

Abstract
Background
Macrophages can polarize to M2 phenotype to decrease inflammation and encourage tissue repair. Nonetheless, its role in sepsis-induced acute lung injury and its effect on endothelial cells (ECs) regeneration remains unknown. The aim of the current study was to explore the impact of M2 macrophages on pulmonary ECs proliferation in sepsis-induced acute lung injury.
Methods
We co-cultured mouse lung microvascular endothelial cells (MLMVECs) with M2 macrophages following LPS challenge. M2 macrophages were intratracheally transplanted into mice subjected to cecal ligation and puncture (CLP). We further performed cytokine array for the supernatant from M2 macrophages and serum from mice subjected with CLP.
Results
We found both co-culture with M2 macrophages and treating with supernatant from M2 macrophages increased ECs viability following LPS challenge. Intratracheal transplantation of M2 macrophages markedly promoted pulmonary ECs proliferation, manifesting as attenuation of lung microvascular permeability and lung tissue edema, as well as improvement of survival rate. We further found that CXCL12, IL-1ra, TIMP-1, IL-4, and CXCL1 were increased in the supernatant of M2 macrophages in vitro. G-CSF and Complement Component 5a (C5/C5a) were increased in the serum of the M2-transplanted mice.
Conclusions
The present study suggested M2 macrophages could promote ECs proliferation in sepsis-induced ALI through secretion of anti-inflammatory cytokines and growth factors.
Keywords: M2 macrophages, sepsis, endothelial cells (EC), proliferation
Introduction
ARDS is an inflammatory process of lungs that develops in response to pulmonary and extra-pulmonary insults, resulting in increased alveolar-capillary permeability and subsequent interstitial or alveolar edema (1). Although enormous efforts have been made to improve the outcome of ARDS, the mortality of severe cases remains greater than 40% due to lack of specific and effective therapy (2). Recently cell therapy emerges as a novel therapeutic approach for ARDS, such as intravenous delivery of mesenchymal stem cells, which interact with injured tissue through the release of multiple soluble bioactive factors (2). Meanwhile, diverse macrophage populations were reported to involve in initiation and resolution of lung inflammation in different phases (3). Nevertheless, whether polarized macrophages have any therapeutic effect on acute lung injury remains unknown.
Over the past century, macrophage research focused primarily on the phagocytosis function of these cells, which plays an essential role in the uptake of microbes and dead cells. It is now clear that the primary day-to-day functions of tissue-resident macrophages go far beyond host defense and removal of dead cells (4). Macrophages also play a central role in subsequent tissue repair (5). Macrophages detect and eliminate the damaged tissue and subsequently promote regeneration. This dichotomy requires the switch of effector functions of macrophages coordinated with other cell types inside the injured tissue (6). Macrophages display remarkable plasticity and can change their phenotype in response to environmental cues. These changes can give rise to different populations of cells with distinct functions (7). There is growing evidence that macrophage polarization dynamics are critical to instruct immune and non-immune tissue-resident cells to both initiate and terminate healing responses (8). Macrophages have the capacity to alter cytokine/chemokine production and various other functions along the spectrum of M1 or M2 activation based on the stimulus detected (9). M1 macrophages can be induced by IFN-γ and LPS, M2a macrophages by IL-4 or IL-13, M2b macrophages by immune complexes in combination with IL-1β or LPS, and M2c macrophages with IL-10, TGF-β or glucocorticoids (10,11). M1 macrophages activation depends on Toll-like receptors (TLRs) and activation of nuclear factor kappa B (nf-κB)/c-Jun N-terminal kinase 1 (JNK1), leading to production of inflammatory cytokines, such as TNF-α and IL-1β and activation of iNOS that results in increased production of nitride oxide (NO). In contrast, M2 macrophages activate PPARγ, PPARδ, or IL-4-STAT6 pathways, leading to alternative, anti-inflammatory phenotype that is associated with upregulation of mannose receptor CD206, and arginase 1 (Arg1) (11-13). PPARγ is transcriptionally active and regulates the growth factor Gdf3 in the macrophages located in regenerating skeletal muscle, forming a paracrine axis connecting macrophages to muscle progenitor fusion (14). In this context, we raised the hypothesis that M2 macrophages might improve acute lung injury through accelerating lung tissue repair. As ECs homeostasis holds the pivotal role in the recovery of acute lung injury, we performed in vitro non-contact co-culture of pulmonary ECs with M2 macrophages and in vivo M2 macrophages transplantation to explore whether M2 macrophages could promote ECs proliferation to facilitate the rehabilitation of lung function.
