

Abstract
Poly (ADP-ribose) polymerase (PARP) inhibitors have emerged as promising targeted therapies for BRCA-mutated cancers by blocking repair of DNA double-strand breaks. However, resistance to PARP inhibitors (PARPi) has been described in some patients lowering the overall response rates. To investigate the underlying mechanisms of PARPi resistance, we developed the adaptive resistant clones in triple-negative breast cancer cell lines. We identified epithelial-mesenchymal transition (EMT) and upregulation of programmed death-ligand 1 (PD-L1) in resistant cells and further demonstrated the important role of Akt S473 phosphorylation in PARPi resistance. In addition, PARPi mediated EMT is independent of PD-L1 upregulation. Blocking the p-Akt S473 axis by metformin reversed EMT and PD-L1 expression which sensitized PARPi-resistant cells to cytotoxic T cells. Thus, a combination of metformin and PARP inhibitors may be a promising therapeutic strategy to increase the efficacy of PARP inhibitors and tumor sensitivity to immunotherapy.
Keywords: PARP, epithelial-mesenchymal transition, PD-L1, triple-negative breast cancer, metformin
Introduction
Breast cancer is the most common cancer type (21% of all new cases) and the leading cause of cancer mortality (414,000 annual deaths, representing 14.1% of female cancer deaths) in women worldwide [1,2]. As a heterogeneous cancer type, it was recently classified into six subgroups, according to the molecular expression: normal-like, luminal A, luminal B, human epidermal growth factor receptor-2 (HER2)-positive, basal-like, and claudin-low [3].
Triple-negative breast cancer (TNBC) is an aggressive subtype of breast cancer that lacks expression of estrogen receptor, progesterone receptor, and HER2, detectable by immunohistochemistry staining or in situ hybridization [4]. In the clinic, TNBC accounts for 15%-20% of breast cancer cases and 25% of breast cancer deaths [5] and is classically divided into four subtypes according to genomic profile: basal-like immune-suppressed, basal-like immune-activated, luminal androgen receptor, and mesenchymal [6]. This subclassification is supported by The Cancer Genome Atlas Program through mRNA, miRNA, DNA, and epigenetic analyses [7]. Similar to the six breast cancer subgroups mentioned above, the subtypes of TNBC are based on a differentiation hierarchy that mimics the developmental cascade of normal epithelial cells [8]. In this developmental process, a luminal progenitor forms the basal-like and HER2-positive subgroups and then differentiated into the luminal A and luminal B subgroups. The claudin subgroup is characterized by the dedifferentiation of cells, resembling the development of more aggressive tumor cells, a process known as epithelial-mesenchymal transition (EMT) [9,10].
EMT has been championed by Weinberg and colleagues as a biological program associated with the transition from stable epithelial cancer cells to mesenchymal-type cells and metastasis as well as the resistance to both traditional chemotherapy and immunotherapy [11,12]. At the cellular level, EMT is accompanied by specific morphologic criteria and disorderly architecture, and at the molecular level, it is characterized by loss of E-cadherin and accompanied by encoding-associated genes, such as vimentin, N-cadherin, fibronectin, and integrins [13]. A significant mediator of EMT is the enrichment of cancer stem cells, also known as tumor-initiating cells, which are characterized by self-renewal, multipotent differentiation, and initiation of invasiveness and proliferation [14]. Moreover, tumor heterogeneity, initiated by cancer stems cells, is a driving force behind tumor relapse, leading to drug resistance, invasiveness, and aggressiveness [15,16]. Tumor cells that progress to EMT are associated with early metastasis and poor prognosis in patients [16].
Various and complex factors are responsible for inducing EMT, which plays a key role in tumor cell resistance of tumor cells to chemotherapy and immunotherapy; among these factors is resistance to poly (ADP-ribose) polymerase (PARP) inhibition. PARPs represent a superfamily of 17 proteins with different cellular functions, such as spindle pole formation, cell cycle regulation, cell death, inflammation, adaptive immunity, and DNA repair. PARPs are the key components of base excision repair, involving the recruitment of repair enzymes at the site of single-strand breaks [17]. Multiple PARP inhibitors (PARPis), have been developed and tested in clinical trials, including breast cancer. In December 2014, the first PARPi (olaparib, or AZD2281) was approved by the U.S. Food and Drug Administration (FDA) for the treatment of advanced BRCA-mutant ovarian cancer [18]. Subsequently, the PARPi, niraparib and rucaparib, were approved as a third-line treatment for advanced ovarian cancer [19]. In June 2018, talazoparib was approved for the treatment of HER2-negative locally advanced or metastatic breast cancer in patients with germline BRCA mutations [20]. Although preclinical and clinical studies indicated that PARP should be an effective target for synthetic lethality, a high percentage of patients with BRCA mutations do not respond to PARPis; instead of developing adaptive resistance [21]. Thus, it is critical to understand detailed mechanisms underlying PARPi resistance and develop strategies to overcome the resistance. In the current study, we focused on the mechanisms of adaptive resistance to newly approved PARPis, olaparib and rucaparib, in particular, their role of EMT.
Materials and methods
Cell cultures and treatments
Cell lines were purchased from American Type Culture Collection. Human TNBC cells (MDA-MB-231, HCC1806, and MDA-MB-468) and Mouse TNBC cells (4T1) were grown in Dulbecco modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 100 mg/ml streptomycin. HCC70 and HCC1937 cells were cultured in RPMI-1640 with 10% fetal bovine serum. SUM149 and SUM190 cells were incubated in HyClone DMEM/high glucose with 15% fetal bovine serum, 100 U/ml penicillin, and 100 mg/ml streptomycin. Human MCF10A cells were cultured in DMEM/F12 medium supplemented with 5% horse serum, 10 mg/ml insulin, 20 ng/ml epidermal growth factor, 100 ng/ml cholera toxin, and 500 ng/ml hydrocortisone. To set up PARPi-treated stable clones in PARPi sensitive cells (HCC70, HCC1937 and SUM190) and PARPi resistant cells (SUM149, MDA-MB-231, MDA-MB-436, MDA-MB-468, HCC1806, 4T1, and MCF10A), we treated the cells with olaparib or rucaparib (5 µM) for at least 5 days.
Reagents
Olaparib, rucaparib, talazoparib, and doxorubicin were purchased from Selleck Chemicals. Daunorubicin was purchased from Calbiochem. N-cadherin, E-cadherin, vimentin, and programmed death ligand-1 (PD-L1) were purchased from Santa Cruz Biotechnology.
Morphology assay
TNBC cells (1 × 105) were seeded in 10 mm dish and treated with PARPis at a concentration of 5 µM in the medium with 10% FBS for 7 days. The concentration of PARPis were selected following literature reports and avoided inducing cytotoxicity throughout the analysis. Microscopic images were obtained with a high-resolution digital charge-coupled camera (AxioCam HRm, Carl Zeiss) and analyzed by ImageJ. Aspect ratio is measured by the major axis/minor axis of about 130 cells. The scatter score is to a method to quantitate the degree of cell scattering, main characteristics of EMT in cells, the numbers of cell islands with cell-cell contacts per microscopic image of at least three independent experiments.
Immunoblottings
Whole-cell extracts were lysed in freshly prepared radioimmunoprecipitation buffer (10 mM Tris-HCl [pH 8.6], 1 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS), 140 mM NaCl, and 1 × protease inhibitor; Complete Mini, Roche), which was freshly added before lysis. To prepare the whole-cell lysates, we added 5 × SDS sample buffer to the cell lysates and sonicated them before they were resolved on SDS-polyacrylamide gel electrophoresis and immunoblotted with primary antibodies at 4°C overnight. The protein concentrations of the lysates were measured using the Bio-Rad protein assay reagent on a Beckman Coulter DU-800 spectrophotometer.
Plasmids and transfection
For stable knockdown of PD-L1 study, breast cancer cells were transfected with pGIPZ shRNA vector (control; Thermo Fisher Scientific, Rockford, IL, USA) and plasmids (ORFeome Core at MD Anderson). Using a pGIPZ-shPD-L1/Flag-PD-L1 dual-expression construct to knock down endogenous PD-L1 expression and reconstitute Flag-PD-L1 simultaneously, endogenous PD-L1-knockdown and WT Flag-PD-L1- or 4NQ mutant (N35Q/N192Q/N200Q/N219Q) expressing cell lines were established [22].
