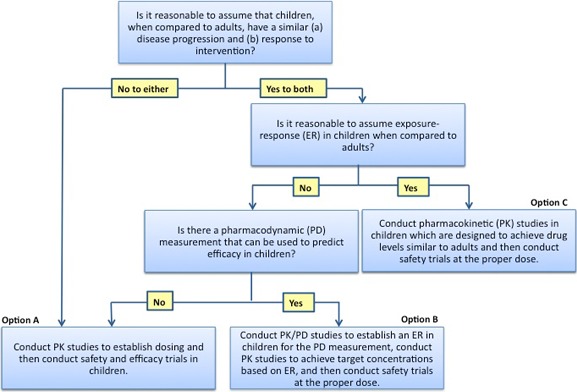
Extrapolation in pediatric drug development, decision tree from the US Food and Drug Administration; image borrowed from https://www.fda.gov/ScienceResearch/SpecialTopics/PediatricTherapeuticsResearch/ucm106614.htm,47 public domain.
Research Infrastructure
The success of a clinical trial not only depends on thoughtful and age‐appropriate study design but also on accessibility to appropriate research facilities to conduct the research. The “right place” for conducting pediatric research encompasses considerations that include the physical space, the professional staff, and a research infrastructure (e.g., IRB, human subjects protection program, professional research education/training) that has the wellbeing of the child at the center of its mission focus and a dedication to creating a safe and positive learning environment at the forefront of its operations.49 Institutions driven by a mission that embraces the core principles of safety, teamwork, compassion, and excellence create the potential for an ideal environment for developing and supporting a successful pediatric clinical trial program. Although, arguably, these core facets are also required of institutions where adult trials are conducted, it is their orientation toward the unique challenges (and opportunities) of pediatrics, encompassing care for infants, children, and adolescents, that makes them distinct.
For example, it is essential that the members of the IRB and professionals who are part of the research administrative infrastructure (e.g., attorneys, grant accountants, pre‐award and post‐award grant/contract specialists) have appropriate knowledge and understanding of the unique federal regulations that impact the study of drugs and devices in infants and children. Clinical research professionals (e.g., investigators, research coordinators, nurses, and laboratory technicians) must understand both the regulatory requirements unique to pediatrics and how the intersection of human development and disease (both expression and course) influence the ability to conduct a clinical trial with minimal disruption to the child's medical and psychosocial needs or the family's daily routine. Finally, the physical environment of the facility where research is to be conducted is critical to the success of the investigation. As recently described, the look, feel, and function of the research facility must be “kid savvy” by creating an environment that is inviting and safe, not threatening, for pediatric patients and their families.50 These accommodating and reassuring features should extend to specialized hospital research units that are designed primarily for conducting nontherapeutic research in subpopulations of infants and children who have a specific medical condition.
Utilizing established networks and research organizations
Established pediatric networks can offer solutions to many of the challenges of conducting pediatric clinical trials. One example of such an organization, well familiar to the authors, is the Eunice Kennedy Shriver National Institute of Child Health and Human Development–funded Pediatric Trials Network (PTN).
Established under the auspices of the Best Pharmaceuticals for Children Act in 2010, the PTN is an alliance of research sites, therapeutic area experts, and thought leaders in key facets of pediatric trials51 that offer research support to new and established investigators. Similar to other pediatric research networks, the PTN strives for excellence in five key components of clinical trials: (i) protocol design and development; (ii) site management and education; (iii) study implementation and execution; (iv) data analysis and interpretation; and (v) regulatory affairs.
Protocol design and development in the PTN is a multidisciplinary and interactive process involving all network stakeholders. By promoting collaboration between clinical therapeutic area experts and thought leaders in clinical trial methodology, innovative protocols that efficiently and safely fill critical knowledge gaps are designed. Examples of protocols designed through collaboration with the PTN include the largest pediatric opportunistic multidrug PK and safety study (POP01), a multidrug study of maternal–infant drug transmission through breast milk (BMS01), and the first ever long‐term safety study of antipsychotics in children and adolescents (LAP01).