Methods
Mice and cecal ligation and puncture (CLP)
C57BL/6 mice were bred and maintained under specific pathogen-free conditions at the animal facility of Zhongshan Hospital Fudan University. Male mice aged 6-8 weeks old were used for all experiments. The Animal Care Committee of the Zhongshan Hospital approved all animal protocols. For in vivo study, C57BL/6 mice were subjected to CLP or sham surgery for 3 hours prior to macrophages transplantation.
Isolation of murine bone marrow-derived macrophages and stimulation medium
Bone marrow-derived macrophages were isolated as reported (15,16). Briefly, femurs and tibia bones from 6–8 weeks old mice were removed and flushed with sterile PBS containing 3% FBS (HyClone™ Fetal Bovine Serum, SH3007103, Fisher Scientific). The cell suspension was centrifuged, treated with red blood cell lysis buffer, washed, and then plated onto sterile Petri dishes in DMEM, containing10% FBS and 15% L929-conditioned media. On day 7, M1macrophages were induced by DMEM containing 10% FBS and 100 ng/mL LPS; M2 macrophages were induced by DMEM containing 10% FBS with 10 ng/mL IL-4.
Cell culture with insert co-culture system and macrophages-conditioned medium
Transwells with 0.4um pore size (Polycarbonate Tissue Culture-Treated Inserts, 29442-106, Corning) was used to non-contact co-culture of two types of cells. Mouse primary endothelial cells (ECs) were isolated as previously described (17) and were cultured in a humidified incubator at 37 °C with 5% CO2. For the Insert experiment, 0.05×106 EC cells were plated in the 24-well plate, treated with LPS for two days. In another 24-well plate with Insert, 0.1×106 macrophages were plated into the Insert, the cells were treated with LPS (for M1) or IL-4 (M2) for 48 hrs. After 48 hours, the insert with M1/M2 macrophages was put into the 24-well plate with LPS-treated ECs and co-culture for three days. Inserts were removed and the MTT were detected for the ECs.
For preparation of macrophages-conditioned medium, macrophages were activated with LPS (100 ng/mL) or IL-4 (10 ng/mL), supernatant was collected and centrifuge to remove the cells. The prepared supernatant was directly added to the injured ECs as mentioned above.
MTT
Five hundred µL of MTT working solution was added to each well with cultured ECs for 3 h. The MTT solution was carefully removed and 500 µL DMSO was added to each well for 30 min. Color development was measured using a spectrophotometer at 570 nm on a plate reader (Bio-Tek Instruments, Winooski, VT, USA).
Flow cytometry
Cell suspension was treated with Blocker (Mouse BD Fc Block™ purified anti-mouse CD16/CD32 mAb, BD Pharmingen™, CN: 553142) for 5 minutes, then incubated with antibody: Alexa Fluor® 488 anti-mouse CD206 (MMR) (BioLgend. CN: 141709); APC anti-mouse CD86 (BioLgend. CN: 105113); PE anti-mouse/human CD11b (BioLgend. CN: 101207); Pacific Blueä anti-mouse F4/80 (BioLgend. CN: 123123) for 1 hour. Propidium iodide (PI) working solution was added to cell suspension in washing buffer for dead cell exclusion. Flow cytometry was performed using CyAn ADP flow cytometer (Dako Cytomation). Data was analyzed by winMDI software.