Quantitative real-time polymerase chain reaction
TNBC cells were washed twice with PBS then immediately lysed in QIAzol lysis reagent. Total RNA was extracted from TNBC cells using a RNeasy Plus Mini Kit (QIAGEN, Venlo, Netherlands) according to the manufacturer’s instructions and sequentially subjected to complementary DNA synthesis via reverse transcription using a SuperScript III kit (Invitrogen). Quantitative real-time polymerase chain reaction analysis was performed using iQ SYBR Green Supermix (Bio-Rad, Hercules, CA, USA) in triplicate with a real-time polymerase chain reaction machine (iQ5; Bio-Rad) and the following primers: N-cadherin-F, 5’-TATGCCCAAGACAA AGAGACC-3’; N-cadherin-R, 5’-CAACTTCTGCTGACTCCTTCA-3’; vimentin-F, 5’-TGAGTACCGGAGACAGGTGCAG-3’; vimentin-R, 5’-TAGCAGCTTCAACGGCAAAGTTC-3’; RAD51-F, 5’-CAACCCATTTCACGGTTAGAGC-3’; RAD51-R, 5’-TTCTTTGGCGATAGGCAACA-3’; PD-L1-F, 5’-TCACTTGGTAATTCTGGGAGC-3’; PD-L1-R, 5’-CTTTGAGTTTGTATCTTGGATGCC-3’; GAPDH-F, 5’-GAAGGTGAAGGTCGGAGTC-3’; GAPDH-R, 5’-GAAGATGGTGATGGGATTTTC-3’. Relative expression to GAPDH was calculated as a 2-ΔCt method, and relative gene expression as fold change was calculated as 2-ΔΔCt.
Sphere assay
Cells were dissociated with trypsin and washed, and cell viability was analyzed with trypan blue exclusion. Cell suspensions were seeded (3,000 viable cells per well) in 24-well ultra-low attachment plates (Corning) in a specified serum-free medium composed of DMEM/F-12, B27 supplement (Invitrogen), 20 ng/ml recombinant basic fibroblast growth factor (BD Biosciences), and 20 ng/ml epidermal growth factor. After 10 days of culturing, spheres numbers were counted (size > 50 µm for MCF10A; size > 100 µm for 4T1).
Flow cytometric analysis
Single-cell suspensions were prepared and resuspended in staining buffer (BD Biosciences). Briefly, 1 × 106 cells were trypsinized and washed in phosphate-buffered saline twice and then fixed in cold 70% ethanol for 30 minutes. The cells were washed with phosphate-buffered saline twice and then resuspended in 400 µl of staining buffer with the following antibodies: PE-CD24 (BD Biosciences, #555428, 1:100), APC-CD44 (BioLegend, #103011, 1:100), PE/Cy7-CD44 (BioLegend, #103029, 1:100), and PD-L1 antibodies (Santa Cruz, #3137, 1:1000). Isotype immunoglobulin G (IgG) or secondary antibody alone was used as a negative control. Stained samples were evaluated by BD FACSCanto II (BD Immunocytometry Systems) and analyzed by FlowJo.
Migration assay
Two cell lines (MCF10A, 4T1) were used to analyze the cell migration of transfectant cells by wound healing assay with IncuCyte ZOOM microscope (Essen BioScience, Ann Arbor, MI). 5,000 cells (control group, pretreated with PARPi for 3 days) were seeded in 96-well plate. Cells were cultured to a confluence of 90% for 24 h then starved with 0.1% FBS overnight. Using the monolayer insert to generate a 0.9 mm wound and cultured in normal medium for an additional 24 h or 48 h. IncuCyte ZOOM microscope was set to record images every 2 hours. The distances invaded by the cells at the front of the wound were measured and analyzed as a percentage of migration. A relative wound density percentage was measured by the IncuCyte ZOOM microscope.
PD-1 binding assay
To evaluate the PD-1 binding ability, single-cell suspensions (1 × 106 cells) were incubated with 5 µg/ml recombinant human PD-1 Fc chimera protein or recombinant mouse PD-1 chimera protein (for 4T1 cells; R&D Systems) at room temperature for 30 minutes. Cells were then stained with fluorescence-conjugated anti-human IgG secondary antibody. A secondary antibody was used as a negative control. After staining, the cells were subjected to analysis or further immunostaining. The immunofluorescence was evaluated by BD FACSCanto II (BD Immunocytometry Systems) and analyzed by FlowJo.
Results
PARPis induce scattering and morphologic changes in TNBC cells
To identify the mechanism underlying adaptive resistance to PARPi, we treated several types of TNBC cells with olaparib and rucaparib for 7 days, PARPi-induced morphologic changes were observed in PARPi-sensitive cells (HCC70, HCC1937) and PARPi-resistant cells (MDA-MB-468, HCC1806) (Figure 1A). In addition, the cell numbers were counted and analyzed after indicated cells (1 × 105) seeding at 10 mm dish (Figure 1B). These morphologic alterations were representative in MDA-MB468 cells, and all determined by significant changes in aspect ratio and scatter score (Figure 1C, 1D). In the HCC70, HCC1937, HCC1806, MDA-MB-436, MDA-MB-468, SUM190, and SUM149 cell lines, the cells were separated and elongated after at least 72 hours of treatment and showed a fibroblastoid shape after 7 days. These morphologic alterations are consistent with EMT features.
Figure 1.
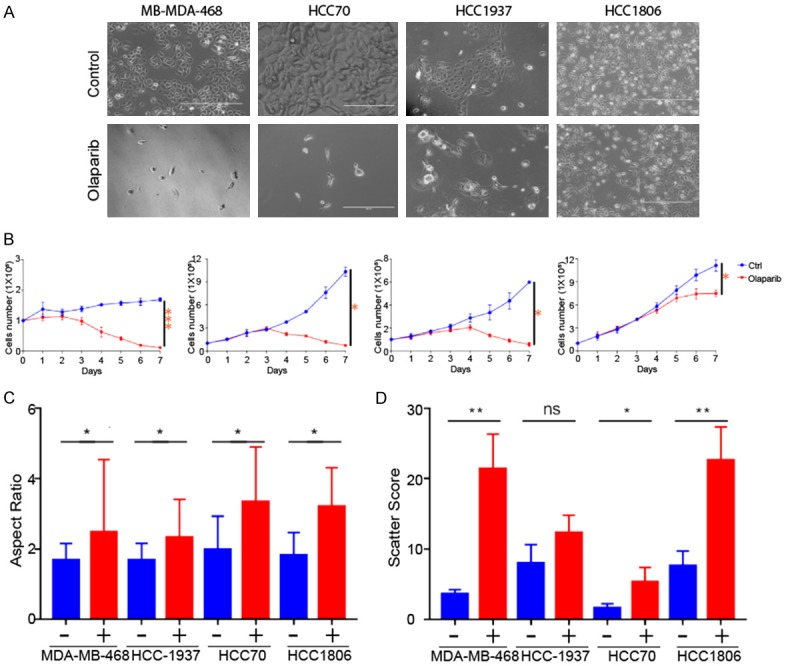
Olaparib induces scattering and morphologic changes in several triple-negative breast cancer (TNBC) cell lines. A. Representative phase-contrast microscopy images of four cell lines treated with olaparib (5 µM) or vehicle for 7 days. Scale bar: 200 µM. B. Cell number counted by ImageJ in PARPi treated group and control group. Two group data were analyzed by GraphPrism 9.0. *P<0.05; ***P<0.001 (Student’s t-test). Error bar represents standard and variation in 3 repeated experiments. C. Quantification of the aspect ratio between the area and circumference of TNBC cell lines, determined 7 days after treatment with or without olaparib. D. Scatter score of TNBC cell lines treated with or without olaparib. *P<0.05; **P<0.01 (Student’s t-test).
PARPis induce EMT markers in TNBC
As the morphology of the treated cells became mesenchymal-like, we assessed the change in the expression levels of EMT markers in a panel of PARPi-treated stable clones. The typical EMT markers N-cadherin and vimentin were enhanced or induced by a PARPi (Figure 2A). We treated both PARPi-sensitive (SUM149) and PARPi-resistant (HCC1806) cell lines with various PARPis (olaparib, rucaparib, and talazoparib). Western blot analysis indicated that both sensitive and resistant cells exhibited EMT following PARPi treatment for 72 h (Figure 2B). We also tested EMT markers in HCC1806 cells treated with olaparib at various doses and durations (Figure 2C), then we found with the increasing dose and duration treated in cells EMT markers exhibited enhanced expression. Immunofluorescence microscopy analysis of morphologic changes showed that the network structure of N-cadherin, and vimentin expression in MDA-MB-468 (PARPi resistant cell line) and HCC1937 (PARPi sensitive cell line) cells were enhanced significantly in PARPi-treated stable clones (Figure 2D). The expression of the typical EMT marker E-cadherin was decreased in olaparib-treated cells. Thus, EMT was induced in both PARPi-sensitive and PARPi-resistant cell lines, suggesting that EMT is a common phenomenon in TNBC cells.
