

Abstract
Inhibin is a heterodimeric TGF-β family ligand that is expressed in many cancers and is a selective biomarker for ovarian cancers, however its tumor-specific functions remain unknown. Here we demonstrate that the α subunit of Inhibin (INHA), which is critical for the functionality of dimeric Inhibin A/B, correlates with microvessel density (MVD) in human ovarian tissues and xenografts and is predictive of poor clinical outcomes in multiple cancers. We demonstrate that Inhibin regulated angiogenesis is necessary for metastasis. While Inhibin had no direct impact on tumor cell signaling, both tumor cell-derived and recombinant Inhibin elicit a strong paracrine response from endothelial cells by triggering SMAD1/5 activation and angiogenesis in vitro and in vivo. Inhibin-induced angiogenesis was abrogated via anti-Inhibinα antibodies. The endothelial-specific TGF-β receptor complex comprising ALK1 and endoglin were crucial mediators of Inhibin signaling, offering a molecular mechanism for Inhibin-mediated angiogenesis. These results are the first to define a role for Inhibin in tumor metastasis and vascularization and offer an antibody-based approach for targeting Inhibin therapeutically
Keywords: Inhibin, Endoglin, ALK1, Angiogenesis, Ovarian cancer, Paracrine
Introduction
Inhibition of angiogenesis - the growth of new blood vessels from pre-existing vasculature is a clinically validated anti-cancer strategy for numerous tumor types. However, while the VEGF/VEGF receptor (VEGFR) signaling axis is widely recognized as the principal target of this therapeutic approach, current FDA-approved anti-VEGF drugs have demonstrated sub-optimal responses in the clinic. In many cases, including in ovarian cancers (OVCA), patient relapse, acquired resistance and cytotoxicity is commonly observed following anti-VEGF therapy. The identification of new vascular targets with potentially fewer adverse effects is therefore critical for improving therapeutic outcomes.
TGF-β family members, particularly TGF-β1 and BMP9, are essential regulators of angiogenesis (1). Targeting these for anti-angiogenic therapy remains a formidable challenge due to their non-endothelial pleiotropic functions. Here, we focus on the unique TGF-β family member Inhibin, an endocrine hormone that sharply declines at the onset of menopause in healthy normal women and remains low (2) unlike other TGF-β family members and prototypical angiogenic factors like VEGF. Importantly, when Inhibin becomes elevated in postmenopausal women, this elevation becomes a diagnostic and prognostic marker for OVCA where along with CA125 detects 95% of ovarian tumors with 95% specificity (3,4).
Inhibin is a heterodimeric member of the TGF-β family composed of an alpha (α) (coded by INHA) and beta (β) subunit (INHBA or INHBB). Combinations of these subunits give rise to either Inhibin A (αβA) or Inhibin B (αβB) (5). While Inhibin null mice (INHA-/-) are viable, they present with alterations to the ovarian vasculature and result in spontaneous gonadal tumors (6). In humans however, Inhibin levels are elevated in multiple cancer types including ovarian, prostate, adrenal, stomach and pancreatic cancers with indications for a role for Inhibin in prostate cancer metastasis (7–11). Despite these findings, the functional consequences of elevated tumor-derived Inhibin have yet to be determined.
Several Inhibin binding proteins/receptors were previously reported (12). However unlike other TGF-β members, whose signal transduction mechanisms have been well studied, the mechanisms of Inhibin signaling remain largely unclear. The best-characterized Inhibin binding protein is the epithelial cell surface TGF-β co-receptor TβRIII/betaglycan (13). Inhibin binding to betaglycan fails to activate any discernable downstream pathways in epithelial cells. Others and we previously demonstrated, several tumor suppressor functions for betaglycan, which is lost in the majority of human cancers (14), but little is known about the impact of elevated Inhibin on non-epithelial cells that do not express significant betaglycan.
Given the urgent need to identify new anti-angiogenic pathways and targets to complement and improve existing therapies, we examined the potential role of Inhibin as a novel regulator of angiogenesis and metastasis. Importantly, we demonstrate Inhibin as an unexpected, clinically relevant, paracrine factor of tumor-induced angiogenesis and define the underlying mechanism of Inhibin action and therapeutic potential.
Materials and Methods
Cell Lines and Reagents:
Ovarian epithelial carcinoma cell lines were obtained either from Duke Gynecology/Oncology Bank (Durham, NC) and ATCC. Authentication was carried out at the University of Colorado (Denver, CO) sequencing facility. HMEC-1 (human dermal microvascular endothelial cells) from ATCC CRL-3243 and MEECs (murine embryonic endothelial cells) ENG+/+ and ENG-/- were as described previously (15). HUVEC (human umbilical vein endothelial cells) was purchased from Lonza, USA. HMEC-1s were grown as per ATCC instructions. Epithelial carcinoma cell lines A2780, HEY, IGROV, OVCA247, M41, OVCA3, OVCA4, OVCA420, OVCA429, OVCA448, SKOV3 and PA1 were cultured in RPMI-1640 (ATCC® 30–2001™) containing L-glutamine, 10% FBS and 100 U of penicillin-streptomycin. All cells lines were maintained at 37°C in a humidified incubator at 5% CO2, routinely checked for mycoplasma 3 times a year and experiments conducted within 3–6 passages depending on the cell line. Antibodies phospho-SMAD1/5 (#9516), phospho-SMAD2/3 (#8828S) and SMAD2/3 (#5678S) were from Cell Signaling Technology (Danvers, CA), SMAD1/5 (#ab75273) from Abcam, Cambridge, MA, USA. Mouse anti-HA antibody, Rabbit anti-HA antibody and mouse anti-Myc antibody were from invitrogen. Monoclonal antibodies to CD31 (#F8402) and Inhibin α (#ab47720) for IHC were purchased from Sigma-Aldrich and Abcam, respectively. Anti-INHA antibody (polyclonal #sc22048, Santa Cruz) and (monoclonal #sc365439, Santa Cruz) were used as indicated. ML347, Dorsomorphin and SB351432 were from Sigma-Aldrich, TRCN105 was a gift from TRACON pharmaceuticals (http://www.traconpharma.com/trc105.php). Inhibin A was from Sigma-Aldrich (# I9149) and R&D Systems (# 8506-AB). Lentiviral particles were generated at the COBRE Center for Targeted Therapeutics Core Facility at SC. For INHA knockdown, SKOV3 cells were infected shRNA lentivirus, selected in 2 μg/ml Puromycin and stable cell lines maintained in 1 μg/ml Puromycin. Transient DNA transfections of HMEC-1 and COS7 were performed using either Targetfect (#HUVEC-01) from Targeting systems (El Cajon, CA) or Lipofectamine 2000 (#11668019) from Life Technologies (Carlsbad, CA).
RNA isolation and Quantitative Polymerase Chain Reaction (qRT-PCR) analysis
Total RNAs was extracted using Trizol and chloroform. RNA was retro-transcribed using iScript™ Reverse Transcription Supermix (#1708841) and Advanced Universal SYBR Green Supermix (#1725271) from Bio-Rad (Hercules, CA). All expression data were normalized to those for RPL13A. qRT-PCR primer sequences are listed in Table 1.
Table 1.
qRT-PCR primer sequences
| Primers | Forward | Reverse |
|---|---|---|
| Human | ||
| RPL13A | AGATGGCGGAGGTGCAG | GGCCCAGCAGTACCTGTTTA |
| ENG | CGCCAACCACAACATGCAG | GCTCCACGAAGGATGCCAC |
| INHA | TTCCACTACTGTCATGGTGGT | AGTGCTGCGTGAGAAGGTTG |
| ALK1 | CATCGCCTCAGACATGACCTC | GTTTGCCCTGTGTACCGAAGA |
| ALK2 | GTGAAGGTCTCTCCTGCGGTA | GCCATCGTTGATGCTCAGTGA |
| ALK3 | TGAAATCAGACTCCGACCAGA | TGGCAAAGCAATGTCCATTAGTT |
| ALK4 | CTTCCCCCTTGTTGTCCTCC | TCTCACACGTGTAGTTGGCC |
| ALK5 | TCCCAAACAGATGGCAGAGC | ATCCGCAATGCTGTAAGCCT |
| ALK6 | ATGGAACTTGCTGTATTGCT | CAACTCGAGTGTTAGGTGGT |
| ALK7 | GATGTGACCGCCTCTGGATC | CCATCTTCCATGCCACACCT |
| Mouse | ||
| RPL13A | CAAGGTTGTTCGGCTGAAGC | GCTGTCACTGCCTGGTACTT |
| ENG | GCTGAAGACACTGACGACCA | AGCCTGACGGGAAACTGATG |
Plasmid Constructs and Stable Cell Lines:
The shRNA sequences for INHA were obtained either from Sigma-Aldrich or Dharmacon. ShINHA1 used in all the experiments was generated using TRCN0000063904: CCGGCCTCGGATGGAGGTTACTCTTCTCGAGAAGAGTAACCTCCATCCGAGGTTTTG in a pLKO1-Puromycin backbone. Scramble vector was used as control (non-targeted control).The second shRNA sequences from Dharmacon: shRNA V3SH11240–226731666: TTCAGTGCTACTCTGTGGC. shRNA for ALK1 was obtained from Sigma-Aldrich TRCN0000000356: CCGGCAGTCCAGAGAAGCCTAAAGTCTCGAGACTTTAGGCTTCTCTGGACTGTTTTT. Human endoglin shRNA was described previously (16).
