

1. INTRODUCTION
Many genetic variants contribute to Alzheimer’s disease (AD); however a small percentage of AD cases have highly penetrant autosomal dominant disease causing mutations in PSEN1, PSEN2 or APP [1]. Over 280 different autosomal dominant pathogenic variants are currently recognized worldwide [2,3]. The clinical phenotype of autosomal dominant AD is often similar to sporadic AD; however some cases have atypical presentations such as spastic paraparesis [4]. Although the age of onset is usually in the fifth to sixth decade the range of onsets is broad from 20 to 70 years [5]. The early onset has allowed investigators to study pre-clinical disease and the transition from pre-clinical to clinical AD [6,7].
The first family with autosomal dominant AD identified in Colombia has a PSEN1 variant resulting in a Glu280Ala amino acid change in PSEN1 [8]. This family is by far the largest family in the world with familial AD consisting of approximately 6000 family members clustered in the state of Antioquia, Colombia. Their age at onset for mild cognitive impairment (MCI) is 44 years (95% CI 43-45) and 49 years for dementia (95% CI 49-50) [9]. We report here a second large family with autosomal dominant AD who also live Antioquia and harbor a novel variant c.1247T>C in codon 416 of PSEN1 resulting in an Ile416Thr substitution. We describe the sociodemographic, clinical characteristics and neuropsychological profile of this family as well as the historical genetic context of their mutation. These data will broaden knowledge of the clinical spectra of autosomal dominant AD, extend the genetic loci in PSEN1 capable of causing disease, reveal patterns of historic gene flow shaping the worldwide map of autosomal dominant AD, and possibly provide the participants an opportunity for prevention trials [10].
2. METHODS
2.1. PARTICIPANT EVALUATION
Sociodemographic, clinical, and genetic data from 93 family members 18 years or older were gathered between 2002 and 2016. The pedigree was built with the information given by the family members (Figure 1). Participant evaluation was done in accordance to The Code of Ethics of the World Medical Association (Declaration of Helsinki) and the individuals signed an informed consent approved by the Institutional Review Board of the School of Medicine of University of Antioquia (Colombia). For cognitively impaired individuals, their proxies signed the informed consent.
Figure 1. PEDRIGREE OF THE FAMILY.
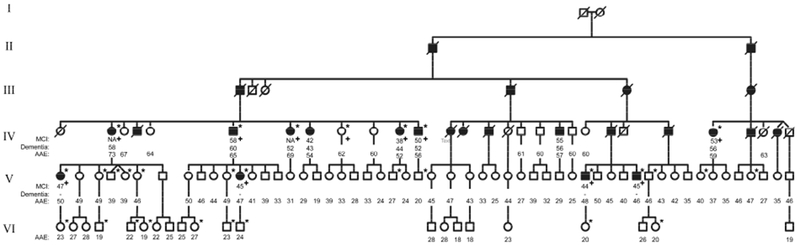
The diagram represents the extended family, square for male and circle for female. Individuals with MCI and dementia have been classified as symptomatic and are represented as filled icons, empty icons represent asymptomatic individuals. Individuals from whom we have information but died before the study were represented as deceased (crossed out). All 93 participants in the study were labeled with age at onset for MCI (if applicable), age at onset for Dementia (if applicable), and age at the clinical evaluation. If the age at onset was unknow the data was labeled as Not Available (NA). The symptomatic individuals from the V generation met the MCI criteria, but none of them met dementia criteria, therefore AAO for dementia was not registered.
MCI: Mild cognitive impairment, AAE: age at examination, NA: data not available
* Individual with whole genome sequencing data, + Individual used for zygosity mapping.
The participants underwent medical, neurological, and neuropsychological evaluations including the CERAD (Consortium to establish a registry for Alzheimer’s disease) test battery with additional neuropsychological tests and dementia functional scales, as previously described [11,12]. Diagnosis of Mild Cognitive Impairment (MCI) and dementia was done without knowledge of the carrier status according to Petersen 2011 [13] and DSM-IV criteria [14] respectively. Collected data were stored in medical records software (SISNE V 2.0). Statistical analyses of the clinical data were done with Stata Statistical Software (Release 15, StataCorp 2017). Sociodemographic, clinical and neuropsychological characteristics were presented using frequency and percentage (%) for categorical variables, and median and interquartile range (25th percentile-75th percentile) for quantitative variables.
2.2. DNA SEQUENCING AND BIOINFORMATIC ANALYSIS
A DNA sample from the proband was sent to a genetic laboratory (Athena Diagnostics Massachusetts, USA) for PSEN1 mutation testing. The analysis of PSEN1 was performed by PCR amplification and automated uni-directional DNA sequencing of the coding region (10 exons, 1204 base pairs), plus 20 additional bases of intronic DNA flanking every exon. All abnormal sequence variants were confirmed by bi-directional sequencing. Peripheral blood samples from the 93 participants were obtained in EDTA tubes. Restriction fragment length polymorphism (RFLP) variation was used to identify the Ile416Thr carriers; DNA was extracted through a modified salting-out technique (Gentra Puregene Blood Kit by Qiagen), PSEN1 Exon 11 was amplified by PCR (forward primer ACAGCAGCATCTACAGTTA, reverse primer TCAGGGCAGAGCTTATAGTT) and the amplicon was digested with VspI to detect NM_000021.3: c.1247T>C (p.Ile416Thr). In addition, whole genome sequencing of 31 individuals (9 asymptomatic non-carriers, 12 asymptomatic carriers and 10 symptomatic carriers, Figure 1.) was completed using the Illumina HiSeqX platform (Human Longevity Inc., San Diego, CA). Samples were sequenced to reach an average 30X coverage with single indexed paired-end 150 base pair reads. Raw sequencing data was processed using the Broad Institute’s Genome Analysis Toolkit (GATK v3.2.2) bestpractices pipeline [15]. Reads were aligned to the hg19 reference assembly of the human genome using the Burrows-Wheeler Aligner (BWA-MEM v0.78)[16]. Joint single nucleotide polymorphism (SNP) variant calling was performed in all 31 samples using GATK HaplotypeCaller. After joint-calling, SNPs were filtered to retain those with genotype quality (GQ) scores greater than 30 and read depth (DP) scores greater than 20. Heterozygous sites were also filtered to retain SNPs with an allelic balance (AB) greater than 0.25 and less than 0.75. In PLINK [17], SNPs in linkage equilibrium (r2 < 0.2) with genotyping rates above 99% and minor allele frequencies (MAF) greater than 5% were retained for estimating identity-by-state (IBS) and relatedness (Pi-hat) by calculating genome-wide IBS distances for all pairs of individuals. Multi-dimensional scaling (MDS) analysis of the IBS pairwise distances was conducted in PLINK utilizing overlapping SNPs from the 31 participants and individuals from the 1000 Genomes Project (phase 3 data release) that represent European (CEU, n = 103), African (YRI, n = 111), and East Asian/Native American (CHB, n= 108) populations [18]. To identify a shared haplotype among the 22 carriers of the pathogenic PSEN1 Ile416Thr variant, SNPs were phased using Beagle v4.1[19] with 1000 Genomes Project populations used as a reference panel. The frequency of the Ile416Thr-associated haplotype in reference populations was determined using phased genotype data for chromosome 14, and all haplotype frequencies were confirmed using LDlink [20,21]. Recombination rates were obtained from the hg19 genetic map provided within Eagle v2.4 [22].