Site management in the PTN is performed in collaboration with the Data Coordinating Center, which offers education and support to participating study sites and helps investigators identify appropriate study sites for their investigation. Site selection is a multistep process, involving network‐wide and study‐specific questionnaires, individual site contacts, and review of prior performance metrics. Sites are trained and supported throughout the study by actively engaged study investigators and operational personnel. High‐enrolling sites are routinely rewarded with coauthorship on study‐derived publications, and study data are made publicly available for secondary analysis to investigators after study completion.
Study implementation and execution is overseen by a core team of faculty and operational staff. Such teams support sites through educational materials and manuals of study procedures, scheduled study calls, and ad hoc consultation for study‐related issues. Monitoring and safety, facilitated by the team, is risk based and includes remote and in‐person site visits, conducted by the Data Coordinating Center.
Data analysis and interpretation is facilitated by experts in biostatistics, PK/PD modeling, clinical pharmacology, and other fields at multiple academic institutions in the United States and worldwide, brought together under the network's umbrella. After completion of the primary end‐point analysis, trial data are quickly and easily made accessible to the public to promote the conduct of secondary analyses.
Regulatory rigor is ensured and maintained by the network, with investigators and regulatory scientists involved in all key phases of the study. For example, all PTN studies are conducted under an FDA Investigational New Drug application. Study databases are 21 CFR compliant and data analysis facilities, including biological laboratories, are routinely reviewed by Quality Assurance/Quality Control teams, facilitating submission of study results to regulatory bodies (e.g., the FDA) to support potential label change negotiations with drug label holders. Thus, new and established investigators are offered an opportunity to learn relevant regulatory elements of conducting pediatric trials, with appropriate oversight and guidance from the network.
Network leadership is committed to training the next generation of pediatric clinical trialists by allowing junior scientists from across network sites to support and/or lead PTN trials, under the mentorship of a senior trialist. Scientists are encouraged to submit study proposals via a brief and easy‐to‐complete sheet, available on the network's website ( www.pediatrictrials.org), which also provides a rapid online survey to encourage participation of new study sites. Although the authors are most familiar with the PTN, similar qualified research networks and organizations are available as valuable resources to investigators around the globe (Table 2).
Table 2.
Select examples of resources available to investigators through established research networks and organizations across the globe
EMA, European Medicines Agency; FDA, US Food and Drug Administration.
Helpful Hints
Taking under advisement the practical considerations, combined research experiences, and support systems outlined in this tutorial, you should feel empowered to take on a pediatric clinical trial at your institution, but before you enroll your first research participant, pause and implement a mock trial run.
Mock trial run
The dogma in research coordinator training clearly states that the data collected on the first study participant will almost certainly be discarded. To avoid this common pitfall, always perform a mock run of the study visit, from consent/assent to post‐study follow‐up telephone call. A mock run of your investigation, in the actual physical space where the investigation will take place, allows you to assess the study flow, enabling you to identify unforeseen gaps in study logistics/procedures before you enroll your first patient. We have captured, and corrected, oversights in case report forms, feasibility of time‐sensitive study steps (e.g., travel time for getting biologic specimens to and from a processing laboratory), availability of equipment (e.g., age‐appropriate blood pressure cuffs, centrifuges, and examination rooms), and/or ancillary staff (e.g., pharmacy dispensing services), particularly relevant if research facilities are shared among investigators or compete for resources with clinical services.
In addition, a trial run ensures consistency in informed consent/assent language used by different members of the study team and confirms that research procedures do not interfere with clinic flow. If any research procedures (e.g., patient recruitment in a busy clinic, timing of the research consent/assent, utilization of clinic examination rooms to confirm study inclusion/exclusion criteria, etc.), even tangentially, encroach on patient care, we recommend an open discussion with the clinical team to look for compromises and solutions. Performing this exercise of a mock trial allows you to make any necessary changes to the protocol, or the manual of operations, before you start collecting precious real data for your investigation.
We also recommend allowing a small time buffer, after the enrollment of your first research participant, to adjust any research procedures (if needed) not captured by the trial run. This approach negates the need for rescheduling subsequent participants if any unforeseen circumstances are encountered during the research experience of your first study participant.
