

Abstract
Ferroptosis is emerging as a new form of regulated cell death driven by oxidative injury promoting lipid peroxidation in an iron-dependent manner. High mobility group box 1 (HMGB1) plays an important role in leukemia pathogenesis and chemotherapy resistance. The mechanisms of ferroptosis in tumor pathogenesis and treatment have been a recent research focus but the role of HMGB1 in regulating ferroptosis especially in leukemia still remains largely unknown. Here, we shown that HMGB1 is a critical regulator of eratin-induced ferroptosis in HL-60 cell line expressing NRASQ61L (HL-60/NRASQ61L). Erastin enhanced ROS levels, thereby promoting cytosolic translocation of HMGB1 and enhancing cell death. Knockdown of HMGB1 decreased erastin-induced ROS generation and cell death in an iron-mediated lysosomal pathway in HL-60/NRASQ61L cells. Knockdown of HMGB1 or rat sarcoma (RAS), or pharmacological inhibition of JNK and p38 decreased TfR1 levels in HL-60/NRASQ61L cells. Importantly, these data were further supported by our in vivo experiment, in which xenografts formed by HMGB1 knockdown HL-60/NRASQ61L cells had lower PTGS2 and TfR1 expression than that in control mice. Taken together, these results suggest that HMGB1 is a novel regulator of ferroptosis via the RAS-JNK/p38 pathway and a potential drug target for therapeutic interventions in leukemia.
Keywords: HMGB1, ferroptosis, MAPK, transferrin receptor 1, acute myeloid leukemia
Introduction
Chemoresistance has become a major obstacle to the successful treatment of leukemia. Although various mechanisms of drug resistance have been proposed to date, an exact mechanism has still not been established [1]. Programmed cell death (PCD) is regulated by an intricate mechanism that is closely related to anticancer therapy and drug resistance [2]. A novel type of PCD called ferroptosis has been recognized to be involved in cancer cell death [3]. Ferroptosis is morphologically, biochemically and genetically distinct from apoptosis, autophagy, and various forms of necrosis [3,4]. It is characterized by the iron-dependent accumulation of reactive oxygen species (ROS) and lipid peroxidation. It can be suppressed by iron chelators, lipophilic antioxidants, and inhibitors of lipid peroxidation [3]. We previously found that erastin-induced ferroptosis enhanced sensitivity of acute myeloid leukemia (AML) cells (HL-60/NRASQ61L) to chemotherapeutic agents [5]. This previous report indicated that inducing ferroptosis may be a new therapeutic anticancer strategy for treating tumors. However, the exact molecular mechanism by which ferroptosis induces cancer cells death, specifically in leukemia cells, has not been clearly defined.
High-mobility group box 1 (HMGB1) is a transcription factor that is involved in chromatin remodelling and DNA recombination and repair processes [6]. HMGB1 occurs inside the cytosol is translocated to be expressed on cell surface membranes, or is diffused into extracellular spaces by multiple cellular stressors (e.g. protein aggregates, radiation, chemotherapy and intracellular pathogens) [7,8]. During tumor development and in cancer therapy, HMGB1 has been reported to play paradoxical roles in promoting both cell survival and death by regulating multiple signaling pathways, including those associated with inflammation, immunity, genome stability, proliferation, metastasis, metabolism, apoptosis, and autophagy [7,9]. Our early studies have shown that HMGB1 plays an important role in leukemia pathogenesis and chemotherapy resistance [10-14]. For example, HMGB1 is a negative regulator of apoptosis in leukemia cells through regulation of Bcl-2 expression and caspase-3 activity [10,12]. As a positive regulator of autophagy, intracellular HMGB1 interacts with Beclin-1 in leukemia cells leading to autophagosome formation, which is an alternative mechanism of resistance to leukemia therapies [14]. While HMGB1 has been found to play an important role in apoptosis and autophagy in leukemia cells, its role in the regulation of ferroptosis has not been clearly defined.
In this study, we demonstrate that HMGB1 is a critical regulator of ferroptosis, as HMGB1 translocation requires a ROS-dependent signal. Knockdown of HMGB1 decreased erastin-induced ROS generation and cell death in an iron-mediated lysosomal pathway. The data suggest that HMGB1 is required for erastin-induced ferroptosis, the regulation of transferrin receptor protein 1 (TfR1) mRNA levels, and c-Jun N-terminal kinase (JNK) and p38 phosphorylation in the rat sarcoma (RAS)-JNK/p38 pathway. Hence, HMGB1 could be a potential drug target for therapeutic interventions in leukemia.
