

SUMMARY
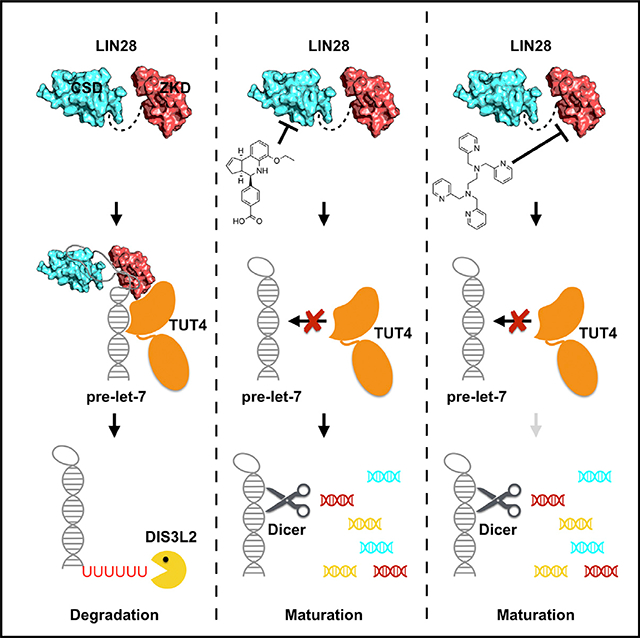
LIN28 is an RNA-binding protein that regulates the maturation of the let-7 family of microRNAs by bipartite interactions with let-7 precursors through its two distinct cold shock and zinc-knuckle domains. Through inhibition of let-7 biogenesis, LIN28 functions as a pluripotency factor, as well as a driver of tumorigenesis. Here, we report a fluorescence polarization assay to identify small-molecule inhibitors for both domains of LIN28 involved in let-7 interactions. Of 101,017 compounds screened, six inhibit LIN28:let-7 binding and impair LIN28-mediated let-7 oligouridylation. Upon further characterization, we demonstrate that the LIN28 inhibitor TPEN destabilizes the zinc-knuckle domain of LIN28, while LI71 binds the cold shock domain to suppress LIN28’s activity against let-7 in leukemia cells and embryonic stem cells. Our results demonstrate selective pharmacologic inhibition of individual domains of LIN28 and provide a foundation for therapeutic inhibition of the let-7 biogenesis pathway in LIN28-driven diseases.
In Brief
LIN28 is an oncogenic protein that promotes transformation in malignancy by suppressing the maturation of let-7 microRNAs. Wang et al. developed a high-throughput screening strategy and identified inhibitors of LIN28 that restore mature let-7 levels.
Graphical Abstract
INTRODUCTION
The LIN28 family of proteins was first discovered in Caenorhabditis elegans as regulators of developmental timing (Ambros and Horvitz, 1984). On a molecular level, LIN28 is an RNA-binding protein that modulates levels of mature let-7 microRNAs by blockade of the Microprocessor and Dicer processing steps necessary for let-7 maturation (Rybak et al., 2008; Viswanathan et al., 2008). Recent studies have revealed that LIN28 also induces oligouridylation of let-7 precursors by recruitment of terminal uridylyltransferases (TUTases), which triggers let-7 degradation by the Dis3L2 exonuclease (Chang et al., 2013; Hagan et al., 2009; Heo et al., 2008). Mammals possess two LIN28 paralogs, LIN28A and LIN28B, which are aberrantly expressed in ~15% of human cancer cell lines and are associated with advanced disease and poor prognosis (Viswanathan et al., 2009). Aberrant expression of LIN28 has been shown to induce T cell lymphoma, neuroblastoma, Wilms tumor, and hepatoblastoma, as well as intestinal and colorectal adenocarcinomas (Beachy et al., 2012; Molenaar et al., 2012; Nguyen et al., 2014; Tu et al., 2015; Urbach et al., 2014). Knocking down LIN28 or restoring let-7 levels effectively suppresses Wilms tumor and hepatoblastoma in mice (Nguyen et al., 2014; Urbach et al., 2014). The mechanism of LIN28’s function in tumor maintenance seems to be as an epigenetic switch involving a negative-feedback loop between LIN28 and let-7 (Iliopoulos et al., 2009; Yang et al., 2010). Given that high let-7 levels are generally tumor suppressive, it is, therefore, theoretically possible to attenuate the malignant state by inverting the LIN28/let-7 switch in LIN28-driven tumors. The normal developmental suppression of LIN28 expression during the transition from fetal to adult life-such that little, if any, LIN28 is normally found in adult tissues—potentially limits adverse effects of specific LIN28-targeting therapies (Rowe et al., 2016).
There are rare cases of successful inhibition of RNA-binding proteins using small molecules; however, the RNA-binding surface of LIN28 offers a unique opportunity. As demonstrated by our published crystal structure (Nam et al., 2011), LIN28 contains two domains that contribute to let-7 suppression. The N-terminal cold shock domain (CSD) remodels the conformation of the let-7 precursors (pre-element; pre-element of let-7f-1 [preE-let-7]) by binding to their terminal loops (Mayr et al., 2012), and the C-terminal zinc-knuckle domain (ZKD; CCHCx2 domain) binds to a highly conserved GGAG sequence motif with high specificity (Loughlin et al., 2011; Nam et al., 2011). Deletion of the GGAG motif renders LIN28 ineffective in suppression of let-7 maturation (Nam et al., 2011). Due to a relatively larger RNA-binding surface, the CSD contributes to most of the LIN28:let-7 binding affinity; however, we found that the ZKD is crucial for both blockage of Dicer and recruitment of TUTases (Wang et al., 2017). We previously reported that point mutations of either CSD or ZKD could significantly restore let-7 processing and maturation (Nam et al., 2011), suggesting that inhibition of either domain could modulate let-7 levels. Nevertheless, the disparities between the affinities of CSD and ZKD for RNA limit screening strategies to identify ZKD inhibitors.
Here, we report a fluorescence polarization (FP) assay sufficiently sensitive to identify small-molecule inhibitors of both domains of LIN28. We performed a high-throughput screen of 101,017 compounds from commercially available libraries. Among those, six compounds, including TPEN and LI71, were found to effectively inhibit LIN28:let-7 binding as well as LIN28-mediated let-7 oligouridylation. Our data revealed that TPEN induces conformational changes of the ZKD, while LI71 functions by competing for the RNA-binding site of the CSD. These studies demonstrate the ability to selectively target binding domains of RNA-binding proteins, which may prove of value in therapy of cancers driven by aberrant LIN28 activity.
RESULTS
Design of a Fluorescence Polarization Assay to Identify LIN28 Inhibitors
To probe the interaction between LIN28 and let-7, we developed a fluorescence polarization assay with recombinant human LIN28Δ (residues 16–187 of wild-type human LIN28A), a LIN28 construct optimized for bacterial expression that retains full biological function (Figures 1A, S1A, and S1B) (Nam et al., 2011). Based on the crystal structure of human LIN28: preE-let-7f-1 complex (Wang et al., 2017), we designed a FAM-labeled human preE-let-7 (Figure 1A). In the fluorescence polarization assay, LIN28Δ was titrated into solutions containing 2 nM FAM-labeled preE-let-7f-1 probes; and after incubation, we measured fluorescence polarization (Figure 1B). In agreement with previous studies, the LIN28Δ:preE-let-7f-1 complex showed a dissociation constant of ~20 nM (Mayr et al., 2012; Nam et al., 2011). We then used unlabeled preE-let-7f-1 to compete with the labeled probe and measured IC50 (inhibitory concentration of 50% substrate binding). The concentration of LIN28Δ was set to be the dissociation constant obtained from the earlier experiment for maximum signal. As shown in Figure 1C, the addition of unlabeled preE-let-7f-1 resulted in a significant decrease in fluorescence polarization (mP) with an IC50 of 44 nM, indicating that this assay is capable of identifying inhibition of the binding between LIN28 and RNA.
