

Abstract
Objective
To describe the features of a new, pathologically distinctive, acquired myopathy with an unusual pattern of scattered necrotic muscle fibers that are neighbored, surrounded, or invaded, by large, often multinucleated, histiocytic cells.
Methods
Retrospective review of records and muscle pathology of 4 patients.
Results
Clinical features common to our patients included muscle pain and proximal, symmetric, moderate to severe, weakness in the arms and legs progressing over 1–4 weeks. Patients had other associated systemic disorders, including anemia in all, and hemophagocytic lymphohistiocytosis, hepatic disease, Raynaud phenomenon, metastatic cancer, and cardiomyopathy, in 1 patient each. Serum creatine kinase (CK) levels at presentation were very high, ranging from 10,000 to 102,000 U/L. Three patients improved within 3 months after treatment. Muscle pathology included scattered necrotic muscle fibers with cytoplasm that stained for C5b-9 complement, especially around fiber peripheries, pale on nicotinamide adenine dinucleotide and often dark on hematoxylin & eosin. Large, often multinucleated, cells with features of histiocytes, including anatomical features on electron microscopy and immunostaining for major histocompatibility complex Class I and histiocyte markers (HAM56, CD68, CD163, and S100), were usually closely apposed to the surface of, or invaded, necrotic myofibers.
Conclusions
Patients with large-histiocyte-associated myopathy (LHIM) had a subacute onset of proximal predominant weakness, associated systemic disorders, very high serum CK, and a pathologically distinctive pattern of large histiocyte-associated muscle fiber necrosis. LHIM may be caused by an autoimmune, histiocyte-mediated attack directed against muscle fibers.
Acquired immune and inflammatory myopathies (IIM) are a heterogeneous group of disorders.1,2 Myopathologic features, including patterns of cellular and humoral immune features, and types of pathology in muscle fibers, connective tissues, and vessels, can be used to characterize IIM.3–7 It has been proposed that one pathologic category of IIM may be “necrotic myopathies.” 2,3,8,9 However, histochemical identification of necrosis can be imprecise, often resting on subjective observations of pale muscle fiber cytoplasm on hematoxylin & eosin (H&E) or Gomori trichrome tinctorial stains, changes that may also result from processing artefact. Pathologic features that may be more specific and sensitive for necrosis include a loss of myofiber oxidative enzyme staining, especially well-observed as reduced nicotinamide adenine dinucleotide (NADH) staining,9 and staining of muscle fiber cytoplasm for the complement membrane attack complex (C5b-9 complement).10,11 In the process of reviewing muscle biopsies using these stains, we identified 4 patients with acquired myopathies who had muscle biopsies with an unusual pattern of muscle fiber necrosis. Necrotic muscle fibers were progressively replaced by individual, or clusters of, unusual, large histiocytic cells with large, sometimes multiple, nuclei and abundant cytoplasm. Damage was selective to muscle fibers. Connective tissue and vessels were unremarkable. No foci of lymphocyte inflammation were observed. We describe the clinical and laboratory features of our 4 patients with this distinctive myopathologic syndrome of large-histiocyte-related immune myopathy (LHIM).
Methods
Patient selection criteria
Our neuromuscular pathology database at Washington University School of Medicine in St. Louis included 15,015 muscle biopsies that were initially evaluated between 1996 and 2016, of which 2,199 were interpreted as having possible immune or inflammatory myopathies. We identified, and reviewed, 4 biopsies that had an unusual pattern of scattered necrotic muscle fibers neighbored, or surrounded, by large cells with large nuclei and abundant cytoplasm (table 1). This myopathology pattern differs from most previously described immune “necrotic” myopathies that have individual muscle fibers in varying stages of necrosis and regeneration but no neighboring large cells during early stages of necrosis. Patient clinical records and biopsy reports were used to obtain clinical and laboratory data. All 4 patients were evaluated and treated by the Neuromuscular Service at Washington University School of Medicine. All procedures were approved by the human studies committee at Washington University in St. Louis.
Table 1.
Clinical and laboratory characteristics of the 4 patients

Histochemistry and immunohistochemistry
Cryostat sections from rapidly frozen muscle were stained using histochemical methods.12–14 Immunohistochemical stains using antibodies were performed on muscle biopsies from patients and simultaneously stained controls on the same glass slide. Primary antibodies were collagen 4 (Millipore [Burlington, MA] AB748; 1:50), C5b-9 complement (membrane attack complex) (Dako [Glostrup, Denmark] Mo777; 1:25), class I human major histocompatibility complex (MHC-1) (US Biological, Swampscott, MA), CD68 (Abcam [Cambridge, UK] ab31630; 1:100), HAM56 (MyBiosource [San Diego, CA] 370085; 1:50), CD1a (Thermo Fisher [Waltham, MA] MA5-12526; 1:200), CD163 (ABD Serotec [Hercules, CA] MCA 1853; 1:100), CD207 (Novus Biologicals [Littleton, CO] DDX0361P-100; 1:50), S100 (Abcam ab14849; 1:200), VE1 (Abcam ab228461 1:100), and Phospho-ERK (Invitrogen [Carlsbad, CA] 14-9109-80 5 μg/mL). Secondary antibodies were conjugated to green or red fluorescent markers (anti-mouse Alexa Fluor 488; anti-rabbit Alexa Fluor 594 [Invitrogen]).
