

Abstract
Background:
Heart failure (HF) is a morbid and heritable disorder for which the biological mechanisms are incompletely understood. We therefore examined genetic associations with HF in a large national biobank, and assessed whether refined phenotypic classification would facilitate genetic discovery.
Methods:
We defined all-cause HF among 488,010 participants from the UK Biobank and performed a genome-wide association analysis. We refined the HF phenotype by classifying individuals with left ventricular dysfunction and without coronary artery disease (CAD) as having nonischemic cardiomyopathy (NICM), and repeated a genetic association analysis. We then pursued replication of lead HF and NICM variants in independent cohorts, and performed adjusted association analyses to assess whether identified genetic associations were mediated through clinical HF risk factors. In addition, we tested rare, loss-of-function mutations in 24 known dilated cardiomyopathy (DCM) genes for association with HF and NICM. Finally, we examined associations between lead variants and left ventricular structure and function among individuals without HF using cardiac magnetic resonance imaging (n=4,158) and echocardiographic data (n=30,201).
Results:
We identified 7,382 participants with all-cause HF in the UK Biobank. Genome-wide association analysis of all-cause HF identified several suggestive loci (P < 1×10−6), the majority linked to upstream HF risk factors, i.e. CAD (CDKN2B-AS1 and MAP3K7CL) and atrial fibrillation (PITX2). Refining the HF phenotype yielded a subset of 2,038 NICM cases. In contrast to all-cause HF, genetic analysis of NICM revealed suggestive loci that have been implicated in DCM (BAG3, CLCNKA-ZBTB17). DCM signals arising from our NICM analysis replicated in independent cohorts, persisted after HF risk factor adjustment, and were associated with indices of left ventricular dysfunction in individuals without clinical HF. Additionally, analyses of loss-of-function variants implicated BAG3 as a disease-susceptibility gene for NICM (loss-of-function variant carrier frequency=0.01%, OR=12.03, P=3.62×10−5).
Conclusions:
We found several distinct genetic mechanisms of all-cause HF in a national biobank that reflect well-known HF risk factors. Phenotypic refinement to a NICM subtype appeared to facilitate the discovery of genetic signals that act independent of clinical HF risk factors, and which are associated with subclinical left ventricular dysfunction.
Keywords: Heart failure, nonischemic cardiomyopathy, dilated cardiomyopathy, genome-wide association study
INTRODUCTION
Heart failure (HF) is a complex clinical syndrome that affects over 30 million individuals worldwide with a projected ~ 40% increase in prevalence by 2030.1–3 Despite considerable advances in HF management, nearly 50% of affected individuals die within 5 years of a first diagnosis.4 The rising global burden of HF and its apparent heritability – estimated at ≥ 18% by epidemiological studies – have prompted the study of genetic determinants to inform new preventive strategies and novel therapeutics.5–8
Significant strides have been made in understanding rare, Mendelian forms of HF.9 Furthermore, genetic association studies of upstream HF risk factors such as coronary artery disease (CAD), atrial fibrillation, and hypertension have yielded numerous susceptibility loci.10–13 Yet, genetic analyses of common, complex HF have achieved limited success, potentially owing to insufficient power and disease heterogeneity.14 Indeed, more recent analyses limited to recruited cohorts of specific HF subpopulations such as nonischemic dilated cardiomyopathy (DCM), the leading global cause of heart transplantation, have identified susceptibility loci that have been replicated.15–18
The emergence of large population-based biobanks with extensive phenotypic and genotypic data enables rigorous investigation of genetic influences on cardiovascular health and disease.19 Yet as these biobanks grow and increasingly rely on efficient electronic phenotyping, the achievement of phenotypic precision may remain a critical challenge that limits genetic discovery and downstream interpretation of findings.20 We therefore conducted a phenotype-driven genetic analysis of HF in the general population. Specifically, we conducted a genetic association analysis of all-cause HF and then of the more precise definition of nonischemic cardiomyopathy (NICM) to determine whether phenotypic refinement improves genetic discovery in a population-based biobank. Given the heterogeneous etiologies of HF, we then characterized putative HF loci by examining associations with relevant risk factors and intermediate traits of left ventricular structure and function.
METHODS
Summary level genetic association results cited in this manuscript are available through the Broad Institute Cardiovascular Disease Knowledge Portal (http://broadcvdi.org) and through the UK Biobank.21
Study subjects
In total, 488,010 individuals from the UK Biobank – a large, prospective population-based cohort – were considered when assessing epidemiological relationships of HF and associated risk factors. In primary genetic analyses, we included 394,156 participants of European ancestry from the UK Biobank. Analysis of the UK Biobank data was approved by the Partners Health Care institutional review board (protocol 2013P001840; application 7089). Informed consent was obtained from all participants by the UK Biobank.
For replication of genetic association results, we studied 1,060 participants from the Genetic Risk Assessment of Defibrillator Events (GRADE) Study, a recruited cohort of pre-defined cardiomyopathy patients with defibrillators, and up to 9,432 participants from the Vanderbilt University Biobank (BioVU), a prospective, hospital-based cohort (Supplemental Methods).
Phenotyping
Disease phenotypes in UK Biobank were defined using a combination of self-reported questionnaire data (confirmed by a trained healthcare professional) and linked hospital-admission and death registry data. Detailed definitions for all disease phenotypes are provided in Supplemental Table S1.
We defined “all-cause” HF as the presence of self-reported “HF/pulmonary edema” or “cardiomyopathy” at any visit; or an International Classification of Diseases (ICD)-10 or ICD-9 billing code indicative of heart/ventricular failure or a cardiomyopathy of any cause. Of note, individuals with a diagnosis of hypertrophic cardiomyopathy – as ascertained by self-report or by pertinent ICD-10 codes – were excluded from the HF and NICM phenotypes even if they met the above criteria due to the substantial Mendelian inheritance pattern of hypertrophic cardiomyopathy.
Among all-cause HF patients, we defined “nonischemic cardiomyopathy” (NICM) on the basis of left ventricular dysfunction and absence of coronary artery disease (CAD). A priori, individuals were considered to have “left ventricular dysfunction” if they carried ICD-10 diagnoses of “dilated cardiomyopathy” or “left ventricular failure,” or an ICD-9 diagnosis of “left heart failure.” Indicators for CAD included myocardial infarction or coronary revascularization, as described previously.10 Myocardial infarction was defined as a self-report of “heart attack” or an ICD-10 code of acute myocardial infarction. Coronary revascularization was defined as the presence of an operative or procedure code for coronary artery bypass surgery or coronary angioplasty (Supplemental Table S1).
Genetic association testing, replication, and meta-analysis
We performed primary genome-wide association testing among UK Biobank participants passing sample quality control by comparing HF or NICM cases against non-HF controls. In total, 6,504 HF cases were compared to 387,652 controls, and 1,816 NICM cases were compared to 388,326 controls. Only variants with minor allele frequency (MAF) > 1% available in the Haplotype Reference Consortium v1.1 panel and imputed with INFO > 0.3 were included (Supplemental Methods).22
Lead variants from the HF and NICM analyses passing a suggestive threshold of P < 1×10−6 were taken forward for replication. For lead HF variants, we pursued replication in two studies: (1) BioVU – comparing 2,982 HF cases against 6,450 controls; and (2) the GRADE Study – comparing 1,060 cases (classified prospectively at the time of recruitment) against an independent sample of 2,327 controls from BioVU genotyped on the same platform as the GRADE samples and selected based on overlapping genetic ancestry. For lead NICM variants, we pursued replication in three studies: (1) BioVU – comparing 226 NICM cases (ascertained retrospectively through application of our NICM phenotyping algorithm to the medical record alongside available echocardiographic data, classifying left ventricular dysfunction as LVEF ≤ 40%) against 4,709 controls; (2) the GRADE Study – comparing 260 NICM cases (classified prospectively at the time of recruitment) against 2,327 controls; and (3) publically available summary exome-chip association statistics from a recent study of DCM including 2,796 cases and 6,877 control subjects from six populations of European ancestry.17 When a lead variant was not available in a replication study, the best-available proxy was selected (Supplemental Methods).
Associations between HF and NICM susceptibility variants and HF risk factors
Using individuals free of HF in the UK Biobank, we performed additional association testing of lead HF and NICM variants with 10 binary and 3 continuous risk factors for HF (Supplemental Table S1). Furthermore, to determine whether lead variant associations with HF and/or NICM were independent of HF risk factors, we repeated genetic association analyses for all lead variants adjusting for relevant risk factors.
Associations between HF and NICM susceptibility variants and cardiac structure / function
We further tested lead variants at identified HF and NICM susceptibility loci for association with intermediate traits of left ventricular (LV) structure and function by assessing: (1) individual-level data on LV ejection fraction, LV end diastolic volume, LV end systolic volume, LV stroke volume, cardiac output, and cardiac index in 4,158 individuals without heart failure who underwent cardiac magnetic resonance imaging (MRI) in the UK Biobank, and (2) summary-level data of 16 echocardiographic traits in 30,201 individuals without heart failure in the EchoGen consortium. For cardiac MRI data in the UK Biobank, we excluded individuals with measurements falling outside of 3 standard deviations from the mean for a given trait.
Rare predicted loss-of-function associations
To complement the above common variant analyses, we examined whether rare (minor allele frequency < 1%) predicted loss-of-function (pLOF) variants at known DCM genes are associated with HF or NICM in the UK Biobank.23 Only directly genotyped variants were included in these analyses. In total, 111 genes were considered, but only 24 had more than two qualifying variants and appreciable pLOF carriers for testing (carrier frequency > 0.0001) (Supplemental Table S2). We annotated genotyped variants from the UK Biobank using Ensembl Variant Effect Predictor version 88 using the “--pick_allele” option to select one consequence per variant allele.24 Variants annotated as protein-truncating, premature stops, canonical splice-sites, or frameshift mutations were classified as pLOF using the LOFTEE plugin for VEP.25
Statistical Analysis
Primary genome-wide association testing for HF and NICM in the UK Biobank was performed using logistic regression and adjusting for age at first visit, sex, genotyping array, and the first 10 principal components of ancestry.