LOD score calculation assumed a 100% penetrant association between genotype and phenotype, where and i is the number of individuals. For identifying the genomic region associated with the phenotype (zygosity mapping), we first filtered variants for quality to single nucleotide variants in the PASS tranche (GATK 99%) with a total depth of at least 30, and genotype quality of 99, not in a RepeatMasker region, and present at least once in the 1000 genomes reference. The presence of variants in the 1000 genomes reference was required to identify the haplotype that flanks the mutation.
2.3. BRAIN IMAGING
Pittsburgh Compound B (PiB), flortaucipir (FTP) positron emission tomography (PET) and structural magnetic resonance imaging (MRI) measurements were acquired from three members of the family (2 mutation carriers and 1 non-carrier) at Massachusetts General Hospital, Boston. 11C PiB PET data were expressed as the distribution volume ratio (DVR) with cerebellar grey as reference tissue, using a large cortical ROI aggregate that included frontal, lateral temporal and retrosplenial cortices (FLR) as described previously [23] 18F FTP regional binding was expressed in FreeSurfer ROIs as the standardized uptake value ratio (SUVR) to cerebellum.
3. RESULTS
3.1. INDEX CASE
The proband was a 55-year-old woman who complained of slowly progressive anterograde memory impairment with onset at age 40 (individual IV-7 in Figure 1). She misplaced objects, forgot conversations, and repeated herself. She was no longer able to perform her daily activities without supervision. Her relatives reported she had insomnia, visual and auditory hallucinations, had become irritable and had mood swings. Her family recalled that her father, her grandfather, and other family members had similar symptoms. Palmomental reflex and dysdiadochokinesia were the only findings on her physical examination. Her Mini Mental State Examination (MMSE) score was 18/30, 9 points below expected for her age and schooling; the CERAD neuropsychological battery showed severe compromise of verbal (5 points below expected in the recall of the CERAD word list) and non-verbal memory. The patient also had mild anomia and her verbal fluency was below expected. Her executive function, attention, praxis and visual perceptual skills were also impaired. She wasn’t able to copy the Rey-Osterrieth complex figure. Her score for the Functional Assessment Staging Test (FAST) was 5, equivalent to moderate dementia. The next clinical evaluation was performed when the patient was 62 years old. She scored 9/30 in the MMSE but was unable to comprehend the other tests. Her FAST score was 6e, equivalent to moderately severe dementia. The patient became bedridden when she was 64 years old; she was incontinent, aphasic, and completely dependent for activities of daily living. She had frequent myoclonus and tonic-clonic seizures with partial response to valproic acid. The patient died at the age of 70 due to sepsis from infected pressure ulcers. Genetic testing of the patient identified a Thymine to Cytosine transition in exon 11 NM_000021.3: c.1247T>C (p. Ile416Thr). This missense variant affects a highly conserved residue in the eighth transmembrane domain of Presenilin1 (Figure 2). Every computational prediction algorithm checked predict deleteriousness, which included a CADD PHRED score of 25.9, a 99.8% conservation score by phyloP in 100 vertebrates, a 98.6% rankscore by GERP, and deleterious prediction by SIFT, PolyPhen2 HDIV and HVAR, MutationTaster, FATHMM, MetaSVM, PROVEAN, and REVEL. The high level of conservation at this site, the predicted damage likely due to a change from a hydrophobic to a polar amino acid in the transmembrane domain, compounded by the location of the variant near a splice site all supported pathogenicity. This variant was initially classified as Unknown clinical significance; it had not been registered in the NCBI database and lacked a refSNP (rs) number.
Figure 2.
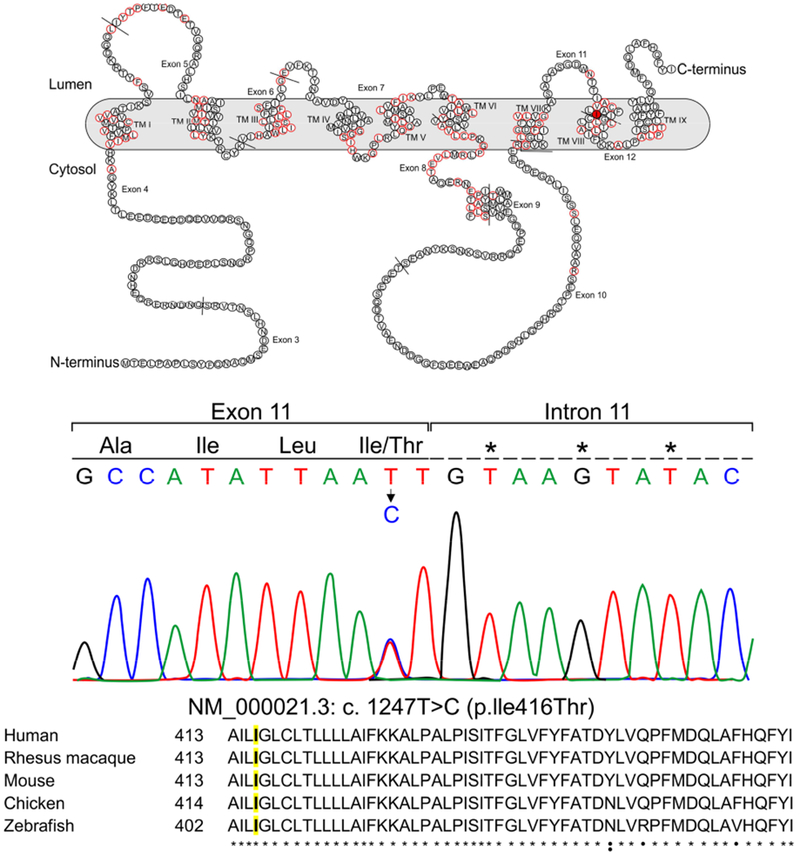
A: schematic representation of Preselenin 1 in the cell membrane. Residues that have variants known to cause Alzheimer’s disease are represented with a red border [3]. Residue 416 in TM VIII is colored in red. TM: Transmembrane
B: Color chromatogram depicting the missense variant in the PSEN1 gene.
C: Alignment of PSEN1 ortologs (uniprot.org). Residue 416 is highlighted in yellow.
* Residues that are conserved among the 5 species.
• Residues that are conserved at least in 4 species.
: Residues that are conserved at least in 3 species.