Electron microscopy
Ultrastructural visualization could be obtained on 3 patients with available tissue. A piece of each muscle biopsy was fixed overnight by immersion in 3% glutaraldehyde in Karnovsky buffer, pH 7.4 at 4°C. Tissue samples were postfixed in phosphate buffered 2% OsO4, dehydrated in graded concentrations of ethanol, and embedded in Embed-812 (Electron Microscopy Sciences, Hatfield, PA), with propylene oxide as an intermediary solvent. One-micrometer-thick plastic sections were examined by light microscopy after staining with toluidine blue. Ultrathin sections of muscle were cut into mesh or Formvar-coated slot grids, which permit visualization of entire fascicular cross sections. Tissues were subsequently stained with uranyl acetate and lead citrate and examined using a JEOL (Tokyo, Japan) 1,200 electron microscope with an AMT (Woburn, MA) digital camera.
Data availability
More detailed, anonymized case reports are available to qualified investigators. Additional images of LHIM myopathology are available on our website.4
Results
Case report
An 18-year-old previously healthy woman (table 1; patient 1) developed fever, diffuse myalgias, sore throat, and fatigue that lasted for 2 weeks, remitted spontaneously, and then recurred 3 weeks later. Examination showed scleral icterus, jaundice, cervical lymphadenopathy, hepatosplenomegaly, and asterixis. Mental status was intact. There was mild bilateral facial weakness. Other cranial nerves were normal. Diffuse proximal (2/5) more than distal (4/5), symmetric weakness involved the neck flexors, arms, and legs with a loss of ability to walk. Sensation was normal. Tendon reflexes were 1+ to 2+. Laboratory studies showed pancytopenia (white blood cells 1.7, hemoglobin 10.9, platelets 37,000), bilirubin 13.1 mg/dL, elevated alkaline phosphatase and transaminases, and positive Monospot and serum Epstein-Barr virus (EBV) PCR testing. Serum testing showed a ferritin level of 59,800 and creatine kinase (CK) of 18,106 U/L. Bone marrow aspiration showed phagocytic histiocytes without malignant features or bone marrow hypoplasia, consistent with EBV-associated hemophagocytic lymphohistiocytosis (HLH). Two weeks after treatment with VP-16, IV methylprednisolone, and G-CSF, strength improved, she regained ambulation, and serum creatine kinase level fell to 86 U/L. Repeat bone marrow biopsy was improved with increased myelopoiesis and reduced erythroid dysplasia and phagocytic histiocytes.
Clinical features
Three women and 1 man developed muscle pain and weakness with onset at ages ranging from 18 to 52 years (table 1). All had a subacute onset of myalgias and proximal-greater-than-distal, symmetric weakness that progressed to moderate disability over 1–4 weeks. Each patient had other associated systemic disorders, including anemia in all, and hemophagocytic syndrome, subacute cardiomyopathy in the setting of a Mycoplasma pneumonia infection, Raynaud syndrome, and metastatic squamous cell carcinoma in 1 each. No patient had a history of exposure to myotoxins, statins, or a family history of muscle disease. Serum CK was very high (>10,000) at presentation in all patients. EMG showed myopathic features in 2 of the 3 patients tested. Myositis-specific antibody panels, including HMGCR and signal recognition particle antibodies, were negative in the 3 patients who were tested. Serum ferritin was high in patient 1, but normal in the 3 others. Other anemia-related laboratory tests were normal except for a low serum vitamin B12 level in patient 3. Three patients (1, 2, and 4) improved to normal strength within 3 months, 1 following standard of care therapy for HLH, 1 with empiric corticosteroids, and 1 with azithromycin. The course of the LHIM was monophasic in these patients with no relapse during observed periods from 1 to 3 years after cessation of treatment. Patient 3 died of complications of his metastatic neoplasm 7 months after presentation.
Muscle pathology
Muscle fiber pathology (figure 1), mainly necrosis, was scattered without preferential anatomic relation to perimysium or vessel locations. On NADH stain, necrotic fibers had pale cytoplasm with loss of internal architecture. H&E, Gomori trichrome, and Verhoeff–van Gieson stains showed dark, or normal, intensity of cytoplasm staining more commonly than pale staining. C5b-9 stained in the cytoplasm of many necrotic muscle fibers, either diffusely, the typical pattern for necrosis in most other disorders, or in a ring around the outside of muscle fibers (figure 2). Necrotic muscle fibers were frequently neighbored by 1 or several large cells. Some of these cells had diameters up to 30 μM (normal <10 μM) and were multinucleated. The cells stained for NADH, acid phosphatase, esterase, MHC-1, HAM56, CD68, S100, and CD163 (figures 1–3), but not CD1a or CD207. The pattern of histiocyte staining in LHIM was similar to staining of cells associated with other types of acute and ongoing muscle fiber necrosis. Some histiocytic disorders have clonal mutations involving genes in the MAPK pathway.15 The VE1 antibody, which selectively stains a mutated BRAF protein (V600E) in fixed tissue,16 was not helpful in our frozen muscle specimens as it stained histiocytes identically to HAM56 in normal, granulomatous, necrotic, and LHIM muscles. In some histiocytoses and other neoplasms, especially with BRAF or MAP2K1 mutations, phospho-ERK expression is increased in histiocytes.17 In our patients, phospho-ERK staining was usually negative in LHIM histiocytes. There was no histiocyte staining in patients 1 and 2, and fewer than 5% positive histiocytic cells in patients 3 and 4. Phospho-ERK mildly stained the cytoplasm of most of the necrotic muscle fibers that also had C5b-9 staining.