Figure 2.

PARP inhibitors (PARPis) induce epithelial-mesenchymal transition (EMT) in several triple-negative breast cancer (TNBC) cell lines. A. Western blot analysis of EMT markers, E-cadherin, N-cadherin, and vimentin, in resting cells after treatment with olaparib (5 µM) for 72 hours. B. Western blot analysis of EMT markers, E-cadherin, N-cadherin, and vimentin, in resting HCC1806 (PARPi-resistant cell lines) and SUM149 (PARPi-sensitive cell lines) cells after treatment with the indicated PARP inhibitor. C. Western blot analysis of EMT markers in HCC1806 cells after treatment with olaparib at the indicated doses and duration. D. Representative images of tumors stained with E-cadherin, N-cadherin, vimentin, and 4’,6-diamidino-2-phenylindole (DAPI). Scale bar: 50 µM. E. mRNA levels of Vimentin in TNBC cells treated with or without PARP inhibitors (quantitative reverse-transcription PCR, qrt-PCR). F. mRNA levels of RAD51 in TNBC cells treated with or without PARP inhibitors (qrt-PCR). G. mRNA levels of N-cadherin in TNBC cells treated with or without PARP inhibitors (qrt-PCR). Error bars represent standard and variation in 3 repeated experiments.
Next, we compared the mRNA expression of the EMT markers N-cadherin, vimentin, and RAD51 (a pivotal DNA double-strand break repair enzyme, also a mediator of PARPi resistance [23]) between olaparib-treated and untreated TNBC cells (Figure 2E-G), and found that mRNA levels of these markers were increased in the treated cells. Taken together, these results indicate that PARPis contribute to tumor resistance and mesenchymal change.
PARPi-treated stable clones exhibit stemness features
Because TNBC cell lines treated with PARPis exhibited mesenchymal-like change, we sought to determine whether PARPi-treated stable clones (MDA-MB-468 and 4T1) would exhibit stemness features and functions. Compared with untreated cell lines, PARPi treatment enriched PD-L1 expression as well as increased EMT marker CD24-/lowCD44+ (Stemness marker) in MDA-MB-468 and 4T1 stable clones (Figure 3A, 3B). We then compared the stemness of PARPi-treated and untreated 4T1, MCF10A, and HCC1806 stable cells, which led to significant increases in stemness function, indicated by enhanced sphere-forming ability (Figure 3C, 3D). These results further illustrated the cells underwent EMT induced by PARPi were functionally stem-like.
Figure 3.
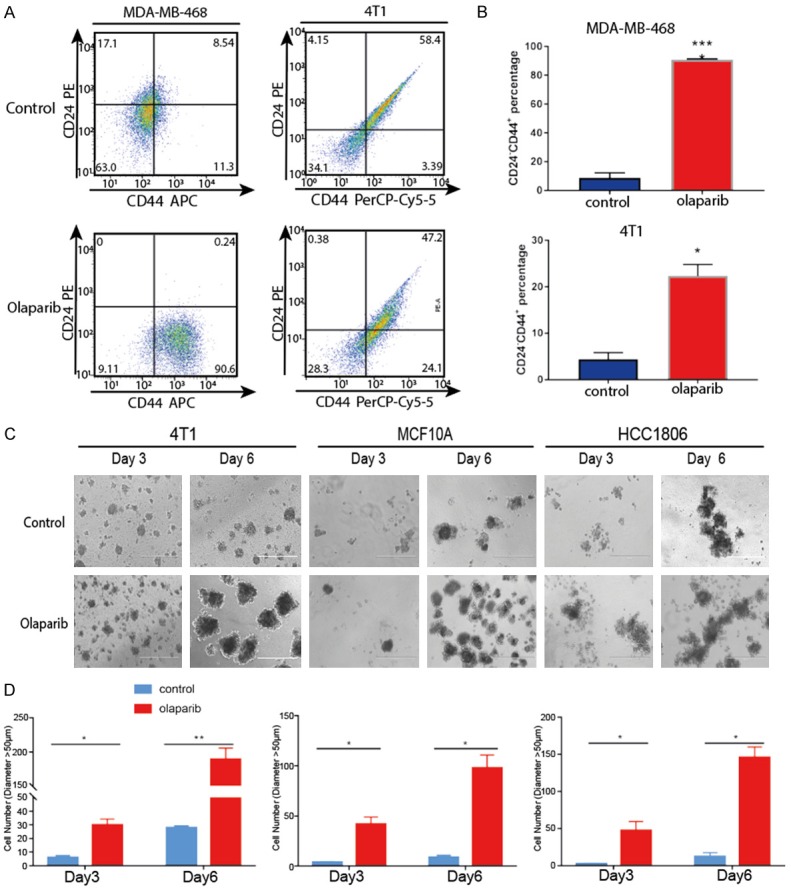
PARP inhibitor-induced tumor cells exhibit stemness features. A. Flow cytometric analysis of stem-like markers (CD44 and CD24) in MDA-MB-468 and 4T1 cells treated with or without olaparib (CD24-/low/CD44+ represents stem-like features). B. Open histograms, isotype IgG negative control. The mean fluorescence intensity of each cell population was quantified by FlowJo for comparison. Experiments were repeated three times. *P<0.05, ***P<0.001, Student’s t-test. C. Representative phase-contrast microscopy images of spheres growing from control cells or cells with epithelial-mesenchymal transition driven by PARP inhibitors. Scale bar: 400 µM. D. Open histograms represent the relative number of spheres (diameter > 50 μM) on the third and sixth day of treatment. *P<0.05, **P<0.01, Student’s t-test.
PARPis promote cell migration and invasion
The results above indicated that PARPi-treated cells underwent morphologic changes, which are generally accompanied by enhanced migration and invasiveness, and decreased adhesion [24]. To validate this, we analyzed cell mobility of MCF10A and 4T1 cells treated with or without olaparib by the IncuCyte 96-well scratch wound cell migration assay. Olaparib-treated cells had much shorter wound distance compared with untreated cells (Figure 4A, 4B). Analysis of relative wound density by IncuCyte, also showed significantly more migration of the treated cells compared with the untreated cells (Figure 4C). To investigate the invasion ability, we analyzed HCC1806 and SUM149 cells treated with or without olaparib by transwell assay. As the representative images showed, olaparib-treated cells were more invasive than the untreated ones (Figure 4D, 4E). These findings suggested inhibition of PARPis increase cell migration and invasion ability.
Figure 4.