Real-time PCR
Total RNA was isolated using Trizol reagent (Life Technologies, Carlsbad, CA, USA) following the manufacturer’s instructions. The cDNA of target gene was synthesized by iScript™ Advanced cDNA Synthesis Kit for RT-qPCR (Bio-Rad, Hercules, CA, USA). qPCR of target gene and internal control were performed by using iTaq™ Universal SYBR® Green Supermix (Bio-Rad, Hercules, CA, USA) according to the instructions of the manual. Cyclophilin expression served as an internal control (18). The following mouse primers were used: IL1b (FW 5'-GAAATGCCACCTTTTGACAGTG-3'; Rev 5'-TGGATGCTCTCATCAGGACAG-3'), PPARγ (FW 5'-ACCTCTGCTGGGGATCTGAA-3'; Rev 5'-TCACCGCTTCTTTCAAATCTACTC-3'), TNF-a (FW 5'-AGCCGATGGGTTGTACCTTG-3'; Rev 5'-ATAGCAAATCGGCTGACGGT-3'), Arg1 (FW 5'-CTCCAAGCCAAAGTCCTTAGAG-3'; Rev 5'-GGAGCTGTCATTAGGGACATCA-3'). The expression (E) of each mRNA relative to internal control was calculated based on the cycle threshold (Ct) as E =2–Δ(ΔCt), in which ΔCt=Ct (target) − Ct (internal control).
ECs proliferation measurement
For labelling of proliferating cells, mice were intraperitoneally injected with 150 mg/kg body of BrdU (Sigma) 24 hours before sacrifice. Mice lung cells were isolated into cell suspension and stained with APC BrdU Flow Kit (BD, CN. 552598) following the manufacturer’s instructions. ECs were labelled by Anti-VE-Cadherin antibody-intercellular junction marker (ab33168, abcam). BrdU was detected by flow cytometry.
Immunofluorescent staining
Cells were fixed with 4% paraformaldehyde for 10 minutes and permeabilized with 0.2% Triton X-100 for 5 minutes. After blocking with 1% BSA for one hour, cells were incubated with the primary antibody anti-F4/80 antibody (BM8) (ab16911, abcam) 1:50 dilution for overnight at 4 °C. After extensive washing with PBS, Alexa Fluor 488 donkey anti-rat IgG (H + L) (A21208, Life technologies) were added in blocking buffer for 1 hour in the dark. Mount cover slips on microscope slides with ProLong® Diamond Antifade Mountant with DAPI (P36966, Fisher) for nuclear staining. Images were obtained with Zeiss LSM 880 Confocal Microscope (Carl Zeiss).
Evans blue albumin pulmonary transvascular efflux measurements
Evans blue albumin extravasation assay was performed as previously described (17). Briefly, Evans blue albumin was injected into anesthetized mice and allowed to circulate in the blood vessels for 30 minutes. Intravascular Evans Blue Dye was washed by PBS perfusion from the right ventricle for 2 minutes. Mouse lungs were homogenized in 1 mL PBS, and extracted in 2 mL formamide for overnight at 60 °C. Evans blue concentration in lung homogenate supernatants was quantified by spectrophotometric method at absorbance of 620 and 740 nm.
Cytokine Array assay
Proteome ProfilerTM Array Mouse Cytokine Array Panel A kit (CN ARY006, R&D) was used to detect the cytokine changes according to the instructions of the manual.
Statistical analysis
Data were analyzed by Student’s t-test for comparisons of two groups or one-way ANOVA of multiple comparisons. Survival analysis was performed by Kaplan-Meier analysis with a log-rank test. All P values were two-sided, and P<0.05 was considered statistically significant.