Research engagement
Whether you are implementing your first or your 100th clinical trial, to build a culture of research at your institution, engage your colleagues and your study participants in the research you are conducting. Identification of potential study participants by ancillary staff, nurses, and clinicians increases if they understand the scientific merit of your clinical trial and its value to children. Do not underestimate the altruism of children and families when they fully understand the future implications (both risks and benefits) of the knowledge they help generate through voluntary participation in research. Finally, make every effort to periodically update your colleagues, research staff, and research participants (to the extent permitted by the study protocol) regarding your study progress and interim data analysis. There is nothing more rewarding than seeing the smile on a child's face when they hear that the research study they participated in was published in a scientific journal, cited on social media, or helped to make a difference for another child. We have successfully used printouts of our featured studies from the PTN website, for example, to update research participants on their contribution to science when they return to the clinic for routine care or to our research unit for another clinical trial. Sharing the outcomes of your clinical trial with the community creates a culture of trust and mutual respect for future research participation at your institution.
Conclusions
In summary, extensive prestudy planning, designed to establish a thorough understanding of the developmental differences in physiology that differentiate the stages of human growth and development from birth to adulthood, their impact on PK and PD, and your study team's ability to accommodate these dynamic changes, is key to successfully implementing a pediatric clinical trial at your institution. Although the study planning process may seem daunting, resources are available to help you. Guidelines are available from regulatory agencies (e.g., the FDA and EMA) regarding PK/PD extrapolations, support from established networks (e.g., PTN) for financial and practical considerations, and education from seasoned trialists and coordinators to help you avoid common pitfalls and fatal mistakes (Figure 4). Equally important are solicitations of feedback and input from your local IRB, clinical colleagues who are familiar with the medical and psychosocial nuances of the pediatric population you are targeting for study participation, and the patient population (children and parents) you are trying to engage in research.
Figure 4.

Checklist of key elements for successful pediatric clinical trial design and execution; pharmacokinetics (PKs), pharmacodynamics (PDs). CFR, Code of Federal Regulations; DME, drug metabolizing enzyme; FDA, US Food and Drug Administration; IRB, institutional review board; PTN, Pediatric Trials Network.
Funding
No funding was received for this work.
Conflict of Interest
All authors, currently or in the past, have received funding support from the Pediatric Trials Network (PTN; www.pediatrictrials.org), funded by the Eunice Kennedy Shriver National Institute of Child Health and Human Development. As an Associate Editor for Clinical and Translational Science, Valentina Shakhnovich was not involved in the review or decision process for this paper.
References
- 1. Tolbert, J. , Golman, J. , Kauffmann, R. & Abdel‐Rahman, S.M. The creating hope act: what is old is new again. Pediatr. Health Med. Ther. 5, 49–57 (2014). [Google Scholar]