Figure 1. Large-histiocyte-related myofiber necrosis: histochemistry.

(A) Large, multinucleated, histiocytic cells (arrow) have basophilic cytoplasm and are closely apposed to necrotic muscle fibers. The necrotic muscle fiber has moderately dark, but featureless, cytoplasm compared to surrounding intact myofibers (hematoxylin & eosin stain); bar = 10 μm. (B) Large histiocytic cells have large, variably shaped nuclei, often with pale centers (Congo red stain); bar = 10 μm. (C) The necrotic muscle fiber is pale on nicotinamide adenine dinucleotide (NADH) stain. Large neighboring histiocytic cells have dark stained cytoplasm compared to surrounding intact myofibers (NADH stain); bar = 10 μm. (D, E) Large histiocytic cells apposed to, and invading, muscle fibers have characteristics of histiocytic cells with staining for acid phosphatase (D) (arrow), HAM56 (E), and other histiocytic markers (figures 2 and 3). Bars = 20 μm.
Figure 2. Histiocytic cells: relationship to C5b-9 stained muscle fiber cytoplasm.

(A, B). Histiocytes stained for HAM56 (red), a pan-histiocyte marker, are present around, and within, damaged, C5b-9 stained (green) cytoplasm in the periphery of muscle fibers. Bars = 50 μm (A) and 20 μm (B). (C) Large histiocytes stained for HAM56 (red) surround a necrotic muscle fiber with diffusely C5b-9 stained (green) cytoplasm. This diffuse pattern of C5b-9 staining of the cytoplasm of necrotic muscle fibers is the typical pattern observed in most necrotic fibers in other disorders. Bar = 20 μm.
Figure 3. Histiocytic cells: relation to collagen 4–stained muscle fiber basal lamina.

(A) Early pathology: histiocytes stained for S100 (green) are present outside, and over, collagen 4–stained muscle fiber basal lamina (red). Bar = 50 μm. (B) Later pathology: histiocytes stained for S100 (green) are present inside the collagen 4–stained muscle fiber basal lamina (red), and within areas occupied by muscle fibers, but not within the central regions of these areas. Bar = 50 μm. (C) Histiocytes stained for CD68 (green) are present around collagen 4–stained muscle fiber basal lamina (red). Bar = 20 μm. (D) Histiocyte stained for CD163 (green) (arrow) extends through the collagen 4–stained muscle fiber basal lamina (red). Bar = 20 μm. (E) Large histiocytes stained for CD163 (green) inside the collagen 4–stained muscle fiber basal lamina (red). Bar = 20 μm. (F) Large region of 1 or several histiocytes stained for CD163 (green) replacing area of a muscle fiber inside the collagen 4–stained muscle fiber basal lamina (red). Bar = 20 μm.
Histiocytic cells tended to be larger when associated with remnants of necrotic muscle fibers having smaller cross-sectional area (figure 1, A and C). Many of the other necrotic fibers were not invaded, or replaced, by multiple normal-sized histiocytic cells, a pattern different from patterns of myofiber necrosis due to other causes. Toluidine blue–stained plastic sections showed large macrophage-like cells surrounding and invading scattered necrotic muscle fibers. Ultrastructural examination showed that the large cells near myofibers had features of activated histiocytic cells or macrophages (figure 4). The large histiocytic cells featured the following: surface membranes that were wavy and projected thin, irregular pseudopodia; cytoplasm that contained abundant, variably sized, irregularly shaped membrane-bound phagocytic vacuoles containing debris and abundant smooth endoplasmic reticulum and mitochondria; and nuclei with irregular membranes, markedly larger than endomysial macrophages, and prominent nucleoli. Some of the large histiocytic cells contained several nuclei. In early stages of pathology, the histiocytic cells inserted long wavy irregular processes through the muscle fiber basal lamina and then between the basal lamina and the sarcolemma. At some sites, neighboring muscle fiber sarcolemma was damaged or discontinuous. In later stages of muscle fiber pathology (figure 5), changes typical of necrosis were observed with the cytoplasm showing loss of myofibrils and organelles, mitochondria and glycogen being the last to disappear, and replacement of cytoplasm with amorphous granular material. MHC-1 was diffusely upregulated by both histologically normal and abnormal muscle fibers in 3 of the 4 biopsies. Perimysial and endomysial connective tissue and vessels were normal. There were no lymphocyte foci or eosinophils.
Figure 4. Electron microscopy of large-histiocyte-related myofiber necrosis: histiocytic cells and processes.