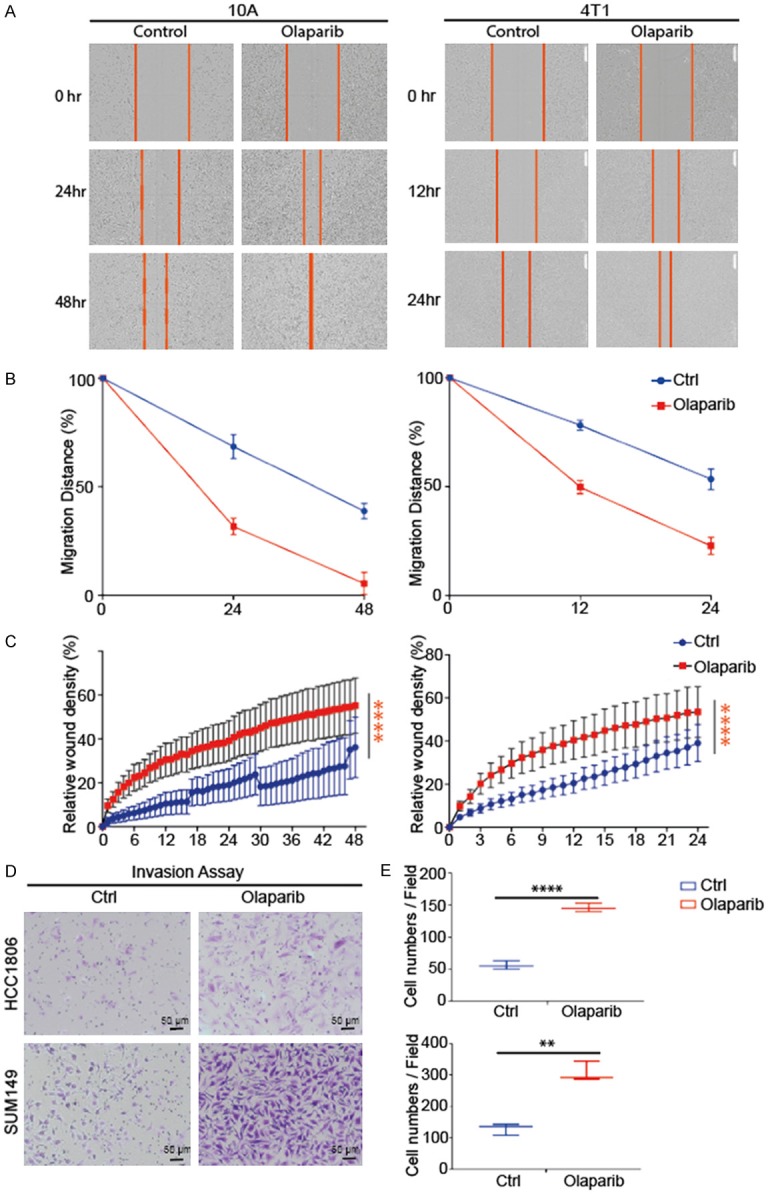
PARPi treated stable cells with mesenchymal function induced by PARP inhibitors. A. The migration in MCF10A and 4T1 cells cultured with or without olaparib was determined using the wound-healing assay (IncuCyte 96-well Scratch Wound Cell Migration assay). Representative phase-contrast microscopic images were obtained by IncuoCyte ZOOM microscope. Red line labels the migration line of cells. B. Comparison of PARPi on cell migration with wound distance percentage. C. Comparison of migration (relative wound density) between primary cells and treated cells (Student’s t-test). *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. D. Transwell Matrigel invasion assay. Representative microscopic images of PARPi-treated stable cells (HCC1806 and SUM149) and untreated cells that invaded through the transwell in the Matrigel invasion assay. (Giemsa stain, magnification × 10). E. The box-and-whisker plot of cell number per visual field was shown and analyzed (magnification × 10) of 3 replicate wells. (Welch’s test, **P<0.01, ****P<0.0001).
Reversal of PARPi-induced upregulation of PD-L1 by metformin and chemotherapy
We showed above that PARPis induce EMT resulting in enhanced migration and invasiveness. EMT has been reported to regulate PD-L1 [25,26], the main immune checkpoint marker in the PD-1/PD-L1 axis. As shown in Figure 5A, PD-L1 protein expression was upregulated in PARPi-treated stable cells (Figure 5A). Consistent with prior studies [27], enhanced levels of PD-L1 were detected on the cell membrane in PARPi-treated MCF10A and HCC1806 cells (Figure 5B). Furthermore, as the previous study mentioned [28], metformin was able to downregulate PD-L1 in PARPi-treated stable (MCF10A) and resistant (HCC1806) clones (Figure 5B). PD-L1 mRNA levels were positively correlated with EMT markers (Figure S1).
Figure 5.
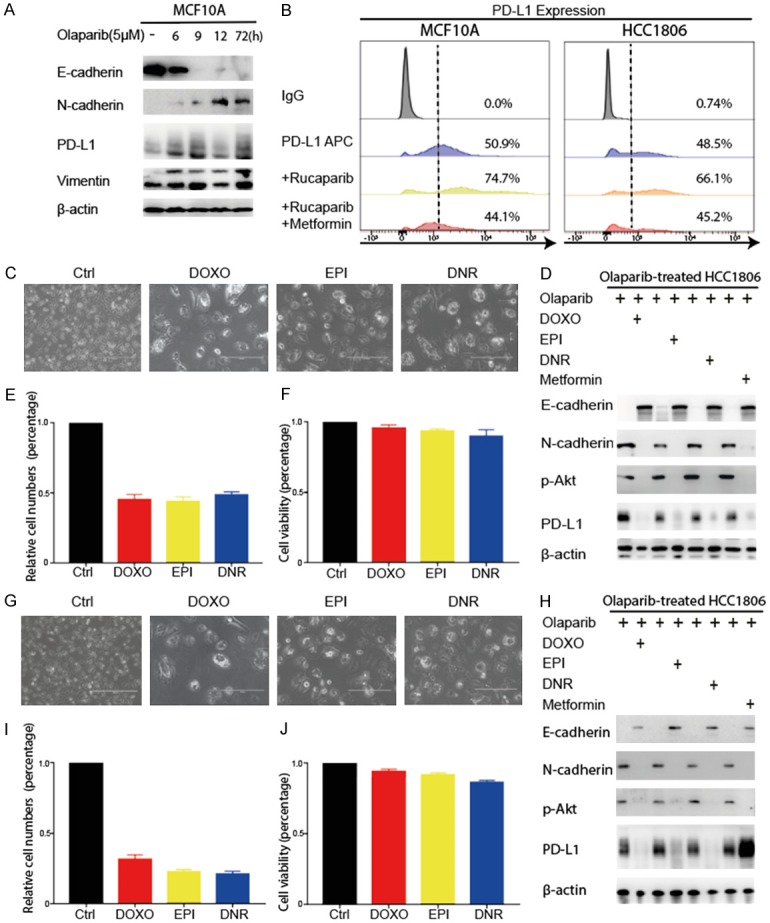
Reversal of PARP inhibitor-mediated PD-L1 upregulation metformin and chemotherapy. A. Western blot analysis of EMT markers (E-cadherin, N-cadherin, and vimentin) and PD-L1 in MCF10A cells at the indicated times upon treatment with 5 µM olaparib. B. Flow cytometry analysis of PD-L1 expression levels in treated populations of MCF10A and HCC1806 cells. The three representative groups shown are IgG, untreated, rucaparib (10 µM), and metformin (10 µM). C. Representative phase-contrast image of PARPi-induced HCC1806 cells with a high dose of doxorubicin, epirubicin and daunorubicin for 24 h. CTRL, control; DOXO, doxorubicin (0.1µM); EPI, epirubicin (1 µM); DNR, daunorubicin (0.5 µM). Scale bars: 400 µM. D. The right panel shows a Western blot analysis of EMT markers (E-cadherin, N-cadherin, and vimentin) in resting cells after treatment with the indicated agents on a high-dose, short-term (24-hour) schedule. E. A relative number of cells after treatment with the indicated agents on a high-dose, short-term schedule. F. Cell viability was analyzed by trypan blue exclusion. G. Representative phase-contrast image of PARPi-induced HCC1806 cells with a low dose of doxorubicin, epirubicin and daunorubicin for 96 h. CTRL, control; DOXO, doxorubicin (0.01 µM); EPI, epirubicin (0.01 µM); DNR, daunorubicin (0.01 µM). Scale bars: 400 µM. H. The right panel shows Western blot analysis of EMT markers (E-cadherin, N-cadherin, and vimentin) in resting cells after treatment with the indicated agents on a low-dose, long-term schedule. I. A relative number of cells after treatment with the indicated agents on a low-dose, long-term (0.01 µM, 96-hour) schedule. J. Cell viability was also analyzed by trypan blue exclusion. Error bars represent standard and variation in 3 repeated experiments. *P<0.05, **P<0.01.
The finding that the PARPi/EMT signaling axis is crucial for mediating PD-L1 induction in breast cancer cells suggests that the regulation of PD-L1 expression and increased sensitivity to immunotherapy may be accomplished by reversing EMT. However, no effective EMT inhibitors are currently available for clinical use. We previously showed that topoisomerase II (TOP2) poisons, e.g., doxorubicin, epirubicin, or daunorubicin, as well as metformin, can cause the reversal of EMT to mesenchymal-epithelial transition (MET) in PARPi-treated stable clones [29]. On this basis, we focused on TOP2 poisons and metformin that inhibit EMT. PARPi-treated stable clones treated with TOP2 poisons exhibited increased expression of the MET marker, E-cadherin [29]. Because those three chemotherapeutic agents markedly reduced cell number, we assessed the number and utilized two different treatment strategies: 1) continuous low-dose, long-term treatment, and 2) high-dose, short-term treatment to determine the viability of the treated cells. As expected, we observed non-fusiformed morphology in the PARPis, and TOP2 inhibitors treated tumor cells (Figure 5C, 5G). Doxorubicin, epirubicin, and daunorubicin induced MET in PARPi-induced stable clones (Figure 5D). Low-dose treatment got higher viability (Figure 5J) with the same efficiency in reversing EMT (Figure 5H) compared with high-dose, short-term treatment (Figure 5E, 5F). Metformin downregulated PD-L1 and reversed EMT in PARPi-treated resistant stable clones (Figures 5B, 5D, 5H, S2). Together, these results demonstrated that the TOP2 inhibitors tested and metformin are effective in promoting MET ability [30], thereby sensitizing TNBC cells to PARPis.