- 2. Connor, E.M. et al Meeting the demand for pediatric clinical trials. Sci. Transl. Med. 6, 227fs11 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Joseph, P.D. , Craig, J.C. & Caldwell, P.H.Y. Clinical trials in children. Br. J. Clin. Pharmacol. 79, 357–369 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pica, N. & Bourgeois, F. Discontinuation and nonpublication of randomized clinical trials conducted in children. Pediatrics 138, e20160223 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Abdel‐Rahman, S.M. , Reed, M.D. , Wells, T.G. & Kearns, G.L. Considerations in the rational design and conduct of phase I/II pediatric clinical trials: avoiding the problems and pitfalls. Clin. Pharm. Ther. 81, 483–494 (2007). [DOI] [PubMed] [Google Scholar]
- 6. Kearns, G.L. , Abdel‐Rahman, S.M. , Alander, S.W. , Blowey, D.L. , Leeder, J.S. & Kauffman, R.E. Developmental pharmacology‐drug disposition, action, and therapy in infants and children. N. Engl. J. Med. 349, 1157–1167 (2003). [DOI] [PubMed] [Google Scholar]
- 7. Shakhnovich, V. & Abdel‐Rahman, S.M. General considerations for pediatric oral drug formulations In Pediatric Formulations: A Roadmap (eds. Bar‐Shalom D. & Rose K.) 489–504 (American Association of Pharmaceutical Scientists/Springer, Switzerland, 2014). [Google Scholar]
- 8. Kearns, G.L. et al Cisapride disposition in neonates and infants: in vivo reflection of cytochrome P450 3A4 ontogeny. Clin. Pharmacol. Ther. 74, 312–325 (2003). [DOI] [PubMed] [Google Scholar]
- 9. Shakhnovich, V. et al Obese children require lower doses of pantoprazole than non‐obese peers to achieve equal systemic drug exposures. J. Pediatr. 193, 102–108 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Eidelman, C. & Abdel‐Rahman, S.M. Pharmacokinetic considerations when prescribing in children. Int. J. Pharmacokinet. 1, 69–80 (2016). [Google Scholar]
- 11. Lawless, H. Sensory development in children: research in taste and olfaction. J. Am. Diet. Assoc. 85, 577–582 (1985). [PubMed] [Google Scholar]
- 12. Luo, J. et al The pharmacokinetic exposure to fexofenadine is volume‐dependently reduced in healthy subjects following oral administration with apple juice. Clin. Transl. Sci. 9, 201–206 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Huang, N.N. & High, R.H. Comparison of serum levels following the administration of oral and parenteral preparations of penicillin to infants and children of various age groups. J. Pediatr. 42, 657–658 (1953). [DOI] [PubMed] [Google Scholar]
- 14. Mitra, A. & Kesisoglou, F. Impaired drug absorption due to high stomach pH: a review of strategies for mitigation of such effect to enable pharmaceutical product development. Mol. Pharm. 10, 3970–3979 (2013). [DOI] [PubMed] [Google Scholar]
- 15. Gupta, M. & Brans, Y.W. Gastric retention in neonates. Pediatrics 62, 26–29 (1978). [PubMed] [Google Scholar]
- 16. Abdel‐Rahman, S.M. & Kearns, G.L. Single oral dose escalation pharmacokinetics of pleconaril (VP 63843) capsules in adults. J. Clin. Pharmacol. 39, 613–618 (1999). [DOI] [PubMed] [Google Scholar]
- 17. Kearns, G.L. et al Single‐dose pharmacokinetics of a pleconaril in neonates. Pediatr. Infect. Dis. J. 19, 833–839 (2000). [DOI] [PubMed] [Google Scholar]
- 18. Poley, J.R. , Dower, J.C. , Owen, C.A. & Stickler, G.B. Bile acids in infants and children. J. Lab. Clin. Med. 63, 838–846 (1964). [PubMed] [Google Scholar]
- 19. Suchy, F.J. , Balistreri, W.F. , Heubi, J.E. , Searcy, J.E. & Levin, R.S. Physiologic cholestasis: elevation of the primary serum bile acid concentrations in normal infants. Gastroenterology 80, 1037–1041 (1981). [PubMed] [Google Scholar]
- 20. Filer, L.J. , Mattson, F.H. & Fomon, S.J. Triglyceride configuration and fat absorption by the human infant. J. Nutr. 99, 293–298 (1969). [DOI] [PubMed] [Google Scholar]
- 21. Lee, P.C. , Borysewicz, R. , Struve, M. , Raab, K. & Werlin, S.L. Development of lipolytic activity in gastric aspirates from premature infants. J. Pediatr. Gastroenterol. Nutr. 17, 291–297 (1993). [DOI] [PubMed] [Google Scholar]