ELISA:
The enzyme linked immunosorbent assay (ELISA) was from AnshLabs (#AL-134; Webster, TX) and was performed according to manufacturer’s instructions to quantitatively measure total Inhibin (Inhibin A, Inhibin B and Inhibinα (alpha)). Cells were serum starved for 24 h before CM was collected.
Co-patch immunofluorescence:
Antibody mediated immunofluorescence co-patching was performed as described previously (17,18). Lipofectamine HA and Myc tags at the cell surface were immunolabeled at 4 °C for 45 min using anti-HA and anti-Myc antibodies, to allow only surface labeling and to reduce internalization. Alexa 488 and Alexa 594 conjugated to anti-HA and anti-Myc were used as secondary antibodies respectively. Imaging was performed using an Olympus IX81 inverted microscope (Shinjuku, Tokyo, Japan). Images were exported to image J. The number of superimposed (red and green) yellow patches were counted manually on flat cell regions as described previously (17,18). >50 patches/cell were counted.
Endothelial capillary sprouting and tube formation assay:
Tube formation in HMEC-1, HUVEC and MEECs was performed as described previously (16,19) using endothelial cells plated between two layers of Matrigel except in the case of MEEC’s which were plated on a single layer of Matrigel. Briefly, 24 well plates were coated with 200 μl of Matrigel matrix (#354230, BD Biosciences). 30 min following the addition of Matrigel, HMEC-1 cells (70×103), HUVEC (50×103) or MEEC (50×103) were plated onto the matrix. For HMEC-1’s, a second layer of 200 μl Matrigel was added after 1 h of plating cells. After a 30 min incubation at 37 °C, 300 μl of growth medium was growth factors (300 pM Inhibin A, TGF-β, BMP9, or Activin as indicated) or conditioned media collected from shControl or shINHA stable cell lines as indicated. 16 h later images were taken using an Olympus IX81 inverted microscope (4× magnification).
For all tube formation experiments, quantification presented represents the two commonly used angiogenesis parameters in vitro (20): Number of Meshes: that are defined as regions enclosed by segments (tubes delimited by two junctions) and/or closed polygons; Nodes/Branches: defined as individual junctions/branching points. The Angiogenesis Analyzer plugin in ImageJ was used for the analysis. For Spheroid-based sprout assays: Endothelial spheroids were prepared as previously reported (21). Briefly, 1X103 HMEC-1 cells were cultured in hanging drops of 25 μl medium containing 20% methocel and 80% culture medium, and allowed to aggregate as spheroids. After 24h, the spheroids were collected using and plated on 24-well plates coated with growth factor reduced Matrigel and treated as indicated. Sprouts were digitally imaged after the indicated times and the number and length of sprouts per spheroid quantitated. For all experiments, a minimum of two biological trials, with each trial containing three technical replicates were analyzed by counting a minimum of 3 fields/ technical replicate.
Study Approval:
All animal experimental protocols were performed in accordance with the Institutional Animal Care and Use Committee (IACUC) at the University of South Carolina under an approved Protocol (AUP 2329–101161-121916).
In vivo Matrigel plug assay:
Matrigel plug assays were carried out by using Matrigel (BD Biosciences, #354230) mixed with 100 pM Inhibin A or mixed with CM from shControl/shINHA SKOV3 in a ratio of 2:3 (total volume of 0.2 ml) and injected subcutaneously into the right flank of BALB/c female mice aged 5–6 weeks. n=3 mice per group were used (22,23) with each experiment conducted two independent times. Plugs were harvested 12 days after injection and hemoglobin content was determined according to Drabkin’s method.
Orthotopic xenografts:
Stable 5×106 of shControl or shINHA SKOV3 cells were intraperitoneally injected into 7-week-old female NCr nude mice (Taconic Biosciences) athymic nude mice that were housed under pathogen-free conditions on a 12-hour-light/dark cycle. Animals were monitored closely for tumor growth and signs of illness and sacrificed at humane end points. Cells were pathogen free and suspended in 200 μl HBSS. Two independent biological trials were conducted (first with 5 mice per group and the second with 8 mice per group) and monitored weekly with Girth measurements in triplicates. At 7 weeks (or if mice lost 20% of body weight), incisions were made in the mice abdomen, ascites collected followed by necropsy for metastatic burden quantitation.
DAB based immunohistochemistry.
The tissue micro array of OVCA tumor cores was purchased from Protein Biotechnologies Inc. (#TMA-2213) Ramona, CA). The formalin fixed, paraffin-embedded tissue arrays were deparaffinized by sequential washing with xylene, 100% ethanol, 90% ethanol, 80% ethanol, 70% ethanol and distilled water for 10 min each. For antigen retrieval the tissue sections were heated for 10 min in Sodium citrate buffer (pH 6.0) or Tris EDTA buffer (pH 9.0) (anti-INHA (Inhibin α) or anti-CD31, respectively). Endogenous peroxidases were blocked with 3% H2O2 for 10 min followed by block with 10% normal goat serum in TBST for 1 h RT, followed by incubation with primary antibody overnight at 4 °C in a humidified chamber. Appropriate secondary antibody conjugated to horseradish peroxidase (HRP) in blocking solution was added for 30 min at room temperature. HRP was detected with 3,3′-diaminobenzidine (DAB; ThermoFisher Scientific, Columbia, SC) substrate for 5 minutes, washed and counterstained with hematoxylin. Monoclonal antibodies to CD31 (#F8402, Sigma) and Inhibin α (#ab47720) from Abcam were used. Stained specimens were examined using a phase-contrast microscope (Model IX70; Olympus). For Inhibinα levels, semi-quantitative analysis was performed independently by 2 blind investigators (one pathologist) using a 3-tiered scoring system (none/trace/low, medium, or high being dark to high staining). Discrepancies between the 2 investigators were discussed and reconciled (<10 samples). For CD31 analysis, images were processed using ImageJ (NIH) to quantify CD31-positive areas. To quantify microvessel density (MVD), microvessel-like structures consisting of endothelial cells that were stained with anti-CD31 antibody were counted in similar fields in the entire core by setting a constant threshold and presented as number of vessels/mm2 of tissue section. Five random fields of each section were analyzed.
Statistics:
All clinical and xenograft data were analyzed using nonparametric statistics. Survival curves were analyzed with log-rank statistics. In vitro experiments were analyzed using parametric statistics (ANOVA global test with Bonferroni-corrected 2-tailed Student’s t tests as post-hoc tests) and presented as mean ± SEM. In cases where data were normalized to control, 1-sample Student’s t test was used with an expected value of 1 or 100% in order to decrease the likelihood of a type I error. All statistical analyses were conducted with GraphPad Prism Software.