PARPis induce EMT and upregulate PD-L1 by activation of phospho-Akt (p-Akt)
To identify the underlying mechanism which upregulates PD-L1 and EMT, we examined EMT-related gene expression by Western blot analysis. Using a tyrosine kinase inhibitor microarray screening [27], we found p-Akt levels were elevated in PARPi-resistant cell lines. To identify the signaling pathway responsible for PARPi-mediated EMT, we examined PARPi-treated and untreated cells by Western blot analysis using the p-Akt S473 primary antibody. PARPi-treated stable cells were treated with two Akt inhibitors (MK2206, 1 µM; LY290004, 1 µM), a Mek1/2 inhibitor (U0126, 1 µM), and metformin (10 µM), and representative mesenchymal markers and p-Akt S473 were analyzed. The results showed the p-Akt S473 expression was upregulated by olaparib (Figure 6A), whereas the Akt inhibitors, Mek1/2 inhibitor, and metformin attenuated PARPi-mediated upregulation of PD-L1 (Figure 6B). These results indicated that PARPi-mediated EMT and upregulation of membrane-bound PD-L1 are regulated through p-Akt S473 activation, suggesting that PARPi activates the Akt pathway to induce EMT.
Figure 6.
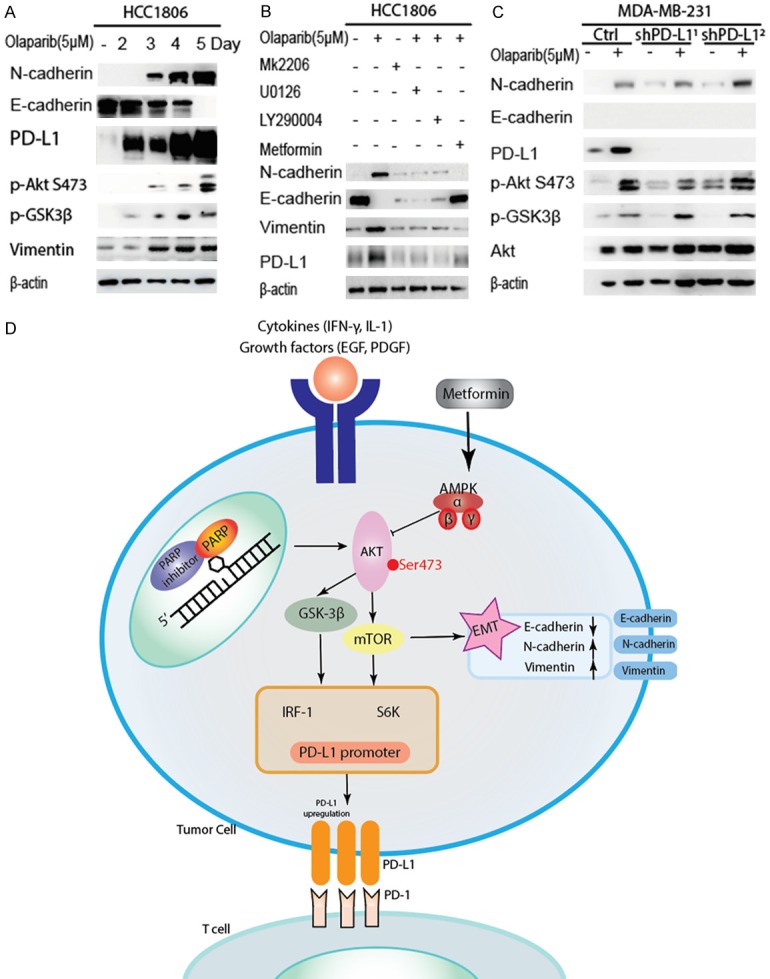
PARP inhibitors induce EMT and PD-L1 upregulation by enhancing p-Akt S473 activation. A. Western blot analysis of N-cadherin, E-cadherin, p-GSK3β, p-Akt S473, PD-L1 and β-actin in olaparib (5 μM) treated HCC1806 cells at indicated time points. B. Western blot analysis of EMT markers (E-cadherin, N-cadherin, and vimentin) and PD-L1 in resting cells after treatment with indicated agents. C. Western blot analysis of E-cadherin, N-cadherin, p-GSK3β, p-Akt, and PD-L1 in MDA-MB-231 shPD-L1 cells. D. A proposed working model. PARPi induces EMT through activation of p-Akt S473/mTOR axis pathway, and independently, transcriptional activation of PD-L1 responds to that pathway. Both can be blocked by metformin and sensitized to T cell. TNBC, triple-negative breast cancer; PARPi, PARP inhibitors.
Since PARPis both induced EMT and enhanced membrane-bound PD-L1 expression, we further investigated the relationship between EMT and membrane-bound PD-L1. PD-L1-knockdown MDA-MB-231 (by short hairpin RNA) cells were treated with olaparib followed by Western blot analysis for EMT marker expression (Figure 6C). Both the parental and PD-L1 knockdown cells exhibited similar expression levels of EMT markers upon PARPi treatment. Thus, the results suggested that EMT and PD-L1 upregulation are independent.
Discussion
PARPis have been widely utilized as single-agent targeted therapy or in combination with Cediranib Maleate in clinical trials for many cancer types, but there is increasing evidence indicating resistance to PARPi is common in the clinic [31]. Olaparib was the first U.S. FDA-approved PARPi for recurrent breast cancer [32], and was later approved PARPi for platinum-sensitive relapsed ovarian cancer [33]. Studies on resistance to PARPi revealed several mechanisms are involved, including mutations of the BRCA1 C-terminal domain [34], re-activation of BRCA1 transcription through epigenetic regulation [35], replication fork stabilization [36,37], miR-622-mediated suppression of the non-homologous end-joining pathway [38], overexpression of the HOX family [39], increases in the P-glycoprotein drug efflux transporter, and restoration of hormone receptor activity [40]. In addition, deregulation of kinases, e.g., the MET proto-oncogene, EGFR, VEGFR, AXL, have been reported to regulate PARP enzymes and further induce acquired resistance in TNBC [41-44]. Despite priors studies indicating that impairing PARP1 induced transforming growth factor-β and Smads, which are correlated with EMT in prostate cancer [45], the current study focused on the newly FDA-approved PARP1 inhibitors, olaparib and rucaparib, with results, showing no difference between these two agents in inducing EMT in breast cancer cells and mediating the adaptive resistance.
In addition, PARPi-sensitive (HCC1937, HCC70) and PARPi-resistant (MDA-MB-468, HCC1806) breast cancer cells treated with PARPi for 72 hours had significant fusiform morphologic changes. The change corresponded with significantly enhanced expression of EMT markers, such as N-cadherin and vimentin, indicating that EMT was induced by PARPis [41,46]. Another study demonstrated that the proportion of cancer stem cells was elevated as indicated by high aldehyde dehydrogenase activity after 7-day PARPi treatment in BRCA1-mutant cell lines, SUM149 and HCC 1937 [23]. The results suggested that adaptive resistance to targeted therapy may be attributed to EMT in both BRCA1-mutant and BRCA1-wild type cells. Recently, Gogola et al. [47] demonstrated that loss of poly (ADP-ribose) glycohydrolase (PARG) contributed to PARPi resistance independently of BRCA1/2 and its depletion was pre-existing in a subset of human serous ovarian cancers and TNBC. PARG loss could be an indicator of PARPi treatment, but some researchers posited that this effect was dependent on the cell line and the degree of PARG suppression [48,49]. The current findings showed that PARPi-induced EMT is a process associated with the adaptive resistance whereas PARG loss may represent a general mechanism for initial PARPi resistance.