3.2. DEMOGRAPHICS AND MEDICAL EVALUATION
A total of 93 family members, including the proband, were evaluated (Figure 1). Twenty-six (27.9%) participants were identified as carriers of the Ile416Thr variant. The majority of them were women (59.2%), but the male:female ratio was similar in carriers compared to non-carriers. Most members of the kindred (71%) were born in Girardota, a small town located in the state of Antioquia (6°22 ‘37”N 75°26 ‘46’O), northwest of Colombia. Most of the population had at least one year of formal education (73.1%), but only 21.5% had completed high school, which affected their proficiency in some neuropsychological tests (complete demographics are in supplemental table 1).
After medical and neuropsychological evaluation four participants were diagnosed with MCI and eight participants with dementia; 37.5% were in early stages (CDR 1), 25% were in moderate stage (CDR 2) and 37.5% were severely demented (CDR 3), all of them were later confirmed as heterozygous Ile416Thr carriers. Symptomatic carriers had a mean age at onset of 42.35 years old (S.D. 6.28) for memory complaints, 47.6 years old (S.D. 5.83) for MCI, and the mean age of onset of dementia was 51.6 years old (S.D. 5.03) (Figure 3). Eight older healthy controls (≥55 years) did not carry the variant. As the variant co-segregated with the disease in three or more cases within the family it was classified as definite pathogenic according to the Guerreiro algorithm [24]. Two cognitively normal Ile416Thr carriers had PET-Amyloid and PET-Tau scans with evidence of preclinical amyloid plaques and tau accumulation while an age-matched non-carrier control did not show this pathology (Figure 4). The cerebral pattern of beta-amyloid deposition in the carriers resembled that found in clinically affected individuals who are at risk for late-onset Alzheimer’s disease and other cases of AD-causing PSEN1 mutations [6] and the PET-Tau scans resembled those previously reported [23]. The findings include preferential PiB binding in posterior cingulate, precuneus, parietotemporal, frontal, and basal ganglia regions. Elevated levels of FTP binding in medial temporal lobe regions were evident within the context of increased amyloid pathology, before estimated years to symptom onset [23].
Figure3. AGE DEPENDANT COGNITIVE DECLINE IN CARRIERS AND NON-CARRIERS.
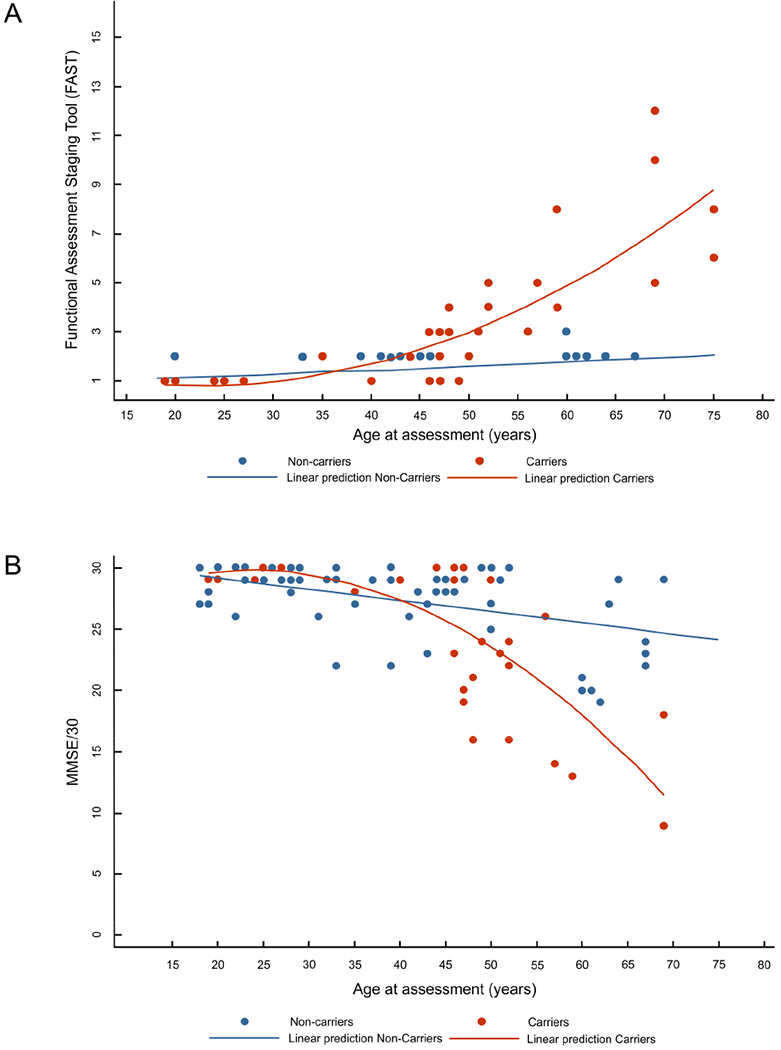
FAST: Functional Assesment Staging Tool. FAST score was converted into a linear scale, where 6a corresponds to 6, 6b to 7 and so on until 16.
Lineal and curvilinear models were used to represent, in non-carriers and carriers, respectively, the relation between the FAST (A) and the Minimental State Examination (MMSE) score (B) and the age at assessment (in years) centered at 40 years (mean of all data). Individuals with overlapping ages and scores were represented as a single point.
Model for non-carriers FAST: Expected mean FAST= 1.43+0.016agecentered. CI95% Intercept (1.314 – 1.546). CI95% agecentered coefficient (0.008-0.023).
Model for carriers FAST: Expected mean FAST= 1.69+0.16agecenterd+0.0064agecenterd2. CI95% Intercept (0.651 – 2.723). CI95% agecentered coefficient (0.099-0.223). CI95% agecentered2 coefficient (0.002-0.010).Model for non-carriers MMSE: Expected mean MMSE= 27.3 to 0.099agecenterd. CI95% Intercept (26.68 to 27.90). CI95% agecentered coefficient (−0.139 to −0.058). Model for carriers MMSE: Expected mean MMSE= 27.2-0.46agecenterd-0.02agecentered2. CI95% Intercept (24,778 to 29.580). CI95% agecentered coefficient (−0.603 to −0.307). CI95% agecentered2 coefficient (−0.028 to −0.006).
Figure 4. SPATIAL PATTERNS OF [11C] PIB AND [18F] FTP BINDING IN ILE416THR PSEN1 MUTATION CARRIERS.