Results
Inhibinα analysis in ovarian cancer and impact on patient survival
Prior reports on analysis of Inhibinα (encoded by INHA) in tumors, have indicated INHA and Inhibinα in circulation, including its utility as a biomarker for ovarian cancers (OVCA). To assess parameters in tumor tissues that could reveal Inhibin function in tumors, we first evaluated Inhibinα expression in human normal and primary OVCA tissues using immunohistochemistry on an ovarian TMA (tissue microarray) with 75 distinct tissue cores spanning different OVCA subtypes. We find that, while Inhibinα was detectable at low levels in the tissue cores from normal patients, 46 % of endometrial, 51 % of serous and 44 % of mucinous tumors expressed high Inhibinα (Fig 1A,B, FigS1i,ii) suggesting a broad increase in Inhibinα across all OVCA subtypes. The levels of Inhibinα were further elevated in higher tumor grades in all subtypes (Fig 1Bii). These results are in agreement with prior reports. Using this TMA, we noted Inhibinα localization exclusively in epithelial cells in 20% of tumors and in both epithelial and stromal cells in the remaining 80% of tumors (Table S1), suggesting potential stromal Inhibin functions. We next examined changes in microvessel density (MVD) using CD31 staining in the same tissues. A total of 52 cores were scorable for both Inhibinα and CD31 based on quality of cores. We find that tumor cores that expressed either medium and/or high levels of Inhibinα (Inhibinαmed/high, N=29) exhibited 2.5 times more microvessels per mm2 as compared to Inhibinαlow tumor cores (N=23) (Fig 1C and Fig S1iii). Regression analysis revealed that higher microvessel density (MVD) correlated with higher Inhibinα levels, with a Spearman’s correlation of 0.337(P<0.05).
Figure 1: Inhibinα expression in ovarian carcinoma and RCC and association with angiogenesis and overall survival.
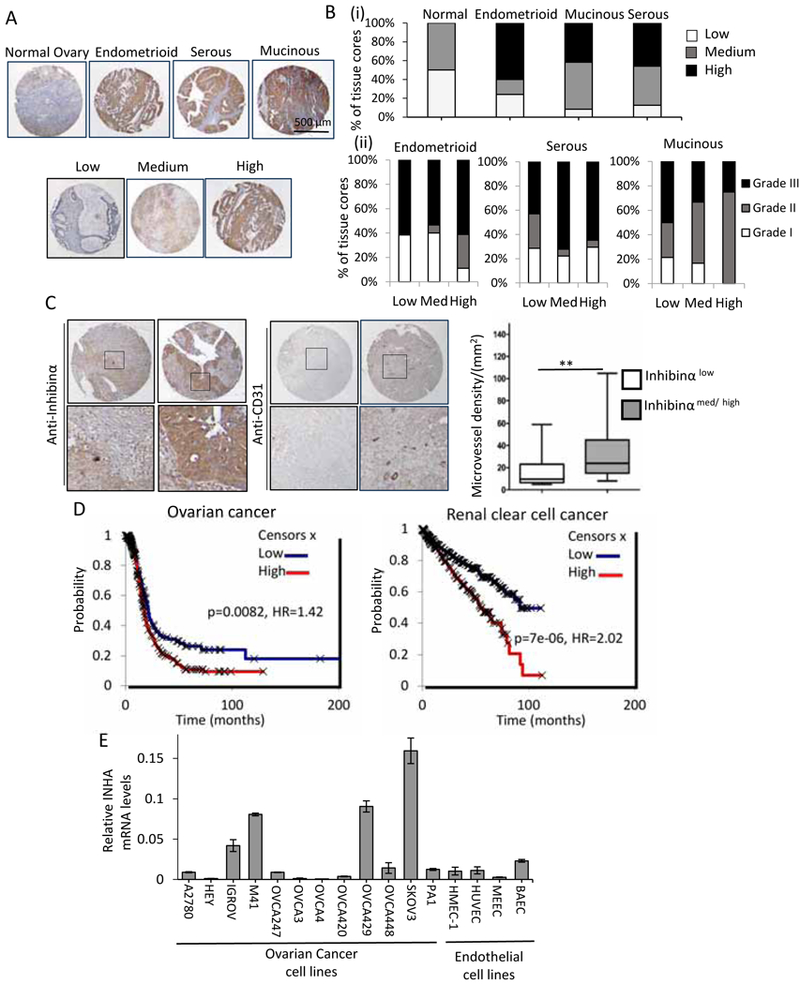
(A) Representative images from immunohistochemistry (IHC) from a human ovarian cancer tissue microarray of either normal tissue or different ovarian cancer subtypes with high Inhibinα staining as determined by immunolabeling with anti-Inhibinα antibody and IgG control (Fig S1i). Top, representative high staining cores across subtypes as shown. Zoomed in examples in Fig S1ii. Bottom, representative cores scored using a three-tier system (Methods) as none/ trace/ low, medium, or high staining as shown across subtypes. (B) Quantitation represented as percentage of cores with Inhibinα immunoreactivity scored as none/ trace/ low, medium, or high staining for (i) each subtype and (ii) relative to tumor grade (I–III) as indicated. (C) IHC of tissue arrays immunolabeled with anti-Inhibinα or anti-CD31 antibody. Representative images of CD31 staining with the corresponding Inhibinα levels from the same cores. Additional examples in Fig S1iii. Chart represents MVD quantitation in tumor cores with respect to Inhibinα levels quantified as in A and B and described in Methods. **P<0.01 (Mann Whitney test). Medium and high cores were combined as indicated. (D) Overall patient survival with low or high Inhibinα (INHA) mRNA in either ovarian cancer or renal clear cell carcinomas (E) INHA mRNA expression by qRT-PCR to detect endogenous INHA mRNA in a panel of ovarian cancer cell lines and endothelial cells as indicated.
Using Cox proportional hazards regression to analyze the impact of INHA expression on patient survival we find that higher Inhibinα mRNA (INHA) was associated with shorter overall survival (OS) in TP53 mutated OVCA’s (P<0.05, HR=1.42, Fig 1D). Since INHA levels correlate with CD31 in ovarian tumors, we examined whether INHA level in tumors would be a stronger predictor of survival in renal clear cell carcinomas that are highly angiogenic and for which anti-angiogenic therapies are the mainstay of treatment. We find that renal clear cell carcinoma patients with high INHA had significantly worse OS, with a median OS of 91.7 versus 54.2 months for tumors with low and high INHA, respectively (P<0.005, HR=2.02, Fig 1D). Based on these observations, to examine whether INHA expression and Inhibinα secretion by tumors was causal to the increased vascular density and to identify model systems for examining the role of Inhibin in tumors, we first examined INHA mRNA levels across a panel of OVCA and endothelial cells. We find a spectrum of secreted Inhibinα and INHA mRNA expression in epithelial cells (Fig S1iv, Fig 1E) in contrast to low to no INHA expression in all four commonly used-endothelial cell lines tested (Fig 1E). Although these data do not rule out endothelial cells as potential sources of INHA in the tumor, they suggest epithelial cells as the likely source of Inhibin in OVCA tissues, and point to a potential correlative impact of Inhibinα expression on tumor vasculature.
Tumor cell-produced Inhibinα and recombinant InhibinA induce specifically endothelial cell responses.
Given the correlation between Inhibinα expression and MVD in tumors (Fig 1C), we examined the impact of epithelial produced Inhibin on endothelial cell biology by, using shRNA-mediated suppression of INHA mRNA in epithelial cancer cells. We used SKOV3 cancer cells, (high INHA mRNA) and two independent shRNAs to reduce INHA mRNA (Fig 2A and Fig S2: 2nd shRNA). Reduction in INHA mRNA correlated with reduced secretion of Inhibinα and total dimeric Inhibin (A/B) (from here on referred to as Inhibin), as confirmed using an ELISA to detect total Inhibin (Fig 2Aii). We find that endothelial cells incubated with conditioned media (CM) from scramble PLKO1 shRNA (shControl) cells robustly induced tube formation of endothelial cells 4.5 times more than CM from shINHA cells (Fig 2B), which secrete less Inhibin (Fig 2Aii). This finding was confirmed using CM from a second independent shRNA to INHA (FigS2i). Since TGF-β members besides Inhibin could be altered in shINHA SKOV3 cells, we aimed to determine whether the CM effects were specific to Inhibin by examining endothelial cell tubulogenesis in the presence of an anti-Inhibinα antibody. We find that incubating CM with two independent anti-Inhibinα antibodies (polyclonal and monoclonal) suppresses CM induced tube formation 2.5 times more than controls (Fig 2B,Fig S2ii). The effect of anti-Inhibinα was not tumor cell line specific as anti-Inhibinα also suppressed CM induced tube formation from a second OVCA cell line M41 (Fig S2iii). These findings are the first to demonstrate the use of an anti-Inhibinα antibody in the suppression of any tumor promoting phenotype. We next used recombinant Inhibin (Inhibin A) to examine Inhibin potency in tube formation alongside equimolar amounts of other TGF-β members that are known angiogenesis regulators: BMP9, Activin and TGF-β, which either suppress or promote angiogenesis in different contexts (24,25). We find that Inhibin induced tube formation 9 and 4.5 times more effectively than TGF-β or Activin, respectively, and to a similar extent as BMP9 (Fig 2C). Combining Inhibin with either Activin or TGF-β resulted in a significant increase in tube formation with no further increase observed upon combining Inhibin with BMP9 (Fig 2C). A second commercial source of Inhibin (Fig S3i) and an antibody to block recombinant Inhibin (Fig S3ii) confirmed the specificity of Inhibin’s impact on tube formation in multiple endothelial cell types (Fig 2D). To examine if the response of endothelilal cells to Inhibin was restricted to tube formation on matrigel, we tested chemotactic migration towards Inhibin, endothelial cell spheroid sprouting (Methods), and cell proliferation. No effect of Inhibin on endothelial cell proliferation was observed (Fig S4i). However Inhibin increased endothelial cell migration towards recombinant Inhibin and increased endothelial spheroid sprouting ((Fig S4ii,iii). Notably, no effect of shRNA to Inhibin was observed on tumor cell growth (Fig S4iv) or matrigel invasion in vitro (Fig S4v). These data suggest a likely endothelial specific response to Inhibin.