Results
Differentially polarized macrophages exhibit distinct phenotype
Macrophages hold the capability of polarization into two subtypes: M1 and M2, which display different genes expression and morphology (19-22). To induce M1 macrophages polarization, we treated bone marrow derived macrophages (BMDMs) with LPS for 24 h. In consistence with studies mentioned above, M1 macrophages expressed high abundance of specific surface marker CD86 (Figure 1A) and proinflammatory cytokines: TNF-α and IL-1β (Figure 1B). Meanwhile, we induced M2 macrophages polarization with IL-4 for 48 h. Specific surface marker CD206 (Figure 1C), as well as PPARγ and Arg1 (Figure 1D) were highly expressed in M2 macrophages. All of BMDMs express F4/80 (Figure 1E), M1-polarizing stimuli (LPS) induced a rounded cell shape whereas M2-polarizing stimuli (IL-4) induced an elongated cell shape (3) (Figure 1E).
Figure 1.

M2 macrophages display different surface marker, gene signature and morphology compared with M1 macrophages. (A,B) LPS (100 ng/mL, 24 hours) induced M1-type macrophages express abundant CD86 detected by flow cytometry (A) and pro-inflammatory cytokines by qPCR (B). Plots show mean ± SEM. The data points depict individual experiment; n=4. (C,D) IL-4 (10 ng/mL, 48 hours) induced M2-type macrophages exhibit upregulated expression of CD206 measured by flow cytometry (C) and genes of PPARγ as well as Arg1 by qPCR (D). Plots show mean ± SEM. The data points depict individual experiment; n=4. (E) Confocal imaging of F4/80 (green) and DAPI (blue) presents the morphology of M0, M1 and M2 macrophages. Scale bar: 10 µM. **P<0.01, ***P<0.001, ****P<0.0001 by 2-tailed t-test.
M2 macrophages improve the EC proliferation following LPS-induced injury in vitro
M2 macrophage was proven to be activated accordingly at later recovery phrase of acute lung injury, playing a crucial role in tissue repair. In this context, we raised the hypothesis that M2 macrophage may promote injured pulmonary EC regeneration, which is pivotal for lung recovery following sepsis-induced injury. First we co-cultured LPS-treated ECs with M2 macrophages to test the role of M2 on ECs proliferation. We found the cell viability was remarkedly increased in M2-co-cultured ECs (Figure 2A). Furthermore, we treated ECs with M2-conditioned medium to testify whether M2 promotes ECs proliferation through secreted factors. We observed the similar results with co-culture assay (Figure 2B), which suggested that cytokine or growth factors secreted from the M2 macrophages might play a role in improving the ECs restoration in vitro.
Figure 2.

Co-culture with M2 macrophages or treatment with M2 macrophage supernatant promotes pulmonary microvascular ECs restoration following LPS challenge in vitro. (A) Co-culture of LPS-challenged (1 µg/mL, 24 h) ECs with M0/M2 macrophages for 72 h. M2, not M0 macrophage markedly promotes ECs proliferation detected by MTT cell viability assay kit. (B) LPS-challenged ECs were treated with supernatant from M0/M2 macrophages (M0S/M2S) for 72 h. M2S, not M0S regenerates ECs detected by MTT cell viability assay kit. Plots show mean ± SEM. The data points depict individual experiment; n=4. *P<0.05, ***P<0.001 by ANOVA.
M2 macrophages accelerate lung ECs proliferation and improve survival in mice with sepsis
Based on the in vitro results, we further investigated whether M2 macrophages could promote lung ECs proliferation in the recovery phrase of sepsis, which is crucial for lung microvascular to recovery barrier function and facilitate survival. Macrophages (M0/M2) or vehicle were intratracheally delivered to mice 3 h after CLP or sham surgery. We found M2 macrophages remarkably improved lung ECs proliferation 3d post-CLP (Figure 3A). To testify whether increased ECs proliferation led to recovery of pulmonary microvascular barrier function, we tested transvascular albumin efflux and lung wet/dry 3 d after CLP. In consistence with BrdU assay, M2 macrophages significantly improved lung microvascular permeability (Figure 3B) and lung tissue edema (Figure 3C). Moreover, we observed M2 macrophages improved survival rate of late phrase, not early phrase after CLP, in mice subjected to CLP (Figure 3D). Combined with aforementioned results, we concluded that M2 macrophage facilitate the recovery from sepsis through boosting ECs proliferation.