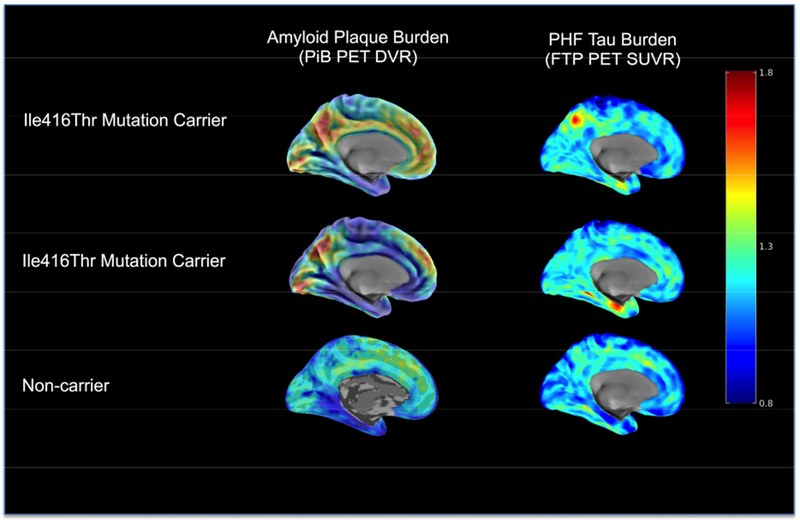
PET maps are shown for two cognitively unimpaired carriers (MMSE>27) and one age-matched non-carrier family member. Sagittal PiB DVR maps are shown on the left, and sagittal FTP SUVR maps are presented on the right. Images are displayed in standardized atlas space, along with whole-brain surface renderings, with a left hemisphere view. Row A) A cognitively unimpaired carrier with high cortical amyloid (DVR=1.39) and with FTP binding in entorhinal cortex (SUVR=1.35). B) A cognitively unimpaired carrier with higher cortical amyloid (DVR=1.41), and FTP binding in entorhinal cortex (SUVR=1.12). C) An age-matched unimpaired non-carrier with low amyloid by PiB PET (DVR=1.10) and low FTP binding in entorhinal cortex (SUVR=1.11).
PET: positron emission tomography, MMSE: Minimental State Exammination, PiB: Pittsburgh Compound B , DVR: Distribution volume ratio, FTP: Flortaucipir, SUVR: Standardized uptake value ratio.
Depression and anxiety were more frequent in carriers than in non-carriers, and more prevalent in those with MCI and dementia when compared to asymptomatic carriers (Table 1). Other neuropsychiatric symptoms such as delusions, hallucinations and insomnia were also reported and the neuropsychiatric inventory scores [25] were higher as the disease progressed. Myoclonus and bilateral tonic-clonic seizures were frequent in early stages as well as in patients with severe dementia (CDR 3). Neuropsychological assessment identified amnestic as the most common type of MCI. Patients with dementia had a relatively better performance in language and attention than praxis and executive function (supplemental table 2).
Table 1: CLINICAL CHARACTERISTICS OF ILE416THR CARRIERS AND NON-CARRIERS.
Age at evaluation and age at onset are given as Mean and Standard Deviation. NPI-Q: Cummings Neuropsychiatric Inventory-Questionnaire IR: interquartile range CDR: clinical dementia rating. MCI mild cognitive impairment
| Non-carriers | Ile416Thr carriers | ||||
|---|---|---|---|---|---|
| Total n= 67 | Total n= 26 | Asymptomatic n=14 | MCI n=4 | Dementia n=8 | |
| Age at onset | NA | NA | NA | 47.6 (5.8) | 51.6 (5.0) |
| Age at evaluation | 36.5 (14.3) | 46.0 (14.4) | 36.5 (12.0) | 50.8 (3.8) | 59.1 (8.7) |
| CDR | |||||
| 0 (None) | 63 (94%) | 14 (53,9%) | 14 (100%) | 0 | 0 |
| 0.5 (Questionable) | 4 (6%) | 4 (15.4%) | 0 | 4 (100%) | 0 |
| 1 (Mild) | 0 | 3 (11.5%) | 0 | 0 | 3 (37.5%) |
| 2 (Moderate) | 0 | 2 (7.7%) | 0 | 0 | 2 (25%) |
| 3 (Severe) | 0 | 3 (11.5%) | 0 | 0 | 3 (37.5%) |
| Depression symptoms | 7 (10.4%) | 8 (30,8%) | 1 (7.1%) | 1 (25%) | 6 (75%) |
| Anxiety symptoms | 0 | 6 (23,1%) | 1 (7.1%) | 1 (25%) | 4 (50%) |
| Delusions | 0 | 3 (11.5%) | 0 | 1 (25%) | 2 (25%) |
| Hallucinations | 0 | 3 (11.5%) | 0 | 1 (25%) | 2 (25%) |
| Insomnia | 0 | 4 (15.4%) | 0 | 1 (25%) | 3 (37.5%) |
| NPI-Q median (IR) | 0 (4) | 2,00 (10,50) | 0,00 (6,25) | 9,5 (21,00) | 41(NA) |
| Myoclonus | 0 | 5 (19.2%) | 0 | 0 | 5 (62.5%) |
| Seizures | 0 | 4 (15.4%) | 0 | 0 | 4 (50%) |
| Traumatic Brain Injury | 9 (13,4%) | 2 (7.7%) | 1 (7.1%) | 1 (25%) | 0 |
| Headache | 0 | 4 (15.4%) | 1 (7.1%) | 2 (50%) | 1 (12.5%) |
| Alcoholism | 2 (3%) | 4 (15.4%) | 1 (7.1%) | 0 | 3 (37.5%) |
| Tobacco use | 14 (20,9%) | 11 (42.3%) | 5 (35.7%) | 2 (50%) | 4 (50%) |
| Drug abuse | 6 (9%) | 2 (7.7%) | 2 (14.3%) | 0 | 0 |
| Learning difficulties | 11 (16.4%) | 4 (15.4%) | 3 | 1 | 0 |
3.3. ZYGOSITY MAPPING
To assess genetic variants associated with the phenotype in an unbiased manner, we performed zygosity mapping and calculated a LOD score assuming 100% penetrance and 100% concordance between genotype and phenotype (see methods for formula) using whole genome data filtered strictly for quality (see methods for filtering criteria) for 10 symptomatic individuals and one asymptomatic family member past the latest age of onset (≥55 years). These 11 individuals yielded 215,030 variants meeting strict quality filters spread approximately evenly across the genome. The goal in selecting very high-quality variants (including presence in 1000 genomes at least once) was not to identify the mutation itself, but rather to identify ancestral variants in the haplotype that flank the mutation that are high quality reliable indicators. We filtered for variants with a genotype consistent with phenotype, i.e. a homozygous reference in the asymptomatic individual past the age of onset, and a heterozygous variant in all 10 symptomatic individuals (consistent with our assumption that the causative variant is dominant and 100% penetrant). This filter yielded 249 variants with 223 variants on chromosome 14 and with 222 variants lying between 62 MB and 76 MB, which is highly enriched by Fisher’s exact for variants on chromosome 14 (*p<0.0001, OR=223, OR 95% CI = 112-∞) when compared to 249 variants randomly distributed across the genome. On average, 9 out of 249 variants would be expected on chromosome 14 if a set of 249 variants were distributed at the same ratios of numbers of variants per chromosome for the unfiltered set of 215,030 variants meeting the original criteria (Figure. S1). When the calculation was restricted to the 11 individuals with whole genome sequencing data available, the LOD score was 3.3 at concordant sites (equivalent to a p value of 0.00049). As the Ile416Thr carrier status was also known in two additional symptomatic individuals and seven additional healthy individuals beyond the latest age of onset without whole genome sequencing data available, a total of 20 informative individuals could be used for LOD calculation, which yielded a LOD score of 6.0 given the stated assumptions for LOD calculation in the methods (equivalent to a p value of 9.5×10−7), providing strong evidence for linkage of this site to phenotype (3 orders of magnitude above the typical genome-wide LOD score cutoff of 3). No disease-associated variants in TREM2, SORL1, or ABCA7 were observed in the 10 carriers past age of onset with WGS data, and there was no detectable contribution of APOE E4 alleles to modify age at onset (Supplemental table 3).