Figure 2: Inhibin increases endothelial cell tubulogenesis.
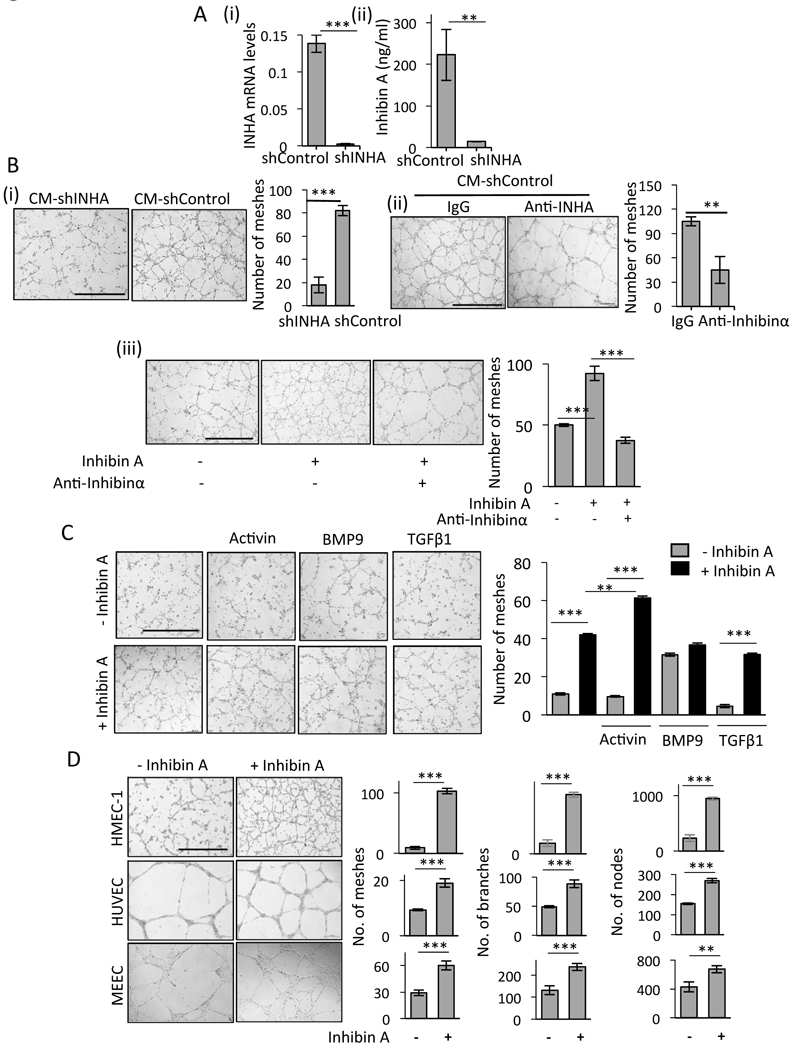
(A) (i) INHA mRNA levels in stable SKOV3 shControl and shINHA cancer cells (Methods). (ii) ELISA for total Inhibin (Inhibinα, InhibinA/B) from shControl and shINHA SKOV3 CM. (B) Tube formation of HMEC-1 cells in the presence of (i) CM from shControl SKOV3 cancer cells or shINHA SKOV3 cells (ii) and 10 μg/ml anti- Inhibinα or IgG control or (iii) treated with 300 pM recombinant Inhibin A alone or in the presence of anti-Inhibinα antibody (10 μg/ml). Bar graphs represent the average number of meshes quantified and represents an average of independent trials (Methods). mean ± SEM, ***P<0.001 and **P<0.01, Student’s t-test. Scale bar=500 μm. (C) HMEC-1 treated with equimolar amounts (300pM) of indicated TGF-β members either alone or in combination with Inhibin A. Bar graph represents quantitation as in B. One way ANOVA, Tukey’s multiple comparison test ***P<0.001 and **P<0.01. (D) Tube formation of HMEC-1, HUVEC and MEEC cells upon treatment with 300 pM Inhibin A for 16 h. Graph represents number of meshes, branches and nodes (Methods). mean ± SEM, ***P<0.001 and **P<0.01, Student’s t-test. Scale bar=500 μm
Effect of Inhibin on tumor angiogenesis and metastasis
Given the SKOV3-derived and recombinant Inhibin-dependent effects on tube formation in vitro (Fig 2) we examined the impact of paracrine produced and recombinant Inhibin in an in vivo Matrigel plug assay. Compared to endothelial cell growth supplement (ECGS, positive control) and CM from shControl SKOV3 cells (Fig 3A), CM from shINHA SKOV3 cells which secrete less Inhibin (Fig 2A) had substantially reduced functional blood flow into the Matrigel plug, as measured using hemoglobin content as a surrogate marker (23,26) (Fig 3A, 40% decrease, **p<0.01). Since components in conditioned media besides Inhibin could impact angiogenesis into the Matrigel plug in vivo, we next examined if recombinant Inhibin alone was sufficient to increase blood flow in the matrigel plug assay using 100ng/mL ECGS as a positive control. We find that compared to PBS, 100 ng/mL Inhibin led to twice as much hemoglobin content with visibly distinct vasculature in the matrigel plug (Fig 3B, 50% increase relative to PBS, *p<0.05). These data together indicate that Inhibin induces blood vessel formation in vivo.
Figure 3: Inhibin increases blood vessel formation, tumor angiogenesis and metastatic burden in vivo.
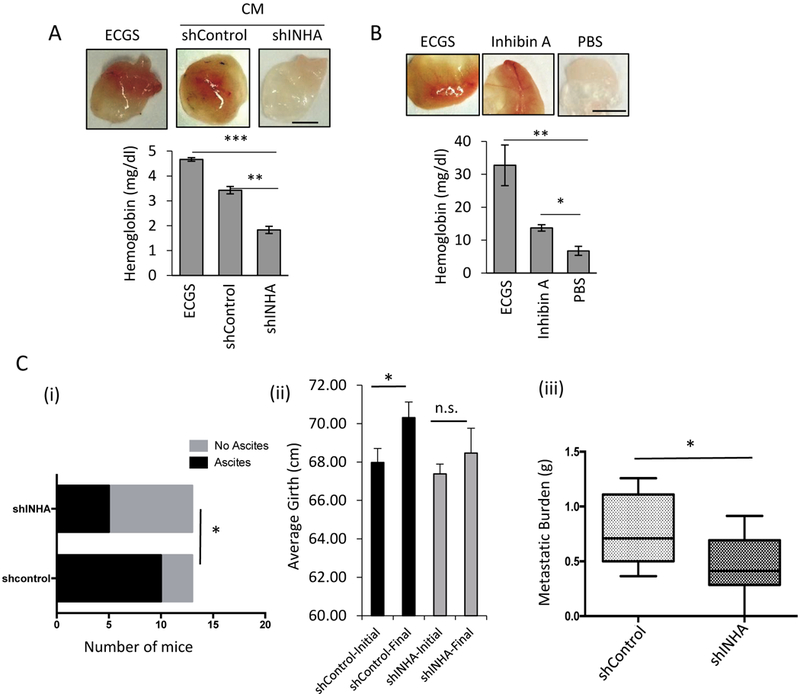
(A-B) Representative images (above) and hemoglobin quantification using Drabkins reagent (below) of matrigel plugs 12 days post subcutaneous injection of growth factor reduced matrigel mixed with either 100 ng/ml ECGS (positive control), (A) shControlCM or shINHA CM from SKOV3 stable cells or (B) recombinant Inhibin A (100 ng/ml) or PBS (negative control). mean ± SEM, (***P<0.001, **P<0.01 and *P<0.05, ANOVA with Holm-Sidak post hoc test) (n=3 mice per condition). Scale bar = 20 μm. (C) Nude mice were injected intraperitoneally with 5×106 shControl or shINHA SKOV3 cells, animals were sacrificed at 7 weeks and/or if body weight loss reached 20% as a humane endpoint. Ascites and metastatic burden were examined at necropsy: (i) number of mice per group that had ascites, (ii) abdominal girth measurements (in triplicate) at the beginning and end points, (iii) overall weight of tumors/lesions found in the peritoneal cavity. *p<0.05, Chi square test for (i) and Mann Whitney U for (ii) and (iii). n=13 mice per group (2 independent trials combined). Sample mouse images are provided in Fig S4vi.