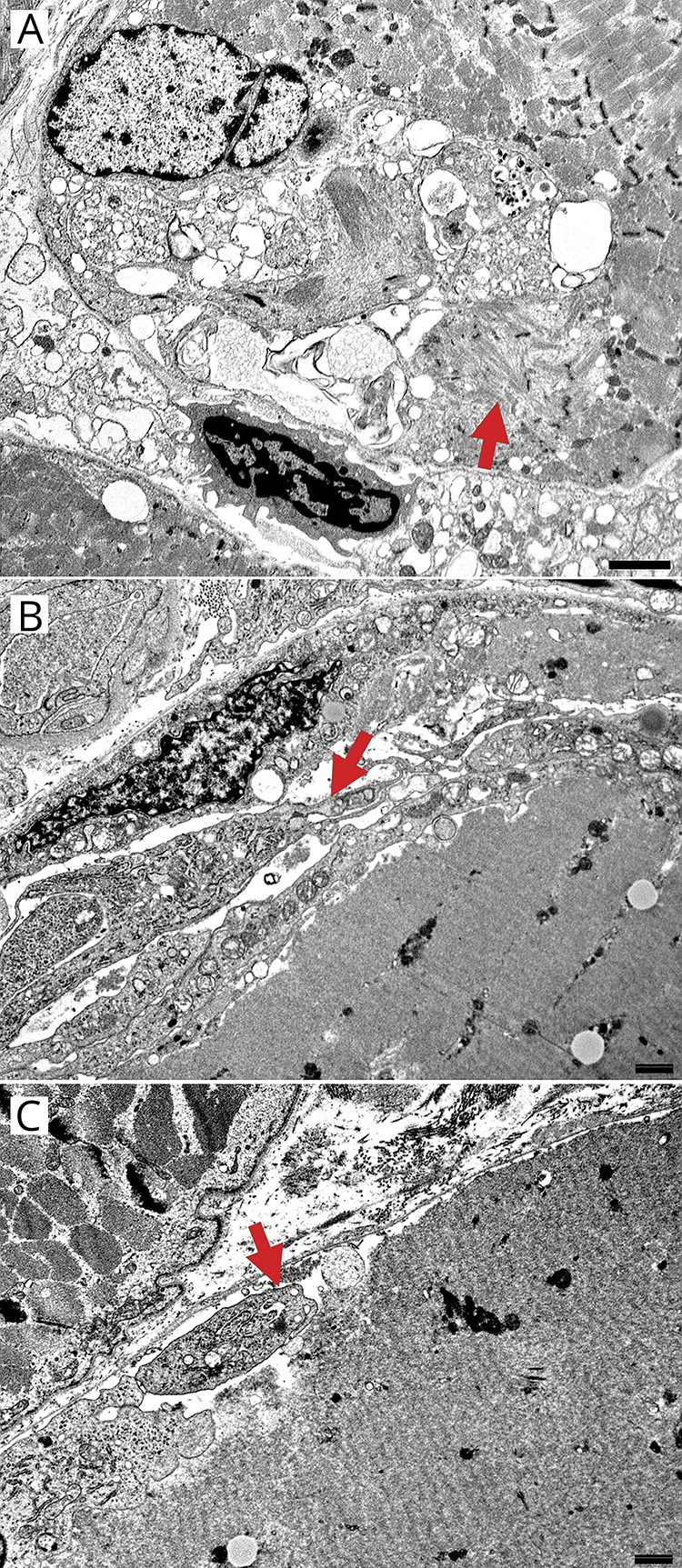
Large cells near myofibers have features of activated histiocytes or macrophages with the following: surface membranes that are wavy and project thin, irregular pseudopodia that project between the muscle fiber basement membrane and surface sarcolemmal membrane; cytoplasm that contains abundant, variably sized, irregularly shaped membrane-bound phagocytic vacuoles containing debris, and abundant smooth endoplasmic reticulum and mitochondria; and nuclei with irregular membranes, markedly larger size than endomysial macrophages, and prominent nucleoli. Some of large histiocytic cells contain several nuclei. (A) Large histiocytic cell apposed to, and invading, a muscle fiber. The histiocyte contains variably sized, and shaped, membrane-bound cytoplasmic vacuoles and a large nucleus (compare large histiocyte nucleus at top left to endomysial cell nucleus at bottom of image). A small region of myofibrillar dissolution neighbors the histiocytic cell (arrow). Bar = 2 μm. (B) Histiocyte processes between basal lamina and necrotic muscle fiber cytoplasm. Histiocyte processes (arrow) are rich in organelles, including smooth endoplasmic reticulum, mitochondria, and vacuoles that are clear or contain granular debris The associated necrotic muscle fiber has pale, featureless cytoplasm with dissolution of myofibrils and sarcoplasmic organelles. Bar = 100 nm. (C) Histiocytic cell process (arrow) between basal lamina and muscle fiber surface contains cytoplasmic vacuoles of varied sizes and irregular shapes. Muscle fiber surface sarcolemma under histiocyte process is disrupted or irregular. Bar = 100 nm.
Figure 5. Electron microscopy of large-histiocyte-related myofiber necrosis: muscle fibers and histiocytic cells.