The Ile416Thr variant had not been documented in disease specific or large population databases such as the Genome Aggregation Database (gnomAD) of 138,632 individuals (17,210 of Latin American ancestry) [26] (gnomAD; http://gnomad.broadinstitute.org), the 1000 Genomes Project [18] (1000G; http://www.internationalgenome.org/), the TOPMed database (62,784 individuals), UK10K, or ESP6500 suggesting the possibility of a founder effect. One caveat is that the admixture in Colombia has rare variants African and Native American ancestries. Combined with the isolation of this group in Antioquia, more rare variants may cluster in this group by chance. However, the absence of this variant in over 200,000 healthy individuals in population databases and segregation of the mutation with the disease phenotype is strong evidence for pathogencity.
The criteria set forth by the American College of Medical Genetics (ACMG) is intended to assess the pathogenicity of gene variants [27]. The segregation of the variant with the illness is scored as strong evidence (PP1S), the absence in population databases is scored as moderate evidence (PM2) and computational deleteriousness prediction is scored as supporting evidence (PP3). As noted in [28] mutations in the transmembrane domain similar to Ile416Thr mutation described here are likely to confer functional impairment.
3.4. ORIGIN OF THE MUTATION
MDS analysis suggested a primarily African and European admixed ancestry for this family (Figure. S2). The haplotype structure around the Ile416Thr variant, after phasing the genotype data for 22 Ile416Thr variant carriers and nine non-carriers (n = 31 family members), identified a 16 SNP, 86.7 kilobase haplotype shared by all carriers spanning most of PSEN1. This haplotype encompassed the PSEN1 disease variant locus and was absent in all non-carriers (Figure 5). Therefore, it seems likely that the mutation occurred on this haplotypic background. All Ile416Thr carriers were heterozygous for 16-SNPs on the same strand as the mutation, which further supports that the pathogenic variant occurs on this haplotypic background. To identify the ethnic ancestry of the shared haplotype, we queried phased genotype data from participants in the reference populations of the 1000 Genomes Project. This background haplotype was observed in 25 individuals of African or admixed African ancestry in the 1000 Genomes Project (Supplemental Table 4), all of whom did not carry the mutation. The haplotype occurred most frequently in Gambian individuals (n = 6; 2.7% frequency in Gambian population). Interestingly, this haplotype was not observed in any European or Asian reference populations. Furthermore, the three SNPs most proximal to Ile416Thr in the shared haplotype (rs362375, rs177413, and rs17126104) occur most frequently in African populations, with rs362375 and rs17126104 found almost exclusively in these populations. Together, these results suggest that the Ile416Thr pathogenic variant occurs on a haplotypic background that is African in origin.
Figure 5. IDENTIFICATION OF AN ILE416THR-ASSOCIATED HAPLOTYPE IN THE COPACABANA FAMILY.
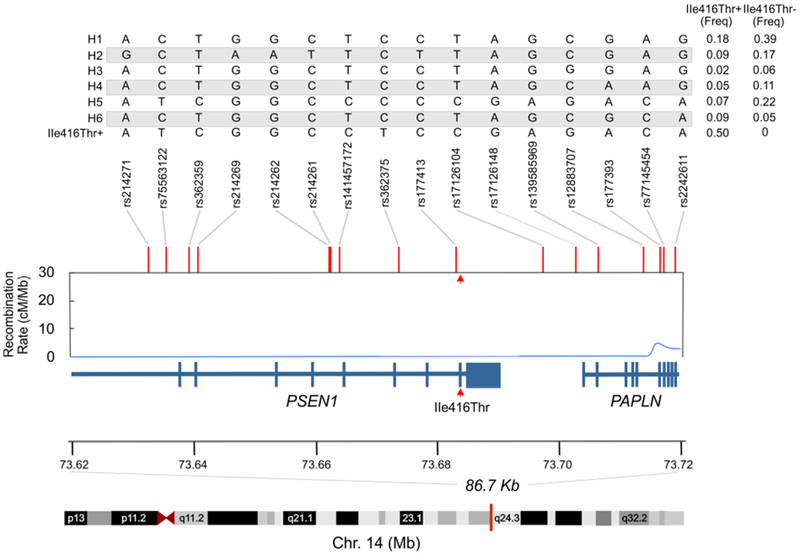
Haplotype analysis of phased genotypes from 22 Ile416Thr variant carriers and 9 non-carriers (n = 31) identified seven haplotypes composed of 16 single nucleotide polymorphism (SNPs) spanning 86,664 base pairs on chromosome 14. This haplotype spanned exons 4 – 12 of PSEN1, including the Ile416Thr variant (last amino acid of exon 11, and exons 1 - 10 of PAPLN. The combination of alleles for all 16 SNPs in each of the seven identified haplotypes are provided in the table in the top panel. A single copy of one haplotype was found exclusively in all variant carriers and was absent from all non-variant carriers (I416T+, last row of top panel table). Recombination rates are represented in centimorgans per megabase, and were obtained from the hg19 genetic map provided as part of the Eagle v2.4 package and. The red arrowheads point to the site of the Ile416Thr mutation. Freq: frequency.
4. DISCUSSION
This study describes a private PSEN1 (Ile416Thr) mutation leading to early onset clinical Alzheimer’s disease in a large family. So far, we have registered 26 carriers of the Ile416Thr variant, 14 of them were in pre-symptomatic stages of the illness. Individuals with MCI had an amnestic profile and depressive symptoms are frequent. In the dementia stage, most of the patients have myoclonus and seizures at late stages of disease. As of today more than 200 variants in PSEN1 gene have been described [3], including another private mutation, Glu280Ala, which affects 25 related families with a total of 6000 individuals. Interestingly, both families come from different but nearby towns in the state of Antioquia, Colombia.
A founder from the Iberian peninsula was reported as the origin of the Glu280Ala mutation [29], and as described here, a founder from Africa gave rise to the family with the Ile416Thr mutation. These vastly distinct geographic origins highlight the complex genetic admixture of contemporary Colombians. Whereas the Colombian population is a three-way admixture of European, Amerindian and African populations [30], the proportions of the admixture show great variation over relatively small geographic sectors [31]. Therefore, the fine structure of the population, defined as the mapping of familial genetic features onto the geographic locales where families reside, reflects specific population histories. Because this admixture occurred over a relatively short time period—less than 500 years—the historical genetic imprint has remained readily detectable. Further marking the historical genetic trace was the tendency, until very recently, for low geographic mobility among the progeny of the founder populations.