Angiogenesis is critical for peritoneal metastasis manifested by accumulation of ascites in >70% of advanced human OVCA(27,28). To mimic the widespread intra-abdominal metastasis observed in the peritoneal cavity during OVCA progression (29,30), we injected nude mice intraperitoneally (i.p. injection) and used this in vivo model to determine whether the reduced angiogenesis observed in shINHA SKOV3 cells in matrigel plugs and in invitro analysis (Fig 2) translated to alterations in peritoneal burden and ascites accumulation. We i.p injected either shControl or shINHA SKOV3 cells into 7-week-old athymic female nude mice. 7 weeks after tumor cell injections, mice were sacrificed and ascites and tumor burden quantified (Methods). We find that all mice contained ovarian tumors (FigS4vi, dotted yellow circles). However, while 10/13 mice injected with shControl SKOV3 cells accumulated ascites, only 5/13 mice injected with shINHA cells had ascites (Fig 3Ci). Consistently, only mice receiving shControl SKOV3 cells showed an increase in abdominal girth (initial girth v. final girth) (Fig 3Cii). Necropsy confirmed that tumor burden in the abdominal cavity was significantly higher in mice recieving shControl cells as compared to mice receiving shINHA SKOV3 cells (Fig 3Ciii). Together, these findings are the first to link a reduction in Inhibin with suppression of angiogenesis and metastatic burden in vivo.
Inhibin specifically promotes SMAD1/5 activation in endothelial cells using the TGF-β co-receptor endoglin.
Since TGF-β family members transduce signals via SMAD2/3 or SMAD1/5 pathways, we examined the effects of paracrine and recombinant Inhibin on SMAD2/3 and SMAD1/5 phosphorylation in endothelial cells. We find that CM from shControl SKOV3 cells (Fig 2A) specifically increased phosphorylation of SMAD1/5 in endothelial cells (Fig 4A), while no change in phosphorylation of SMAD2/3 was observed (Fig 4A). Reducing Inhibin in the CM of SKOV3 cells by INHA shRNA (Fig 2A) resulted in significantly reduced SMAD1/5 activation in endothelial cell lines (Fig 4A). We further tested the specificity of Inhibin’s effects within CM on SMAD1/5 phosphorylation by incubating CM with an Inhibinα antibody. We find that CM induced SMAD1/5 phosphorylation was suppressed significantly by the antibody to similar extents as shINHA SKOV3 CM (Fig S5). To determine the specificity and kinetics of the SMAD1/5 activation, we used recombinant Inhibin and find that recombinant Inhibin robustly activated only SMAD1/5, and not SMAD2/3 (Fig 4B) in multiple endothelial cells in a time and dose dependent manner (FigS6i). In contrast to the signaling responsiveness of endothelial cells to recombinant Inhibin, SKOV3 epithelial cancer cells did not stimulate phosphorylation of SMAD1/5 in response to recombinant Inhibin (FigS6i).
Figure 4: Endoglin is essential for Inhibin induced signaling and tubulogenesis.
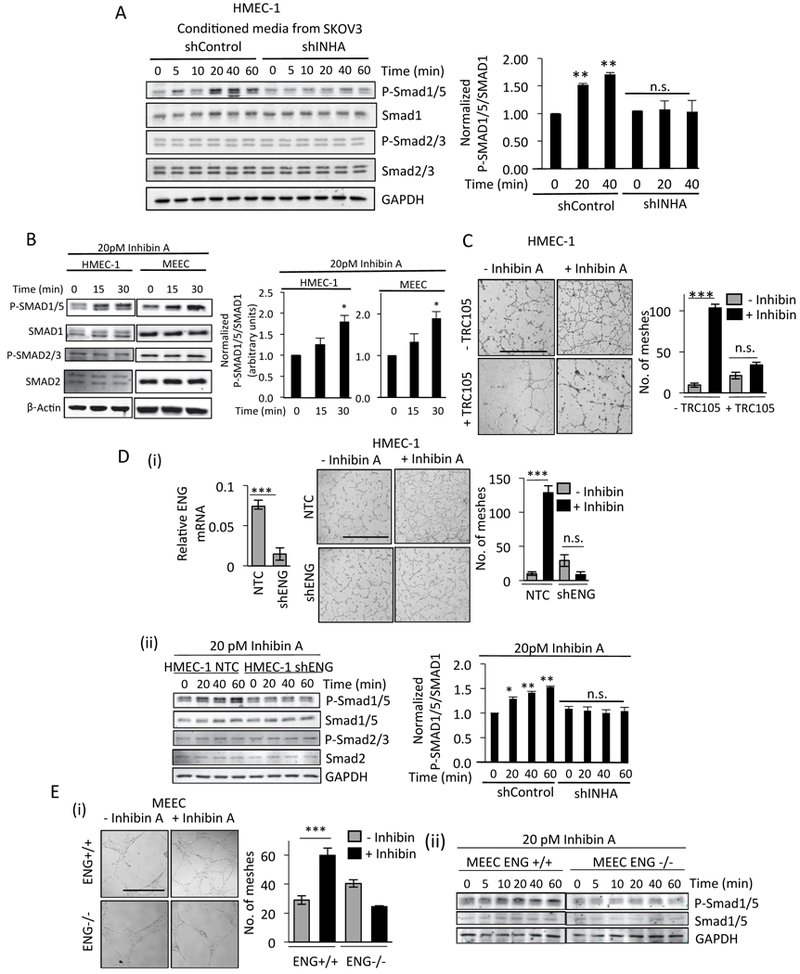
(A) Western blot analysis of lysates from HMEC-1 cells serum starved for 6hr and then incubated with indicated CM for up to 60 min, followed by immunoblotting of lysates. Quantitation of pSMAD1/5 changes (right graph) from two independent biological trials (B) Western blot analysis of lysates from HMEC-1 or MEEC’s serum starved for 6hr and then incubated with 20pM Inhibin A for up to 30 mins, followed by immunoblotting of lysates as indicated. Quantitation of pSMAD1/5 changes (right graph) from two independent biological trials. (C) Tube formation of HMEC-1 cells in the presence or absence of recombinant Inhibin A (300pM) either untreated or pretreated with 100 μg/ml TRC105 for 30 min prior to Inhibin A treatment. Average number of meshes/field quantified 16 h after Inhibin treatment (Methods). Scale bar=500 μm. (D) qRT-PCR analysis of endoglin mRNA after transient knockdown of endoglin using either control shRNA in HMEC-1 (shControl) or shRNA to endoglin followed by (i) tube formation in the absence or presence of recombinant Inhibin A. Graph of average number of meshes as described in Methods. Scale bar=500 μm. (ii) Western blotting for SMAD1/5 phosphorylation in response to 20 pM Inhibin A for up to 60 min. Quantitation of pSMAD1/5 changes (right graph) from two independent biological trials is presented. (E) (i) Tube formation in MEEC ENG+/+ cells and endoglin null ENG-/- MEECs cells in the absence or presence of Inhibin A after 16 h. Graph of average number of meshes as described in Methods. Scale bar=500 μm. (ii) Western blotting for SMAD1/5 phosphorylation in response to 20 pM Inhibin A for up to 60 min in MEEC ENG+/+ or ENG-/- cells as indicated.