To test the association of lead HF and NICM susceptibility variants with HF risk factors, we used a combination of linear and logistic regression adjusing for age at first visit, sex, genotyping array, and the first 10 principal components of ancestry. We considered significant any SNP-risk factor association surpassing a Bonferroni-corrected threshold of P< 5.49×10−4 (0.05 / (13 traits x 7 SNPs)).
To test the association of lead HF and NICM variants with intermediate traits of cardiac structure and function, we performed linear regression adjusted for age at first visit, sex, genotyping array, and the first 10 principal components of ancestry. We considered significant any SNP-trait association surpassing a Bonferroni-corrected threshold of P < 0.0012 (0.05 / (6 traits x 7 SNPs)).
For rare predicted loss-of-function analyses, we performed association testing using a collapsed gene-based test, classifying samples as either carriers or non-carriers of any pLOF variant in a given gene, adjusting for age at baseline, sex, genotyping array, and the first 10 principal components of ancestry. A Bonferroni-corrected p-value significance threshold was set at P = 0.001 (0.05 / (2 phenotypes x 24 genes)).
Primary association analyses for HF and NICM were performed in PLINK2 (https://www.cog-genomics.org/plink/2.0/).26 Association testing with HF risk factors and intermediate cardiac imaging traits was performed in R v3.3.0 (R Foundation, Vienna, Austria). Rare gene-based testing was performed using EPACTS (https://genome.sph.umich.edu/wiki/EPACTS).27
RESULTS
Defining All-Cause HF and Assessing Overlap with HF Risk Factors
The study population was comprised of 488,010 individuals in the UK Biobank with available genotypic and phenotypic data. In total, 7,382 individuals met criteria for the broader classification of all-cause HF. A large proportion of all-cause HF cases had co-morbid HF risk factors, including CAD (47.3%) and atrial fibrillation (43.0%) (Figure 1; Table 1).
Figure 1. Epidemiological overlap between heart failure phenotypes and prominent risk factors in UK Biobank.
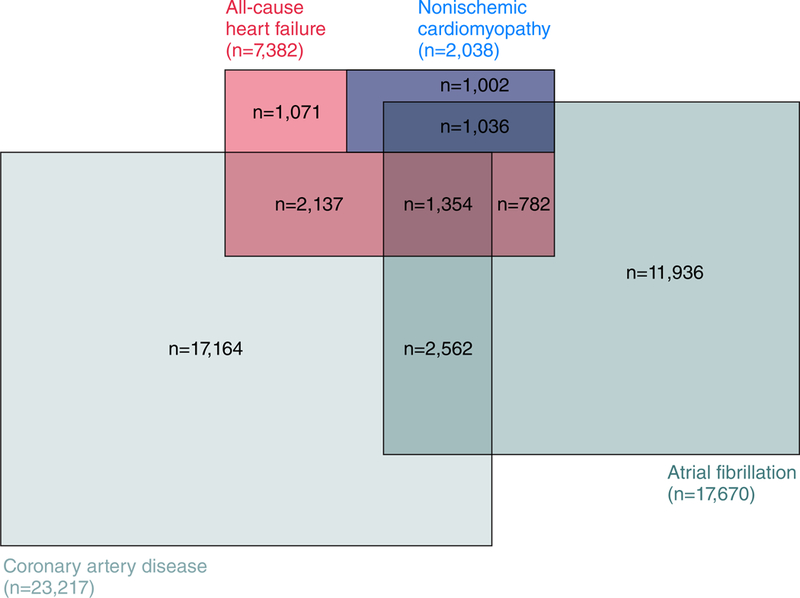
The overlap between all-cause heart failure, nonischemic cardiomyopathy, coronary artery disease, and atrial fibrillation cases are displayed among 488,010 study participants in the UK Biobank. Case counts represent the sum total of disease at baseline and incident cases.
Table 1.
Baseline characteristics of UK Biobank samples by heart failure status.
| All-cause HF (n = 7,382) |
NICM (n = 2,038) |
All-cause HF vs. NICM P-value |
Referents free of all-cause HF (n = 480,628) |
|
|---|---|---|---|---|
| Age at baseline, years | 62.2 (6.3) | 61.6 (6.7) | < 0.001 | 57.0 (8.1) |
| Male gender, n (%) | 5,151 (69.8%) | 1,324 (65.0%) | < 0.001 | 218,203 (45.4%) |
| UK BiLEVE array, n (%) | 1,071 (14.5%) | 274 (13.4%) | 0.12 | 48,830 (10.2%) |
| Height, cm | 170.3 (9.4) | 170.8 (9.6) | 0.003 | 168.5 (9.3) |
| Body mass index, kg/m2 | 29.8 (5.8) | 29.3 (5.9) | < 0.001 | 27.4 (4.8) |
| Waist-hip ratio | 0.94 (0.09) | 0.92 (0.09) | < 0.001 | 0.87 (0.09) |
| Systolic blood pressure, mmHg | 148.5 (22.4) | 147.5 (22.2) | 0.02 | 140.9 (20.7) |
| Diastolic blood pressure, mmHg | 86.5 (12.2) | 86.8 (12.2) | 0.19 | 84.3 (11.3) |
| Coronary Artery Disease, n (%) | 3,491 (47.3%) | 0 (0.0%) | NA | 19,726 (4.1%) |
| Type 2 DM, n (%) | 1,929 (26.1%) | 395 (19.4%) | < 0.001 | 22,515 (4.7%) |
| Atrial Fibrillation, n (%) | 3,172 (43.0%) | 1,036 (50.8%) | < 0.001 | 14,498 (3.0%) |
| Hypertension, n (%) | 5,584 (75.6%) | 1,413 (69.3%) | < 0.001 | 153,369 (31.9%) |
Values are presented as mean (standard deviation) unless otherwise noted. Baseline characteristics of NICM samples (n = 2,038) were compared to all-cause heart failure samples without NICM (n = 5,344) using a standard t-test for continuous measures and chi-square test for dichotomous traits. Abbreviations: HF=heart failure; NICM=nonischemic cardiomyopathy; DM=diabetes mellitus.
Genome-wide association analyses of All-Cause HF in UK Biobank
In the UK Biobank, primary genetic association analyses for all-cause HF (n=6,504 passing sample quality control) yielded one locus that exceeded the threshold for genome-wide statistical significance (rs1906609 upstream of PITX2, OR = 1.15, P = 9.08×10−10) and four other loci with suggestive association signals (P < 1×10−6; rs7857118 near CDKN2B-AS1, OR = 1.10, P = 2.15×10−7; rs12627426 near MAP3K7CL, OR = 1.13, P = 2.63×10−7; rs73839819 near RYBP, OR = 1.33, P = 2.65×10−7; rs2234962 in BAG3, OR = 1.12, P = 3.55×10−7). Most lead signals represented known susceptibility loci for HF risk factors, such as atrial fibrillation (PITX2) and CAD (CDKN2B-AS1 and MAP3K7CL) (Figure 2a; Table 2).12, 28 No meaningful test statistic inflation was detected (Supplemental Figure S1). In a sensitivity analysis in which we repeated genetic association testing after omitting cases of all-cause HF derived solely from self-reported data rather than from ICD codes (n = 197; only 3% of all quality-controlled cases), we observed similar associations and effect estimates, suggesting that our phenotype was not unduly influenced by potential self-report misclassification (Supplemental Table S3).
Figure 2. Manhattan plots of primary genome-wide association discovery analysis in UK Biobank for (a) all-cause heart failure and (b) nonischemic cardiomyopathy.
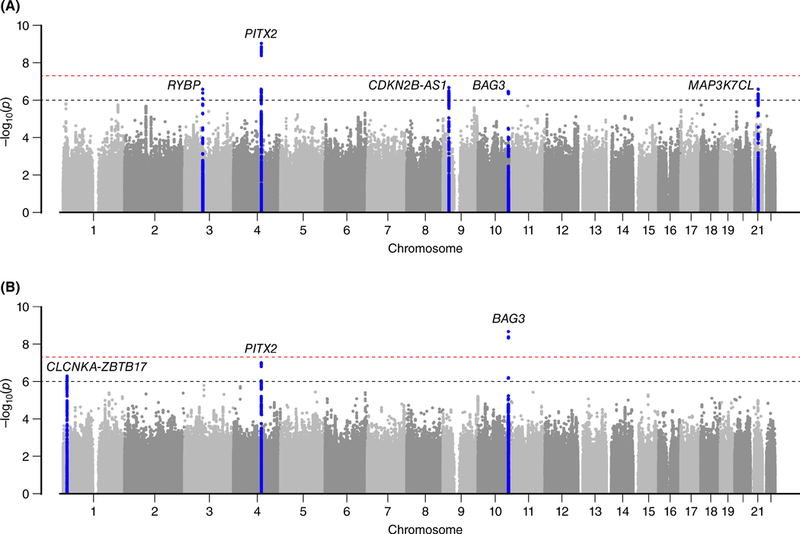
Logistic regression was used to test the association of allelic dosages for all variants with MAF > 1% with both all-cause heart failure and nonischemic cardiomyopathy, adjusting for age at baseline, sex, genotyping chip, and the first 10 principal components of ancestry. Lines are drawn at 1×10−6 and 5×10−8 to denote suggestive and genome-wide significant associations, respectively. Loci demonstrating P-value < 1×10−6 are highlighted in blue and the nearest genes are labeled.