Although some researchers have suggested that EMT regulates PD-L1, others have shown that PARPi induces cross-expression of PD-L1, and the mechanisms of this process remain unclear [27]. Co-expression of PD-L1 and p-Akt has been associated with poor prognosis in diffuse large B-cell lymphoma via the PD-1/PD-L1 axis activating the intracellular Akt/mTOR pathway [50,51]. Inconsistent with the idea, we demonstrated that PARPi-induced upregulation of PD-L1 is correlated with the activation of p-Akt S473 and that both the protein and mRNA levels of the membrane and cytosol-bound PD-L1 increased. Upregulation of PD-L1 has previously been reported to be induced by EMT through the β-catenin/STT3 axis-mediated transcriptional pathway [29]. Others have concluded that EMT epigenetically enriches PD-L1 through miR-200 in the general cell population [52]. Our results illustrated that even after PD-L1 knockout, a PARPi was able to induce EMT in breast cancer cells, which showed PARPi-induced EMT is independent of the upregulation of membranous or cytosolic PD-L1. This finding contrasts with previous studies suggesting that upregulation of PD-L1 is an independent process, which points out anti-PD-L1 monotherapy would not efficiently block the adaptive resistance to PARPi because of the ongoing EMT.
Our findings showed that different chemotherapeutic agents-Akt inhibitors, a MEK1/2 inhibitor, and metformin, induced MET by varying degrees, and are therefore potential candidates for reversing EMT caused by PARPis. Consistent with the literature, the Akt/mTOR pathway mediates many PD-L1 functions, and several inhibitors, e.g., rapamycin, everolimus, were in the clinical trials to circumvent the resistance to immunotherapy [53,54]. However, we found that metformin can efficiently reverse EMT induced by PARPi. Metformin plays a role in the activation of AMPK by inhibiting the mTOR pathway, which is associated with tumor growth, resistance to pharmacotherapy, and poor prognosis [55]. In addition, metformin increases the anti-angiogenic effects by inactivating mTOR and has been shown to inhibit cancer stem cells, which are specifically derived from EMT [56]. The anti-tumor effect of metformin has been reported to occur through its inhibition of the PI3K/Akt/mTOR or Ras/MAPK signaling pathways critical for cancer progression [55,57]. Several clinical studies have demonstrated the therapeutic potential and substantial antitumor effects of metformin in breast cancer patients [58]. A case-control study of breast cancer patients with diabetes showed that after chemotherapy, the pathologic complete response rate was significantly improved by metformin [59]. In addition, neoadjuvant metformin improved the prognosis and overall survival in patients with type 2 diabetes and other cancers [60].
When combined with immunotherapy, metformin blocks the PD-L1/PD-1 pathway and increases tumor cell immune sensitivity to T cells [28]. Here, we also showed that metformin inhibits PARPi-mediated EMT, suggesting that metformin has the potential to both attenuate upregulated PD-L1 and enhanced EMT. Collectively, the addition of metformin may be a promising therapeutic strategy to enhance the efficacy of PARPi in treating TNBC.
Acknowledgements
We sincerely appreciate Dr. Mien-Chie Hung’s great help in developing the project and supporting the experiments both technically and financially. We thank Goodoff Erica A and Bronson Sarah J at the Department of Scientific Publication of M.D. Anderson for the effort in helping us with the manuscript.
Disclosure of conflict of interest
None.
Abbreviations
- HER2
human epidermal growth factor receptor-2
- TNBC
triple-negative breast cancer
- EMT
epithelial-mesenchymal transition
- PARP
poly (ADP-ribose) polymerase
- PARPi
PARP inhibitors
- DMEM
Dulbecco modified Eagle medium
- MET
mesenchymal-epithelial transition
Supporting Information
References
- 1.Torre LA, Trabert B, DeSantis CE, Miller KD, Samimi G, Runowicz CD, Gaudet MM, Jemal A, Siegel RL. Ovarian cancer statistics, 2018. CA Cancer J Clin. 2018;68:284–296. doi: 10.3322/caac.21456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 3.Yersal O, Barutca S. Biological subtypes of breast cancer: prognostic and therapeutic implications. World J Clin Oncol. 2014;5:412–424. doi: 10.5306/wjco.v5.i3.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kumar RV, Panwar D, Amirtham U, Premalata CS, Gopal C, Narayana SM, Patil Okaly GV, Lakshmaiah KC, Krishnamurthy S. Estrogen receptor, progesterone receptor, and human epidermal growth factor receptor-2 status in breast cancer: a retrospective study of 5436 women from a regional cancer center in South India. South Asian J Cancer. 2018;7:7–10. doi: 10.4103/sajc.sajc_211_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee A, Djamgoz MBA. Triple negative breast cancer: emerging therapeutic modalities and novel combination therapies. Cancer Treat Rev. 2018;62:110–122. doi: 10.1016/j.ctrv.2017.11.003. [DOI] [PubMed] [Google Scholar]
- 6.Hubalek M, Czech T, Muller H. Biological subtypes of triple-negative breast cancer. Breast Care (Basel) 2017;12:8–14. doi: 10.1159/000455820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martignetti L, Tesson B, Almeida A, Zinovyev A, Tucker GC, Dubois T, Barillot E. Detection of miRNA regulatory effect on triple negative breast cancer transcriptome. BMC Genomics. 2015;16:S4. doi: 10.1186/1471-2164-16-S6-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kumar B, Prasad M, Bhat-Nakshatri P, Anjanappa M, Kalra M, Marino N, Storniolo AM, Rao X, Liu S, Wan J, Liu Y, Nakshatri H. Normal breast-derived epithelial cells with luminal and intrinsic subtype-enriched gene expression document interindividual differences in their differentiation cascade. Cancer Res. 2018;78:5107–5123. doi: 10.1158/0008-5472.CAN-18-0509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gross K, Wronski A, Skibinski A, Phillips S, Kuperwasser C. Cell fate decisions during breast cancer development. J Dev Biol. 2016;4:4. doi: 10.3390/jdb4010004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pang A, Carbini M, Moreira AL, Maki RG. Carcinosarcomas and related cancers: tumors caught in the act of epithelial-mesenchymal transition. J. Clin. Oncol. 2018;36:210–216. doi: 10.1200/JCO.2017.74.9523. [DOI] [PubMed] [Google Scholar]
- 11.Shibue T, Weinberg RA. EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat Rev Clin Oncol. 2017;14:611–629. doi: 10.1038/nrclinonc.2017.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brabletz T, Kalluri R, Nieto MA, Weinberg RA. EMT in cancer. Nat Rev Cancer. 2018;18:128–134. doi: 10.1038/nrc.2017.118. [DOI] [PubMed] [Google Scholar]
- 13.Pecina-Slaus N, Cicvara-Pecina T, Kafka A. Epithelial-to-mesenchymal transition: possible role in meningiomas. Front Biosci (Elite Ed) 2012;4:889–896. doi: 10.2741/E427. [DOI] [PubMed] [Google Scholar]
- 14.Ye X, Tam WL, Shibue T, Kaygusuz Y, Reinhardt F, Ng Eaton E, Weinberg RA. Distinct EMT programs control normal mammary stem cells and tumour-initiating cells. Nature. 2015;525:256–260. doi: 10.1038/nature14897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mallini P, Lennard T, Kirby J, Meeson A. Epithelial-to-mesenchymal transition: what is the impact on breast cancer stem cells and drug resistance. Cancer Treat Rev. 2014;40:341–348. doi: 10.1016/j.ctrv.2013.09.008. [DOI] [PubMed] [Google Scholar]