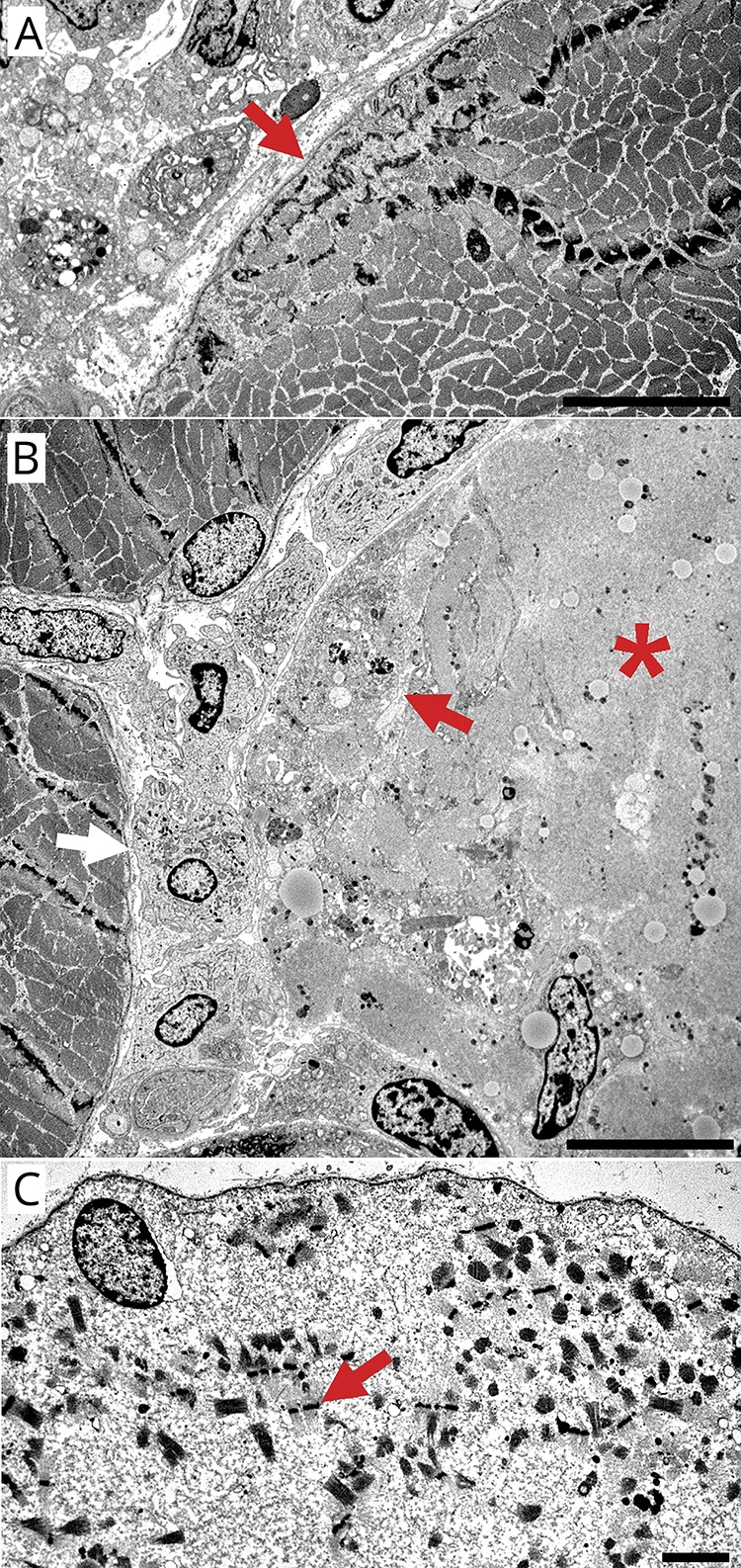
(A) Focal, subsarcolemmal dissolution of myofibrils (arrow) in a muscle fiber. Histiocytic cells are present in the neighboring endomysial connective tissue. Bar = 10 µM. (B) Histiocytic cells surrounding (white arrow), apposed to, and invading (red arrow) a necrotic muscle fiber (*). The necrotic muscle fiber has pale cytoplasm and almost complete dissolution of myofibrils and sarcoplasmic organelles. Non-necrotic muscle fibers are located at the left side in the image. Bar = 10 µM. (C) Necrotic muscle fiber surrounded by intact basal lamina. Misoriented sarcomeres with Z-bands (arrow) and glycogen are scattered, and organelles lost, within amorphous granular material that has replaced muscle fiber cytoplasm. Bar = 2 µM.
Discussion
The pattern of muscle fiber pathology that defined our patients with LHIM included scattered necrotic muscle fibers that were intimately associated with large cells that had histiocytic features (figures 2 and 3). The large LHIM cells were often multinucleated, and closely associated with, or apposed to, muscle fibers (figures 1 and 4). The cells had some histochemical features that were similar to regenerating muscle fibers with their large size, frequent multinucleation, and dark NADH staining (figure 1C). However, the LHIM cells had features that identify them as histiocytes, including staining with multiple histiocyte markers, HAM56, CD68, CD163, and S100 (figures 2 and 3), and ultrastructural features, with cytoplasmic vacuoles and surface membrane processes (figure 4). A common feature of the LHIM cells was finger-like processes that protruded between, and separated, muscle fiber basal lamina and the sarcolemmal plasma membrane. The myofiber necrosis in our patients could be related to sarcolemmal damage produced by the neighboring histiocytic cells and their processes.