Figure 1. Design of a Fluorescence Polarization Assay to Identify LIN28 Inhibitors.

(A) Schematic view of the human LIN28 and preE-let-7f-1 used in our fluorescence polarization assay. CSD and ZKD are colored as cyan and red, respectively. In preE-let-7f-1, residues that bind to the CSD and ZKD are also colored (PDB: 5UDZ).
(B) Fluorescence polarization assay of LIN28 binding to preE-let-7f-1 (n = 3 replicates). Error bars denote mean ± SEM.
(C) Competition fluorescence polarization assay using unlabeled preE-let-7f-1
(D) Competition fluorescence polarization assay using an unlabeled CSD riboprobe. CSD RNA is sufficient to inhibit LIN28/let-7 interaction.
(E and F) Competition fluorescence polarization assay using unlabeled ZKD riboprobe. ZKD RNA (UAGGAGA) is a weak competitor to preE-let-7f-1. LIN28ΔF73A (F) shows more increased sensitivity than LIN28Δ (E) for detecting ZKD inhibitions by UAGGAGA RNA.
The green star in (B)-(F) represents 3’ FAM label. The black arrow in (B) and (F) indicates the F73A mutation site.
Next, we sought to test the inhibition of individual domains of LIN28 using this assay, since small-molecule inhibitors are expected to target either of the domains of human LIN28Δ. We first designed CSD-binding (GGGGUAGUGAUUUUACCCU GUUUA) and ZKD-binding (UAGGAGA) riboprobes based on the crystal structure of the human LIN28:preE-let-7f-1 complex and measured their IC50s using our assay (Wang et al., 2017) (Figure 1C). We found that the CSD-binding RNA could effectively inhibit LIN28:let-7f-1 at an IC50 of 157 nM (Figure 1D), while the ZKD-binding RNA has a significantly higher IC50 of 3.5 μM (Figure 1E), indicating a relatively higher affinity CSD:RNA interaction that might favor identification of CSD inhibitors over ZKD inhibitors in screening studies, as reported previously (Nam et al., 2011; Wang et al., 2017).
To improve the potential sensitivity of the fluorescence polarization assay for detecting ZKD inhibitors, we introduced a phenylalanine-73-to-alanine (F73A) mutation in the LIN28 protein. F73 directly contributes to let-7 binding in the CSD, and we have previously shown that an F73A mutation has a particularly strong effect on binding of RNAs bearing a mutation in the GGAG motif (GGAG deletion or GGAG-to-GGAU substitution) (Nam et al., 2011). Therefore, we hypothesized that, in our screening assay, this mutation would increase sensitivity to detecting small molecules that bind in the ZKD pocket. To test this, we performed a similar competitive fluorescence polarization assay using LIN28ΔF73A and found that the uaGGAGa competitor showed an IC50 of 1.2 μM, 3-fold less than the IC50 for LIN28Δ (Figure 1F).
To further demonstrate the sensitivity of the human LIN28ΔF73A fluorescence polarization assay, we generated a double-mutant LIN28ΔF73A/Y140A construct. Y140 was previously shown to be critical for ZKD:RNA binding (Nam et al., 2011). The rationale for this approach was to replace a large hydroxyphenyl side chain with a small methyl group to mimic a small molecule binding the ZKD pocket. The dissociation constant (Kd) of preE-let-7fFAM to this modified target increases by 8.1-fold, from 72.41 nM (LIN28ΔF73A) to 588.6 nM (LIN28ΔF73A/Y140A); however, the increase between the wild-type (WT) and Y140A mutation is only 2.7-fold, from 28.52 nM (LIN28Δ) to 78.50 nM (LIN28ΔY140A) (Table S1; Figures S1C and S1D). Interpreting these data as the Y140A mutation simulating the effect of a small molecule binding in the ZKD pocket, the LIN28ΔF73A construct provides a screening target that we predict to support detection of both CSD and ZKD inhibitors in fluorescence polarization screenings.
Based on our results, human LIN28ΔF73A was used to develop a high-throughput screening (HTS) assay in a 384-well format. UAGGAGA RNA and buffer alone were used as positive and negative controls, respectively. We evaluated pre-elements from five family members of let-7 and calculated their Z-factors (Table S2). preE-let-7f-1FAM had the highest Z score (0.83). Moreover, the existence of a corresponding X-ray structure, which would provide a structural basis for further structure-activity relation (SAR) studies, rendered it the most suitable candidate for HTS.
HTS Assay Identifies Candidate Compounds Inhibiting the LIN28:let-7 Interaction
To identify small-molecule inhibitors of the LIN28:let-7 interaction, we performed HTS using our fluorescence polarization assay (Figure 2A). For each screening session, freshly prepared human LIN28ΔF73A was mixed with 2 nM prE-let-7fFAM and then added to 384-well plates. After a short incubation, a liquid-handling robot transferred library compounds to corresponding wells of the assay plate at a final concentration of 40 μM, followed by overnight incubation. We used the UAGGAGA RNA as positive control, which we manually pipetted to the assay plate with the final concentration of 1 μM. Wells with neither positive controls nor compounds were labeled as negative controls. We recorded fluorescence polarization and total fluorescence intensity (FI) for each plate and calculated Z scores for each plate during screening (Figure 2A).
Figure 2. High-Throughput Screening Assay Identifies a Panel of Candidate Compounds Inhibiting the LIN28:let-7 Interaction.

(A) High-throughput screening assay strategy (left and middle panel). Correlations between duplicate screening results (right panel). Each spot represents a compound screened. The x axis and y axis are the duplicate fluorescence polarization (FP) readings of the same molecule. All compounds lie near the diagonal line, indicating that the screening assay is robust.
(B) Left: dose-response FP assays overlays. Right: distributions of the IC50s for 350 cherry-picked inhibitors.
(C) A selection of oligouridylation assays to identify LIN28 inhibitors that can block LIN28-mediated oligouridylation. *pre-let-7g; **oligouridylated pre-let-7g.
The gray-shaded triangle is an illustration of the selectivity of the described screening pipeline, including the numbers of compounds at each step.
The complete HTS spanned 17 compound libraries (101,017 compounds) in duplicate. We selected hit compounds from the primary screen based on Z score, fluorescence polarization, uniqueness of the chemical fingerprint, and promiscuity indicated by the PubChem screening database (Figures S2A–S2C). In addition, we used a web application developed in house (MightyScreen; http://mightyscreen.net) to visually examine the screening data quality and exclude compounds with outlier fluorescence intensities (Figures S2A and S2B). As a result, 350 compounds were cherry-picked as 2-μL stocks, and dose-response fluorescence polarization titrations were performed using a liquid dispensing robot to measure their IC50s (Figure 2B). We selected 53 compounds with IC50 values ranging from 200 nM to 10 μM (Figure 2C), of which 42 compounds were commercially available and purchased (Table S3). About 70% (27) of the purchased compounds retained their activities when validated in the same dose-response fluorescence polarization assay, which is consistent with the screening facility’s confirmation rate. Cell viability assays were performed on selected compounds (Figures S2D and S2E).