Table 2.
Replication of suggestive signals from genetic association analyses for all-cause heart failure in UK Biobank.
| UK Biobank (n cases=6,504) |
BioVU Study (n cases=2,982) |
GRADE (n cases=1,060) |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||||
| Rsid | Chr | Pos | Nearest Gene |
RA/ NRA |
RAF | OR (95% CI) |
P-value | RAF | OR (95% CI) |
P-value | RAF | OR (95% CI) |
P-value |
| rs1906609 | 4 | 111666451 | PITX2 | T/G | 0.16 | 1.15 (1.10–1.21) | 9.08×10−10 | 0.18 | 1.00 (0.92 −1.09) | 0.95 | 0.18 | 1.14 (0.99–1.33) | 0.08 |
| rs7857118 | 9 | 22124140 | CDKN2B-AS1 | T/A | 0.51 | 1.10 (1.06–1.14) | 2.15×10−7 | 0.52 | 1.05 (0.99–1.12) | 0.11 | 0.53 | 1.07 (0.96–1.20) | 0.24 |
| rs12627426 | 21 | 30519457 | MAP3K7CL | A/T | 0.16 | 1.13 (1.08–1.18) | 2.63×10−7 | 0.15 | 1.02 (0.93–1.11) | 0.70 | 0.15 | 1.20 (1.03–1.40) | 0.02 |
| rs73839819 | 3 | 72579834 | RYBP | G/A | 0.02 | 1.33 (1.19–1.48) | 2.65×10−7 | 0.02 | 0.94 (0.75–1.17) | 0.55 | 0.02 | 0.90 (0.60–1.36) | 0.63 |
| rs2234962 | 10 | 121429633 | BAG3 | T/C | 0.78 | 1.12 (1.07–1.17) | 3.55×10−7 | 0.79 | 1.00 (0.92–1.07) | 0.90 | 0.80 | 1.27 (1.10–1.46) | 0.001 |
Abbreviations: HF=heart failure; Chr=chromosome; Pos=hg19 position; RA=all-cause HF risk allele; NRA=all-cause HF non-risk allele; RAF=all-cause HF risk allele frequency; OR=odds ratio; 95% CI=95% confidence interval
Refining the All-Cause HF phenotype to NICM
We then refined the all-cause HF phenotype to NICM on the basis of left ventricular dysfunction without CAD and identified 2,038 individuals who met phenotypic criteria (Figure 1). There was a higher proportion of females in the NICM group as compared to the all-cause HF group (35.0% v. 30.2%, P < 0.001). Furthermore, compared to the all-cause HF group, individuals in the NICM subset were more likely to have co-morbid atrial fibrillation (50.8% v. 43.0%, P < 0.001), and less likely to have co-morbid type 2 diabetes mellitus (19.4% v. 26.1%, P < 0.001) and hypertension (69.3% v. 75.6%, P < 0.001) (Table 1).
Validation of NICM phenotype
To validate our NICM phenotype, we applied the above phenotyping algorithm to individuals in the Partners HealthCare Biobank (Supplemental Methods) and performed manual chart reviews for 50 individuals who met criteria for NICM. Forty-five of the 50 study participants had evidence of a NICM diagnosis within the medical record (PPV = 0.90), which we considered sufficient validation to support genetic analysis.
Genome-wide association analysis of NICM in UK Biobank
A genome-wide association analysis for our refined NICM phenotype resulted in three signals – one locus reaching genome-wide significance (rs2234962, a missense variant in BAG3, OR = 1.30; P = 2.32×10−9) and two others at suggestive significance (rs12138073, an intronic variant near CLCNKA and ZBTB17, OR = 1.29, P = 5.35×10−7; rs2634071 in high linkage disequilibrium with rs1906609 upstream of PITX2, OR = 1.25, P = 1.06×10−7) – the majority at loci previously implicated in DCM (BAG3 and CLCNKA-ZBTB17)(Figure 2b; Table 3).16, 17, 29–31 Notably, we observed strong association signals at BAG3 for all-cause HF and NICM, although effect estimates were consistently stronger for NICM. No meaningful test statistic inflation was detected (Supplemental Figure S1).
Table 3.
Replication of suggestive signals from genetic association analyses for nonischemic cardiomyopathy in UK Biobank.
| UK Biobank (n cases=1,816) |
BioVU Study (n cases=226) |
GRADE (n cases=260) |
Esslinger et al.
(2017) (n cases=2,796) |
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||||||||
| Rsid | Chr | Pos | Nearest Gene | RA/ NRA | RAF | OR (95% CI) | P-value | RAF | OR (95% CI) | P-value | RAF | OR (95% CI) | P-value | RAF | OR (95% CI) | P-value |
| rs2234962 | 10 | 121429633 | BAG3 | T/C | 0.78 | 1.30 (1.19–1.41) | 2.32×10−9 | 0.80 | 1.49 (1.14–1.94) | 3.12×10−3 | 0.79 | 1.39 (1.08–1.80) | 0.01 | 0.81 | 1.61 (1.48–1.77) | 1.70×10−25 |
| rs2634071 | 4 | 111669220 | PITX2 | T/C | 0.16 | 1.25 (1.15–1.36) | 1.06×10−7 | 0.17 | 1.05 (0.82–1.35) | 0.68 | 0.17 | 1.13 (0.87–1.46) | 0.36 | - | - | - |
| rs6843082* | 4 | 111718067 | PITX2 | G/A | 0.19 | 1.24 (1.14–1.34) | 1.09×10−7 | 0.21 | 0.93 (0.73–1.18) | 0.53 | 0.20 | 1.13 (0.89–1.43) | 0.33 | 0.23 | 1.11 (1.03–1.20) | 7.52×10−3 |
| rs12138073 | 1 | 16354958 | CLCNKA | T/C | 0.10 | 1.29 (1.17–1.42) | 5.35×10−7 | 0.10 | 1.06 (0.78–1.44) | 0.72 | 0.10 | 1.19 (0.88–1.62) | 0.26 | - | - | - |
| rs34471231† | 1 | 16356522 | CLCNKA | G/A | 0.10 | 1.29 (1.17–1.42) | 6.58×10−7 | 0.10 | 1.05 (0.77–1.43) | 0.74 | 0.10 | 1.19 (0.88–1.62) | 0.26 | 0.10 | 1.20 (1.08–1.34) | 9.61×10−4 |
| rs10927875 | 1 | 16299312 | ZBTB17 | C/T | 0.68 | 1.15 (1.07–1.24) | 1.15×10−4 | 0.68 | 1.22 (0.98–1.50) | 0.07 | 0.68 | 1.18 (0.96–1.47) | 0.12 | 0.69 | 1.30 (1.21–1.40) | 8.11×10−13 |
Abbreviations: NICM=nonischemic cardiomyopathy; Chr=chromosome; Pos=hg19 position; RA=NICM risk allele; NRA= NICM non-risk allele; RAF= NICM risk allele frequency; OR=odds ratio; 95% CI=95% confidence interval
rs6843082 is present in the exome chip analysis from Esslinger et al. (2017)17 and in LD with rs2634071 in the UK Biobank (r2 = 0.82)
rs34471231 is present in the exome chip analysis from Esslinger et al. (2017)17 and in LD with rs12138073 in the UK Biobank (r2 = 0.99)
Replication of lead All-cause HF and NICM signals
We sought replication for the all-cause HF and NICM variants surpassing our suggestive significance threshold of P < 1×10−6 – rs1906609/rs2634071 (PITX2), rs7857118 (CDKN2B-AS1), rs12627426 (MAP3K7CL), rs73839819 (RYBP), rs2234962 (BAG3), and rs12138073 (CLCNKA-ZBTB17).
Of the five lead variants associated with all-cause HF, we observed replication in the GRADE cohort for those at BAG3 and MAP3K7CL. Replication was more modest for variants at CDKN2B-AS1 and PITX2, albeit with effect estimates similar to those observed in UK Biobank. In contrast, the association signal at RYBP did not replicate in GRADE, with an effect estimate directionally inconsistent with that observed in UK Biobank. No lead variants associated with all-cause HF replicated in Vanderbilt BioVU (Table 2).
Of the lead NICM variants, we observed strong and consistent replication for the signal at BAG3, which associated with NICM in the Vanderbilt-BioVU, GRADE, and Esslinger et al. cohorts. The lead variant at CLCNKA-ZBTB17 (rs12138073) demonstrated a more modest, but directionally-consistent, association with NICM in GRADE, while a proxy-SNP (rs34471231, r2 = 0.99) associated strongly with NICM in the Esslinger et al. cohort. Of note, an association with NICM has been reported previously for an independent variant (rs10927875, r2 = 0.01) near our lead CLCNKA-ZBTB17 signal.17 As this variant was associated with NICM in UK Biobank (OR = 1.15, p = 1.15×10−4), and demonstrated modest to strong replication in the Vanderbilt-BioVU, GRADE, and Esslinger et al. cohorts, we included it in subsequent follow-up analyses. Finally, the lead variant for PITX2 did not replicate in Vanderbilt-BioVU or GRADE, although a proxy SNP (rs6843082, r2 = 0.82 with rs2634071) did associate strongly with NICM in the Esslinger et al. cohort (Table 3).