Myofiber necrosis was ongoing. Individual necrotic muscle fibers were in different stages of damage with varying degrees of replacement by large histiocyte-like cells. Some histiocytic cells were localized near, or outside, the muscle fiber basal lamina (figure 3A). Other histiocytic cell collections surrounded muscle fibers but within the basal lamina (figure 3B). Individual necrotic muscle fibers commonly had contrasting patterns of histochemical staining of cytoplasm, pale on NADH but dark and featureless (reduced staining of internal architecture) on H&E and Gomori trichrome. The mechanisms underlying dark stained cytoplasm of necrotic muscle fibers may be related to calcium leakage through damaged sarcolemma into the sarcoplasm causing loss of sarcoplasmic reticulum staining or hypercontraction of the myofibrillar apparatus. Similar dark staining of necrotic muscle fibers occurs in dystrophinopathies and other disorders with defects in sarcolemmal membranes.18–20
It seems likely that LHIM is an immune disorder, despite the paucity of lymphocyte foci, based on its subacute onset that was parainfectious/paraneoplastic in 3 patients; variant-histiocyte-associated muscle fiber necrosis; MHC-1 upregulation by muscle fibers in most patients; and absence of the histiocytic neoplasm marker, phospho-ERK, in most LHIM histiocytes. LHIM can be considered in the context of common clinical classifications of immune myopathies, which include polymyositis, dermatomyositis, inclusion body myositis, and necrotic myopathies.1,2 Polymyositis and inclusion body myositis are unlikely because no mononuclear cell inflammatory foci or inclusion bodies were identified in LHIM muscles. Dermatomyositis is excluded clinically, as there were no skin changes, and pathologically as there was no perifascicular predominant pathology in LHIM muscle biopsies, or evidence of damage to vessels or connective tissue.
Necrotic myopathies (tables 2 and 3) are a possible class within which LHIM could be included. General categories of systemic myopathies with abundant necrosis include hereditary, immune, toxic, and vascular (table 2). No features of the history or muscle pathology of our patients with LHIM suggested hereditary, vascular, or toxic disorders. Immune myopathies with myofiber necrosis but little lymphocyte inflammation (table 3) include the signal recognition particle (SRP) antibody-associated myopathy,13 immune myopathies with perimysial pathology (IMPP),5 and regional ischemic immune myopathy (RIIM).7 LHIM has abundant muscle fiber necrosis in common with each of these other syndromes and could be classified as a new type of necrotic myopathy. However, the classification of myopathies based on the presence of myofiber necrosis has several problems. One important issue is that, other than the necrosis, pathologic features vary markedly among the different immune myopathies with abundant necrosis suggesting that these disease syndromes have very different underlying pathogenic mechanisms (tables 2 and 3). SRP myopathy has prominent endomysial connective tissue pathology, with proliferation even early in its course, which is not present in LHIM.13 IMPP have perimysial connective tissue pathology.5 RIIM is likely a vascular disorder.7 Neither perimysial connective tissue nor vessels are damaged in LHIM. Further, patterns of distribution of necrotic myofibers differ among acquired immune myopathies with prominent necrosis. RIIM has clusters of necrotic fibers.7 IMPP has a predominance of necrotic fibers near the edge of fascicles.5 LHIM, like SRP antibody-related immune myopathy, has randomly scattered necrotic fibers,13 but the neighboring large histiocytic cells are distinctive to LHIM, suggesting that cellular, rather than humoral or structural, mechanisms induce the myofiber necrosis. Overall, LHIM adds to the disparity of pathologic features in different immune myopathies with prominent muscle fiber necrosis. LHIM could be considered a histiocytic immune myopathy3 based on its main type of immune cells. However, LHIM differs from other syndromes in this group. Although multinucleated large cells were present, granulomatous myopathy21,22 and inflammatory myopathy with abundant macrophages (IMAM)23 are both characterized by focal histiocytic cell collections that were not observed in LHIM, except associated with necrotic muscle fibers. Further, the abundant muscle fiber necrosis in our patients with LHIM is not typical of granulomatous myopathies or IMAM.
Table 2.
Myopathies with abundant muscle fiber necrosis: comparative pathology

Table 3.
Immune myopathies with prominent myofiber necrosis: comparative muscle pathology, laboratory, and clinical features

Common clinical neuromuscular features in patients with LHIM included a subacute onset of moderately severe muscle pain, predominantly proximal, symmetric weakness, a very high serum CK, and improvement of weakness after treatment. As a group, patients with LHIM have among the highest serum CKs reported in immune myopathies. At presentation, some of our patients were initially diagnosed as having rhabdomyolysis. Muscle pathology showing an ongoing, rather than monophasic, process and the distinctive variant large histiocytic cells adjacent to necrotic muscle fibers distinguish LHIM from rhabdomyolysis. The myopathy in our patients was always associated with concurrent systemic disorders and was parainfectious/paraneoplastic in 3 patients. Anemia was documented during most of the disease course of all patients. With a full syndrome of HLH having been diagnosed in 1 patient, one might speculate that LHIM, with its atypical histiocytes in muscle, could be a subset of macrophage activation syndromes. Myopathies have only rarely been reported in acquired, systemic HLH or macrophage activation syndromes.24–27 The pattern of histiocyte staining in LHIM is the same as that seen in our laboratory for cells associated with muscle fiber necrosis in other acquired and hereditary myopathies. The LHIM cells did not stain for antigens, such as CD1a, CD207, or phospho-ERK, which would support variants of systemic histiocytoses or malignancies.15,28,29 Examination of circulating cytokines could provide information regarding the pathogenesis of LHIM and its histiocytic cells, which have unusual morphology and location.