To investigate effects of the identified inhibitors on let-7 degradation, we developed an assay to test the inhibition of LIN28-mediated let-7 oligouridylation, which is the first step in degradation of let-7 precursors. The assay involved incubation of the mouse LIN28Δ (residues 16–184 of wild-type mouse LIN28A), mouse terminal uridylyltransferase 4 (TUT4, residues 230–1,424), and pre-let-7g at 37°C with 40 μM compound inhibitors. After incubation, we terminated the reaction by adding SDS and subjected the reaction to denaturing PAGE under conditions wherein the pre-let-7g and oligouridylated pre-let-7g are separated (Figure 2C). It is worth noting that monouridylated pre-let-7g can be distinguished from oligouridylated pre-let-7g in this assay, as demonstrated previously (Wang et al., 2017). Since the sequence and structure of the RNA-binding regions of mouse LIN28A and human LIN28A are nearly identical (Figure S1A) (Nam et al., 2011; Wang et al., 2017), we expected LIN28 inhibitors to demonstrate comparable activities on LIN28. As shown in Figures 3, S2F, and S2G, DAQ-B1 (demethylasterriquinone B1, LI2), BVT-948 (LI3), Gossypol (LI11), LI20, TPEN (LI38), and LI71 completely inhibited LIN28-mediated oligouridylation at a concentration of 40 μM, similar to the positive control reactions in which LIN28 is excluded. Interestingly, LI20 and LI71 are the only two compounds that share a benzoic acid moiety and have not been extensively studied. Our results demonstrate that inhibitors identified using the fluorescence polarization assay are biologically active by inhibiting LIN28-mediated oligouridylation of let-7 microRNAs.
Figure 3.

Structure of Identified LIN28 Inhibitors that Can Abolish LIN28-Mediated Oligouridylation
TPEN Is a Potent Inhibitor of the LIN28 ZKD
TPEN (LI38) is a cell-permeable compound that was previously shown to induce apoptosis by chelating Zn2+ in cells (McCabe et al., 1993). In our fluorescence polarization screen, TPEN exhibits high potency with an IC50 of 2.5 μM. Therefore, we hypothesize that TPEN ejects Zn2+ and disrupts the ZKD of LIN28. To test this, we performed a heteronuclear single-quantum coherence (HSQC) experiment using 15N-labeled mouse LIN28Δ in complex with unlabeled preE-let-7d. We collected the H-15N HSQC spectrum of the LIN28Δ:preE-let-7d complex in the presence of TPEN and found that a number of peaks had significant reduction in intensity compared to the spectrum of LIN28Δ:preE-let-7d complex (Figure 4A), indicating line-broadening effects caused by structure rearrangement. Based on the assignments from our previous study (Nam et al., 2011), we identified residues of LIN28 that were disturbed by addition of TPEN and found that these residues were mostly located within the zinc knuckles as well as RNA-binding surface of the ZKD (Figure 4B). As shown in Figure 4C, CSD residues L87 and L104 showed no intensity changes with the addition of TPEN; however, two zinc-knuckle residues, C139 and C161, as well as Y140, a residue critical for the binding between ZKD and GGAG RNA (Nam et al., 2011), showed reduced intensities that correlate with the concentration of TPEN, suggesting drastic structural changes within both zinc knuckles and RNA-binding sites of the ZKD. TPEN shows toxicity to LIN28-expressing mouse embryonic stem cells as well as HeLa cells that do not express LIN28; therefore, the toxicity of TPEN is likely due to nonspecific effects of zinc chelation. Our data show that TPEN can effectively inhibit the ZKD of LIN28, indicating that our assay has adequate sensitivity to detect inhibitors that target the ZKD.
Figure 4. TPEN Is a Potent Inhibitor of the LIN28 ZKD.

(A) H-15N HSQC spectrum of mouse LIN28:preE-let-7d complex with residue assignments.
(B) Model representation of residues affected by TPEN that are evident through intensity changes. Modeling was done based on the crystal structure of mouse LIN28:preE-let-7d complex (PDB: 3TRZ).
(C) Blowup spectrums showing that TPEN selectively affects ZKD but not CSD.
LI71 Inhibits LIN28-Mediated Oligouridylation by Interacting with the CSD of LIN28
We focused on LI71, since this compound shares the same benzoic acid moiety with LI20 but has better water solubility. To investigate the mechanism of inhibition of LI71, we sought to map residues influenced by LI71 using HSQC titrations (LI20 was excluded from this study due to poor solubility in solution). We added LI71 to a solution containing a mouse LIN28Δ:preE-let-7d complex and collected the 2D H-15N HSQC spectrum. We identified minor intensity reductions for a number of residues involved in RNA binding in LIN28 (Figure S3A); however, these residues spread over both the CSD and ZKD of LIN28, without an obvious focused location, suggesting that the LI71 interferes with LIN28:let-7 interactions but without clear identification of the binding site in this assay.
To determine whether LI71 interacts with LIN28, but not RNA, we carried out an fluorescence polarization dose-response assay using pre-elements of four members of the let-7 family—preE-let-7f, preE-let-7a, preE-let-7d, and preE-let-7g—and measured their IC50s for LI71 (Figure 5A). These data show moderate differences in inhibition potencies with their IC50s against human LIN28ΔF73A, all within the low micromolar range (Table S4). Given the diversities of the preE-let-7 sequences, we reasoned that LI71 either acts on LIN28 protein or the conserved regions of let-7 pre-elements.
Figure 5. LI71 Inhibits LIN28 by Competing for the RNA Binding Site in the CSD.

(A) LI71 inhibits the interactions between LIN28 and preE-let-7a, preE-let-7d, preE-let-7f, and preE-let-7g. The IC50 of LI71 in our primary fluorescence polarization assay is ~7 μM. The IC50 of LI71 in oligouridylation assay is ~27 μM.
(B) Surface presentation of CSD and saturation transfer difference (STD) spectrum of LI71 in the presence of LIN28, preE-let-7, and LIN28 variants. When LI71 is in close contact with an irradiated bio-molecule, saturation transfer difference peaks can be observed.
(C) Dose-response oligouridylation assay for LI71. *pre-let-7g; **oligouridylated pre-let-7g. The bottom band intensity is measured and plotted. LI71 shows an IC50 of 27 μM.
(D) LI71 variants and their IC50s determined from dose-response fluorescence polarization assay.