Figure 3.

Intra-tracheally transplanted M2 macrophages boost lung ECs proliferation and improve survival in mice with sepsis. (A) C57B mice subjected to sham surgery or cecal ligation and puncture (CLP) were treated with intratracheal M0/M2 macrophages (2×106/mouse) 3 h post-surgery. Representative figures of lung ECs proliferation (VE-Cadherin+/BrdU+) were obtained by flow cytometry 72 h post-surgery. M2 macrophages remarkably increase ECs proliferation in mice with sepsis. Plots show mean ± SEM. The data points depict individual mice; n=4. (B,C) Pulmonary transvascular albumin efflux (B) and lung tissue wet-dry ratio (C) were measured 72 h post-surgery. M2 macrophages noteworthy attenuate sepsis-induced lung microvascular permeability. Plots show mean ± SEM. The data points depict individual mice; n=5. **P<0.01, ***P<0.001 by ANOVA. (D) Survival of mice with M0 or M2 macrophages was accessed by Kaplan Meier plots. M2 macrophages significantly improve survival rate in mice with sepsis.
M2 macrophages release anti-inflammatory and pro-growth cytokines both in vitro and in vivo
To explore the mechanism under which M2 macrophages improve ECs proliferation, we performed cytokine array to screen the M2-secretting factors which is associated with promotion of ECs regeneration. Comparing the supernatant of M2 with M0, there are totally 12 genes which changed more than 2-fold (Figure 4A). CXCL12 increased 5.4-fold; IL-1ra 6.4-fold; TIMP-1 6.5-fold, IL-4 7.7-fold, and CXCL1 9.6-fold (Table 1). In similarity with in vitro result, we observed in the serum from mice subjected with CLP, 5 genes have more than 2-fold change (Figure 4B), G-CSF increased 6.48-fold and Complement Component 5a (C5/C5a) increased 41.88-fold (Table 2).
Figure 4.
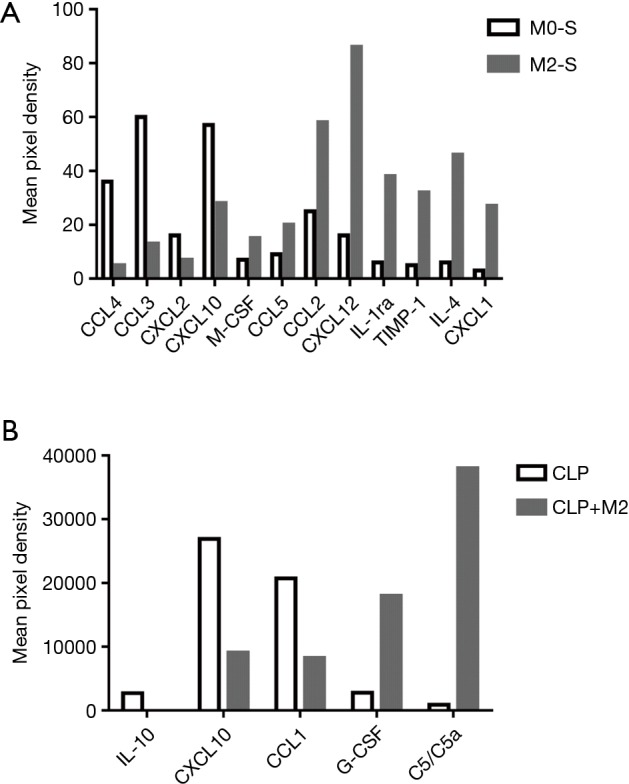
M2 macrophages express anti-inflammatory and proliferation-promoting cytokines both in vitro and in vivo. (A,B) Cytokine array in supernatant from M0/M2 macrophages (A) or serum from mice subjected to CLP/sham surgery (B) were assessed by Proteome ProfilerTM Array Mouse Cytokine Array Panel A kit. Proliferation-associated cytokines were upregulated in M2 macrophages.