- 22. Hamosh, M. et al Gastric lipolysis and fat absorption in preterm infants: effect of medium‐chain triglyceride or long‐chain triglyceride‐containing formulas. Pediatrics 83, 86–92 (1989). [PubMed] [Google Scholar]
- 23. Johnson, T.N. , Tanner, M.S. , Taylor, C.J. & Tucker, G.T. Enterocytic CYP3A4 in a paediatric population: developmental changes and the effect of coeliac disease and cystic fibrosis. Br. J. Clin. Pharmacol. 51, 451–460 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gibbs, J.P. , Liacouras, C.A. , Baldassano, R.N. & Slattery, J.T. Up‐regulation of glutathione S‐transferase activity in enterocytes of young children. Drug Metab. Dispos. 27, 1466–1469 (1999). [PubMed] [Google Scholar]
- 25. Fakhoury, M. et al Localization and mRNA expression of CYP3A and P‐glycoprotein in human duodenum as a function of age. Drug Metab. Dispos. 33, 1603–1607 (2005). [DOI] [PubMed] [Google Scholar]
- 26. Shakhnovich, V. et al Decreased pregnane X receptor expression in children with active Crohn's disease. Drug Metab. Dispos. 44, 1066–1069 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Brouwer, K.L. et al Human ontogeny of drug transporters: review and recommendations of the Pediatric Transporter Working Group. Clin. Pharm. Ther. 98, 266–287 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ginsberg, G. et al Pediatric pharmacokinetic data: implications for environmental risk assessment for children. Pediatrics 113, 973–983 (2004). [PubMed] [Google Scholar]
- 29. Mahmood, I. Developmental pharmacology: impact on pharmacokinetics and PD of drugs In Pediatric Pharmacology and Pharmacokinetics (ed.Mahmood I.) 68–107 (Pine House Publishers, Rockville, MD, 2008). [Google Scholar]
- 30. Strachunsky, L.S. , Nazarov, A.D. , Firsov, A.A. & Petrachenkova, N.A. Age dependence of erythromycin rectal bioavailability in children. Eur. J. Drug Metab. Pharmacokinet. 3, 321–323 (1991). [PubMed] [Google Scholar]
- 31. Hines, R.H. The ontogeny of drug metabolism enzymes and implications for adverse drug events. Pharmacol. Ther. 118, 250–267 (2008). [DOI] [PubMed] [Google Scholar]
- 32. Kearns, G.L. et al Single dose pharmacokinetics of linezolid in infants and children. Pediatr. Infect. Dis. J. 19, 1178–1184 (2000). [DOI] [PubMed] [Google Scholar]
- 33. Siegel, M. Gastric emptying time in premature and compromised infants. J. Pediatr. Gastroenterol. Nutr. 2(suppl. 1), S136–S140 (1983). [DOI] [PubMed] [Google Scholar]
- 34. Bharucha, A.E. et al Delayed gastric emptying is associated with early and long‐term hyperglycemia in type 1 diabetes mellitus. Gastroenterology 149, 330–339 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zani‐Ruttenstock, E. , Zani, A. , Paul, A. & De‐Ajayi, N. Interstitial cells of Cajal are decreased in patients with gastroschisis associated intestinal dysmotility. J. Pediatr. Surg. 10, 750–754 (2015). [DOI] [PubMed] [Google Scholar]
- 36. Feichter, S. , Meier‐Ruge, W.A. & Bruder, E. The histopathology of gastrointestinal motility disorders in children. Semin. Pediatr. Surg. 18, 206–211 (2009). [DOI] [PubMed] [Google Scholar]
- 37. Burckart, G.J. General pharmacology considerations for drugs and biological products. US Food and Drug Administration; < http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm425885.pdf> (2014). Accessed May 2, 2018. [Google Scholar]
- 38. European Medicines Agency Committee for Medicinal Products for Human Use . Guideline on the investigation of medicinal products in the term and preterm neonate. European Medicines Agency; < http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003754.pdf> (2007). Accessed May 2, 2018. [Google Scholar]
- 39. European Medicines Agency Committee for Medicinal Products for Human Use . Guidelines on the role of pharmacokinetics in the development of medicinal products in the paediatric population. < http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003066.pdf> (2006). Accessed May 2, 2018.