To validate that LI71 directly binds to LIN28 and its target domain, we performed saturation transfer difference (STD) spectroscopy experiments separately on human LIN28Δ and preE-let-7f, in which the protein or RNA, but not LI71, is excited; therefore, LI71 peaks are only present when there is energy transfer between the macromolecule and LI71 due to close contact. The experiments were carried out in deuterated buffer to minimize the background noise and the irradiation frequency was set to excite only protein. After irradiation and a 2-s delay, we collected the H spectra. As shown in Figure 5B, saturation transfer difference peaks of LI71 occur in the presence of LIN28Δ but not preE-let-7f, indicating a direct interaction between LIN28 and LI71, in agreement with our previous results showing LI71 is likely targeting LIN28 but not RNA. The interaction of LIN28:LI71 is also evident by line-broadening effects induced by LIN28. We then collected the 1D H and COSY spectra of LI71 and carried out assignments of the saturation transfer difference peaks for LI71 (Figure S3B). Based on the assignment and saturation transfer difference spectrum, we identified hydrogen atoms that are present in saturation transfer difference spectra, which revealed that the benzoic acid head, the edge of the cyclopentaquinoline body, and the ethoxy tail were all in close contact with LIN28Δ (Figure S3C). We next performed the same saturation transfer difference experiments using CSD (residues 16–136) and ZKD (residues 120–181) and observed that the CSD, but not the ZKD, of LIN28 induces the LI71 saturation transfer difference signals, suggesting that the binding site of LI71 located at the CSD of LIN28 (Figure 5B). The crystal structure of LIN28 reveals a pocket that binds to the 0 position residue of the pre-element RNA (Nam et al., 2011; Wang et al., 2017). The pocket includes residue K102, which shows intensity changes in the HSQC titration of LI71. To validate the role of K102 in LI71’s inhibition, we carried out saturation transfer difference experiments using a mutant protein LIN28ΔK102. Intriguingly, the K102A mutation increases the saturation transfer difference signal of LI71, indicating an alteration of the energy exchange rate between LIN28 and LI71 (Figure 5B). Thus, it is likely that K102 contributes to the binding of LI71 to LIN28.
To investigate LI71’s inhibition of LIN28’s biological function, we first performed a dose-response assay and showed that LI71 has moderate activity with an IC50 of 27 μM for inhibition of LIN28-mediated oligouridylation of let-7 (Figures 5C and S4A). LI71 does not abolish catalytic activity of TUT4 when LIN28 is not present (Figure S4B). The IC50 of LI71 in oligouridylation assay is in range with an fluorescence polarization assay using LIN28Δ instead of LIN28ΔF73A (Figure S4C).
We next examined the stability of the LIN28:preE-let-7f complex upon addition of LI71, using a differential scanning fluorimetry (DSF) assay. We mixed the human LIN28Δ:preE-let-7f complex with SYPRO Orange dye and steadily heated the mixture to 95°C while the emitted fluorescence was collected. The derivative of fluorescence against temperature was plotted to measure the melting temperatures (Tms). As shown in Figures S4D and S4E, LI71 destabilized the LIN28Δ:preE-let-7f complex by decreasing the melting temperature from 63°C to 60°C.
To further characterize the structure-activity relation of LI71 based on our saturation transfer difference results, we evaluated 4 commercially available derivatives of LI71 with alterations on the benzoic ring and tail group (compounds LI71D1–4; Figure 5D). We measured their IC50s in the dose-response fluorescence polarization assay. This experiment revealed that the carboxyl head group was critical for LI71’s function. Any derivatives with a carboxyl group modifications resulted in complete loss of activity. Mutations of the ethoxy tail group result in decreased activity. We next tested the orientation of the carboxyphenyl group and compared the IC50s and saturation transfer difference spectroscopies of LI71 and its enantiomer (compound L171enan; Figure S5A). The enantiomer shows a moderate increase of IC50 in fluorescence polarization assay and is able to induce saturation transfer difference signals (Figures S5B and S5C), indicating that the orientation of the carboxyphenyl is important for LIN28:LI71 interaction. Although more systematic SAR studies are required to fully characterize the structure-activity relationship of LI71 to LIN28, based on existing saturation transfer difference and SAR studies we conclude that LI71 specifically inhibits LIN28 and that the carboxyphenyl head group is essential for this inhibition, with contributing structural features from the cyclopentaquinoline body and the ethoxy tail group.
LI71 Suppresses LIN28 in Leukemia Cells and Embryonic Stem Cells
To test the ability of LI71 to inhibit LIN28 activity in cells, we developed a dual luciferase reporter assay utilizing a Renilla luciferase cassette with eight tandem let-7 recognition sites in the 3′ UTR, which confers sensitivity to let-7 microRNA levels (Figure 6A). This cassette was coexpressed with a constitutively active firefly luciferase as an internal control. We stably transduced these expression cassettes into HeLa cell lines ectopically expressing LIN28A and LIN28B (HeLaLIN28A and HeLaLIN28B; Figures S6A and S6B), or GFP as controls, as parental HeLa cells do not express LIN28A or LIN28B. LI71 could significantly reduce the relative Renilla luciferase activity in cells expressing LIN28A and LIN28B at 50–100 μM (Figures 6B and 6C). However, LI71 did not show an effect in control cells expressing GFP and not LIN28 at a concentration of up to 100 μM, consistent with specific inhibition of LIN28 blockade of let-7 maturation in these cells (Figure 6B). LI71 did not increase apoptosis at 100 μM (Figure S6C).
Figure 6. LI71 Inhibits LIN28 in Embryonic and Cancer Cell Lines.

(A) Schematic of the let-7-sensitive dual luciferase reporter assay.
(B) HeLa clones expressing the dual luciferase let-7 sensor were incubated with either vehicle (DMSO) or 100 μM LI71 for 48 hr, at which time luciferase activity was measured (n = 5 biologic replicates; *p < 0.0001).
(C) HeLa cells expressing LIN28A (A1) were treated with vehicle (DMSO) or the indicated concentrations of LI-71 for 48 hr, at which time luciferase activity was assayed (n = 3 biologic replicates; *p < 0.05, by Student’s t test compared to vehicle).
(A-C) Error bars denote mean ± SEM.
(D) K562 leukemia cells were incubated with 100 μM LI-71 or vehicle for 48 hr, at which time let-7 species were measured by qPCR (n = 6 replicates over two independent experiments; *p < 0.05).
(E) DKO+A mESCs were incubated with 100 μM LI-71 or vehicle for 48 hr, at which time let-7 species were measured by qPCR (n = 6 replicates over two independent experiments; *p < 0.0001).
Human K562 leukemia cells express LIN28B, and disruption of LIN28B activity has been shown to increase endogenous let-7 levels (Viswanathan et al., 2009). We treated K562 cells with 100 μM LI71 or vehicle control (DMSO) for 48 hr and observed a significant increase in several let-7 species, although we did observe a small but significant effect on the non-LIN28-regulated microRNA miR-16 (Figure 6D). However, we did not observe significant effects on the non-LIN28-regulated micro-RNAs miR-93, miR-99a, and miR-151 (Figure S6D). In K562 cells, the induction of mature let-7 species is not correlated with increased levels of let-7 precursor species (Figure S6E). We also tested the effects of LI71 on mouse embryonic stem cells (mESCs). We used Lin28a/Lin28b double-knockout cells expressing exogenous wild-type LIN28A (DKO + A) (Tsanov et al., 2017), cultured in the leukemia inhibitory factor/2 inhibitor (LIF/2i) system (Ying et al., 2008). We found that treatment of DKO+A cells with 100 μM LI71 for 48 hr increased levels of mature let-7 species compared to vehicle-treated cells (Figure 6E). Together, these results demonstrate that, consistent with our in vitro data, LI71 inhibits LIN28’s activity on let-7 micro-RNAs in LIN28-dependent cancer and embryonic stem cells.