Materials and methods
Cell culture
HL-60/NRASQ61L cells (non-APL AML, NRAS_Q61L, #CCL-240) was obtained from the American Type Culture Collection (Manassas, VA, USA). NB4 (acute promyelocytic leukemia (APL), RAS wild type), HL-60 (non-APL AML, RAS wild type), KG-1 (myeloid leukemia cell line, RAS wild type), U937 (myeloid leukemia cell line, RAS wild type) and THP-1 (acute monocytic leukemia, RAS wild type) were obtained from Xiangya School of Medicine Type Culture Collection (Changsha, China). Cells were cultured with RPMI 1640 medium (Invitrogen, Carlsbad, CA, USA) with the addition of 10% fetal bovine serum (FBS; Gibco, Thermo Fisher Scientific Inc., Waltham, MA) and 1% antibiotics (100 U/mL penicillin G and 100 mg/mL streptomycin) at 37°C with 5% CO2 in a cell incubator (Thermo Fisher Scientific Inc. USA).
Lentivirus, plasmid transfection, and RNA interference
Following the procedures outlined in our previous study [15], HMGB1 small hairpin RNA (shRNA) lentiviral knockdown (GeneCopoeia, Guangzhou, China) or shRNA non-silencing control were packaged with HIV-based packaging mix (GeneCopoeia) for infecting HL-60/NRASQ61L cells to establish cells that constitutively repress HMGB1. Stable clones were selected using puromycin. The HMGB1 shRNA oligonucleotide sequences were as follows: HMGB1 shRNA1: 5’-CCGGGCAGATGACAAGCAGCCTTATCTCGAGATAAGGCTGCTTGTCATCTGCTTTTT-3’; HMGB1 shRNA2: 5’-CCGGGATGCAGCTTATACGAAATAACTCGAGTTATTTCGTATAAGCTGCATCTTTTT-3’; HMGB1 shRNA3: 5’-CCGGCCGTTATGAAAGAGAAATGAACTCGAGTTCATTTCTCTTTCATAACGGTTTTT-3’. Non-silencing shRNA (control shRNA) were used as mock-transfected controls (target sequence: TTCTCCGAACGTGTCACGT). Then HMGB1 expression was verified by RT-PCR and Western blot (Figure S1). Two of HMGB1 shRNA (HMGB1 shRNA1 and HMGB1 shRNA2) that proved the most effective for knockdown of gene expression were selected. The pEGFP-N1-HMGB1 expression vector was a gift from Rui Kang (University of Pittsburgh, Pittsburgh, PA) and was used in our previous study [16]. HMGB1 expression vector transfection was performed using Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. Human SOD1-siRNA (Target sequence: GGUGGAAAUGAAGAAAGUAC), human NRAS-siRNA (Target sequence: GUGUGAUUUGCCAACAAGG) and human TfR1-siRNA (Target sequence: CTGACTGCTCTCAGCTC) (GeneCopoeia, Guangzhou, China) were transfected into cells using Lipofectamine RNAiMAX reagent (for siRNA) according to the manufacturer’s instructions (Invitrogen, Carlsbad, CA, USA). After RNA interference (RNAi) incubation for 48 h, the medium over the cells was changed before subsequent treatments.
Reagents and cell viability assay
Z-VAD-FMK (#V116), deferoxamine (DFO; #D9533), 3-methyladenine (3-MA; #M9281), necrostatin-1 (NEC-1; #N9037), FeCl3 (#157740), SP600125 (#S5567), SB202190 (#S8307), N-acetylcysteine (NAC; #A0737), Neopterin (#N3386), Diphenyleneiodonium chloride (DPI; #D2926), ethyl pyruvate (EP; #W245712), H2O2 (#323381) and cytarabine (Ara-C; #C1768) were obtained from Sigma-Aldrich (St. Louis, MO, USA). Ferrostatin-1 (Fer-1; #S7243) and erastin (#S7242) were obtained from Selleck Chemicals (Houston, TX, USA).
Cell viability was evaluated using a cell counting kit-8 (CCK-8; #CK04, Dojindo Molecular Technologies, Tokyo, Japan) according to the manufacturer’s instructions. Briefly, cells (5 × 104/well/100 μL) were seeded into a 96-well plate and treated with different drugs at various concentrations for the indicated times. After the addition of 10 μL of CCK-8 solution to each well, cells were incubated at 37°C for another 3 h and the absorbance was determined at 450 nm using a microplate reader. All experiments were performed in triplicate.