Limitations of our study include the small number of patients. LHIM is likely to be an unusual syndrome. Pathologic reexamination of disorders previously diagnosed as idiopathic ongoing necrotic myopathies may lead to identification of additional patients with LHIM. Our study stained the histiocytes in frozen muscle samples, while standard characterization of cells in the histiocytoses is performed in fixed tissue.15 This study emphasizes the utility of pathologic analysis of muscle biopsies in characterization and evaluation of acquired immune and inflammatory myopathies. The target of the immune process in LHIM appears to be muscle fibers. However, the molecular nature of the antigenic targets on muscle fibers is uncertain. In any case, the identification of LHIM pathology seems to be useful as it characterizes myopathies that may improve or remit after treatment and predicts systemic features that are associated with the myopathy syndrome.
Glossary
- CK
creatine kinase
- EBV
Epstein-Barr virus
- H&E
hematoxylin & eosin
- HLH
hemophagocytic lymphohistiocytosis
- IIM
immune and inflammatory myopathies
- IMAM
inflammatory myopathy with abundant macrophages
- IMPP
immune myopathies with perimysial pathology
- LHIM
large-histiocyte-related immune myopathy
- MHC-1
class I human major histocompatibility complex
- NADH
nicotinamide adenine dinucleotide
- RIIM
regional ischemic immune myopathy
- SRP
signal recognition particle
Author contributions
A. Pestronk: study concept and design, acquisition of data, analysis and statistical analysis and interpretation, critical revision of the manuscript for important intellectual content, study supervision. N. Sinha: acquisition of data, critical revision of the manuscript for important intellectual content. Z. Alhumayyd: acquisition of data, critical revision of the manuscript for important intellectual content. C. Ly: acquisition of data, critical revision of the manuscript for important intellectual content. R. Schmidt: analysis and interpretation, critical revision of the manuscript for important intellectual content. R. Bucelli: study concept and design, acquisition of data, analysis, critical revision of the manuscript for important intellectual content, study supervision.
Study funding
Neuromuscular Research Fund.
Disclosure
The authors report no disclosures relevant to the manuscript. Go to Neurology.org/N for full disclosures.
References
- 1.Mammen A. Autoimmune muscle disease. Handb Clin Neurol 2016;133:467–484. [DOI] [PubMed] [Google Scholar]
- 2.Allenbach Y, Benveniste O, Goebel HH, Stenzel W. Integrated classification of inflammatory myopathies. Neuropathol Appl Neurobiol 2017;43:62–81. [DOI] [PubMed] [Google Scholar]
- 3.Pestronk A. Acquired immune and inflammatory myopathies: pathologic classification. Curr Opin Rheumatol 2011;23:595–604. [DOI] [PubMed] [Google Scholar]
- 4.Pestronk A. Immune and inflammatory myopathies: Neuromuscular Disease Center. Available at: neuromuscular.wustl.edu/antibody/infmyop.htm. Accessed March 2019.
- 5.Bucelli RC, Pestronk A. Immune myopathies with perimysial pathology (IMPP): clinical and laboratory features. Neurol Neuroimmunol Neuroinflamm 2018;5:e434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pestronk A, Kos K, Lopate G, Al-Lozi MT. Brachio-cervical inflammatory myopathies: clinical, immune, and myopathologic features. Arthritis Rheum 2006;54:1687–1696. [DOI] [PubMed] [Google Scholar]
- 7.Cai C, Alshehri A, Choksi R, Pestronk A. Regional ischemic immune myopathy: a paraneoplastic dermatomyopathy. J Neuropathol Exp Neurol 2014;73:1126–1133. [DOI] [PubMed] [Google Scholar]
- 8.Pinal-Fernandez I, Mammen AL. Spectrum of immune-mediated necrotizing myopathies and their treatments. Curr Opin Rheumatol 2016;28:619–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pestronk A. Muscle fiber necrosis: Neuromuscular Disease Center. Available at: neuromuscular.wustl.edu/pathol/necrosis.htm. Accessed March 2019.
- 10.Engel AG, Biesecker G. Complement activation in muscle fiber necrosis: demonstration of the membrane attack complex of complement in necrotic fibers. Ann Neurol 1982;12:289–296. [DOI] [PubMed] [Google Scholar]
- 11.Allenbach Y, Arouche-Delaperche L, Preusse C, et al. Necrosis in anti-SRP+ and anti-HMGCR + myopathies: role of autoantibodies and complement. Neurology 2018;90:e507–e517. [DOI] [PubMed] [Google Scholar]
- 12.Mozaffar T, Pestronk A. Myopathy with anti-Jo-1 antibodies: pathology in perimysium and neighbouring muscle fibres. J Neurol Neurosurg Psychiatry 2000;68:472–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miller T, Al-Lozi MT, Lopate G, Pestronk A. Myopathy with antibodies to the signal recognition particle: clinical and pathological features. J Neurol Neurosurg Psychiatry 2002;73:420–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pestronk A. Muscle fiber necrosis: Neuromuscular Disease Center. Available at: neuromuscular.wustl.edu/lab/mbiopsy.htm#stain. Accessed March 2019.