Association of lead All-cause HF and NICM variants with HF risk factors
To assess whether lead variants for all-cause HF and NICM confer increased risk of disease through upstream risk factors, we first performed an association scan of 13 HF risk factors in UK Biobank. We observed robust associations between rs7857118 (CDKN2B-AS1) and CAD (OR per HF/NICM risk allele = 1.23, P = 1.24×10−72), and rs1906609 (PITX2) and atrial fibrillation (OR per HF/NICM risk allele = 1.49, P = 3.61×10−143), both well beyond a Bonferroni-corrected level of statistical significance [P < 0.05/(7 variants x 13 traits) = 5.49×10−4]. We also noted the following, more modest risk factor associations surpassing the statistical threshold for multiple testing: rs2234962 (BAG3) and hypertension (OR per HF/NICM risk allele = 1.02, P = 4.23×10−4), systolic blood pressure (Effect per HF/NICM risk allele = 0.19 mmHg, P = 3.52×10−4) and diastolic blood pressure (Effect per HF/NICM risk allele = 0.19 mmHg, P =1.07×10−10); rs10927875 (CLCNKA-ZBTB17) and hypertension (OR per HF/NICM risk allele = 1.02, P = 1.02×10−4), systolic blood pressure (Effect per HF/NICM risk allele = 0.20, P = 1.12×10−5), and diastolic blood pressure (Effect per HF/NICM risk allele = 0.09, P = 5.58×10−4) (Supplemental Table S4).
Coronary Artery Disease
Since CDKN2B-AS1 on chromosome 9 represents a well-known CAD susceptibility locus, and the lead variant at this locus (rs7857118) associated strongly with CAD in the UK Biobank (above), we performed an association analysis for all-cause HF adjusted for baseline CAD to test whether this locus increased HF risk independent of CAD. Adjustment for baseline CAD resulted in marked attenuation of the association between rs7857118 and all-cause HF (OR = 1.04, P = 0.03). Moreover, rs7857118 showed no association with NICM (OR = 1.04, P = 0.25), further suggesting that the link between the CKDN2B-AS1 locus and all-cause HF is largely mediated by CAD. Adjustment for baseline CAD did not significantly influence the strength of association between other lead variants and all-cause HF (Figure 3; Supplemental Table S5).
Figure 3. Association of suggestive all-cause heart failure and nonischemic cardiomyopathy variants adjusted for known heart failure risk factors.
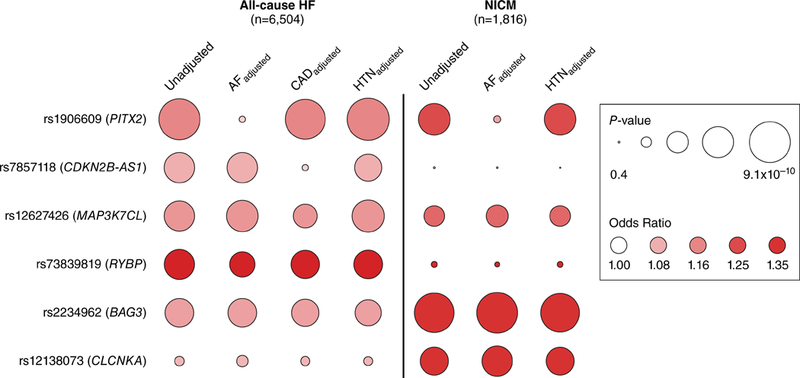
Logistic regression was used to test the association of lead variants identified at suggestive loci (P < 1×10−6) for either all-cause heart failure or nonischemic cardiomyopathy against both endpoints adjusted for baseline atrial fibrillation, baseline coronary artery disease, and baseline hypertension. Nonischemic cardiomyopathy testing was not adjusted for coronary artery disease as coronary artery disease was an exclusion criteria. All analyses were additionally adjusted for age at baseline, sex, genotyping array, and the first 10 principal components of ancestry. Circle size denotes P-value and shading represents the odds ratio for a 1-allele increase of the all-cause heart failure/nonischemic cardiomyopathy risk allele. Abbreviations: HF=heart failure; NICM=nonischemic cardiomyopathy; AF=atrial fibrillation; CAD=coronary artery disease; HTN=hypertension.
Atrial Fibrillation
PITX2 on chromosome 4 is a recognized risk locus for atrial fibrillation, which may mediate the observed association between this gene region and all-cause HF/NICM. However, since the link between atrial fibrillation and HF is bidirectional, we first examined the prevailing temporal relationship between incident atrial fibrillation and incident all-cause HF/NICM in the UK Biobank.32, 33 Of the 1,536 individuals who developed both incident all-cause HF and incident atrial fibrillation, 1,237 (81%) carried a diagnosis of atrial fibrillation at or prior to a first diagnosis of HF. A similar pattern was observed for NICM, as 436 of 513 individuals with co-incident disease (85%) had evidence of prior or concurrent atrial fibrillation (Supplemental Figure S2).
We therefore performed genetic association testing for all-cause HF and NICM in UK Biobank adjusted for baseline atrial fibrillation. Adjustment for baseline atrial fibrillation resulted in marked attenuation of the association between rs1906609 and all-cause HF (OR = 1.05, P = 0.04), and between rs2634071 and NICM (OR = 1.11, P = 0.02), suggesting that the association between PITX2 and all-cause HF/NICM in UK Biobank is largely mediated by co-incident or antecedent atrial fibrillation. Adjustment for baseline atrial fibrillation did not significantly influence the strength of association between other lead variants and all-cause HF or NICM (Figure 3; Supplemental Table S6 and Supplemental Table S7).
Hypertension
Neither BAG3 nor CLCNKA-ZBTB17 is an established susceptibility locus for hypertension, but variants at each were associated with hypertension and systolic/diastolic blood pressure in the UK Biobank (above). We therefore pursued genetic association testing for all-cause HF and NICM in UK Biobank adjusted for prevalent hypertension, systolic blood pressure, or diastolic blood pressure. Adjusted analyses demonstrated persistently strong signals at rs2234962 (BAG3) and rs10927875 (CLCNKA-ZBTB17) with minimal attenuation of the allelic effect size, suggesting that variation at BAG3 and CLCNKA-ZBTB17 confer risk of all-cause HF and NICM independent of elevated blood pressure (Figure 3; Supplemental Table S8 and Supplemental Table S9).
Association of lead All-cause HF and NICM variants with intermediate traits of LV structure and function in individuals without clinical heart failure
To evaluate the relationship between lead all-cause HF and NICM variants with quantitative measures of LV structure and function in the general population, we queried available imaging data in individuals without clinical HF from two sources: (1) cardiac MRI data from 4,158 participants in the UK Biobank (Supplemental Figure S3), and (2) summary-level data on 16 echocardiographic parameters in 30,081 individuals from a recent genome-wide association study (EchoGen Consortium).34 Clinical characteristics of HF-free UK Biobank participants who did and did not undergo cardiac MRI are presented in Supplemental Table S10. Individuals who underwent cardiac MRI in the UK Biobank were generally healthier than their counterparts, as evidenced by younger mean age, lower mean BMI, and lower rates of CAD, atrial fibrillation and type 2 diabetes mellitus.
Of the seven lead all-cause HF and NICM variants assessed, only those at BAG3 and CLCNKA-ZBTB17 associated with cardiac MRI measures of LV structure and function in the UK Biobank at a Bonferroni-corrected level of statistical significance [P < 0.05 / (6 traits x 7 SNPs) = 0.0012]. Specifically, we observed associations between rs2234962 (BAG3) and reduced LVEF (Effect per NICM risk allele = −0.58%, P = 5.68×10−5) and increased LVESV (Effect per NICM risk allele = 1.53ml, P = 3.41×10−4) (Figure 4a, Supplemental Table S11); these associations replicated in analogous, summary-level data from the EchoGen Consortium, where rs2234962 associated with reduced fractional shortening (Effect per NICM risk allele = −0.30%, P = 6.05×10−8) and increased LV diastolic diameter (Effect per NICM risk allele = 0.017cm, P = 6.59×10−5).34 In addition, rs10927875 (CLNCKA-ZBTB17) was significantly associated with reduced LVEF (Effect per NICM risk allele = −0.42%, P = 1.08×10−3) and increased LVESV (Effect per NICM risk allele = 1.31ml, P = 6.49×10−4) in UK Biobank (Figure 4b; Supplemental Table S11).
Figure 4. Association of suggestive all-cause heart failure and nonischemic cardiomyopathy variants with selected cardiac MRI traits of left ventricular structure and function in UK Biobank.
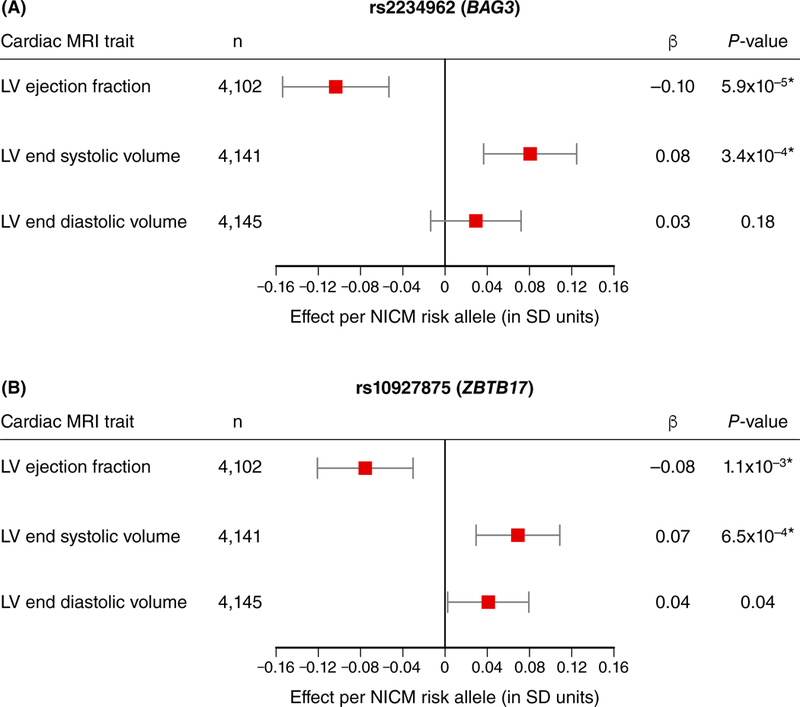
Linear regression was used to test the association of suggestive signals for all-cause heart failure and nonischemic cardiomyopathy variants with measured cardiac MRI traits in up to 4,158 individuals free of clinical heart failure in the UK Biobank. Testing was performed using allelic dosages, adjusting for age at baseline, sex, genotyping chip, and the first 10 principal components of ancestry. Results are displayed for (a) rs2234962 near BAG3 and (b) rs10927875 near ZBTB17 against three selected cardiac MRI traits as no other variants had associations reaching statistical significance. Points represent the effect in SD units of each respective cardiac MRI trait and error bars denote 95% confidence intervals. Significant associations passing Bonferroni significance (P < 0.05 / 42 = 1.19×10−3) are denoted with a star (*). Abbreviations: NICM=nonischemic cardiomyopathy; β=effect per NICM risk allele in SD units of the cardiac MRI trait; SD=standard deviation.