Measurements of HMGB1 release
HL-60/NRASQ61L cells were pretreated with Fer-1 (1 μM) and then erastin (5 μM) for 24, 48 and 72 h. Supernatants from different groups of HL-60/NRASQ61L cells were collected and used to determine the concentration of HMGB1. The release of HMGB1 into cell culture supernatants was evaluated with enzyme-linked immunosorbent assay (ELISA) kits from Shino-Test Corporation (Sagamihara-shi, Kanagawa, Japan) according to the manufacturer’s instructions. All experiments were performed in triplicate.
Lactate dehydrogenase release assay
The HMGB1 shRNA and control shRNA vector transfected HL-60/NRASQ61L cells were treated with dimethyl sulfoxide (DMSO), erastin (5 μM) and H2O2 (50 mM) for 48 h. Supernatants were collected and used to determine lactate dehydrogenase (LDH) concentrations. LDH release was assessed using an LDH assay kit (#ab65393, Abcam, Cambridge, MA) according to the manufacturer’s instructions. All experiments were performed in triplicate.
Determination of ROS generation
The intracellular alterations of ROS were determined by measuring the oxidative conversion of cell-permeable 2’,7’-dichlorofluorescein diacetate (DCFH-DA) into fluorescent dichlorofluorescein (DCF) on a fluorospectrophotometer (F4500, Hitachi, Tokyo, Japan) according to the methods described in our previous study [16]. In brief, cells exposed to different treatments were collected, rinsed with D-Hank’s buffer, and incubated with DCFH-DA at 37°C for 20 min. Then, the DCF fluorescence of 20,000 cells was detected using a fluorospectrophotometer at an excitation wavelength of 488 nm and an emission wavelength of 535 nm. The incremental production of ROS was expressed as a percentage of the control.
Malondialdehyde assay
The relative malondialdehyde (MDA) concentration in cell lysates was assessed using a lipid peroxidation assay kit (#ab118970, Abcam) according to the manufacturer’s instructions. Briefly, the MDA in the sample reacted with thiobarbituric acid (TBA) to generate an MDA-TBA adduct. The MDA-TBA adduct was quantified fluorometrically (at a 532 nm excitation wavelength and a 553 nm emission wavelength).
Flow cytometry for mitochondrial membrane permeabilization and mitochondrial membrane potential detection
The mitochondrial membrane permeabilization (MMP) and mitochondrial membrane potential (Δψm) of the treated cells were measured using MitoTracker Deep Red FM (M22462, Invitrogen, Carlsbad, CA, USA) and DIOC6 staining (D273, Invitrogen, USA). In brief, the culture medium was aspirated after treatment, and the cells were collected and incubated with MitoTracker Deep Red FM (100 nM in phosphate-buffered saline [PBS]), Dioc6 (1 μM in PBS), LysoTracker (50 nM) for 15 min at 37°C. The fluorescence emission was analyzed by flow cytometry using a FACS Vantage system (Becton Dickinson Inc., Franklin Lakes NJ).
Reverse transcription polymerase chain reaction
Following the procedures outlined in our previous study [13]. Total RNA was isolated from BMMCs using TRIzol (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. RNA concentration and purity were measured with a spectrophotometer at A260 and A260/280, respectively. RNA was reverse-transcribed into cDNA using PrimeScriptTM RT Master Mix (Takara Biomedical Technology Co., Ltd, Beijing, China) according to the manufacturer’s instructions. The sequences of primers used were as follows: β-actin forward: 5’-TCCTTCCTGGGCATGGAGTC-3’, β-actin reverse: 5’-GTAACGCAACTAAGTCATAGTC-3’. HMGB1 forward, 5’-TTTCAAACAAAGATGCCACA-3’, and HMGB1 reverse, 5’-GTTCCCTAAACTCCTAAGCAGATA-3’. β-actin was used as an internal control to evaluate the relative expressions of HMGB1. The conditions for polymerase chain reaction (PCR) to HMGB1 were: denaturation at 94°C for 2 min, followed by 30 cycles of 94°C for 30 s, 56°C for 30 s (β-actin: 50°C for 30 s), 72°C for 30 s, and then by a 5 min elongation at 72°C. PCR products were analyzed with 1.0% agarose gel electrophoresis, ethidium bromide (EB) stained, photographed and scanned using Band Leader software for gray-scale semiquantitative analysis.