Table 1. Relative result of cytokine array of M2 macrophages supernatant.
| SN | ID/gene | M2/M0 |
|---|---|---|
| 1 | CCL4 | 0.1 |
| 2 | CCL3 | 0.2 |
| 3 | CXCL2 | 0.4 |
| 4 | CXCL10 | 0.5 |
| 5 | M-CSF | 2.0 |
| 6 | CCL5 | 2.2 |
| 7 | CCL2 | 2.3 |
| 8 | CXCL12 | 5.4 |
| 9 | IL-1ra | 6.4 |
| 10 | TIMP-1 | 6.5 |
| 11 | IL-4 | 7.7 |
| 12 | CXCL1 | 9.6 |
Table 2. Relative result of cytokine array of serum from mice receiving M2 macrophages transplantation.
| SN | ID/gene | M2/M0 |
|---|---|---|
| 1 | IL-10 | 0.00 |
| 2 | CXCL10 | 0.34 |
| 3 | CCL1 | 0.40 |
| 4 | G-CSF | 6.48 |
| 5 | C5/C5a | 41.88 |
Discussion
It’s widely accepted that the nature of sepsis has two phrases: early pro-inflammatory phrase and later anti-inflammatory phrase (23). Macrophages play a central role in both of two phrases in a sequential manner to activate inflammation to remove the pathogen as M1-type, then resolve the inflammation and repair the damaged tissue as M2-type (3). Macrophages are recognized to engage the M1 phenotype in the initiation of infection to orchestrate host immunity, and then obtain the M2 phenotype to restrain inflammation and facilitate tissues repair. As sepsis is a dysregulated host response to infection and ECs is the primary target of over-inflammation, we hypothesized that M2 macrophages could attenuate sepsis via improving ECs regeneration. Our study proved that M2 macrophage can improve pulmonary ECs proliferation both in vitro and in vivo, resulting in recovery of sepsis-induced lung ECs dysfunction, which provides a clue that M2 macrophage transplantation may be a novel therapeutic approach for sepsis-elicited acute lung injury.
Carlos et al found lung surfactant protein A enhanced IL-4-dependent macrophages, which accelerates lung repair after infection (24). Meanwhile, endothelium and M2 macrophages are in complex reciprocal relationship: expansion of M2 macrophages needs instruction of endothelium, on the other side, M2 macrophages promotes angiogenesis (25). Anti-inflammatory M2, but not pro-inflammatory M1 macrophages promote angiogenesis in vivo (26). Our results showed M2 macrophages could restore pulmonary ECs viability in vitro and in vivo, which contributes to attenuation of microvascular permeability and improvement of survival of mice with bacteria sepsis. In similarity with our results, Matrigel plug supplemented with macrophage subsets experiment showed increased numbers of ECs and tubular structures were observed in M2-enriched plugs compared to control and other subsets. Additionally, more tubular structures formed in the presence of M2 macrophages-conditioned medium (26).