Rare, loss-of-function variants in DCM genes and risk of All-cause HF or NICM in UK Biobank
We next investigated whether rarer mutations with predicted functional consequences might be differentially associated with all-cause HF and NICM in the UK Biobank. Rare mutations of larger effect size have been identified previously for DCM. We tested whether rare, predicted loss-of-function variants in 24 known DCM genes with carrier frequency > 0.0001 associated with our phenotypes for all-cause HF or NICM in the UK Biobank. Only the association between predicted loss-of-function mutations at BAG3 and NICM surpassed a Bonferroni-corrected significance threshold (P < 0.05 / (2 phenotypes x 24 genes) = 0.001): 0.165% of NICM cases carried a rare, loss-of-function mutation in BAG3 whereas 0.014% of controls did (OR = 12.03, P = 3.62×10−5). There were no statistically significant associations between any predicted loss-of-function mutations at the tested DCM genes and all-cause HF (Supplemental Table S12).
DISCUSSION
Genome-wide association analysis of all-cause HF in the UK Biobank identified multiple known loci for HF risk factors – i.e. CAD and atrial fibrillation – highlighting major genetic determinants of this common disease. By comparison, refinement of all-cause HF to a specific, NICM phenotype yielded strong genetic signals at loci for DCM that were independent of HF risk factors and associated with intermediate traits of LV structure and function in individuals without clinical HF.
These results permit several conclusions. First, our genetic analysis of all-cause HF underscores the complexity of this condition and points to several etiologic subtypes, driven partly by a genetic predisposition to prominent HF risk factors. For instance, we found that PITX2, a known susceptibility locus for atrial fibrillation, and both CDKN2B-AS1 and MAP3K7CL, known CAD loci, were strongly associated with all-cause HF.
Atrial fibrillation and HF are established risk factors for one another and frequently co-exist.32 A recent study noted that > 50% of all HF patients have co-incident atrial fibrillation, and that atrial fibrillation is more likely to precede rather than follow a diagnosis of HF.33 Among all-cause HF and NICM patients in the UK Biobank, we observed comparable rates of antecedent and comorbid atrial fibrillation. Furthermore, the attenuation of the PITX2 association signal after adjustment for prevalent atrial fibrillation indicates that the observed association between PITX2 and HF likely reflects co-incident disease. Inconsistent replication of PITX2 in our independent cohorts may reflect the exclusion of patients with tachycardia-induced cardiomyopathy in recruited, hospital-based registries, highlighting the phenotypic precision offered by recruited cohorts capable of disentangling the complex HF-atrial fibrillation relationship a priori. In contrast, population-based approaches are complementary and enable the analysis of HF in the context of prominent risk factors.
Similarly, the association signal at CDKN2B-AS1 was diminished after adjusting for prevalent CAD and in the NICM sample, underscoring the importance of CAD as a driver of HF. Notably, we observed only modest attenuation of the association between the MAP3K7CL locus and all-cause HF, and a stronger effect estimate for the association of this locus with NICM, suggesting that variation at MAP3K7CL may influence HF risk via mechanisms independent of CAD. Further analyses are needed to determine how the MAP3K7CL locus might mediate HF beyond its contribution to CAD risk.
Second, phenotypic refinement of HF within a large, population-based biobank is feasible and may facilitate genetic discovery. Prior efforts to uncover the genetics of common, complex HF have been hindered by marked disease heterogeneity. Although recent advances have come from a small number of genetic analyses of selected heart failure subpopulations, there has been limited consideration to date of such disease subtypes and/or HF with preserved versus reduced ejection fraction.13, 16–18 Whereas large sample sizes enhance power for discovery, our data suggest that precise phenotyping is important for the discovery of subtype-specific HF susceptibility loci. Compared to the all-cause HF phenotype, the more precise NICM definition yielded stronger genetic association signals at known loci for DCM (i.e., BAG3 and CLCNKA-ZBTB17) despite three-fold fewer cases. Moreover, lead NICM variants demonstrated more consistent replication in our independent cohorts than did the lead all-cause HF variants, likely due to the heterogeneity of the all-cause HF phenotype. Also, whereas our analysis of loss-of-function variation corroborates prior data implicating BAG3 as a bona-fide disease susceptibility gene for NICM, the association with all-cause heart failure was not significant, further underscoring the importance of phenotype for specificity of associations.16,17,29 Finally, our algorithm for ascertaining NICM in the UK Biobank utilized standard self-reported and billing-code data and may therefore be portable to other electronic health systems and forthcoming population-based biobanks.
Third, genetic drivers of DCM – best identified by the NICM phenotype – may mediate a subclinical cardiomyopathic process that predisposes to clinical HF. Here, we demonstrate that common genetic variants associated with clinical HF also associate with intermediate traits of LV structure and function in individuals without clinical disease. Prior epidemiological studies have noted that subtle, preclinical abnormalities in LV chamber size and function may herald progression to overt HF, prompting subsequent genetic association studies of intermediate echocardiographic traits.34–37 Consistent associations between a genetic variant, an intermediate imaging trait, and clinical HF therefore imply a causal mechanistic pathway. In our analyses of cardiac MRI and echocardiographic traits, two lead NICM variants previously linked to DCM – at BAG3 and CLCNKA-ZBTB17 – associated significantly with reduced LV systolic function and increased LV chamber size. Importantly, these associations were observed among those without clinical HF, suggesting a subclinical process that may portend a genetic predisposition to clinical heart failure. Whether such genetically mediated cardiomyopathies confer a prognosis similar to that of other cardiomyopathies – i.e. with respect to the risk of sudden cardiac death – remains unclear and requires further study.
Of note, ample functional data support the mechanistic roles of both BAG3 and CLCNKA-ZBTB17 in the pathogenesis of HF. For example, recent studies have suggested an anti-apoptotic function for BAG3 in cardiomyocytes, with morpholino knockdown in zebrafish resulting in cardiac enlargement and systolic dysfunction. 31, 38–45 Similarly, recent in vitro and in vivo analyses of ZBTB17 have identified an anti-apoptotic gene product critical for the adaptation of cardiomyocytes to biomechanical stress.46 Alongside our human genetic observations at these two loci, the data advocate for the pursuit and prioritization of other DCM signals with similar prognostic and therapeutic implications to advance current understanding of HF genetics.
Several limitations should be acknowledged. First, quantitative measures of LV structure and function were unavailable for most UK Biobank participants preventing classification of heart failure with reduced versus preserved ejection fraction and precluding a robust genetic association study of intermediate imaging traits. Forthcoming cardiac MRI data on 100,000 individuals in the UK Biobank will soon enable categorization of many more study participants based on LV systolic function. Second, in the absence of cohort-wide cardiac imaging data to permit morphologic classifications of disease, phenotyping was predicated on data from self-reports and the medical record, which carry the potential for disease misclassification. Furthermore, our refinement of all-cause HF focuses on the NICM subset, but not on the remainder of the HF population, which remains a heterogeneous group. However, we submit that our phenotyping approach serves only as an initial strategy for addressing the heterogeneity of HF. Future study using more sophisticated phenotyping strategies, including the integration of clinical, laboratory, and imaging data, may provide more nuanced classifications of HF and further facilitate genetic discovery. Third, temporal disease associations in the UK Biobank relied on hospitalization-based health registry data and periodic study examinations; disease status may have gone clinically unrecognized during interval periods. Fourth, analyses of rare, loss-of-function mutations were limited to those variants available on the genotyping array, and were unable to detect novel and private mutations. Fifth, our analyses were limited to participants of European ancestry; as these findings may not apply to individuals of other ancestries, validation of these results in ancestries outside of Europe is required.
In conclusion, we found evidence for distinct genetic mechanisms of HF, including those that operate through known HF risk factors. Phenotypic refinement of all-cause HF to a specific NICM subset appears to facilitate genetic discovery by better identifying genetic signals for cardiomyopathy that operate independent of HF risk factors and associate with clinical and subclinical disease. Future studies are warranted to investigate the prognostic and therapeutic implications of these findings for the prevention and management of HF.
Supplementary Material
CLINICAL PERSPECTIVE
What is New?
We performed a population-based genetic association study of “all-cause heart failure,” which yielded multiple genetic signals for known heart failure risk factors, such as coronary artery disease and atrial fibrillation.