Quantitative polymerase chain reaction
TRIzol (Invitrogen, USA) was used to isolate the total RNA of tissues, and cDNA was synthesized with PrimeScriptTM RT Master Mix (Takara Biomedical Technology Co., Ltd, Beijing, China). A 7900 Real-Time PCR System (Applied Biosystems, Foster City, CA) with AceQ qPCR SYBR Green Master Mix (High ROX Premixed Vazyme Biotech Co., Ltd, Nanjing, China) was used to perform quantitative PCR (qPCR). Relative mRNA expression was standardized using the housekeeping gene β-actin (forward primer 5’-ATTGCCGACAGGATGCAGAA-3’, and reverse primer 5’-ACATCTGCTGGAAGGTGGACAG-3’). The following human primers were used in this study: PTGS2 forward (5’-CGGTGAAACTCTGGCTAGACAG-3’) and reverse (5’-GCAAACCGTAGATGCTCAGGGA-3’); TfR1 forward (5’-ACCCATTCGTGGTGATCAAT-3’) and reverse (5’-CGTTTCCAACTGCCCTATGA-3’). The procedures were performed in triplicate.
Antibodies and Western blot
The following commercially available antibodies were used. Mouse anti-HMGB1 (#H00003146-M08) was obtained from Novus (Littleton, CO). Rabbit anti-glutathione peroxidase 4 (GPX4; #ab125066), rabbit anti-SOD1 (ab13498), goat anti-NRAS (ab77392), rabbit anti-tubulin (ab18251), mouse anti-fibrillarin (ab4575) and mouse anti-actin (ab3280) were obtained from Abcam (Cambridge, MA, USA). Rabbit anti-CD71/TfR1 (#13113), rabbit anti-JNK (#9252), rabbit anti-P-JNK (#9251), rabbit anti-p38 (#9212), rabbit anti-P-p38 (#9215), and anti-rabbit/mouse IgG, horseradish peroxidase (HRP)-linked antibody (#7074/#7076) were obtained from Cell Signaling Technology (Danvers, MA).
Cells were washed with PBS, collected, resuspended in lysis buffer (Beyotime, Beijing, China), and maintained on ice for 15 min. Cell extracts were cleared by microcentrifugation at 14,000 × g for 30 min at 4°C. The whole cell lysates were separated by 8%, 10%, or 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and electrophoretically transferred onto polyvinylidene difluoride (PVDF) blotting membranes (Beyotime, Beijing, China). The membranes were blocked with 5% non-fat dry milk in Tris-buffered saline containing Tween 20 (TBST; 50 mM Tris [pH 7.5], 100 mM NaCl, 0.15% Tween-20), incubated with diluted primary antibodies for 12 h at 4°C, and washed three times with TBST for 10 min. Then, the membranes were incubated for 12 h at 4°C with different secondary antibodies, and hybridization was detected using enhanced chemiluminescence reagents (Pierce, Waltham, MA) after three rinses with TBST for 10 min. Membranes were exposed to X-ray film and the expression levels of the targeted proteins were quantified. The BandScan 5.0 system was used to quantify and analyze each specific western blot band.
Immunofluorescence analysis
Cells were fixed in 4% formaldehyde for 30 min at room temperature before cell permeabilization with 0.1% Triton X-100 (4°C, 10 min). Cells were saturated with PBS containing 2% bovine serum albumin for 1 h at room temperature and processed for immunofluorescence with anti-HMGB1 antibody followed by Alexa Fluor 488-conjugated immunoglobulin and Hoechst 33258 (Invitrogen, Carlsbad, CA, USA). Between all incubation steps, cells were washed three times for 3 min with PBS containing 0.2% bovine serum albumin. Fluorescence signals were analyzed using an Olympus Fluoview 1000 confocal microscope (Olympus Corp, Tokyo, Japan).