SMAD1/5 activation positively regulates angiogenesis (31,32) and our data indicate Inhibin specifically induces SMAD1/5 activation in endothelial cells. These data suggest a requirement of endothelial specific receptors for the observed Inhibin response. Endoglin/CD105 is a well-established endothelial specific TGF-β co-receptor that we, and several others, have shown to have key roles in transducing angiogenic signals (33). To test if Inhibin induced tube formation and SMAD1/5 phosphorylation in endothelial cells are dependent on endoglin, we employed three approaches, 1) a humanized monoclonal antibody to endoglin TRC105 that we and others have shown to neutralize endoglin function (34,35), 2) a previously characterized endoglin knockout mouse embryonic endothelial cell line (MEECs, ENG-/- and corresponding wild type MEECs (15,36)), and 3) endoglin shRNA. We find that Inhibin-induced tube formation was significantly reduced by TRC105 compared to cells treated with Inhibin alone (Fig 4C). Both endoglin shRNA in HMEC-1s and endoglin knockout in MEEC ENG-/- abrogated Inhibin induced tube formation and SMAD1/5 activation (Fig 4D,E respectively). These data demonstrate that endoglin is a key mediator of Inhibin responses.
Inhibin-induced signaling and tube formation require ALK1 and promote endoglin-ALK1 interactions in endothelial cells.
SMAD1/5 is phosphorylated by specific serine threonine kinase Type I receptors (37). We thus sought to identify the Type I TGF-β receptor in Inhibin induced SMAD1/5 signaling and angiogenesis. We first determined the mRNA levels of different TGF-β Type I receptors (ALK1–7) in HMEC-1’s as compared to epithelial SKOV3 cells, since Inhibin-induced signaling was specific to endothelial cells (FigS6i). QRT-PCR analysis (Fig S7i) indicated higher ALK1 and ALK3 mRNA levels compared to ALK2, 4, 5, 6,7 in endothelial cells with limited ALK1 expression in SKOV3 cells (Fig S7i, right graph). We thus used the ALK1/2 inhibitor ML347, to block ALK1 and partially block ALK2/3 (IC50 for ALK1,2,3 are 46 nM, 32 nM and 10 μM, respectively). SB431542 and Dorsomorphin were used to block ALK5, 4,7 and ALK2, 3,6, respectively (38). We find that ML347 was able to suppress Inhibin induced tube formation (Fig 5A, 1.2 fold, ***P<0.001), and completely blocked Inhibin induced SMAD1/5 activation (Fig 5B). SB431542 and dorsomorphin had no significant impact on Inhibin induced tube formation (Fig S7ii). These data implicate ALK1 kinase activity in Inhibin induced tube formation and signaling responses. To confirm a role for ALK1, shRNA to ALK1 (shALK1) was used in HMEC-1’s and compared to control HMEC-1’s. shALK1 cells were unable to mount an Inhibin mediated tubulogenesis response (Fig 5C). shAlk1 HMEC-1 cells were also unable to increase SMAD1/5 phosphorylation in response to Inhibin, to the same extent as shControl HMEC-1 cells (Fig 5D). These data implicate ALK1 in mediating Inhibin signaling in endothelial cells.
Figure 5: Inhibin A induced tube formation and SMAD1/5 phosphorylation is mediated via ALK1 receptor kinases.
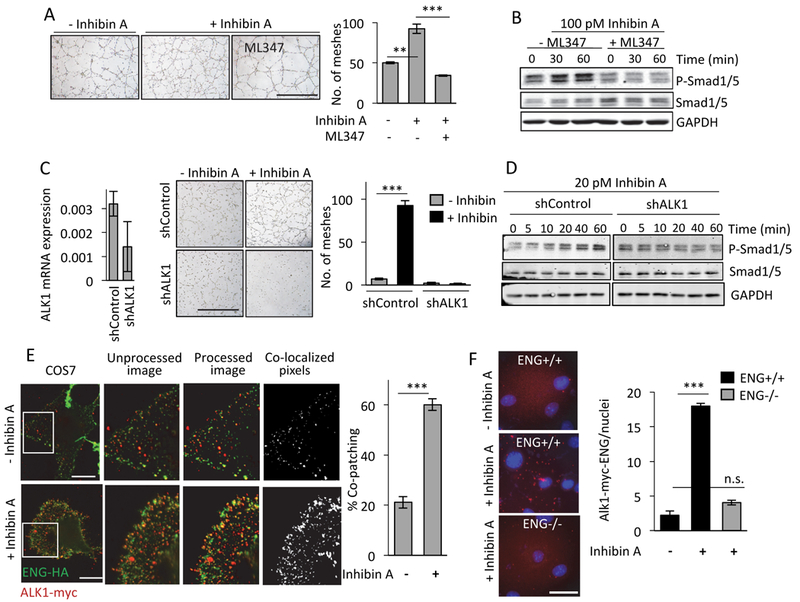
(A-B) (A) Tube formation and (B) SMAD1/5 phosphorylation in HMEC-1 cells in response to Inhibin A in the absence and presence of 5 μM of ML347 (ALK1,2 inhibitor (62), (89) (63)) added 30 min prior to treatment with Inhibin A. Graph of average number of meshes as described in Methods. (C-D) qRT-PCR analysis of ALK1 mRNA in HMEC-1 cells transfected with either control (shControl) or ALK1 shRNA (shALK1) for 48 hrs. before (C) tube formation assays in the absence or presence of recombinant Inhibin A (D) Western blotting for SMAD1/5 phosphorylation in response to 20 pM Inhibin A treatments for up to 60 min. Graph of average number of meshes as described in Methods. mean ± SEM, ***P<0.001 and **P<0.01, Student’s t-test. Scale bar=500 μm. (E) Endoglin-HA and ALK1-Myc receptor complex formation at the cell surface of COS7 cells either untreated or stimulated with 100 pM Inhibin A followed by immunolabeling with anti-Myc and anti-HA antibodies at 4°C to capture cell surface interactions (Methods). The unprocessed images are raw images. Processed images represent a 20% reduction in intensity and co-localized images show only those pixels where red (ALK1-Myc) and green (ENG-HA) punctum overlap. Bar graph shows percentage of co-patched puncta of ALK1-Myc/Endoglin HA complexes ***P<0.001 (n=5 fields per condition), ANOVA with Holm-Sidak post hoc test. Scale bar=500 μm. (F) Endogenous endoglin interactions with Myc tagged Alk1 determined using a proximity ligation assay in MEEC ENG+/+ and MEEC ENG-/- either untreated or upon Inhibin A treatment for 30 mins. Scale bar = 20 μm. Bar graph shows ALK1-ENG interactions normalized to nuclei (n=10/condition).
ALK1, a primarily endothelial specific receptor (39), forms stable complexes with endoglin, and this interaction is required for SMAD1/5 signaling (40). To test whether stable, cell surface ALK1- endoglin interactions could be regulated by Inhibin, we used a live cell copatching method (41). We transiently expressed HA-tagged Endoglin (ENG-HA) and Myc-tagged ALK1 (ALK1-Myc) in COS7 cells and determined the change in percentage of co-patched receptors at the cell surface upon Inhibin treatment. We find a 20% baseline interaction between ALK1-Myc and Endoglin-HA, which increased significantly to 60% upon Inhibin treatment (Fig 5E, ***P<0.001), suggesting Inhibin-dependent increases in ALK1-endoglin complexes at the cell surface. To complement the COS7 cell surface co-patch studies and confirm the effect of Inhibin in the endothelial environment, we used a proximity ligation assay (42) in MEECs. We examined and quantified the proximity of ALK1-HA and endogenous endoglin in unstimulated and Inhibin stimulated MEECs and find a significant increase in the ALK1 proximity interactions with endogenous endoglin in response to a 30 min Inhibin treatment. ENG-/- MEECs were used as controls (Fig 5F, ***P<0.001). Together, these findings strongly suggest a novel endoglin-ALK1 response to Inhibin, a TGF-β family ligand with previously unknown functions in cancer and in endothelial cells. Taken together, we demonstrate a novel signaling ligand for angiogenesis in cancer.