- 16.Hong D, Fritz AJ, Zaidi SK, van Wijnen AJ, Nickerson JA, Imbalzano AN, Lian JB, Stein JL, Stein GS. Epithelial-to-mesenchymal transition and cancer stem cells contribute to breast cancer heterogeneity. J Cell Physiol. 2018;233:9136–9144. doi: 10.1002/jcp.26847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang X, Shi Y, Huang D, Guan X. Emerging therapeutic modalities of PARP inhibitors in breast cancer. Cancer Treat Rev. 2018;68:62–68. doi: 10.1016/j.ctrv.2018.05.014. [DOI] [PubMed] [Google Scholar]
- 18.Ledermann JA. PARP inhibitors in ovarian cancer. Ann Oncol. 2016;27(Suppl 1):i40–i44. doi: 10.1093/annonc/mdw094. [DOI] [PubMed] [Google Scholar]
- 19.Evans T, Matulonis U. PARP inhibitors in ovarian cancer: evidence, experience and clinical potential. Ther Adv Med Oncol. 2017;9:253–267. doi: 10.1177/1758834016687254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sulai NH, Tan AR. Development of poly (ADP-ribose) polymerase inhibitors in the treatment of BRCA-mutated breast cancer. Clin Adv Hematol Oncol. 2018;16:491–501. [PubMed] [Google Scholar]
- 21.Freimund AE, Beach JA, Christie EL, Bowtell DDL. Mechanisms of drug resistance in high-grade serous ovarian cancer. Hematol Oncol Clin North Am. 2018;32:983–996. doi: 10.1016/j.hoc.2018.07.007. [DOI] [PubMed] [Google Scholar]
- 22.Li CW, Lim SO, Xia W, Lee HH, Chan LC, Kuo CW, Khoo KH, Chang SS, Cha JH, Kim T, Hsu JL, Wu Y, Hsu JM, Yamaguchi H, Ding Q, Wang Y, Yao J, Lee CC, Wu HJ, Sahin AA, Allison JP, Yu D, Hortobagyi GN, Hung MC. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat Commun. 2016;7:12632. doi: 10.1038/ncomms12632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu Y, Burness ML, Martin-Trevino R, Guy J, Bai S, Harouaka R, Brooks MD, Shang L, Fox A, Luther TK, Davis A, Baker TL, Colacino J, Clouthier SG, Shao ZM, Wicha MS, Liu S. RAD51 mediates resistance of cancer stem cells to PARP inhibition in triple-negative breast cancer. Clin Cancer Res. 2017;23:514–522. doi: 10.1158/1078-0432.CCR-15-1348. [DOI] [PubMed] [Google Scholar]
- 24.Paul CD, Mistriotis P, Konstantopoulos K. Cancer cell motility: lessons from migration in confined spaces. Nat Rev Cancer. 2017;17:131–140. doi: 10.1038/nrc.2016.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dongre A, Rashidian M, Reinhardt F, Bagnato A, Keckesova Z, Ploegh HL, Weinberg RA. Epithelial-to-mesenchymal transition contributes to immunosuppression in breast carcinomas. Cancer Res. 2017;77:3982–3989. doi: 10.1158/0008-5472.CAN-16-3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lou Y, Diao L, Cuentas ER, Denning WL, Chen L, Fan YH, Byers LA, Wang J, Papadimitrakopoulou VA, Behrens C, Rodriguez JC, Hwu P, Wistuba II, Heymach JV, Gibbons DL. Epithelial-mesenchymal transition is associated with a distinct tumor microenvironment including elevation of inflammatory signals and multiple immune checkpoints in lung adenocarcinoma. Clin Cancer Res. 2016;22:3630–3642. doi: 10.1158/1078-0432.CCR-15-1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jiao S, Xia W, Yamaguchi H, Wei Y, Chen MK, Hsu JM, Hsu JL, Yu WH, Du Y, Lee HH, Li CW, Chou CK, Lim SO, Chang SS, Litton J, Arun B, Hortobagyi GN, Hung MC. PARP inhibitor upregulates PD-L1 expression and enhances cancer-associated immunosuppression. Clin Cancer Res. 2017;23:3711–3720. doi: 10.1158/1078-0432.CCR-16-3215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cha JH, Yang WH, Xia W, Wei Y, Chan LC, Lim SO, Li CW, Kim T, Chang SS, Lee HH, Hsu JL, Wang HL, Kuo CW, Chang WC, Hadad S, Purdie CA, McCoy AM, Cai S, Tu Y, Litton JK, Mittendorf EA, Moulder SL, Symmans WF, Thompson AM, Piwnica-Worms H, Chen CH, Khoo KH, Hung MC. Metformin promotes antitumor immunity via endoplasmic-reticulum-associated degradation of PD-L1. Mol Cell. 2018;71:606–620. e7. doi: 10.1016/j.molcel.2018.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hsu JM, Xia W, Hsu YH, Chan LC, Yu WH, Cha JH, Chen CT, Liao HW, Kuo CW, Khoo KH, Hsu JL, Li CW, Lim SO, Chang SS, Chen YC, Ren GX, Hung MC. STT3-dependent PD-L1 accumulation on cancer stem cells promotes immune evasion. Nat Commun. 2018;9:1908. doi: 10.1038/s41467-018-04313-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aguirre-Alvarado C, Segura-Cabrera A, Velazquez-Quesada I, Hernandez-Esquivel MA, Garcia-Perez CA, Guerrero-Rodriguez SL, Ruiz-Moreno AJ, Rodriguez-Moreno A, Perez-Tapia SM, Velasco-Velazquez MA. Virtual screening-driven repositioning of etoposide as CD44 antagonist in breast cancer cells. Oncotarget. 2016;7:23772–23784. doi: 10.18632/oncotarget.8180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim Y, Kim A, Sharip A, Sharip A, Jiang J, Yang Q, Xie Y. Reverse the resistance to PARP inhibitors. Int J Biol Sci. 2017;13:198–208. doi: 10.7150/ijbs.17240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Turk AA, Wisinski KB. PARP inhibitors in breast cancer: bringing synthetic lethality to the bedside. Cancer. 2018;124:2498–2506. doi: 10.1002/cncr.31307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, Scott C, Meier W, Shapira-Frommer R, Safra T, Matei D, Macpherson E, Watkins C, Carmichael J, Matulonis U. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N Engl J Med. 2012;366:1382–1392. doi: 10.1056/NEJMoa1105535. [DOI] [PubMed] [Google Scholar]
- 34.Johnson N, Johnson SF, Yao W, Li YC, Choi YE, Bernhardy AJ, Wang Y, Capelletti M, Sarosiek KA, Moreau LA, Chowdhury D, Wickramanayake A, Harrell MI, Liu JF, D’Andrea AD, Miron A, Swisher EM, Shapiro GI. Stabilization of mutant BRCA1 protein confers PARP inhibitor and platinum resistance. Proc Natl Acad Sci U S A. 2013;110:17041–17046. doi: 10.1073/pnas.1305170110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang Y, Bernhardy AJ, Cruz C, Krais JJ, Nacson J, Nicolas E, Peri S, van der Gulden H, van der Heijden I, O’Brien SW, Zhang Y, Harrell MI, Johnson SF, Candido Dos Reis FJ, Pharoah PD, Karlan B, Gourley C, Lambrechts D, Chenevix-Trench G, Olsson H, Benitez JJ, Greene MH, Gore M, Nussbaum R, Sadetzki S, Gayther SA, Kjaer SK kConFab Investigators. D’Andrea AD, Shapiro GI, Wiest DL, Connolly DC, Daly MB, Swisher EM, Bouwman P, Jonkers J, Balmaña J, Serra V, Johnson N. The BRCA1-Delta11q alternative splice isoform bypasses germline mutations and promotes therapeutic resistance to PARP inhibition and cisplatin. Cancer Res. 2016;76:2778–2790. doi: 10.1158/0008-5472.CAN-16-0186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ray Chaudhuri A, Callen E, Ding X, Gogola E, Duarte AA, Lee JE, Wong N, Lafarga V, Calvo JA, Panzarino NJ, John S, Day A, Crespo AV, Shen B, Starnes LM, de Ruiter JR, Daniel JA, Konstantinopoulos PA, Cortez D, Cantor SB, Fernandez-Capetillo O, Ge K, Jonkers J, Rottenberg S, Sharan SK, Nussenzweig A. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature. 2016;535:382–387. doi: 10.1038/nature18325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chaudhuri AR, Callen E, Ding X, Gogola E, Duarte AA, Lee JE, Wong N, Lafarga V, Calvo JA, Panzarino NJ, John S, Day A, Crespo AV, Shen B, Starnes LM, de Ruiter JR, Daniel JA, Konstantinopoulos PA, Cortez D, Cantor SB, Fernandez-Capetillo O, Ge K, Jonkers J, Rottenberg S, Sharan SK, Nussenzweig A. Erratum: replication fork stability confers chemoresistance in BRCA-deficient cells. Nature. 2016;539:456. doi: 10.1038/nature19826. [DOI] [PubMed] [Google Scholar]