Consistent with the historic record, the family harboring the Ile416Thr mutation arose from a mini-population bottleneck. Girardota is located in a distinct geographic sector known as “Tierras del Río Abajo” or Lands of the Lower River, just north of the modern Antioquia capital, Medellín. The region, where currently the towns Girardota and Copacabana are located, was legally designated Río Abajo in 1598 and the town which came to be known as Girardota (originally Hatogrande) was founded in 1620. This location created a degree of geographic isolation from surrounding population groups. In 1665, 103 slaves imported from Guinea, Angola, Congo and Cabo Verde were recorded in treasury of the Municipality of Girardota [32]. As the anti-slave movement took root, many of these individuals fled to a mountainous location in the region and were further isolated. The genetic findings, in which the mutation lies within a unique haplotype, fit well with the historical record of the region. This novel PSEN1 mutation supports the view that founder effects and genetic drift can explain the contemporary geographic dispersion of these rare early-onset Alzheimer mutations.
The well-defined age at onset and the moderately large size of the family are ideal for future prevention trials as appropriate pharmacological interventions emerge. Both the Ile416hr family reported here and the very large Glu280Ala family have similar ages at onset. The progression of disease is similar to the extensively studied progression of patients with Glu280Ala mutation, the largest known family with genetic AD, and for which there is extensive data about the neuropsychological performance in different stages of the illness [9,33].
Supplementary Material
Supplemental figure 1: ZYGOSITY MAPPING OF 11 FAMILY MEMBERS PAST THE AGE OF ONSET WITH WGD
Whole genome data (WGD) from 11 individuals was filtered to high quality variants with zygosity matching phenotype (heterozygous alternate for 10 affected individuals and homozygous reference for 1 healthy individual past the latest age of onset). Variants are highly enriched on chromosome 14 (*p<0.0001 by Fisher’s exact test, OR=223, OR 95% CI = 112-∞). Note the location of the candidate mutation in the block of variants with zygosity matching phenotype between 62 MB and 76 MB on chromosome 14.
{kind=link}
Supplemental figure 2: MULTIDIMENSIONAL (MDS) SCALING ANALYSIS OF 31 ILE416THR FAMILY MEMBERS.
Population substructure in 22 Ile416Thr variant carriers (o) and nine non-carriers (o) was assessed by multidimensional scaling analysis and visualized by plotting the first two dimensions. Individuals from the CEU (o), YRI (o), and CHB (o) populations in the 1000 Genomes Project were used as references.
(CEU: Utah Residents with Northern and Western European Ancestry. CHB: Han Chinese in Beijing, China. YRI: Yoruba in Ibadan, Nigeria)
{kind=link}
Supplemental table 1: SOCIODEMOGRAPHIC CHARACTERISTICS OF CARRIERS AND NON-CARRIERS OF ILE416THR IN ANTIOQUIA, COLOMBIA
SD: Standard deviation
Supplemental table 2: NEUROPSYCHOLOGICAL PROFILE OF CARRIERS AND NON-CARRIERS OF ILE416THR.
*4 patients with dementia (1 with CDR 2 and 3 with CDR 3) were not able to understand the tests. Hence, were excluded from this table.
IR. Interquartile Range. CDR: Clinical Dementia Rating. MCI mild cognitive impairment CERAD-WL: The Consortium to Establish a Registry for Alzheimer’s Disease Word List task. ROCF: The Rey–Osterrieth complex figure test. TMT A: Trail making test A. Phonemic fluency was evaluated with letters F-A-S WCST: Wisconsin Card Sorting Test. SBV-15 Shortened Boston Version of15 images. Semantic fluency was evaluated by naming animals. GDS-s: Shortened version of the Geriatric Depression scale. SDS: Zung Self-Rating Depression Scale. IADL: Lawton And Brody Instrumental Activities Of Daily Living Scale
Supplemental table 3: APOE ALLELE INFORMATION OF THE 10 AFFECTED INDIVIDUALS WITH WGS
The table represents the 10 symptomatic carriers with whole genome sequence data (WGS), with their respective age at onset for MCI, age at onset for dementia and age at the clinical examination. If the age at onset for MCI was unknown it was represented as Not Available. If the individual met MCI criteria but not dementia criteria the age at onset for dementia was left blank.
AAO: age at onset, MCI: Mild cognitive impairment, AAE: age at examination, NA: data not available
Supplemental Table 4 FREQUENCY OF ILE416THR-ASSOCIATED HAPLOTYPE IN ALL 1000 GENOMES PROJECT REFERENCE POPULATIONS.
The frequency of the Ile416Thr pathogenic variant harboring haplotype in all individuals of a given super population is reported in parenthesis after each super population code. AFR, African; ACB, African Caribbeans in Barbados; ASW, Americans of African Ancestry in Southwest USA; ESN, Esan in Nigeria; GWD, Gambian in Western Divisions of Gambia; LWK, Luhya in Webuye, Kenya; MSL, Mende in Sierra Leone; YRI, Yoruba in Ibadan, Nigeria; AMR, Ad Mixed American; CLM, Colombians from Medellin; MXL, Mexican ancestry from Los Angeles, PUR, Puerto Ricans from Puerto Rico; PEL, Peruvians from Lima, Peru; EAS, East Asian; CHB, Han Chinese in Beijing, China; JPT, Japanese in Tokyo, Japan; CHS, Southern Han Chinese; CDX, Chinese Dai in Xishuangbanna, China; KHV, Kinh in Ho Chi Minh City, Vietnam; EUR, European, CEU, Utah Residents (CEPH) with Northern and Western European Ancestry; TSI, Toscani in Italia; FIN, Finnish in Finland; GBR, British in England and Scotland; IBS, Iberian Population in Spain; SAS, South Asian; GIH, Gujarati Indian from Houston, Texas; PJL, Punjabi from Lahore, Pakistan; BEB, Bengali from Bangladesh; STU, Sri Lankan Tamil from the UK; ITU, Indian Telugu from the UK.
HIGHLIGHTS.
PSEN1 variant Ile416Thr is pathogenic and causes early onset Alzheimer’s Disease
Dementia in PSEN1 Ile416Thr carriers occurs in the 6th decade of life
This variant likely originated as consequence of a mini-population bottleneck
The family haplotype supports the historical African influence in the region
Founder effects and genetic drift can explain the geographic dispersion of rare mutations
Systematic Review
The full extent of familial Alzheimer’s disease (AD) remains poorly delineated and large families with early onset familial disease have been particularly informative. PSEN1 Ile416Thr variant has not been reported in PubMed or human genome databases.
Interpretation
PSEN1 Ile416Thr is a novel variant that is likely pathogenic by ACMG criteria and definitely pathogenic according to the Guerreiro algorithm. The phenotype of this family supports the recognized clinical description of families with genetic AD. Their genetic background extends the world-wide haplotypic backgrounds on which these mutations occur, in this case, to an uncommon African haplotype. The geographic location of the family and the genetic observations are consistent with historical records of former slave settlements.