5-(Methylamino)nicotinic Acid Presents a Minimal Scaffold to Inhibit LIN28
To further investigate the benzoic acid group’s function in inhibiting LIN28, we performed a search of the carboxyl group in all cherry-picked compounds, and we identified a minimal structure 5-(methylamino)nicotinic acid (MNA; LI101), with a benzene ring replaced by pyridine, that could inhibit LIN28 in our fluorescence polarization assay at an IC50 of 76 μM (Figure 7A). 5-(Methylamino)nicotinic acid was originally excluded from further examination due to high IC50. To test whether 5-(methylamino)nicotinic acid could block LIN28-mediated oligouridylatinon, we performed our previously described oligouridylation assay with 5-(methylamino)nicotinic acid at a concentration of 80 μM. As shown in Figure 7B, 5-(methylamino)nicotinic acid shows strong inhibition of LIN28-mediated let-7 oligouridylation compared to DMSO negative control.
Figure 7. 5-(Methylamino)nicotinic Acid Presents a Minimal Scaffold to Inhibit LIN28.

(A) Dose-response fluorescence polarization assay of 5-(methylamino)nicotinic acid (MNA) (LI101) shows an IC50 of 76 μM (n = 3 replicates). Error bars denote mean ± SEM.
(B) Oligouridylation assay with positive control (RNA), negative control (DMSO), and 5-(methyl-amino)nicotinic acid. *Oliouridylated pre-let-7g.
(C) HeLa clones expressing the dual luciferase let-7 sensor were incubated with either vehicle (DMSO) or 5-(methylamino)nicotinic acid at various concentrations (50 μM, 100 μM, 150 μM, 250 μM, and 500 μM) for 48 hr, at which time luciferase activity was measured (n = 5 replicates; *p < 0.0001). Error bars denote mean ± SEM.
We then asked whether 5-(methylamino)nicotinic acid inhibits LIN28 in cells, since 5-(methylamino)nicotinic acid is predicted to be cell permeable with a logP (octanol-water partition coefficient) of 0.28. We performed the dual luciferase reporter assay with various concentrations of 5-(methylamino)nicotinic acid. As shown in Figure 7C, 5-(methylamino)nicotinic acid induces significant reduction of the relative Renilla luciferase activity at 100 μM or at higher concentrations, demonstrating specific inhibition of LIN28 in HeLaLIN28A cells. Our results highlight 5-(methylamino)nicotinic acid as a structurally minimal inhibitor of the LIN28/let-7 interaction both in vitro and in cells.
DISCUSSION
In this study, we developed a fluorescence polarization assay that is capable of identifying inhibitors for either the CSD or the ZKD of LIN28. Using this assay, we screened 101,017 compounds and identified 6 compounds that could inhibit both LIN28:let-7 binding and LIN28-mediated oligouridylation. We defined the inhibition mechanism of TPEN and LI71 and showed that, by chelating the Zn2+ atoms of zinc knuckles, TPEN is a potent inhibitor of the ZKD domain of LIN28. In contrast, LI71 directly binds to the CSD of LIN28 and disrupts the RNA binding through its head and tail groups. Our study demonstrates methods and tools that are useful for identifying and characterizing small-molecule inhibitors of protein/RNA interactions, which provides a basis for further discovery of inhibitors of LIN28 proteins.
There is a rapidly growing body of studies that highlight the validity of LIN28 as a target in various cancers (Zhou et al., 2013). As a result, significant effort has focused on the discovery of inhibitors of LIN28. Recent studies have described small-molecule LIN28 inhibitors discovered by fluorescence-resonance-energy-transfer (FRET)-based and fluorescence polarization-based assays (Lightfoot et al., 2016; Lim et al., 2016; Roos et al., 2016). However, these assays were performed with a small set of screening compounds with minimal elucidation of the mechanisms of inhibition of the identified inhibitors, which may hinder the future improvement of the potency and potential bioavailability of these chemicals. In this study, we describe an fluorescence polarization-based assay that is amenable to general studies of LIN28. We identified a set of LIN28 inhibitors that abolish LIN28-mediated oligouridylation of let-7 microRNAs and demonstrate their activity in vitro and in cells. We also describe several methods such as DSF, saturation transfer difference, and HSQC assays to further elucidate the mechanisms of inhibition of LIN28 inhibitors. We posit that, using these approaches, we have defined the mechanisms of action of the inhibitors of LIN28. More generally, these methodologies are potentially applicable for the identification and characterization of inhibitors of protein/RNA interfaces.
Importantly, we show that the ZKD of LIN28 plays a central role in LIN28 regulation of let-7 and contains a cavity that is suitable for targeting small-molecule inhibitors (Wang et al., 2017). In this study, we found that, due to the disparities of binding affinities between the CSD and ZKD, it is difficult to detect ZKD inhibitors with assays using native full-length LIN28. By introducing the point mutation F73A, which weakens the CSD binding to RNA, we were able to improve the sensitivity of detection of ZKD inhibitors in our assay. However, there are limitations to this method. First, the sensitivity for ZKD inhibitors, although improved, is still not sensitive for detecting weak ZKD inhibitors. Second, the CSD RNA-binding surface has been altered and may prevent the detection of CSD inhibitors that target the mutated site. It is worth noting that we discovered two CSD mutations, G90A and G119A, that are not located at the CSD RNA-binding surface but, instead, weaken the CSD/RNA interaction through allosteric effects (Figure S7), which could be considered in designing future screening assays.
Our data identify LI71 as a LIN28 CSD inhibitor, and we show that LI71 directly binds to the LIN28 CSD and inhibits the LIN28:let-7 interaction as well as the LIN28-mediated oligouridylation of let-7. LI71 has ample solubility in solution and low cellular toxicity, even at high micromolar concentrations, but it inhibits LIN28 with relatively low potency, which makes it a candidate for future improvements through medicinal chemistry. Our data highlight the benzoic acid motif, since it is shared in both LI71 and LI20, and loss of this motif causes loss of function of LI71. Changing the chirality of either the cyclopentatequinoline group or the ethoxy tail group results in reduced activity, suggesting that these groups are important but not essential for LI71’s function. Interestingly, our screenings also identified 5-(methylamino)nicotinic acid as a weak LIN28 inhibitor with an IC50 of 76 μM in the fluorescence polarization assay and weak activities in the oligouridylation assay as well as our cell-based let-7 reporter assay (Figure 6). 5-(methylamino)nicotinic acid features a carboxyl group, the same group that is essential for LI71’s activity but with a molecular weight of merely 152 Da, presenting opportunities for fragment-based drug development.
Our screen also identified TPEN, a cell-permeable, high-affinity, heavy-metal chelator with a preference of Zn2+ (McCabe et al., 1993), as a potent LIN28 inhibitor. We demonstrated that TPEN targets only the ZKD domain of LIN28 and causes drastic structural changes of the zinc knuckles and RNA-binding residues in the ZKD, confirming that our fluorescence polarization assay is sufficiently sensitive to identify a ZKD inhibitor. Although TPEN is unlikely to be specific to LIN28, it may be feasible to design Zn2+ chelators with high specificity to LIN28. Since LIN28 is highly expressed in a number of tumors relative to normal tissue (Viswanathan et al., 2009), it could be speculated that TPEN may exhibit more toxicity to LIN28-expressing cancer cells relative to normal cells. A recent study revealed that TPEN inhibits the growth of pancreatic cancer cells (Donadelli et al., 2008), where LIN28 was reported to be highly expressed (Hamada et al., 2012).