Refining the heart failure phenotype to a “nonischemic cardiomyopathy” subset enhanced the detection of genetic loci associated with dilated cardiomyopathy, which appear to operate independent of traditional heart failure risk factors.
Genetic variants associated with nonischemic cardiomyopathy were also associated with subclinical traits of left ventricular dysfunction.
What are the clinical implications?
Phenotypic refinement aids in the discovery of novel genetic signals that reflect distinct etiologic heart failure subtypes.
The BAG3 locus is a principal nonischemic cardiomyopathy susceptibility locus, and future functional characterization of this and other genetic loci may inform therapeutic development.
Common genetic variants associated with both clinical and subclinical heart failure may be leveraged to improve heart failure risk prediction and prevention.
Acknowledgments
The authors would like to thank all UK Biobank participants, staff, and investigators. This research has been conducted using the UK Biobank Resource under Application Numbers 7089 and 17488.
Sources of Funding
This work was funded by the National Institutes of Health (R01 HL139731 to S.A.L. and R01 HL127564 to S.K.) and the Doris Duke Charitable Foundation (Clinical Scientist Development Award 2014105 to S.A.L.). Vanderbilt University Medical Center’s BioVU projects are supported by numerous sources: institutional funding, private agencies, and federal grants. These include the NIH funded Shared Instrumentation Grant S10RR025141; CTSA grants UL1TR002243, UL1TR000445, and UL1RR024975. Genomic data are also supported by investigator-led projects that include U01HG004798, R01NS032830, RC2GM092618, P50GM115305, U01HG006378, U19HL065962, R01HD074711; and additional funding sources listed at https://victr.vanderbilt.edu/pub/biovu/?sid=229. The funding agencies had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
Appendix
Genetic Risk Assessment of Defibrillator Events (GRADE) Investigators: Heather L. Bloom, MD (Division of Cardiology, Emory University and Atlanta VA Medical Center, Atlanta, GA), Samuel C. Dudley, MD, PhD (Cardiovascular Division and Lillehei Heart Institute, University of Minnesota, Minneapolis, MN), Patrick T. Ellinor, MD, PhD (Massachusetts General Hospital, Boston, MA), Alaa A. Shalaby, MD (Division of Cardiology, University of Pittsburgh, PA; Cardiology Division, Pittsburgh VA Healthcare System, Pittsburgh, PA), Raul Weiss, MD (Division of Cardiovascular Medicine, The Ohio State University, Columbus, OH), Rebecca Gutmann, RN, BSN, CCRC (Division of Cardiovascular Medicine and Abboud Cardiovascular Research Center, University of Iowa, Iowa City), Samir Saba, MD (Division of Cardiology, University of Pittsburgh, PA), Barry London, MD, PhD (Division of Cardiovascular Medicine and Abboud Cardiovascular Research Center, University of Iowa, Iowa City).
Footnotes
Disclosures
K.G.A. is supported by an award from the American Heart Association Institute for Precision Cardiovascular Medicine (17IFUNP33840012). M.B.S is supported by NIH K23 HL127704. M.E.L. is supported by the National Heart, Lung, and Blood Institute of the National Institutes of Health (R01 HL130113) and by the Fredman Fellowship, the Toomey Fund for Aortic Dissection Research, and the Hassenfeld Fellowship. J.G.S. is supported by the Wallenberg Center for Molecular Medicine in Lund and grants from the Swedish Heart-Lung Foundation (2016–0134 and 2016–0315), the Swedish Research Council (2017–02554), the European Research Council (ERC-STG-2015–679242), the Crafoord Foundation, Skåne University Hospital, the Scania county, and governmental funding of clinical research within the Swedish National Health Service. C.N.C. is supported by the National Heart, Lung, and Blood Institute of the National Institutes of Health (R01HL113933 and R01HL124262). B.L. is supported by the National Heart, Lung, and Blood Institute of the National Institutes of Health (R01 HL77398). Q.S.W. is supported by the National Heart, Lung, and Blood Institute of the National Institutes of Health (R01HL140074). P.T.E. is supported by the Foundation Leducq (14CVD01) and by awards from the National Heart, Lung, and Blood Institute (RO1HL092577, R01HL128914, K24HL105780). S.K. is supported by an Ofer and Shelly Nemirovsky Research Scholar Award from Massachusetts General Hospital, and by awards from the National Heart, Lung, and Blood Institute (RO1 HL127564), and the National Human Genome Research Institute (5UM1HG008895). S.A.L. is supported by the National Heart, Lung, and Blood Institute (RO1HL092577) and the Doris Duke Charitable Foundation (Clinical Scientist Development Award 2014105), and receives sponsored research support from Bristol Myers Squibb, Bayer HealthCare, Biotronik, and Boehringer Ingelheim, and has consulted for Abbott, Quest Diagnostics, and Bristol Myers Squibb. All other authors have reported that they have no relationships relevant to the contents of this paper to disclose.
REFERENCES
- 1.Writing Group M, Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, Das SR, de Ferranti S, Despres JP, Fullerton HJ, Howard VJ, Huffman MD, Isasi CR, Jimenez MC, Judd SE, Kissela BM, Lichtman JH, Lisabeth LD, Liu S, Mackey RH, Magid DJ, McGuire DK, Mohler ER 3rd, Moy CS, Muntner P, Mussolino ME, Nasir K, Neumar RW, Nichol G, Palaniappan L, Pandey DK, Reeves MJ, Rodriguez CJ, Rosamond W, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Woo D, Yeh RW, Turner MB, American Heart Association Statistics C and Stroke Statistics S. Heart Disease and Stroke Statistics-2016 Update: A Report From the American Heart Association. Circulation. 2016;133:e38–360. [DOI] [PubMed] [Google Scholar]
- 2.Heidenreich PA, Albert NM, Allen LA, Bluemke DA, Butler J, Fonarow GC, Ikonomidis JS, Khavjou O, Konstam MA, Maddox TM, Nichol G, Pham M, Pina IL, Trogdon JG, American Heart Association Advocacy Coordinating C, Council on Arteriosclerosis T, Vascular B, Council on Cardiovascular R, Intervention, Council on Clinical C, Council on E, Prevention and Stroke C. Forecasting the impact of heart failure in the United States: a policy statement from the American Heart Association. Circ Heart Fail. 2013;6:606–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ziaeian B and Fonarow GC. Epidemiology and aetiology of heart failure. Nat Rev Cardiol. 2016;13:368–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roger VL, Weston SA, Redfield MM, Hellermann-Homan JP, Killian J, Yawn BP and Jacobsen SJ. Trends in heart failure incidence and survival in a community-based population. JAMA. 2004;292:344–350. [DOI] [PubMed] [Google Scholar]
- 5.Lee DS, Pencina MJ, Benjamin EJ, Wang TJ, Levy D, O’Donnell CJ, Nam BH, Larson MG, D’Agostino RB and Vasan RS. Association of parental heart failure with risk of heart failure in offspring. N Engl J Med. 2006;355:138–47. [DOI] [PubMed] [Google Scholar]
- 6.Savarese G and Lund LH. Global Public Health Burden of Heart Failure. Card Fail Rev. 2017;3:7–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ambrosy AP, Fonarow GC, Butler J, Chioncel O, Greene SJ, Vaduganathan M, Nodari S, Lam CSP, Sato N, Shah AN and Gheorghiade M. The global health and economic burden of hospitalizations for heart failure: lessons learned from hospitalized heart failure registries. J Am Coll Cardiol. 2014;63:1123–1133. [DOI] [PubMed] [Google Scholar]