Future Directions
Genetic analyses of smaller families with familial AD that live throughout this region will provide a comprehensive picture of the genetic burden of neurodegeneration in the region and the influence of the genetic background in admixed populations.
5. ACKNOWLEDGEMENTS
We thank the patients, their families and caregivers, who have generously given their time to make this research possible. Thanks to Caroline Ackley and Gabriel Luna for digital artwork.
FUNDING
This work was supported by “Comité para el Desarrollo de la Investigatión (CODI)”, Universidad de Antioquia, Medellin, Colombia. Additional support was provided by the Rainwater Charitable Foundation (KSK, JSY), Bluefield Project to Cure FTD (JSY), Larry L. Hillblom Foundation 2016-A-005-SUP (JSY), National Institute on Aging K01 AG049152 (JSY), and John Douglas French Alzheimer’s Foundation (JSY). National Institute of Aging R01 AG054671, National Institute of Health Office of the Director DP5OD019833, MGH ECOR Clafin Distinguished Scholar Award, and MGH Physician/Scientist Development Award (YTQ).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest on the research, authorship, and/or publication of this study. Francisco Lopera, M.D. and Margarita Giraldo, M.D. are investigators at API-Colombia clinical trial for genetic Alzheimer’s Disease (NCT01998841).
8. REFERENCES:
- [1].Naj AC, Schellenberg GD. Genomic variants, genes, and pathways of Alzheimer’s disease: An overview. Am J Med Genet Part B Neuropsychiatr Genet 2017; 174:5–26. doi: 10.1002/ajmg.b.32499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Cruts M, Theuns J, Van Broeckhoven C. Locus-specific mutation databases for neurodegenerative brain diseases. Hum Mutat 2012;33:1340–4. doi: 10.1002/humu.22117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Cruts M Alzheimer Disease & Frontotemporal Dementia Mutation Database n.d. http://www.molgen.vib-ua.be/ADMutations (accessed July 19, 2018). [Google Scholar]
- [4].Ryan NS, Nicholas JM, Weston PSJ, Liang Y, Lashley T, Guerreiro R, et al. Clinical phenotype and genetic associations in autosomal dominant familial Alzheimer’s disease: a case series. Lancet Neurol 2016;15:1326–35. doi: 10.1016/S1474-4422(16)30193-4. [DOI] [PubMed] [Google Scholar]
- [5].Ryman DC, Acosta-Baena N, Aisen PS, Bird T, Danek A, Fox NC, et al. Symptom onset in autosomal dominant Alzheimer disease: A systematic review and meta-analysis. Neurology 2014;83:253–60. doi: 10.1212/WNL.0000000000000596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Fleisher AS, Chen K, Quiroz YT, Jakimovich LJ, Gomez MG, Langois CM, et al. Florbetapir PET analysis of amyloid-β deposition in the presenilin 1 E280A autosomal dominant Alzheimer’s disease kindred: A cross-sectional study. Lancet Neurol 2012;11:1057–65. doi: 10.1016/S1474-4422(12)70227-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Fleisher AS, Chen K, Quiroz YT, Jakimovich LJ, Gutierrez Gomez M, Langois CM, et al. Associations Between Biomarkers and Age in the Presenilin 1 E280A Autosomal Dominant Alzheimer Disease Kindred. JAMA Neurol 2015;72:316. doi: 10.1001/jamaneurol.2014.3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Lopera F Clinical features of early-onset Alzheimer disease in a large kindred with an E280A presenilin-1 mutation. JAMA J Am Med Assoc 1997;277:793–9. doi: 10.1001/jama.277.10.793. [DOI] [PubMed] [Google Scholar]
- [9].Acosta-Baena N, Sepulveda-Falla D, Lopera-Gómez CM, Jaramillo-Elorza M, Moreno S, Aguirre-Acevedo D, et al. Pre-dementia clinical stages in presenilin 1 E280A familial early-onset Alzheimer’s disease: A retrospective cohort study. Lancet Neurol 2011;10:213–20. doi: 10.1016/S1474-4422(10)70323-9. [DOI] [PubMed] [Google Scholar]
- [10].Moulder KL, Snider BJ, Mills SL, Buckles VD, Santacruz AM, Bateman RJ, et al. Dominantly inherited alzheimer network: Facilitating research and clinical trials. Alzheimer’s Res Ther 2013;5:48. doi: 10.1186/alzrt213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ardila A, Lopera F, Rosselli M, Moreno S, Madrigal L, Arango-Lasprilla JC, et al. Neuropsychological profile of a large kindred with familial Alzheimer’s disease caused by the E280A single presenilin-1 mutation. Arch Clin Neuropsychol 2000; 15:515–28. doi : 10.1016/S0887-6177(99)00041-4. [DOI] [PubMed] [Google Scholar]
- [12].Aguirre-Acevedo DC, Gómez RD, Moreno S, Henao-Arboleda E, Motta M, Muñoz C, et al. Validez y fiabilidad de la batería neuropsicológica CERAD-Col. Rev Neurol 2007;45:655–60. [PubMed] [Google Scholar]
- [13].Petersen RC. Mild Cognitive Impairment. N Engl J Med 2011;364:2227–34. 10.1056/NEJMcp0910237. [DOI] [PubMed] [Google Scholar]
- [14].American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. American Psychiatric Association; 2013. doi: 10.1176/appi.books.9780890425596. [DOI] [Google Scholar]
- [15].Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, del Angel G, Levy-Moonshine A, et al. From fastQ data to high-confidence variant calls: The genome analysis toolkit best practices pipeline. Curr Protoc Bioinforma 2013;43:11.10.1–33. 10.1002/0471250953.bi1110s43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009;25:1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am J Hum Genet 2007;81:559–75. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Gibbs RA, Boerwinkle E, Doddapaneni H, Han Y, Korchina V, Kovar C, et al. A global reference for human genetic variation. Nature 2015;526:68–74. 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Browning SR, Browning BL. Rapid and Accurate Haplotype Phasing and Missing-Data Inference for Whole-Genome Association Studies By Use of Localized Haplotype Clustering. Am J Hum Genet 2007;81:1084–97. 10.1086/521987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Machiela MJ, Chanock SJ. LDlink: A web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics 2015;31:3555–7. doi: 10.1093/bioinformatics/btv402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Machiela MJ, Chanock SJ. LDassoc: An online tool for interactively exploring genome-wide association study results and prioritizing variants for functional investigation. Bioinformatics 2018;34:887–9. 10.1093/bioinformatics/btx561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Loh PR, Danecek P, Palamara PF, Fuchsberger C, Reshef YA, Finucane HK, et al. Reference-based phasing using the Haplotype Reference Consortium panel. Nat Genet 2016;48:1443–8. 10.1038/ng.3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Quiroz YT, Sperling RA, Norton DJ, Baena A, Arboleda-Velasquez JF, Cosio D, et al. Association Between Amyloid and Tau Accumulation in Young Adults With Autosomal Dominant Alzheimer Disease. JAMA Neurol 2018. 10.1001/jamaneurol.2017.4907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Guerreiro RJ, Baquero M, Blesa R, Boada M, Brás JM, Bullido MJ, et al. Genetic screening of Alzheimer’s disease genes in Iberian and African samples yields novel mutations in presenilins and APP. Neurobiol Aging 2010;31:725–31. 10.1016/j.neurobiolaging.2008.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Cummings JL, Mega M, Gray K, Rosenberg-Thompson S, Carusi DA, Gornbein J. The Neuropsychiatric Inventory: Comprehensive assessment of psychopathology in dementia. Neurology 1994;44:2308–2308. 10.1212/WNL.44.12.2308. [DOI] [PubMed] [Google Scholar]