We identified three additional LIN28 inhibitors—gossypol, DAQ-B1, and BVT-948—of which extensive studies were not performed due to their complex and unfavorable chemical structures. Interestingly, gossypol—a natural phenol derived from the cotton plant—has been shown to suppress the growth of a number of LIN28-expressing tumors (Jarzabek et al., 2014; Sadahira et al., 2014; Volate et al., 2010). It has been suggested that gossypol functions by inhibiting oncoproteins such as Bcl-1, BcI-XL and MSI1 (Lan et al., 2015; Oliver et al., 2005). In our study, we demonstrate that gossypol also inhibits LIN28, although further efforts are needed to verify LIN28 as a valid target of gossypol in LIN28-expressing tumors.
Overall, we have used a fluorescence polarization assay optimized for cell-free, HTS to identify inhibitors of LIN28. We have validated candidate LIN28-targeting compounds as inhibitors of let-7 oligouridylation in cell-free systems and let-7 repression in three distinct cell types. We have used a combination of biophysical approaches to dissect the mechanisms by which these inhibitors impair LIN28 function. We propose that these findings lay the groundwork and identify an approach for further development and validation of clinically useful LIN28 inhibitory compounds.
EXPERIMENTAL PROCEDURES
Cloning, Protein Expression, and Purification
The cloning, expression, and purification of LIN28 was previously described (Nam et al., 2011). All LIN28 mutants were induced using the QuickChange method. Briefly, LIN28 constructs were overexpressed in E. coli BL21(DE3) Rosetta pLysS, followed by nickel nitrilotriacetic acid (Ni-NTA) affinity chromatography. His-tags were removed using tobacco etch virus (TEV) protease. Purified LIN28 was obtained from sequential cation exchange chromatography and size exclusion chromatography. LIN28 mutants CSD and ZKD were prepared using the same protocol.
Fluorescence Polarization Assay
Fluorescence polarization titration assay was carried out by titrating LIN28 protein into FAM-labeled preE-let-7 at a 2-nM final concentration and buffer M (100 mM sodium chloride, 20 mM Tris-HCl [pH 7.0], 5 mM magnesium chloride, 10% v/v glycerol, 5 mM DTT, 0.1% v/v NP-40). The assay plate was briefly vortexed, and air bubbles were removed by spinning at 1,000 rpm. An EnVision plate reader (PerkinElmer) was used to quantify the fluorescence polarization using the following equation:
where P is the fluorescence polarization reading, Fparallel is the fluorescence intensity parallel to the excitation plane, and Fperpendicular is the fluorescence intensity perpendicular to the excitation plane. The total fluorescence intensity is estimated using the following equation:
The fluorescence polarization competition assay was carried out in a similar fashion, except compounds were titrated into buffer M containing LIN28 and FAM-labeled preE-let-7. The final concentration of LIN28 is the dissociation constant of LIN28 and preE-let-7 determined through fluorescence polarization titration assay. Fluorescence polarization readings were transferred into Prism 6.0 (GraphPad). The dissociation constant and IC50 were acquired by curve fitting using 1:1 binding/inhibition model. All fluorescence polarization assays were repeated at least two times.
Primary HTS and Data Analysis
The primary screening was performed in the ICCB-Longwood screening facility at Harvard Medical School. 25 μL master mix (150 nM recombinant LIN28ΔF73A, 2 nM preE-let-7 probe with FAM label in buffer M: 100 mM sodium chloride, 20 mM Tris-HCl [pH 7.0], 5 mM magnesium chloride, 10% v/v glycerol, 5 mM DTT, 0.1% v/v NP-40) was added to each well of a 384-well plate (Corning 3820) using a Matrix Wellmate (Thermo Fisher Scientific). Each plate also included 16 positive-control wells with 10 μM UAGGAGA oligonucleotide and 16 negative-control wells containing only master mix. Positive controls were added with an Eppendorf repeater. Assay plates were briefly vortexed and then centrifuged at 1,500 rpm for 5 min. Plates were then sealed with plastic covers and protected from light before the pin transfer. 100 nL library compound was then added via stainless-steel pin array. Plates were then covered again with plastic seal and incubated overnight at room temperature before both fluorescence polarizations and estimated fluorescence intensities were collected for each well. The screening was performed in duplicate to increase the precision. MightyScreen (Wang et al., 2018) was used for data visualization and assay quality control. Z-factor was also used for quality control purposes:
Plates with a Z-factor higher than 0.5 are considered to be a robust assay. To eliminate false-positives caused by autofluorescence, a filter was set to exclude compounds of which the total fluorescence intensity is an outlier (5 • SD) in the fluorescence intensity distribution (approximately normal) of all compounds. Z-scores were used for hit selection:
The compounds with highest Z scores were then manually examined. Structure, partition coefficient (logP), molecular polar surface area (PSA), molecular weight, and active rate in previous reported biochemical assays were used as the criteria to finalize the hit list. The data analysis was performed using Python 2.7, including two modules: pandas and matplotlib.
Dose-response assays were performed using the small volume dispenser (Hewlett-Packard D300) with the same fluorescence polarization assay. 1 μL compound was dispensed to 12 assay wells with the final concentration ranging from 50 μM to 20 nM. The fluorescence polarization was read by EnVision, and the IC50s of compound inhibitors were acquired using Prism (GraphPad).
LIN28 inhibitors were purchased from commercial vendors. The structure and purity of purchased inhibitors were assessed using mass spectrometry and 1D nuclear magnetic resonance (NMR).
In Vitro Uridylation Assay
We adopted a previously described uridylation assay to measure the inhibition of LIN28-mediated uridylation by LIN28 inhibitors (Wang et al., 2017). Recombinant full-length mouse LIN28A and truncated but fully active recombinant mouse TUT4 (230–1424) were purified as previously described (Wang et al., 2017). Pre-let-7g was transcribed in vitro (Walker et al., 2003). Pre-let-7g was radiolabeled with using T4 polynucleotide kinase. Mouse LIN28, mouse TUT4, and radiolabeled pre-let-7g were mixed and incubated for 40 min at 37°C in buffer containing 20 mM Tris 7.5, 5% glycerol, 6 mM magnesium chloride, 6 mM DTT, 50 μM zinc chloride, 40 mM KCl, and 200 μM UTP. The reactions were stopped by adding 30 mM EDTA, 1% SDS, and 0.1 mg/mL protease K and incubating at 50°C for 30 min. Pre-let-7g and uridylated pre-let-7g were separated through denaturing PAGE electrophoresis. For secondary screening, uridylation assays were performed in the presence of 40 μM test compound.