Discussion
Using a combination of antibody and shRNA based approaches coupled with in vivo experimental models; we have identified and defined a novel paracrine regulator of tumor angiogenesis. While Inhibin has long been known to be elevated in multiple cancers no specific mechanism of action and outcome of elevated Inhibin has been previously established. Our findings provide the first evidence of Inhibin’s ability to transduce signals and act as a pacracrine tumor angiogenic factor. Likewise, we provide the first use of an anti- inhibinα antibody to suppress angiogenesis. In accordance with prior studies, we observed Inhibinα overexpression in a spectrum of ovarian cancers (Fig 1) and observed increased Inhibinα levels in tumors correlating with MVD in patients and xenograft tumors. Increased Inhibinα also correlated with poor overall patient survival, particularly in high-grade serous ovarian cancer patients with alterations in TP53 and in renal clear cell carcinomas that are highly angiogenic. Notably, we demonstrate a novel function for Inhibin that is dependent on endothelial specific TGF-β receptors endoglin and ALK1, which are upregulated during tumor angiogenesis and explored as angiogenic targets (43). Surprisingly, suppressing Inhibinα expression and Inhibin secretion in tumor cells had marginal effects on epithelial (autocrine) cell growth (Fig S4iv). However in the intraperitoneal model that mimics human disease leading to ascites accumulation and peritoneal spread (44),(30), Inhibinα shRNA significantly reduced ascites accumulation and peritoneal tumor burden in vivo (Fig3C) indicating not just a significant role for Inhibin in metastasis but also the potential significance of Inhibin induced angiogenesis in this process. While our findings using Inhibinα antibodies (Fig 2) and recombinant Inhibin in in vitro (Fig 2,4,5) and in vivo Matrigel plug assays (Fig 3A) indicate a direct role for Inhibin on angiogenesis that is reduced upon INHA shRNA, one alternate mechanism for reduced metastasis observed using shINHA tumor cells could be alterations in pro-angiogenic factors secreted from the tumor cells that warrants future investigation.
Previous studies on Inhibin signaling, emphasize the non-signaling interactions with the TGF-β co-receptor betaglycan via the α subunit in epithelial cells, highlighting a competitive antagonist model for Inhibin with TGF-β members (13,45). However, some outcomes of Inhibin including effects of Inhibin in prostate cancer metastasis (11) and systems where Inhibin does not antagonize Activin (46), appear inconsistent with this model as Inhibin’s only mode action. Supporting the possibility of additional modes of action for Inhibin, we find that Activin primarily activates SMAD2/3 in endothelial cells (FigS8), contrasting with Inhibin, which primarily activates SMAD1/5 exclusively in endothelial cells (Fig 4B and FigS6ii). Since our findings demonstrate the critical role for endoglin (Fig 4) in Inhibin induced angiogenesis and signaling (Fig4), it is likely that the established model of Inhibin’s functional antagonism utilizing betaglycan, may not be the same in endothelial cells that express endoglin.
In addition to Inhibin, the ligands that utilize these receptors include TGF-β1, and BMP9/GDF2 that can activate both ALK1/SMAD1/5/8 and ALK5/Smad2/3 (1). We find that Activin significantly activates Smad2/3 with limited activation of SMAD1/5 (FigS8). The SMAD2/3 pathway is an established anti-angiogenic mechanism contrasting with SMAD1/5 as a pro- angiogenic mechanism (1). Consistently, Activin did not support angiogenesis in vitro (Fig 2) and was reported to suppress angiogenesis in vivo (47). Combining Inhibin with other TGF-β members in vitro (Fig 2), reveal that Inhibin promotes angiogenesis to the same extent as BMP9 and can override both TGF-β and Activin in inducing angiogenesis. It is likely that the specificity of SMAD1/5 activation in response to Inhibin (unlike TGF-β,BMP9) represents an important distinction between Inhibin and other TGF-β members in angiogenesis that necessitate future in depth analysis.
In summary, we provide new insights for Inhibin in cancer, a hormone and TGF-β family member under investigation for over 90 years and provide a molecular and mechanistic basis for clinical outcomes observed in these cancers based on Inhibin expression. Inhibin can have pronounced effects on angiogenesis and metastasis with broad therapeutic implications for other cancers as well. We thus propose Inhibin as an attractive and safe target for ovarian cancer and other cancers prevalent in postmenopausal women where Inhibin levels are normally very low thereby limiting potential on target side effects. Further investigation into anti-Inhibin therapeutic strategies, as a result, is warranted.
Supplementary Material
Significance:
Inhibin is a predictor of poor patient survival in multiple cancers and is a potential target for anti-angiogenic therapies.
Acknowledgements
We thank Drs. Shannon Davis and Ioluia Chatzistamou for help with mouse tissue processing and analysis of tissue cores, Andy Nixon and Miao at Duke University with optimizing Inhibin ELISAs and the “University of South Carolina Center for Targeted Therapeutics Functional Genomics Core Facility” for lentiviral preparations and the “University of South Carolina Center for Targeted Therapeutics Microscopy and Flow Cytometry Core Facility” for help with imaging. This work was funded by the National Institutes of Health (NIH) Grant 5P20-GM109091 and a grant from the Marsha Rivkin Foundation to Karthikeyan Mythreye and the USC SPARC Graduate Research Grant, AAUW Dissertation Fellowship and NIH Predoctoral F31 Fellowship (GM122379–01) to L.M. Jenkins
Footnotes
Authors declare no conflict of interest.
References
- 1.Pardali E, ten Dijke P. Transforming growth factor-beta signaling and tumor angiogenesis. Front Biosci (Landmark Ed) 2009;14:4848–61 [DOI] [PubMed] [Google Scholar]
- 2.Overlie I, Morkrid L, Andersson AM, Skakkebaek NE, Moen MH, Holte A. Inhibin A and B as markers of menopause: a five-year prospective longitudinal study of hormonal changes during the menopausal transition. Acta Obstet Gynecol Scand 2005;84:281–5 [DOI] [PubMed] [Google Scholar]
- 3.Walentowicz P, Krintus M, Sadlecki P, Grabiec M, Mankowska-Cyl A, Sokup A, et al. Serum inhibin A and inhibin B levels in epithelial ovarian cancer patients. PLoS One 2014;9:e90575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Robertson DM, Pruysers E, Jobling T. Inhibin as a diagnostic marker for ovarian cancer. Cancer letters 2007;249:14–7 [DOI] [PubMed] [Google Scholar]
- 5.Walton KL, Makanji Y, Robertson DM, Harrison CA. The synthesis and secretion of inhibins. Vitamins and hormones 2011;85:149–84 [DOI] [PubMed] [Google Scholar]
- 6.Matzuk MM, Finegold MJ, Su JG, Hsueh AJ, Bradley A. Alpha-inhibin is a tumour-suppressor gene with gonadal specificity in mice. Nature 1992;360:313–9 [DOI] [PubMed] [Google Scholar]
- 7.Shanbhag SA, Sheth AR, Nanivadekar SA, Sheth NA. Immunoreactive inhibin-like material in serum and gastric juice of patients with benign and malignant diseases of the stomach. British journal of cancer 1985;51:877–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Suzuki Y, Sugiyama M, Abe N, Fujioka Y, Atomi Y. Immunohistochemical similarities between pancreatic mucinous cystic tumor and ovarian mucinous cystic tumor. Pancreas 2008;36:e40–6 [DOI] [PubMed] [Google Scholar]
- 9.McCluggage WG, Maxwell P. Adenocarcinomas of various sites may exhibit immunoreactivity with anti-inhibin antibodies. Histopathology 1999;35:216–20 [DOI] [PubMed] [Google Scholar]
- 10.McCluggage WG, Maxwell P, Patterson A, Sloan JM. Immunohistochemical staining of hepatocellular carcinoma with monoclonal antibody against inhibin. Histopathology 1997;30:518–22 [DOI] [PubMed] [Google Scholar]
- 11.Balanathan P, Williams ED, Wang H, Pedersen JS, Horvath LG, Achen MG, et al. Elevated level of inhibin-alpha subunit is pro-tumourigenic and pro-metastatic and associated with extracapsular spread in advanced prostate cancer. British journal of cancer 2009;100:1784–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Farnworth PG, Harrison CA, Leembruggen P, Chan KL, Stanton PG, Ooi GT, et al. Inhibin binding sites and proteins in pituitary, gonadal, adrenal and bone cells. Mol Cell Endocrinol 2001;180:63–71 [DOI] [PubMed] [Google Scholar]