- 15.Emile JF, Abla O, Fraitag S, et al. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood 2016;127:2672–2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ballester LY, Cantu MD, Lim KPH, et al. The use of BRAF V600E mutation-specific immunohistochemistry in pediatric Langerhans cell histiocytosis. Hematol Oncol 2018;36:307–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tran G, Huynh TN, Paller AS. Langerhans cell histiocytosis: a neoplastic disorder driven by Ras-ERK pathway mutations. J Am Acad Dermatol 2018;78:579-e4. [DOI] [PubMed] [Google Scholar]
- 18.Bodensteiner JB, Engel AG. Intracellular calcium accumulation in Duchenne dystrophy and other myopathies: a study of 567,000 muscle fibers in 114 biopsies. Neurology 1978;28:439–446. [DOI] [PubMed] [Google Scholar]
- 19.Karpati G, Carpenter S. Micropuncture lesions of skeletal muscle cells: a new experimental model for the study of muscle cell damage, repair and regeneration. In: Schotland DE, ed. Disorders of the Motor Unit. New York: Wiley; 1982:517–533. [Google Scholar]
- 20.Pestronk A, Parhad IM, Drachman DB, Price DL. Membrane myopathy: morphological similarities to Duchenne muscular dystrophy. Muscle Nerve 1982;5:209–214. [DOI] [PubMed] [Google Scholar]
- 21.Takanashi T, Suzuki Y, Yoshino Y, Nonaka I. Granulomatous myositis: pathologic re-evaluation by immunohistochemical analysis of infiltrating mononuclear cells. J Neurol Sci 1997;145:41–47. [DOI] [PubMed] [Google Scholar]
- 22.Mozaffar T, Lopate G, Pestronk A. Clinical correlates of granulomas in muscle. J Neurol 1998;245:519–524. [DOI] [PubMed] [Google Scholar]
- 23.Bassez G, Authier FJ, Lechapt-Zalcman E, et al. Inflammatory myopathy with abundant macrophages (IMAM): a condition sharing similarities with cytophagic histiocytic panniculitis and distinct from macrophagic myofasciitis. J Neuropathol Exp Neurol 2003;62:464–474. [DOI] [PubMed] [Google Scholar]
- 24.Kumakura S, Murakawa Y. Clinical characteristics and treatment outcomes of autoimmune-associated hemophagocytic syndrome in adults. Arthritis Rheumatol 2014;66:2297–2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ramos-Casals M, Brito-Zerón P, López-Guillermo A, Khamashta MA, Bosch X. Adult haemophagocytic syndrome. Lancet 2014;383:1503–1516. [DOI] [PubMed] [Google Scholar]
- 26.Cron RQ, Davi S, Minoia F, Ravelli A. Clinical features and correct diagnosis of macrophage activation syndrome, Expert Rev Clin Immunol, 2015;11:1043–1053. [DOI] [PubMed] [Google Scholar]
- 27.Azuma K, Tamura M, Kurajoh M, et al. A case of anti-aminoacyl tRNA synthetase antibody syndrome complicated by hemophagocytic syndrome [in Japanese]. Nihon Rinsho Meneki Gakkai Kaishi 2016;39:538–544. [DOI] [PubMed] [Google Scholar]
- 28.Vannella KM, Wynn TA. Mechanisms of organ injury and repair by macrophages. Annu Rev Physiol 2017;79:593–617. [DOI] [PubMed] [Google Scholar]
- 29.Weaver LK, Behrens EM. Hyperinflammation, rather than hemophagocytosis, is the common link between macrophage activation syndrome and hemophagocytic lymphohistiocytosis. Curr Opin Rheumatol 2014;26:562–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hudgson P, Pearce GW, Walton JN. Pre-clinical muscular dystrophy: histopathological changes observed on muscle biopsy. Brain 1967;90:565–576. [DOI] [PubMed] [Google Scholar]
- 31.Mokri B, Engel AG. Duchenne dystrophy: electron microscopic findings pointing to a basic or early abnormality in the plasma membrane of the muscle fiber. Neurology 1975;25:1111–1120. [DOI] [PubMed] [Google Scholar]
- 32.Hardy D, Besnard A, Latil M, et al. Comparative study of injury models for studying muscle regeneration in mice. PLoS One 2016;11:e0147198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Karpati G, Carpenter S, Melmed C, Eisen AA. Experimental ischemic myopathy. J Neurol Sci 1974;23:129–161. [DOI] [PubMed] [Google Scholar]
- 34.Alshehri A, Choksi R, Bucelli R, Pestronk A. Myopathy with anti-HMGCR antibodies: perimysium and myofiber pathology. Neurol Neuroimmunol Neuroinflamm 2015;2:e124. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
More detailed, anonymized case reports are available to qualified investigators. Additional images of LHIM myopathology are available on our website.4