The mechanism under which M2 macrophages accelerate ECs growth may be complex. Our co-culture system with non-contact of the cells suggested that M2 macrophages may impact endothelium proliferation in a secretion manner. Macrophages can stimulate endothelial proliferation and angiogenesis by secreting pro-angiogenic factors and stabilizing tip cell fusion, thereby increasing vascular complexity (27). In our study, we observed that the expression of CXCL12, IL-1ra, TIMP-1, IL-4, and CXCL1 increased more than 2-fold compared with M0. During inflammation, once the inflammatory stimulus or pathogen is eliminated, activation of M1 macrophages will diminishes, instead, M2 macrophages will accumulate. M2 macrophages can promote wound healing and fibrosis through the production of MMPs including MMP12, tissue inhibitor of metalloproteinases 1 (TIMP1), growth factors including platelet-derived growth factor (PDGF) and cytokines (such as transforming growth factor-β1 (TGFβ1). M2 macrophages also promote the resolution of wound healing by antagonizing M1-elicited inflammatory responses, manifested as producing nitric oxide (NO), reactive oxygen species (ROS), interleukin-1 (IL-1) and tumor necrosis factor (TNF-𝛼) (28). Our results showed expression of IL-1ra in M2 increased 6.4-fold compared with M0 macrophages. Members of IL-1 family include IL-1F1 (IL-1α), IL-1F2 (IL-1β), and IL-1F3 (IL-1Rα). IL-1α and IL-1β were initially identified during research on immune defense. Both molecules function as proinflammatory cytokines, while IL-1Rα is an anti-inflammatory cytokine that blocks the pathway downstream of IL-1 signaling and maintains homeostasis by competitively binding IL-1α and IL-1β receptors (29). All these cytokines involved in anti-inflammation and growth promotion are orchestrated to accelerate the tissue repair, which requires ECs proliferation and angiogenesis.
Conclusions
In conclusion, the current study clearly demonstrated that M2 macrophages could accelerate ECs proliferation and promote microvascular barrier function recovery in sepsis-induced acute lung injury, although the underlying mechanism still need more works. Our study suggests that transplantation of M2 macrophages may be a potential therapeutic approach for sepsis-induced acute lung injury.
Acknowledgements
Funding: This work was supported by grants from the National Natural Science Foundation of China (No. 8180943, 81400681) and the China Postdoctoral Science Foundation (2018M631394).
Ethical Statement: The study was approved by Animal Care Committee of the Zhongshan Hospital.
Footnotes
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- 1.Villar J, Blanco J, Kacmarek RM. Current incidence and outcome of the acute respiratory distress syndrome. Curr Opin Crit Care 2016;22:1-6. 10.1097/MCC.0000000000000266 [DOI] [PubMed] [Google Scholar]
- 2.Thompson BT, Chambers RC, Liu KD. Acute Respiratory Distress Syndrome. N Engl J Med 2017;377:562-72. 10.1056/NEJMra1608077 [DOI] [PubMed] [Google Scholar]
- 3.Aggarwal NR, King LS, D'Alessio FR. Diverse macrophage populations mediate acute lung inflammation and resolution. Am J Physiol Lung Cell Mol Physiol 2014;306:L709-25. 10.1152/ajplung.00341.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guilliams M. Macrophage, a long-distance middleman. Science 2017;355:1258-9. 10.1126/science.aam9743 [DOI] [PubMed] [Google Scholar]
- 5.Bouchery T, Harris NL. Specific repair by discerning macrophages. Science 2017;356:1014. 10.1126/science.aan6782 [DOI] [PubMed] [Google Scholar]
- 6.Mills EL, Kelly B, Logan A, et al. Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell 2016;167:457-470.e13. 10.1016/j.cell.2016.08.064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol 2008;8:958-69. 10.1038/nri2448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eming SA, Wynn TA, Martin P. Inflammation and metabolism in tissue repair and regeneration. Science 2017;356:1026-30. 10.1126/science.aam7928 [DOI] [PubMed] [Google Scholar]