- 13.Lewis KA, Gray PC, Blount AL, MacConell LA, Wiater E, Bilezikjian LM, et al. Betaglycan binds inhibin and can mediate functional antagonism of activin signalling. Nature 2000;404:411–4 [DOI] [PubMed] [Google Scholar]
- 14.Gatza CE, Oh SY, Blobe GC. Roles for the type III TGF-beta receptor in human cancer. Cell Signal 2010;22:1163–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pece-Barbara N, Vera S, Kathirkamathamby K, Liebner S, Di Guglielmo GM, Dejana E, et al. Endoglin null endothelial cells proliferate faster and are more responsive to transforming growth factor beta1 with higher affinity receptors and an activated Alk1 pathway. J Biol Chem 2005;280:27800–8 [DOI] [PubMed] [Google Scholar]
- 16.Tian H, Mythreye K, Golzio C, Katsanis N, Blobe GC. Endoglin mediates fibronectin/alpha5beta1 integrin and TGF-beta pathway crosstalk in endothelial cells. The EMBO journal 2012;31:3885–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gilboa L, Wells RG, Lodish HF, Henis YI. Oligomeric structure of type I and type II transforming growth factor beta receptors: homodimers form in the ER and persist at the plasma membrane. J Cell Biol 1998;140:767–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Varadaraj A, Jenkins LM, Singh P, Chanda A, Snider J, Lee NY, et al. TGF-beta triggers rapid fibrillogenesis via a novel TbetaRII dependent fibronectin trafficking mechanism. Mol Biol Cell 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Malinda KM. In vivo matrigel migration and angiogenesis assay. Methods in molecular biology 2009;467:287–94 [DOI] [PubMed] [Google Scholar]
- 20.DeCicco-Skinner KL, Henry GH, Cataisson C, Tabib T, Gwilliam JC, Watson NJ, et al. Endothelial cell tube formation assay for the in vitro study of angiogenesis. J Vis Exp 2014:e51312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Laib AM, Bartol A, Alajati A, Korff T, Weber H, Augustin HG. Spheroid-based human endothelial cell microvessel formation in vivo. Nature protocols 2009;4:1202–15 [DOI] [PubMed] [Google Scholar]
- 22.Wu SY, Rupaimoole R, Shen F, Pradeep S, Pecot CV, Ivan C, et al. A miR-192-EGR1-HOXB9 regulatory network controls the angiogenic switch in cancer. Nature communications 2016;7:11169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stone RL, Baggerly KA, Armaiz-Pena GN, Kang Y, Sanguino AM, Thanapprapasr D, et al. Focal adhesion kinase: an alternative focus for anti-angiogenesis therapy in ovarian cancer. Cancer biology & therapy 2014;15:919–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.David L, Feige JJ, Bailly S. Emerging role of bone morphogenetic proteins in angiogenesis. Cytokine Growth Factor Rev 2009;20:203–12 [DOI] [PubMed] [Google Scholar]
- 25.Jin Y, Kaluza D, Jakobsson L. VEGF, Notch and TGFbeta/BMPs in regulation of sprouting angiogenesis and vascular patterning. Biochem Soc Trans 2014;42:1576–83 [DOI] [PubMed] [Google Scholar]
- 26.Pecot CV, Rupaimoole R, Yang D, Akbani R, Ivan C, Lu C, et al. Tumour angiogenesis regulation by the miR-200 family. Nature communications 2013;4:2427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tan DS, Agarwal R, Kaye SB. Mechanisms of transcoelomic metastasis in ovarian cancer. Lancet Oncol 2006;7:925–34 [DOI] [PubMed] [Google Scholar]
- 28.Ahmed N, Stenvers KL. Getting to know ovarian cancer ascites: opportunities for targeted therapy-based translational research. Frontiers in oncology 2013;3:256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shaw TJ, Senterman MK, Dawson K, Crane CA, Vanderhyden BC. Characterization of intraperitoneal, orthotopic, and metastatic xenograft models of human ovarian cancer. Molecular therapy : the journal of the American Society of Gene Therapy 2004;10:1032–42 [DOI] [PubMed] [Google Scholar]
- 30.Connolly DC, Hensley HH. Xenograft and transgenic mouse models of epithelial ovarian cancer and non-invasive imaging modalities to monitor ovarian tumor growth in situ: applications in evaluating novel therapeutic agents. Current protocols in pharmacology / editorial board, SJ Enna; 2009;Chapter 14:Unit14 2 [DOI] [PubMed] [Google Scholar]
- 31.Goumans MJ, Valdimarsdottir G, Itoh S, Lebrin F, Larsson J, Mummery C, et al. Activin receptor-like kinase (ALK)1 is an antagonistic mediator of lateral TGFbeta/ALK5 signaling. Molecular cell 2003;12:817–28 [DOI] [PubMed] [Google Scholar]
- 32.David L, Mallet C, Mazerbourg S, Feige JJ, Bailly S. Identification of BMP9 and BMP10 as functional activators of the orphan activin receptor-like kinase 1 (ALK1) in endothelial cells. Blood 2007;109:1953–61 [DOI] [PubMed] [Google Scholar]
- 33.Nassiri F, Cusimano MD, Scheithauer BW, Rotondo F, Fazio A, Yousef GM, et al. Endoglin (CD105): a review of its role in angiogenesis and tumor diagnosis, progression and therapy. Anticancer Res 2011;31:2283–90 [PubMed] [Google Scholar]
- 34.Rosen LS, Hurwitz HI, Wong MK, Goldman J, Mendelson DS, Figg WD, et al. A Phase 1 First-in-Human Study of TRC105 (Anti-Endoglin Antibody) in Patients with Advanced Cancer. Clin Cancer Res 2012;18:4820–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kumar S, Pan CC, Bloodworth JC, Nixon AB, Theuer C, Hoyt DG, et al. Antibody-directed coupling of endoglin and MMP-14 is a key mechanism for endoglin shedding and deregulation of TGF-beta signaling. Oncogene 2014;33:3970–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barbara NP, Wrana JL, Letarte M. Endoglin is an accessory protein that interacts with the signaling receptor complex of multiple members of the transforming growth factor-beta superfamily. J Biol Chem 1999;274:584–94 [DOI] [PubMed] [Google Scholar]
- 37.Massague J, Attisano L, Wrana JL. The TGF-beta family and its composite receptors. Trends Cell Biol 1994;4:172–8 [DOI] [PubMed] [Google Scholar]
- 38.Inman GJ, Nicolas FJ, Callahan JF, Harling JD, Gaster LM, Reith AD, et al. SB-431542 is a potent and specific inhibitor of transforming growth factor-beta superfamily type I activin receptor-like kinase (ALK) receptors ALK4, ALK5, and ALK7. Mol Pharmacol 2002;62:65–74 [DOI] [PubMed] [Google Scholar]
- 39.Bertolino P, Deckers M, Lebrin F, ten Dijke P. Transforming growth factor-beta signal transduction in angiogenesis and vascular disorders. Chest 2005;128:585S–90S [DOI] [PubMed] [Google Scholar]
- 40.Pomeraniec L, Hector-Greene M, Ehrlich M, Blobe GC, Henis YI. Regulation of TGF-beta receptor hetero-oligomerization and signaling by endoglin. Mol Biol Cell 2015;26:3117–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gilboa L, Nohe A, Geissendorfer T, Sebald W, Henis YI, Knaus P. Bone morphogenetic protein receptor complexes on the surface of live cells: a new oligomerization mode for serine/threonine kinase receptors. Mol Biol Cell 2000;11:1023–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Soderberg O, Gullberg M, Jarvius M, Ridderstrale K, Leuchowius KJ, Jarvius J, et al. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat Methods 2006;3:995–1000 [DOI] [PubMed] [Google Scholar]
- 43.Jonker L TGF-beta & BMP receptors endoglin and ALK1: overview of their functional role and status as antiangiogenic targets. Microcirculation 2014;21:93–103 [DOI] [PubMed] [Google Scholar]
- 44.Jazaeri AA, Bryant JL, Park H, Li H, Dahiya N, Stoler MH, et al. Molecular requirements for transformation of fallopian tube epithelial cells into serous carcinoma. Neoplasia 2011;13:899–911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Looyenga BD, Wiater E, Vale W, Hammer GD. Inhibin-A antagonizes TGFbeta2 signaling by down-regulating cell surface expression of the TGFbeta coreceptor betaglycan. Molecular endocrinology 2010;24:608–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang QF, Tilly KI, Tilly JL, Preffer F, Schneyer AL, Crowley WF, Jr., et al. Activin inhibits basal and androgen-stimulated proliferation and induces apoptosis in the human prostatic cancer cell line, LNCaP. Endocrinology 1996;137:5476–83 [DOI] [PubMed] [Google Scholar]
- 47.Kaneda H, Arao T, Matsumoto K, De Velasco MA, Tamura D, Aomatsu K, et al. Activin A inhibits vascular endothelial cell growth and suppresses tumour angiogenesis in gastric cancer. British journal of cancer 2011;105:1210–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.