Saturation Transfer Difference Spectroscopy
LI71 was dissolved in deuterated DMSO (DMSO-d6, Cambridge Isotope Laboratories). The 1D H spectrum and the correlation spectroscopy (COSY) of compounds were collected using the Agilent 700 MHz NMR system. LIN28 sample was dialyzed against 20 mM Bis-Tris (pH 6.0), 100 mM sodium chloride, 10% v/v glycerol-d8 (Chembridge Isotope Laboratories), 5 mM DTT, and D2O the night before the saturation transfer difference experiment. The saturation transfer difference spectrum was then collected using sample containing 100 μM LI71 and 5 μM LIN28 in dialysis buffer. The excitation was set at −2 ppm, and transfer delay was set to be 2 s.
HSQC Spectroscopy
The heteronuclear single-quantum coherence (HSQC) 2D spectrum of LIN28:preE-let-7d complexes was carried out as previously described.(Nam et al., 2011). LI71 was added as powder to minimize solvent and dilution effects. CcpNmr was used to visualize and analyze HSQC spectra (Skinner et al., 2015). The intensity change was measured after normalizing the intensities of the two spectra. The chemical changes were calculated using the following equation:
The structure representations was created using PyMOL (DeLano, 2002).
Differential Scanning Fluorimetry
QuantStudio 7 Flex (Thermo Fisher Scientific) was used to measure the melting temperature of LIN28:preE-let-7 complexes. LIN28Δ:preE-let-7 complex samples were purified as previously described and then dialyzed with 50 mM HEPES (pH 7.0), 100 mM sodium chloride, and 5% v/v glycerol. Samples were mixed with 2× SYPRO Orange (Thermo Fisher Scientific) and equilibrated at 25°C for 2 min and then gradually heated to 95°C at a rate of 0.2°C/min. The melting temperature (Tm) was calculated using the QuantStudio software suite.
Cell Culture
To generate the HeLa lines used in this study, HeLa cells were infected with retrovirus produced from pBABE-puro containing N-FLAG-tagged LIN28A, LIN28B, or GFP cDNAs. Cells were selected in puromycin for 2 days and then sorted into single cells using fluorescence-activated cell sorting. Stable clones with low levels of LIN28 proteins adequate to repress let-7 microRNA maturation were selected for subsequent study. Puromycin-resistant HeLa lines stably expressing GFP, LIN28A, or LIN28B were lentivirally transduced with pLenti-8x-let-7 dual luciferase, and stable polyclonal populations were isolated with G418 (Life Technologies).
Reporter Assays
The Renilla luciferase-8x-let-7-HSV TK-Firefly luciferase cassette from psiCHECK2-let-7 8× (a gift from Yukihide Tomari (Addgene plasmid #20931) was cloned into pLenti cytomegalovirus (CMV) GFP Neo (657–2) (a gift from Eric Campeau (Addgene plasmid #17447). The CMV promoter was removed from the resulting construct due to silencing of this promoter in stable HeLa lines, and so Renilla expression was driven by the lentiviral long terminal repeats (pLenti-8x-let-7 dual luciferase). HeLa lines stably expressing GFP, LIN28A, or LIN28B, as well as pLenti-8x-let-7 dual luciferase, were exposed to up to 100 μM LI-71 for 24 hr. Following drug treatment, cells were lysed, dual luciferase activity was monitored using the Dual Luciferase Assay System (Promega), and results were normalized to blank samples.
qPCR
Whole RNA was isolated from cells using TRIzol (Life Technologies), and cDNA was synthesized using the miScript system (QIAGEN). MicroRNA specific primer assays (QIAGEN) were used to quantify species of mature and precursor microRNAs relative to the endogenous control U6 microRNA.
Flow Cytometry
Cells were treated with 100 μM LI71 or vehicle for 48 hr, at which time cells were harvested and stained with annexin V-APC (BD Biosciences) according to the manufacturer’s instructions.
Supplementary Material
Highlights.
A screening pipeline developed to identify inhibitors targeting both domains of LIN28
Six compounds effectively inhibit LIN28-mediated oligouridylation of let-7
TPEN destabilizes the zinc-knuckle domain, while LI71 modulates the cold shock domain
MNA represents a minimal scaffold as a LIN28 inhibitor targeting the cold shock domain
ACKNOWLEDGMENTS
This work was supported by a grant to P.S. from the National Cancer Institute (R01CA163647). R.G.R. and G.Q.D. were supported by an Innovation Award from Alex’s Lemonade Stand Foundation. R.G.R. was supported by the National Institute of Diabetes and Digestive and Kidney Diseases (K08-DK114527-01). We thank members of ICCB-Longwood facility for their support in performing HTS screening and related data analysis. We thank Kelly Arnett from CMI-HMS for the support in performing DSF thermo-stability assays.
Footnotes
DECLARATION OF INTERESTS
G.Q.D. and P.S. hold options and intellectual property related to 28/7 Therapeutics, a company seeking to develop inhibitors of the LIN28/let-7 pathway.
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures and four tables and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.04.116.
REFERENCES
- Ambros V, and Horvitz HR (1984). Heterochronic mutants of the nematode Caenorhabditis elegans. Science 226, 409–416. [DOI] [PubMed] [Google Scholar]
- Beachy SH, Onozawa M, Chung YJ, Slape C, Bilke S, Francis P, Pineda M, Walker RL, Meltzer P, and Aplan PD (2012). Enforced expression of Lin28b leads to impaired T-cell development, release of inflammatory cytokines, and peripheral T-cell lymphoma. Blood 120, 1048–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang H-M, Triboulet R, Thornton JE, and Gregory RI (2013). A role for the Perlman syndrome exonuclease Dis3l2 in the Lin28-let-7 pathway. Nature 497, 244–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano WL (2002). PyMOL: an open-source molecular graphics tool. CCP4 Newsletter on Protein Crystallography, March (40), http://www.ccp4.ac.uk/newsletters/newsletter40/11_pymol.pdf.