- 8.Tayal U, Prasad S and Cook SA. Genetics and genomics of dilated cardiomyopathy and systolic heart failure. Genome Med. 2017;9:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.MacRae CA. Mendelian forms of structural cardiovascular disease. Curr Cardiol Rep. 2013;15:399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Klarin D, Zhu QM, Emdin CA, Chaffin M, Horner S, McMillan BJ, Leed A, Weale ME, Spencer CCA, Aguet F, Segre AV, Ardlie KG, Khera AV, Kaushik VK, Natarajan P, Consortium CAD and Kathiresan S. Genetic analysis in UK Biobank links insulin resistance and transendothelial migration pathways to coronary artery disease. Nat Genet. 2017;49:1392–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Warren HR, Evangelou E, Cabrera CP, Gao H, Ren M, Mifsud B, Ntalla I, Surendran P, Liu C, Cook JP, Kraja AT, Drenos F, Loh M, Verweij N, Marten J, Karaman I, Lepe MP, O’Reilly PF, Knight J, Snieder H, Kato N, He J, Tai ES, Said MA, Porteous D, Alver M, Poulter N, Farrall M, Gansevoort RT, Padmanabhan S, Magi R, Stanton A, Connell J, Bakker SJ, Metspalu A, Shields DC, Thom S, Brown M, Sever P, Esko T, Hayward C, van der Harst P, Saleheen D, Chowdhury R, Chambers JC, Chasman DI, Chakravarti A, Newton-Cheh C, Lindgren CM, Levy D, Kooner JS, Keavney B, Tomaszewski M, Samani NJ, Howson JM, Tobin MD, Munroe PB, Ehret GB, Wain LV, International Consortium of Blood Pressure GA, Consortium B, Lifelines Cohort S, Understanding Society Scientific g, Consortium CHDE, Exome BPC, Consortium TDG, Go TDC, Cohorts for H, Ageing Research in Genome Epidemiology BPEC, International Genomics of Blood Pressure C and group UKBCCBw. Genome-wide association analysis identifies novel blood pressure loci and offers biological insights into cardiovascular risk. Nat Genet. 2017;49:403–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Christophersen IE, Rienstra M, Roselli C, Yin X, Geelhoed B, Barnard J, Lin H, Arking DE, Smith AV, Albert CM, Chaffin M, Tucker NR, Li M, Klarin D, Bihlmeyer NA, Low SK, Weeke PE, Muller-Nurasyid M, Smith JG, Brody JA, Niemeijer MN, Dorr M, Trompet S, Huffman J, Gustafsson S, Schurmann C, Kleber ME, Lyytikainen LP, Seppala I, Malik R, Horimoto A, Perez M, Sinisalo J, Aeschbacher S, Theriault S, Yao J, Radmanesh F, Weiss S, Teumer A, Choi SH, Weng LC, Clauss S, Deo R, Rader DJ, Shah SH, Sun A, Hopewell JC, Debette S, Chauhan G, Yang Q, Worrall BB, Pare G, Kamatani Y, Hagemeijer YP, Verweij N, Siland JE, Kubo M, Smith JD, Van Wagoner DR, Bis JC, Perz S, Psaty BM, Ridker PM, Magnani JW, Harris TB, Launer LJ, Shoemaker MB, Padmanabhan S, Haessler J, Bartz TM, Waldenberger M, Lichtner P, Arendt M, Krieger JE, Kahonen M, Risch L, Mansur AJ, Peters A, Smith BH, Lind L, Scott SA, Lu Y, Bottinger EB, Hernesniemi J, Lindgren CM, Wong JA, Huang J, Eskola M, Morris AP, Ford I, Reiner AP, Delgado G, Chen LY, Chen YI, Sandhu RK, Li M, Boerwinkle E, Eisele L, Lannfelt L, Rost N, Anderson CD, Taylor KD, Campbell A, Magnusson PK, Porteous D, Hocking LJ, Vlachopoulou E, Pedersen NL, Nikus K, Orho-Melander M, Hamsten A, Heeringa J, Denny JC, Kriebel J, Darbar D, Newton-Cheh C, Shaffer C, Macfarlane PW, Heilmann-Heimbach S, Almgren P, Huang PL, Sotoodehnia N, Soliman EZ, Uitterlinden AG, Hofman A, Franco OH, Volker U, Jockel KH, Sinner MF, Lin HJ, Guo X, ISGC MCot, Neurology Working Group of the CC, Dichgans M, Ingelsson E, Kooperberg C, Melander O, Loos RJF, Laurikka J, Conen D, Rosand J, van der Harst P, Lokki ML, Kathiresan S, Pereira A, Jukema JW, Hayward C, Rotter JI, Marz W, Lehtimaki T, Stricker BH, Chung MK, Felix SB, Gudnason V, Alonso A, Roden DM, Kaab S, Chasman DI, Heckbert SR, Benjamin EJ, Tanaka T, Lunetta KL, Lubitz SA, Ellinor PT and Consortium AF. Large-scale analyses of common and rare variants identify 12 new loci associated with atrial fibrillation. Nat Genet. 2017;49:946–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rau CD, Lusis AJ and Wang Y. Genetics of common forms of heart failure: challenges and potential solutions. Curr Opin Cardiol. 2015;30:222–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smith NL, Felix JF, Morrison AC, Demissie S, Glazer NL, Loehr LR, Cupples LA, Dehghan A, Lumley T, Rosamond WD, Lieb W, Rivadeneira F, Bis JC, Folsom AR, Benjamin E, Aulchenko YS, Haritunians T, Couper D, Murabito J, Wang YA, Stricker BH, Gottdiener JS, Chang PP, Wang TJ, Rice KM, Hofman A, Heckbert SR, Fox ER, O’Donnell CJ, Uitterlinden AG, Rotter JI, Willerson JT, Levy D, van Duijn CM, Psaty BM, Witteman JC, Boerwinkle E and Vasan RS. Association of genome-wide variation with the risk of incident heart failure in adults of European and African ancestry: a prospective meta-analysis from the cohorts for heart and aging research in genomic epidemiology (CHARGE) consortium. Circ Cardiovasc Genet. 2010;3:256–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Everly MJ. Cardiac transplantation in the United States: an analysis of the UNOS registry. Clin Transpl. 2008:35–43. [PubMed] [Google Scholar]
- 16.Villard E, Perret C, Gary F, Proust C, Dilanian G, Hengstenberg C, Ruppert V, Arbustini E, Wichter T, Germain M, Dubourg O, Tavazzi L, Aumont MC, DeGroote P, Fauchier L, Trochu JN, Gibelin P, Aupetit JF, Stark K, Erdmann J, Hetzer R, Roberts AM, Barton PJ, Regitz-Zagrosek V, Cardiogenics C, Aslam U, Duboscq-Bidot L, Meyborg M, Maisch B, Madeira H, Waldenstrom A, Galve E, Cleland JG, Dorent R, Roizes G, Zeller T, Blankenberg S, Goodall AH, Cook S, Tregouet DA, Tiret L, Isnard R, Komajda M, Charron P and Cambien F. A genome-wide association study identifies two loci associated with heart failure due to dilated cardiomyopathy. Eur Heart J. 2011;32:1065–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Esslinger U, Garnier S, Korniat A, Proust C, Kararigas G, Muller-Nurasyid M, Empana JP, Morley MP, Perret C, Stark K, Bick AG, Prasad SK, Kriebel J, Li J, Tiret L, Strauch K, O’Regan DP, Marguiles KB, Seidman JG, Boutouyrie P, Lacolley P, Jouven X, Hengstenberg C, Komajda M, Hakonarson H, Isnard R, Arbustini E, Grallert H, Cook SA, Seidman CE, Regitz-Zagrosek V, Cappola TP, Charron P, Cambien F and Villard E. Exome-wide association study reveals novel susceptibility genes to sporadic dilated cardiomyopathy. PLoS One. 2017;12:e0172995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meder B, Ruhle F, Weis T, Homuth G, Keller A, Franke J, Peil B, Lorenzo Bermejo J, Frese K, Huge A, Witten A, Vogel B, Haas J, Volker U, Ernst F, Teumer A, Ehlermann P, Zugck C, Friedrichs F, Kroemer H, Dorr M, Hoffmann W, Maisch B, Pankuweit S, Ruppert V, Scheffold T, Kuhl U, Schultheiss HP, Kreutz R, Ertl G, Angermann C, Charron P, Villard E, Gary F, Isnard R, Komajda M, Lutz M, Meitinger T, Sinner MF, Wichmann HE, Krawczak M, Ivandic B, Weichenhan D, Gelbrich G, El-Mokhtari NE, Schreiber S, Felix SB, Hasenfuss G, Pfeufer A, Hubner N, Kaab S, Arbustini E, Rottbauer W, Frey N, Stoll M and Katus HA. A genome-wide association study identifies 6p21 as novel risk locus for dilated cardiomyopathy. Eur Heart J. 2014;35:1069–1077. [DOI] [PubMed] [Google Scholar]
- 19.Littlejohns TJ, Sudlow C, Allen NE and Collins R. UK Biobank: opportunities for cardiovascular research. Eur Heart J. 2017. doi: 10.1093/eurheartj/ehx254. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smith J Molecular epidemiology of heart failure: translational challenges and opportunities. J Am Coll Cardiol Basic Trans Science 2:757–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Broad Institute. Cardiovascular Disease Knowledge Portal. http://broadcvdi.org. August 2018. Accessed September 11, 2018.
- 22.McCarthy S, Das S, Kretzschmar W, Delaneau O, Wood AR, Teumer A, Kang HM, Fuchsberger C, Danecek P, Sharp K, Luo Y, Sidore C, Kwong A, Timpson N, Koskinen S, Vrieze S, Scott LJ, Zhang H, Mahajan A, Veldink J, Peters U, Pato C, van Duijn CM, Gillies CE, Gandin I, Mezzavilla M, Gilly A, Cocca M, Traglia M, Angius A, Barrett JC, Boomsma D, Branham K, Breen G, Brummett CM, Busonero F, Campbell H, Chan A, Chen S, Chew E, Collins FS, Corbin LJ, Smith GD, Dedoussis G, Dorr M, Farmaki AE, Ferrucci L, Forer L, Fraser RM, Gabriel S, Levy S, Groop L, Harrison T, Hattersley A, Holmen OL, Hveem K, Kretzler M, Lee JC, McGue M, Meitinger T, Melzer D, Min JL, Mohlke KL, Vincent JB, Nauck M, Nickerson D, Palotie A, Pato M, Pirastu N, McInnis M, Richards JB, Sala C, Salomaa V, Schlessinger D, Schoenherr S, Slagboom PE, Small K, Spector T, Stambolian D, Tuke M, Tuomilehto J, Van den Berg LH, Van Rheenen W, Volker U, Wijmenga C, Toniolo D, Zeggini E, Gasparini P, Sampson MG, Wilson JF, Frayling T, de Bakker PI, Swertz MA, McCarroll S, Kooperberg C, Dekker A, Altshuler D, Willer C, Iacono W, Ripatti S, Soranzo N, Walter K, Swaroop A, Cucca F, Anderson CA, Myers RM, Boehnke M, McCarthy MI, Durbin R and Haplotype Reference C. A reference panel of 64,976 haplotypes for genotype imputation. Nat Genet. 2016;48:1279–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McNally EM and Mestroni L. Dilated Cardiomyopathy: Genetic Determinants and Mechanisms. Circ Res. 2017;121:731–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GR, Thormann A, Flicek P and Cunningham F. The Ensembl Variant Effect Predictor. Genome Biol. 2016;17:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25..Karczewski KJ. GitHub. LOFTEE (Loss-Of-Function Transcript Effect Estimator). https://github.com/konradjk/loftee. 2015. Accessed September 11, 2018.
- 26.Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM and Lee JJ. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015;4:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27..EPACTS: Efficient and Parallelizable Association Container Toolbox. https://github.com/statgen/EPACTS. Dec 15th, 2016. Accessed September 11, 2018.