- [26].Lek M, Karczewski KJ, Minikel E V., Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016;536:285–91. 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–24. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Shen J, Kelleher RJ. The presenilin hypothesis of Alzheimer’s disease: Evidence for a loss-of-function pathogenic mechanism. vol. 104 2007. doi: 10.1073/pnas.0608332104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Lalli MA, Cox HC, Arcila ML, Cadavid L, Moreno S, Garcia G, et al. Origin of the PSEN1 E280A mutation causing early-onset Alzheimer’s disease. Alzheimer’s Dement 2014;10:S277–83. 10.1016/j.jalz.2013.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Rishishwar L, Conley AB, Wigington CH, Wang L, Valderrama-Aguirre A, Jordan IK, et al. Ancestry, admixture and fitness in Colombian genomes. Sci Rep 2015;5:12376. doi : 10.1038/srep12376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Ossa H, Aquino J, Pereira R, Ibarra A, Ossa RH, Pérez LA, et al. Outlining the Ancestry Landscape of Colombian Admixed Populations. PLOSONE 2016;11:e0164414 10.1371/journal.pone.0164414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Correa Bustamante CM. De Hatogrande a Girardota. Universidad de Antioquia, 2002. [Google Scholar]
- [33].Aguirre-Acevedo DC, Lopera F, Henao E, Tirado V, Muñoz C, Giraldo M, et al. Cognitive decline in a colombian kindred with autosomal dominant Alzheimer disease a retrospective cohort study. JAMA Neurol 2016;73:431–8. 10.1001/jamaneurol.2015.4851. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental figure 1: ZYGOSITY MAPPING OF 11 FAMILY MEMBERS PAST THE AGE OF ONSET WITH WGD
Whole genome data (WGD) from 11 individuals was filtered to high quality variants with zygosity matching phenotype (heterozygous alternate for 10 affected individuals and homozygous reference for 1 healthy individual past the latest age of onset). Variants are highly enriched on chromosome 14 (*p<0.0001 by Fisher’s exact test, OR=223, OR 95% CI = 112-∞). Note the location of the candidate mutation in the block of variants with zygosity matching phenotype between 62 MB and 76 MB on chromosome 14.
Supplemental figure 2: MULTIDIMENSIONAL (MDS) SCALING ANALYSIS OF 31 ILE416THR FAMILY MEMBERS.
Population substructure in 22 Ile416Thr variant carriers (o) and nine non-carriers (o) was assessed by multidimensional scaling analysis and visualized by plotting the first two dimensions. Individuals from the CEU (o), YRI (o), and CHB (o) populations in the 1000 Genomes Project were used as references.
(CEU: Utah Residents with Northern and Western European Ancestry. CHB: Han Chinese in Beijing, China. YRI: Yoruba in Ibadan, Nigeria)
Supplemental table 1: SOCIODEMOGRAPHIC CHARACTERISTICS OF CARRIERS AND NON-CARRIERS OF ILE416THR IN ANTIOQUIA, COLOMBIA
SD: Standard deviation
Supplemental table 2: NEUROPSYCHOLOGICAL PROFILE OF CARRIERS AND NON-CARRIERS OF ILE416THR.
*4 patients with dementia (1 with CDR 2 and 3 with CDR 3) were not able to understand the tests. Hence, were excluded from this table.
IR. Interquartile Range. CDR: Clinical Dementia Rating. MCI mild cognitive impairment CERAD-WL: The Consortium to Establish a Registry for Alzheimer’s Disease Word List task. ROCF: The Rey–Osterrieth complex figure test. TMT A: Trail making test A. Phonemic fluency was evaluated with letters F-A-S WCST: Wisconsin Card Sorting Test. SBV-15 Shortened Boston Version of15 images. Semantic fluency was evaluated by naming animals. GDS-s: Shortened version of the Geriatric Depression scale. SDS: Zung Self-Rating Depression Scale. IADL: Lawton And Brody Instrumental Activities Of Daily Living Scale
Supplemental table 3: APOE ALLELE INFORMATION OF THE 10 AFFECTED INDIVIDUALS WITH WGS
The table represents the 10 symptomatic carriers with whole genome sequence data (WGS), with their respective age at onset for MCI, age at onset for dementia and age at the clinical examination. If the age at onset for MCI was unknown it was represented as Not Available. If the individual met MCI criteria but not dementia criteria the age at onset for dementia was left blank.
AAO: age at onset, MCI: Mild cognitive impairment, AAE: age at examination, NA: data not available
Supplemental Table 4 FREQUENCY OF ILE416THR-ASSOCIATED HAPLOTYPE IN ALL 1000 GENOMES PROJECT REFERENCE POPULATIONS.
The frequency of the Ile416Thr pathogenic variant harboring haplotype in all individuals of a given super population is reported in parenthesis after each super population code. AFR, African; ACB, African Caribbeans in Barbados; ASW, Americans of African Ancestry in Southwest USA; ESN, Esan in Nigeria; GWD, Gambian in Western Divisions of Gambia; LWK, Luhya in Webuye, Kenya; MSL, Mende in Sierra Leone; YRI, Yoruba in Ibadan, Nigeria; AMR, Ad Mixed American; CLM, Colombians from Medellin; MXL, Mexican ancestry from Los Angeles, PUR, Puerto Ricans from Puerto Rico; PEL, Peruvians from Lima, Peru; EAS, East Asian; CHB, Han Chinese in Beijing, China; JPT, Japanese in Tokyo, Japan; CHS, Southern Han Chinese; CDX, Chinese Dai in Xishuangbanna, China; KHV, Kinh in Ho Chi Minh City, Vietnam; EUR, European, CEU, Utah Residents (CEPH) with Northern and Western European Ancestry; TSI, Toscani in Italia; FIN, Finnish in Finland; GBR, British in England and Scotland; IBS, Iberian Population in Spain; SAS, South Asian; GIH, Gujarati Indian from Houston, Texas; PJL, Punjabi from Lahore, Pakistan; BEB, Bengali from Bangladesh; STU, Sri Lankan Tamil from the UK; ITU, Indian Telugu from the UK.