- 38.Choi YE, Meghani K, Brault ME, Leclerc L, He YJ, Day TA, Elias KM, Drapkin R, Weinstock DM, Dao F, Shih KK, Matulonis U, Levine DA, Konstantinopoulos PA, Chowdhury D. Platinum and PARP inhibitor resistance due to overexpression of microRNA-622 in BRCA1-mutant ovarian cancer. Cell Rep. 2016;14:429–439. doi: 10.1016/j.celrep.2015.12.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Esposito MT, Zhao L, Fung TK, Rane JK, Wilson A, Martin N, Gil J, Leung AY, Ashworth A, So CW. Synthetic lethal targeting of oncogenic transcription factors in acute leukemia by PARP inhibitors. Nat Med. 2015;21:1481–1490. doi: 10.1038/nm.3993. [DOI] [PubMed] [Google Scholar]
- 40.Durmus S, Sparidans RW, van Esch A, Wagenaar E, Beijnen JH, Schinkel AH. Breast cancer resistance protein (BCRP/ABCG2) and P-glycoprotein (P-GP/ABCB1) restrict oral availability and brain accumulation of the PARP inhibitor rucaparib (AG-014699) Pharm Res. 2015;32:37–46. doi: 10.1007/s11095-014-1442-z. [DOI] [PubMed] [Google Scholar]
- 41.Du Y, Yamaguchi H, Wei Y, Hsu JL, Wang HL, Hsu YH, Lin WC, Yu WH, Leonard PG, Lee GRt, Chen MK, Nakai K, Hsu MC, Chen CT, Sun Y, Wu Y, Chang WC, Huang WC, Liu CL, Chang YC, Chen CH, Park M, Jones P, Hortobagyi GN, Hung MC. Blocking c-Met-mediated PARP1 phosphorylation enhances anti-tumor effects of PARP inhibitors. Nat Med. 2016;22:194–201. doi: 10.1038/nm.4032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Balaji K, Vijayaraghavan S, Diao L, Tong P, Fan Y, Carey JP, Bui TN, Warner S, Heymach JV, Hunt KK, Wang J, Byers LA, Keyomarsi K. AXL inhibition suppresses the DNA damage response and sensitizes cells to PARP inhibition in multiple cancers. Mol Cancer Res. 2017;15:45–58. doi: 10.1158/1541-7786.MCR-16-0157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ivy SP, Liu JF, Lee JM, Matulonis UA, Kohn EC. Cediranib, a pan-VEGFR inhibitor, and olaparib, a PARP inhibitor, in combination therapy for high grade serous ovarian cancer. Expert Opin Investig Drugs. 2016;25:597–611. doi: 10.1517/13543784.2016.1156857. [DOI] [PubMed] [Google Scholar]
- 44.Pfaffle HN, Wang M, Gheorghiu L, Ferraiolo N, Greninger P, Borgmann K, Settleman J, Benes CH, Sequist LV, Zou L, Willers H. EGFR-activating mutations correlate with a Fanconi anemia-like cellular phenotype that includes PARP inhibitor sensitivity. Cancer Res. 2013;73:6254–6263. doi: 10.1158/0008-5472.CAN-13-0044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pu H, Horbinski C, Hensley PJ, Matuszak EA, Atkinson T, Kyprianou N. PARP-1 regulates epithelial-mesenchymal transition (EMT) in prostate tumorigenesis. Carcinogenesis. 2014;35:2592–2601. doi: 10.1093/carcin/bgu183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Aiello NM, Brabletz T, Kang Y, Nieto MA, Weinberg RA, Stanger BZ. Upholding a role for EMT in pancreatic cancer metastasis. Nature. 2017;547:E7–E8. doi: 10.1038/nature22963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gogola E, Duarte AA, de Ruiter JR, Wiegant WW, Schmid JA, de Bruijn R, James DI, Guerrero Llobet S, Vis DJ, Annunziato S, van den Broek B, Barazas M, Kersbergen A, van de Ven M, Tarsounas M, Ogilvie DJ, van Vugt M, Wessels LFA, Bartkova J, Gromova I, Andujar-Sanchez M, Bartek J, Lopes M, van Attikum H, Borst P, Jonkers J, Rottenberg S. Selective loss of PARG restores PARylation and counteracts PARP inhibitor-mediated synthetic lethality. Cancer Cell. 2018;33:1078–1093. e1012. doi: 10.1016/j.ccell.2018.05.008. [DOI] [PubMed] [Google Scholar]
- 48.Gravells P, Grant E, Smith KM, James DI, Bryant HE. Specific killing of DNA damage-response deficient cells with inhibitors of poly(ADP-ribose) glycohydrolase. DNA Repair (Amst) 2017;52:81–91. doi: 10.1016/j.dnarep.2017.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fathers C, Drayton RM, Solovieva S, Bryant HE. Inhibition of poly (ADP-ribose) glycohydrolase (PARG) specifically kills BRCA2-deficient tumor cells. Cell Cycle. 2012;11:990–997. doi: 10.4161/cc.11.5.19482. [DOI] [PubMed] [Google Scholar]
- 50.Morrison C, Pabla S, Conroy JM, Nesline MK, Glenn ST, Dressman D, Papanicolau-Sengos A, Burgher B, Andreas J, Giamo V, Qin M, Wang Y, Lenzo FL, Omilian A, Bshara W, Zibelman M, Ghatalia P, Dragnev K, Shirai K, Madden KG, Tafe LJ, Shah N, Kasuganti D, de la Cruz-Merino L, Araujo I, Saenger Y, Bogardus M, Villalona-Calero M, Diaz Z, Day R, Eisenberg M, Anderson SM, Puzanov I, Galluzzi L, Gardner M, Ernstoff MS. Predicting response to checkpoint inhibitors in melanoma beyond PD-L1 and mutational burden. J Immunother Cancer. 2018;6:32. doi: 10.1186/s40425-018-0344-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dong L, Lv H, Li W, Song Z, Li L, Zhou S, Qiu L, Qian Z, Liu X, Feng L, Meng B, Fu K, Wang X, Pan-Hammarstrom Q, Wang P, Wang X, Zhang H. Co-expression of PD-L1 and p-AKT is associated with poor prognosis in diffuse large B-cell lymphoma via PD-1/PD-L1 axis activating intracellular AKT/mTOR pathway in tumor cells. Oncotarget. 2016;7:33350–33362. doi: 10.18632/oncotarget.9061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen L, Gibbons DL, Goswami S, Cortez MA, Ahn YH, Byers LA, Zhang X, Yi X, Dwyer D, Lin W, Diao L, Wang J, Roybal J, Patel M, Ungewiss C, Peng D, Antonia S, Mediavilla-Varela M, Robertson G, Suraokar M, Welsh JW, Erez B, Wistuba II, Chen L, Peng D, Wang S, Ullrich SE, Heymach JV, Kurie JM, Qin FX. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat Commun. 2014;5:5241. doi: 10.1038/ncomms6241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xie J, Wang X, Proud CG. mTOR inhibitors in cancer therapy. F1000Res. 2016:5. doi: 10.12688/f1000research.9207.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hirayama Y, Gi M, Yamano S, Tachibana H, Okuno T, Tamada S, Nakatani T, Wanibuchi H. Anti-PD-L1 treatment enhances antitumor effect of everolimus in a mouse model of renal cell carcinoma. Cancer Sci. 2016;107:1736–1744. doi: 10.1111/cas.13099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rena G, Hardie DG, Pearson ER. The mechanisms of action of metformin. Diabetologia. 2017;60:1577–1585. doi: 10.1007/s00125-017-4342-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang Y, Wu Z, Hu L. Epithelial-mesenchymal transition phenotype, metformin, and survival for colorectal cancer patients with diabetes mellitus II. Gastroenterol Res Pract. 2017;2017:2520581. doi: 10.1155/2017/2520581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lei Y, Yi Y, Liu Y, Liu X, Keller ET, Qian CN, Zhang J, Lu Y. Metformin targets multiple signaling pathways in cancer. Chin J Cancer. 2017;36:17. doi: 10.1186/s40880-017-0184-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Camacho L, Dasgupta A, Jiralerspong S. Metformin in breast cancer - an evolving mystery. Breast Cancer Res. 2015;17:88. doi: 10.1186/s13058-015-0598-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hajjar J, Habra MA, Naing A. Metformin: an old drug with new potential. Expert Opin Investig Drugs. 2013;22:1511–1517. doi: 10.1517/13543784.2013.833604. [DOI] [PubMed] [Google Scholar]
- 60.Morales DR, Morris AD. Metformin in cancer treatment and prevention. Annu Rev Med. 2015;66:17–29. doi: 10.1146/annurev-med-062613-093128. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.