- Donadelli M, Dalla Pozza E, Costanzo C, Scupoli MT, Scarpa A, and Palmieri M (2008). Zinc depletion efficiently inhibits pancreatic cancer cell growth by increasing the ratio of antiproliferative/proliferative genes. J. Cell. Biochem 104, 202–212. [DOI] [PubMed] [Google Scholar]
- Hagan JP, Piskounova E, and Gregory RI (2009). Lin28 recruits the TUTase Zcchc11 to inhibit let-7 maturation in mouse embryonic stem cells. Nat. Struct. Mol. Biol 16, 1021–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamada S, Masamune A, Takikawa T, and Suzuki N (2012). Pancreatic stellate cells enhance stem cell-like phenotypes in pancreatic cancer cells. Biochem. Biophys. Res. Commun 421, 349–354. [DOI] [PubMed] [Google Scholar]
- Heo I, Joo C, Cho J, Ha M, Han J, and Kim VN (2008). Lin28 mediates the terminal uridylation of let-7 precursor MicroRNA. Mol. Cell 32, 276–284. [DOI] [PubMed] [Google Scholar]
- Iliopoulos D, Hirsch HA, and Struhl K (2009). An epigenetic switch involving NF-kappaB, Lin28, Let-7 microRNA, and IL6 links inflammation to cell transformation. Cell 139, 693–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarzabek MA, Amberger-Murphy V, and Callanan JJ (2014). Interrogation of gossypol therapy in glioblastoma implementing cell line and patient-derived tumour models. Br. J. Cancer 111, 2275–2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan L, Appelman C, Smith AR, Yu J, Larsen S, Marguez RT, Liu H, Wu X, Gao P, Roy A, et al. (2015). Natural product (−)-gossypol inhibits colon cancer cell growth by targeting RNA-binding protein Musashi-1. Mol. Oncol 9, 1406–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lightfoot HL, Miska EA, and Balasubramanian S (2016). Identification of small molecule inhibitors of the Lin28-mediated blockage of pre-let-7g processing. Org. Biomol. Chem 14, 10208–10216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim D, Byun WG, Koo JY, Park H, and Park SB (2016). Discovery of a small-molecule inhibitor of protein-microRNA interaction using binding assay with a site-specifically labeled Lin28. J. Am. Chem. Soc 138, 13630–13638. [DOI] [PubMed] [Google Scholar]
- Loughlin FE, Gebert LFR, Towbin H, Brunschweiger A, Hall J, and Allain FH-T (2011). Structural basis of pre-let-7 miRNA recognition by the zinc knuckles of pluripotency factor Lin28. Nat. Struct. Mol. Biol 19, 84–89. [DOI] [PubMed] [Google Scholar]
- Mayr F, Schütz A, Döge N, and Heinemann U (2012). The Lin28 cold-shock domain remodels pre-let-7 microRNA. Nucleic Acids Res. 40, 7492–7506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCabe MJ Jr., Jiang SA, and Orrenius S (1993). Chelation of intracellular zinc triggers apoptosis in mature thymocytes. Lab. Invest 69, 101–110. [PubMed] [Google Scholar]
- Molenaar JJ, Domingo-Fernández R, Ebus ME, Lindner S, Koster J, Drabek K, Mestdagh P, van Sluis P, Valentijn LJ, van Nes J, et al. (2012). LIN28B induces neuroblastoma and enhances MYCN levels via let-7 suppression. Nat. Genet 44, 1199–1206. [DOI] [PubMed] [Google Scholar]
- Nam Y, Chen C, Gregory RI, Chou JJ, and Sliz P (2011). Molecular basis for interaction of let-7 microRNAs with Lin28. Cell 147, 1080–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen LH, Robinton DA, Seligson MT, Wu L, Li L, Rakheja D, Comerford SA, Ramezani S, Sun X, Parikh MS, et al. (2014). Lin28b is sufficient to drive liver cancer and necessary for its maintenance in murine models. Cancer Cell 26, 248–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver CL, Miranda MB, Shangary S, Land S, Wang S, and Johnson DE (2005). (−)-Gossypol acts directly on the mitochondria to overcome Bcl-2- and Bcl-X(L)-mediated apoptosis resistance. Mol. Cancer Ther 4, 23–31. [PubMed] [Google Scholar]
- Roos M, Pradère U, Ngondo RP, Behera A, Allegrini S, Civenni G, Zagalak JA, Marchand J-R, Menzi M, Towbin H, et al. (2016).A small-molecule inhibitor of Lin28. ACS Chem. Biol 11, 2773–2781. [DOI] [PubMed] [Google Scholar]
- Rowe RG, Wang LD, Coma S, Han A, Mathieu R, Pearson DS, Ross S, Sousa P, Nguyen PT, Rodriguez A, et al. (2016). Developmental regulation of myeloerythroid progenitor function by the Lin28b-let-7-Hmga2 axis. J. Exp. Med 213, 1497–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rybak A, Fuchs H, Smirnova L, Brandt C, Pohl EE, Nitsch R, and Wulczyn FG (2008). A feedback loop comprising lin-28 and let-7 controls pre-let-7 maturation during neural stem-cell commitment. Nat. Cell Biol 10, 987–993. [DOI] [PubMed] [Google Scholar]
- Sadahira K, Sagawa M, Nakazato T, Uchida H, Ikeda Y, Okamoto S, Nakajima H, and Kizaki M (2014). Gossypol induces apoptosis in multiple myeloma cells by inhibition of interleukin-6 signaling and Bcl-2/Mcl-1 pathway. Int. J. Oncol 45, 2278–2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner SP, Goult BT, Fogh RH, Boucher W, Stevens TJ, Laue ED, Vuister GW, and Cr IU (2015). Structure calculation, refinement and validation using CcpNmr Analysis. Acta Crystallogr. D Biol. Crystallogr 71,154–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsanov KM, Pearson DS, Wu Z, Han A, Triboulet R, Seligson MT, Powers JT, Osborne JK, Kane S, Gygi SP, et al. (2017). LIN28 phosphorylation by MAPK/ERK couples signalling to the post-transcriptional control of pluripotency. Nat. Cell Biol 19, 60–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu H-C, Schwitalla S, Qian Z, LaPier GS, Yermalovich A, Ku Y-C, Chen S-C, Viswanathan SR, Zhu H, Nishihara R, et al. (2015). LIN28 cooperates with WNT signaling to drive invasive intestinal and colorectal adenocarcinoma in mice and humans. Genes Dev. 29, 1074–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbach A, Yermalovich A, Zhang J, Spina CS, Zhu H, Perez-Atayde AR, Shukrun R, Charlton J, Sebire N, Mifsud W, et al. (2014). Lin28 sustains early renal progenitors and induces Wilms tumor. Genes Dev. 28, 971–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanathan SR, Daley GQ, and Gregory RI (2008). Selective blockade of microRNA processing by Lin28. Science 320, 97–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanathan SR, Powers JT, Einhorn W, Hoshida Y, Ng TL, Toffanin S, O’Sullivan M, Lu J, Phillips LA, Lockhart VL, et al. (2009). Lin28 promotes transformation and is associated with advanced human malignancies. Nat. Genet 41, 843–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volate SR, Kawasaki BT, Hurt EM, Milner JA, Kim YS, White J, and Farrar WL (2010). Gossypol induces apoptosis by activating p53 in prostate cancer cells and prostate tumor-initiating cells. Mol. Cancer Ther 9, 461–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker SC, Avis JM, and Conn GL (2003). General plasmids for producing RNA in vitro transcripts with homogeneous ends. Nucleic Acids Res. 31, e82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Nam Y, Lee AK,Yu C, Roth K, Chen C, Ransey EM, and Sliz P (2017). LIN28 zinc knuckle domain is required and sufficient to induce let-7 oligouridylation. Cell Rep. 18, 2664–2675. [DOI] [PubMed] [Google Scholar]
- Wang L, Yang Q, Jaimes A, Wang T, Strobelt H, Chen J, and Sliz P (2018). MightyScreen: an open-source visualization application for screening data analysis. SLAS Discov. 23, 218–223. [DOI] [PubMed] [Google Scholar]
- Yang X, Lin X, Zhong X, Kaur S, Li N, Liang S, Lassus H, Wang L, Katsaros D, Montone K, et al. (2010). Double-negative feedback loop between reprogramming factor LIN28 and microRNA let-7 regulates aldehyde dehydrogenase 1-positive cancer stem cells. Cancer Res. 70, 9463–9472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying Q-L, Wray J, Nichols J, Batlle-Morera L, Doble B, Woodgett J, Cohen P, and Smith A (2008). The ground state of embryonic stem cell self-renewal. Nature 453, 519–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Ng S-B, and Chng W-J (2013). LIN28/LIN28B: an emerging oncogenic driver in cancer stem cells. Int. J. Biochem. Cell Biol 45, 973–978. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.