- 28.van der Harst P and Verweij N. Identification of 64 Novel Genetic Loci Provides an Expanded View on the Genetic Architecture of Coronary Artery Disease. Circ Res. 2018;122:433–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cappola TP, Li M, He J, Ky B, Gilmore J, Qu L, Keating B, Reilly M, Kim CE, Glessner J, Frackelton E, Hakonarson H, Syed F, Hindes A, Matkovich SJ, Cresci S and Dorn GW 2nd. Common variants in HSPB7 and FRMD4B associated with advanced heart failure. Circ Cardiovasc Genet. 2010;3:147–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cappola TP, Matkovich SJ, Wang W, van Booven D, Li M, Wang X, Qu L, Sweitzer NK, Fang JC, Reilly MP, Hakonarson H, Nerbonne JM and Dorn GW 2nd . Loss-of-function DNA sequence variant in the CLCNKA chloride channel implicates the cardio-renal axis in interindividual heart failure risk variation. Proc Natl Acad Sci U S A. 2011;108:2456–2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Norton N, Li D, Rieder MJ, Siegfried JD, Rampersaud E, Zuchner S, Mangos S, Gonzalez-Quintana J, Wang L, McGee S, Reiser J, Martin E, Nickerson DA and Hershberger RE. Genome-wide studies of copy number variation and exome sequencing identify rare variants in BAG3 as a cause of dilated cardiomyopathy. Am J Hum Genet. 2011;88:273–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang TJ, Larson MG, Levy D, Vasan RS, Leip EP, Wolf PA, D’Agostino RB, Murabito JM, Kannel WB and Benjamin EJ. Temporal relations of atrial fibrillation and congestive heart failure and their joint influence on mortality: the Framingham Heart Study. Circulation. 2003;107:2920–2925. [DOI] [PubMed] [Google Scholar]
- 33.Santhanakrishnan R, Wang N, Larson MG, Magnani JW, McManus DD, Lubitz SA, Ellinor PT, Cheng S, Vasan RS, Lee DS, Wang TJ, Levy D, Benjamin EJ and Ho JE. Atrial Fibrillation Begets Heart Failure and Vice Versa: Temporal Associations and Differences in Preserved Versus Reduced Ejection Fraction. Circulation. 2016;133:484–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wild PS, Felix JF, Schillert A, Teumer A, Chen MH, Leening MJG, Volker U, Grossmann V, Brody JA, Irvin MR, Shah SJ, Pramana S, Lieb W, Schmidt R, Stanton AV, Malzahn D, Smith AV, Sundstrom J, Minelli C, Ruggiero D, Lyytikainen LP, Tiller D, Smith JG, Monnereau C, Di Tullio MR, Musani SK, Morrison AC, Pers TH, Morley M, Kleber ME, Aragam J, Benjamin EJ, Bis JC, Bisping E, Broeckel U, Cheng S, Deckers JW, Del Greco MF, Edelmann F, Fornage M, Franke L, Friedrich N, Harris TB, Hofer E, Hofman A, Huang J, Hughes AD, Kahonen M, Investigators K, Kruppa J, Lackner KJ, Lannfelt L, Laskowski R, Launer LJ, Leosdottir M, Lin H, Lindgren CM, Loley C, MacRae CA, Mascalzoni D, Mayet J, Medenwald D, Morris AP, Muller C, Muller-Nurasyid M, Nappo S, Nilsson PM, Nuding S, Nutile T, Peters A, Pfeufer A, Pietzner D, Pramstaller PP, Raitakari OT, Rice KM, Rivadeneira F, Rotter JI, Ruohonen ST, Sacco RL, Samdarshi TE, Schmidt H, Sharp ASP, Shields DC, Sorice R, Sotoodehnia N, Stricker BH, Surendran P, Thom S, Toglhofer AM, Uitterlinden AG, Wachter R, Volzke H, Ziegler A, Munzel T, Marz W, Cappola TP, Hirschhorn JN, Mitchell GF, Smith NL, Fox ER, Dueker ND, Jaddoe VWV, Melander O, Russ M, Lehtimaki T, Ciullo M, Hicks AA, Lind L, Gudnason V, Pieske B, Barron AJ, Zweiker R, Schunkert H, Ingelsson E, Liu K, Arnett DK, Psaty BM, Blankenberg S, Larson MG, Felix SB, Franco OH, Zeller T, Vasan RS and Dorr M. Large-scale genome-wide analysis identifies genetic variants associated with cardiac structure and function. J Clin Invest. 2017;127:1798–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mahon NG, Murphy RT, MacRae CA, Caforio AL, Elliott PM and McKenna WJ. Echocardiographic evaluation in asymptomatic relatives of patients with dilated cardiomyopathy reveals preclinical disease. Ann Intern Med. 2005;143:108–115. [DOI] [PubMed] [Google Scholar]
- 36.Crispell KA, Hanson EL, Coates K, Toy W and Hershberger RE. Periodic rescreening is indicated for family members at risk of developing familial dilated cardiomyopathy. J Am Coll Cardiol. 2002;39:1503–1507. [DOI] [PubMed] [Google Scholar]
- 37.Vasan RS, Glazer NL, Felix JF, Lieb W, Wild PS, Felix SB, Watzinger N, Larson MG, Smith NL, Dehghan A, Grosshennig A, Schillert A, Teumer A, Schmidt R, Kathiresan S, Lumley T, Aulchenko YS, Konig IR, Zeller T, Homuth G, Struchalin M, Aragam J, Bis JC, Rivadeneira F, Erdmann J, Schnabel RB, Dorr M, Zweiker R, Lind L, Rodeheffer RJ, Greiser KH, Levy D, Haritunians T, Deckers JW, Stritzke J, Lackner KJ, Volker U, Ingelsson E, Kullo I, Haerting J, O’Donnell CJ, Heckbert SR, Stricker BH, Ziegler A, Reffelmann T, Redfield MM, Werdan K, Mitchell GF, Rice K, Arnett DK, Hofman A, Gottdiener JS, Uitterlinden AG, Meitinger T, Blettner M, Friedrich N, Wang TJ, Psaty BM, van Duijn CM, Wichmann HE, Munzel TF, Kroemer HK, Benjamin EJ, Rotter JI, Witteman JC, Schunkert H, Schmidt H, Volzke H and Blankenberg S. Genetic variants associated with cardiac structure and function: a meta-analysis and replication of genome-wide association data. JAMA. 2009;302:168–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rosati A, Graziano V, De Laurenzi V, Pascale M and Turco MC. BAG3: a multifaceted protein that regulates major cell pathways. Cell Death Dis. 2011;2:e141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McCollum AK, Casagrande G and Kohn EC. Caught in the middle: the role of Bag3 in disease. Biochem J. 2009;425:e1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maloyan A, Sayegh J, Osinska H, Chua BH and Robbins J. Manipulation of death pathways in desmin-related cardiomyopathy. Circ Res. 2010;106:1524–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Homma S, Iwasaki M, Shelton GD, Engvall E, Reed JC and Takayama S. BAG3 deficiency results in fulminant myopathy and early lethality. Am J Pathol. 2006;169:761–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Selcen D, Muntoni F, Burton BK, Pegoraro E, Sewry C, Bite AV and Engel AG. Mutation in BAG3 causes severe dominant childhood muscular dystrophy. Ann Neurol. 2009;65:83–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Odgerel Z, Sarkozy A, Lee HS, McKenna C, Rankin J, Straub V, Lochmuller H, Paola F, D’Amico A, Bertini E, Bushby K and Goldfarb LG. Inheritance patterns and phenotypic features of myofibrillar myopathy associated with a BAG3 mutation. Neuromuscul Disord. 2010;20:438–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ellinor PT, Sasse-Klaassen S, Probst S, Gerull B, Shin JT, Toeppel A, Heuser A, Michely B, Yoerger DM, Song BS, Pilz B, Krings G, Coplin B, Lange PE, Dec GW, Hennies HC, Thierfelder L and MacRae CA. A novel locus for dilated cardiomyopathy, diffuse myocardial fibrosis, and sudden death on chromosome 10q25–26. J Am Coll Cardiol. 2006;48:106–111. [DOI] [PubMed] [Google Scholar]
- 45.Arimura T, Ishikawa T, Nunoda S, Kawai S and Kimura A. Dilated cardiomyopathy-associated BAG3 mutations impair Z-disc assembly and enhance sensitivity to apoptosis in cardiomyocytes. Hum Mutat. 2011;32:1481–1491. [DOI] [PubMed] [Google Scholar]
- 46.Buyandelger B, Mansfield C, Kostin S, Choi O, Roberts AM, Ware JS, Mazzarotto F, Pesce F, Buchan R, Isaacson RL, Vouffo J, Gunkel S, Knoll G, McSweeney SJ, Wei H, Perrot A, Pfeiffer C, Toliat MR, Ilieva K, Krysztofinska E, Lopez-Olaneta MM, Gomez-Salinero JM, Schmidt A, Ng KE, Teucher N, Chen J, Teichmann M, Eilers M, Haverkamp W, Regitz-Zagrosek V, Hasenfuss G, Braun T, Pennell DJ, Gould I, Barton PJ, Lara-Pezzi E, Schafer S, Hubner N, Felkin LE, O’Regan DP, Brand T, Milting H, Nurnberg P, Schneider MD, Prasad S, Petretto E and Knoll R. ZBTB17 (MIZ1) Is Important for the Cardiac Stress Response and a Novel Candidate Gene for Cardiomyopathy and Heart Failure. Circ Cardiovasc Genet. 2015;8:643–652. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.