- 9.Hardbower DM, Asim M, Luis PB, et al. Ornithine decarboxylase regulates M1 macrophage activation and mucosal inflammation via histone modifications. Proc Natl Acad Sci U S A 2017;114:E751-60. 10.1073/pnas.1614958114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Greenlee-Wacker MC. Clearance of apoptotic neutrophils and resolution of inflammation. Immunol Rev 2016;273:357-70. 10.1111/imr.12453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ying W, Cheruku PS, Bazer FW, et al. Investigation of Macrophage Polarization Using Bone Marrow Derived Macrophages. J Vis Exp 2013;(76). [DOI] [PMC free article] [PubMed]
- 12.Thomas GD, Hanna RN, Vasudevan NT, et al. Deleting an Nr4a1 Super-Enhancer Subdomain Ablates Ly6Clow Monocytes while Preserving Macrophage Gene Function. Immunity 2016;45:975-87. 10.1016/j.immuni.2016.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity 2010;32:593-604. 10.1016/j.immuni.2010.05.007 [DOI] [PubMed] [Google Scholar]
- 14.Varga T, Mounier R, Patsalos A, et al. Macrophage PPARγ, a Lipid Activated Transcription Factor Controls the Growth Factor GDF3 and Skeletal Muscle Regeneration. Immunity 2016;45:1038-51. 10.1016/j.immuni.2016.10.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang X, Goncalves R, Mosser DM. The isolation and characterization of murine macrophages. Curr Protoc Immunol 2008;Chapter 14:Unit 14.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Han CZ, Juncadella IJ, Kinchen JM, et al. Macrophages redirect phagocytosis by non-professional phagocytes and influence inflammation. Nature 2016;539:570-4. 10.1038/nature20141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cheng KT, Xiong S, Ye Z, et al. Caspase-11-mediated endothelial pyroptosis underlies endotoxemia-induced lung injury. J Clin Invest 2017;127:4124-35. 10.1172/JCI94495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mittal M, Tiruppathi C, Nepal S, et al. TNFα-stimulated gene-6 (TSG6) activates macrophage phenotype transition to prevent inflammatory lung injury. Proc Natl Acad Sci U S A 2016;113:E8151-8. 10.1073/pnas.1614935113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Udalova IA, Mantovani A, Feldmann M. Macrophage heterogeneity in the context of rheumatoid arthritis. Nat Rev Rheumatol 2016;12:472-85. 10.1038/nrrheum.2016.91 [DOI] [PubMed] [Google Scholar]
- 20.Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol 2011;11:723-37. 10.1038/nri3073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ivashkiv LB. Epigenetic regulation of macrophage polarization and function. Trends Immunol 2013;34:216-23. 10.1016/j.it.2012.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Glass CK, Natoli G. Molecular control of activation and priming in macrophages. Nat Immunol 2016;17:26-33. 10.1038/ni.3306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hotchkiss RS, Monneret G, Payen D. Sepsis-induced immunosuppression: from cellular dysfunctions to immunotherapy. Nat Rev Immunol 2013;13:862-74. 10.1038/nri3552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Minutti CM, Jackson-Jones LH, García-Fojeda B, et al. Local amplifiers of IL-4Ralpha-mediated macrophage activation promote repair in lung and liver. Science 2017;356:1076-80. 10.1126/science.aaj2067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.He H, Xu J, Warren CM, et al. Endothelial cells provide an instructive niche for the differentiation and functional polarization of M2-like macrophages. Blood 2012;120:3152-62. 10.1182/blood-2012-04-422758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jetten N, Verbruggen S, Gijbels MJ, et al. Anti-inflammatory M2, but not pro-inflammatory M1 macrophages promote angiogenesis in vivo. Angiogenesis 2014;17:109-18. 10.1007/s10456-013-9381-6 [DOI] [PubMed] [Google Scholar]
- 27.Gerri C, Marín-Juez R, Marass M, et al. Hif-1α regulates macrophage-endothelial interactions during blood vessel development in zebrafish. Nat Commun 2017;8:15492. 10.1038/ncomms15492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol 2011;11:723-37. 10.1038/nri3073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tan Q, Hu J, Yu X, et al. The Role of IL-1 Family Members and Kupffer Cells in Liver Regeneration. Biomed Res Int 2016;2016:6495793. 10.1155/2016/6495793 [DOI] [PMC free article] [PubMed] [Google Scholar]