- 40. Kelly, L.E. , Siha, Y. , Baker, C.I.S. , Standing, J.F. & Offringa, M. Useful pharmacodynamics endpoints in children: selection, measurement, and next steps. Pediatr. Res. 83, 1–9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Burns, L.E. , Hodgman, J.E. & Cass, A.B. Fatal circulatory collapse in premature infants receiving chloramphenicol. N. Engl. J. Med. 261, 1318–1321 (1959). [DOI] [PubMed] [Google Scholar]
- 42. Bavdekar, S.B. Pediatric clinical trials. Perspect. Clin. Res. 4, 89–99 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Caldwell, P.H.Y. , Murphy, S.B. , Butow, P.N. & Craig, J.C. Clinical trials in children. Lancet 364, 803–811 (2004). [DOI] [PubMed] [Google Scholar]
- 44. U.S. Department of Health and Human Services . Additional Protections for Children Involved as Subjects in Research. 2002. 45 CFR 46.406 and 46.407 [PubMed]
- 45. Abdel‐Rahman, S.M. Evaluating the effectiveness of an illustrated permission/assent form. J. Immigr. Minor. Health 17, 1504–1508 (2015). [DOI] [PubMed] [Google Scholar]
- 46. Wendler, D. , Rackoff, J.E. , Emanuel, E.J. & Grady, C. The ethics of paying for children's participation in research. J. Pediatr. 141, 166–171 (2002). [DOI] [PubMed] [Google Scholar]
- 47. US Food and Drug Administration . Pediatric science and research activities. < https://www.fda.gov/ScienceResearch/SpecialTopics/PediatricTherapeuticsResearch/ucm106614.htm> (2018). Accessed January 6, 2019.
- 48. Glasser, S.P. & Howard, G. Clinical trial design issues: at least 10 things you should look for in clinical trials. J. Clin. Pharmacol. 46, 1106–1115 (2006). [DOI] [PubMed] [Google Scholar]
- 49. Britto, M.T. et al Using network organizational architecture to support the development of learning healthcare systems. Br. Med. J. Qual. Saf. 27, 937–946 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Prescott, K. Designing for health: interior design's important role in health care In Arkansas Hospital Association Publications (ed. Vowell, Inc. produces Arkansas Hospitals on behalf of the Arkansas Hospital Association ) 22–24 (Arkansas Hospital Association, Little Rock, AR, 2018). [Google Scholar]
- 51. Zajicek, A. The National Institutes of Health and the Best Pharmaceuticals for Children Act. Pediatr. Drugs 11, 45–47 (2009). [DOI] [PubMed] [Google Scholar]
- 52. Eunice Kennedy Shriver National Institute of Child Health and Human Development . Pediatric Trials Network. < www.pediatrictrials.org> (2018). Accessed January 6, 2019.
- 53. Critical Paths Institute . Pediatric Trials Consortium. < https://c-path.org/programs/ptc/> (2018). Accessed January 6, 2019.
- 54. US Food and Drug Administration . Pediatrics < https://www.fda.gov/ScienceResearch/SpecialTopics/PediatricTherapeuticsResearch/default.htm> (2018). Accessed January 6, 2019.
- 55. US Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER) . General clinical pharmacology considerations for pediatric studies for drugs and biological products. < https://www.fda.gov/downloads/drugs/guidances/ucm425885.pdf> (2014). Accessed January 6, 2019.
- 56. US Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER) Center for Biologics Evaluation and Research (CBER) . Pediatric study plans: content of and process for submitting initial pediatric study plans and amended initial pediatric study plans guidance for industry. < https://www.fda.gov/downloads/drugs/guidances/ucm360507.pdf> (2016). Accessed January 6, 2019.
- 57. European Medicines Agency . Human regulatory: pediatric clinical trials. < https://www.ema.europa.eu/en/human-regulatory/research-development/paediatric-medicines/paediatric-clinical-trials> (2018). Accessed January 6, 2019.
- 58. Canadian Institutes of Health Research . Institute of Human Development, Child and Youth Health. < http://www.cihr-irsc.gc.ca> (2018). Accessed January 6, 2019.
- 59. World Health Organization . International Clinical Trials Registry Platform: clinical trials in children. < https://www.who.int/ictrp/child/en/> (2018). Accessed January 6, 2019.
- 60. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) . ICH Harmonised Tripartite Guideline: clinical investigation of medicinal products in the pediatric population (E11). < https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E11/Step4/E11_Guideline.pdf> (2000). Accessed January 6, 2019.
- 61. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) . ICH Harmonised Guideline: addendum to ICH E11 clinical investigation of medicinal products in the pediatric population (E11R1). < https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E11/E11-R1EWG_Step4_Addendum_2017_0818.pdf> (2017). Accessed January 6, 2019.