Immunohistochemistry
The tumors were fixed in 10% formalin for 24 h followed by dehydration and paraffin embedding. Then, the paraffin-embedded specimens were sliced into 5-μm thick sections by microtome (Leica, Wetzlar, Germany) and placed on glass slides. The expression of CD71/TfR1 was determined by immunostaining. After deparaffinization and rehydrating, sections were pressure cooked in antigen retrieval buffer (0.01 M citrate buffer, pH 6.0) for 2 min to unmask antigens. Then, they were incubated with murine anti-rat CD71 monoclonal antibody (1:200, Cat. No. 113802, Biolegend, San Diego, CA) at 4°C overnight, followed by biotinylated anti-mouse IgG secondary antibody (ZSGB-bio, Beijing, China) for 1 h at 37°C and streptavidin-HRP (ZSGB-bio) for 30 min at 37°C. Further, the HRP substrate DAB (3, 3-diaminobenzidine; ZSGB-bio, China) was used to develop and visualize the immunostaining, whereas the cell nuclei were counter-stained with hematoxylin. Images were acquired with the Olympus BX51 microscope to assess the proportion of positive stained cells.
Electron microscopy
Cells were collected and fixed in 2.5% glutaraldehyde for at least 3 h. Then the cells were treated with 2% paraformaldehyde at room temperature for 60 min and 0.1% glutaraldehyde in 0.1 M sodium cacodylate for 2 h, followed by post-fixing with 1% OsO4 for 1.5 h, After a second washing, cells were dehydrated with graded acetone and finally embedded in Quetol 812. Ultrathin sections were observed under an H7500 electron microscope (Hitachi, Tokyo, Japan).
Monitoring cellular iron level using flow cytometry
Cells were collected and washed with PBS twice and stained with 5 μM of phen green SK (PGSK), diacetate (cat. #P14313, Molecular Probes, Eugene, OR) in PBS by incubating plates for 15 min in a culture incubator. Cells were harvested in 2 mL of PBS and centrifuged at 1000 rpm for 5 min. The cell pellets were resuspended in 1 mL of PBS, the cell suspensions were transferred to disposable FACS tubes, and the fluorescence profiles of the samples were assessed using a FACSCalibur system (BD Biosciences, San Jose, CA).
Xenograft assay in NOD/SCID mice
Seven- to eight-week male NOD/SCID (non-obese diabetic/severe combined immunodeficient) mice that weighed about 20 g were purchased from Xiangya Medical College Animal Laboratory (Changsha, China). All experiments were approved by the Animal Ethics Committee of Xiangya Medical College, Central South University, China. The animals were raised in pathogen-free conditions with a 12-hour light-dark cycle with water and food provided ad libitum. For each experiment, 16 mice were randomly divided into the following four groups: (1) control shRNA model receiving DMSO (vehicle); (2) control shRNA model receiving erastin ; (3) HMGB1 shRNA model receiving DMSO (vehicle) and (4) HMGB1 shRNA model receiving erastin. Indicated HL-60/NRASQ61L cells were subcutaneously injected into the dorsal flanks right of the midline in NOD/SCID mice (weight, approximately 20 g). At day seven, mice were intraperitoneal injected with erastin (20 mg/kg i.v., three times a week) for two weeks. Erastin was dissolved in the vehicle (2% DMSO and 98% PBS) and prepared by Ultrasonic Cleaner (Fisher Scientific, Hampton, NH). A final volume of 300 μL of erastin was applied via intraperitoneal injection. Tumors were measured twice a week. The volumes were calculated using the following formula: volume (mm3) = length × width2 × π/6.
Statistical analysis
All statistical analyses were performed using SPSS 19.0 software (IBM Corp, Armonk, NY, USA). Results are expressed as the mean ± standard deviation (SD). Group means were compared using Student’s t-test for independent data. All P-values are two-tailed, and P < 0.05 was considered to indicate statistical significance.
Results
Erastin promotes ROS-dependent extranuclear HMGB1 translocation
Our former study have demonstrated that erastin selectively induced growth inhibition in HL-60/NRASQ61L cells, but not in Jurkat (RAS wild type), THP-1 (NRAS_G12D), NB4 (KRAS_A18D) and K562 (RAS wild type) cells with an RAS-independent manner [5]. To further determine whether erastin induces growth inhibition in the other common leukemia cell line, we analyzed the sensitivity of HL-60, U937, KG-1, NB4 and THP-1 cells (RAS wild type) against erastin. Contrary to the sensitivity of HL-60/NRASQ61L cells, erastin did not induce the growth inhibition in HL-60 (RAS wild type) (Figure 1A), which is consistent with the results of Yagoda et al [17]. Meanwhile, erastin also did not induce the growth inhibition in U937, KG-1, NB4 and THP-1 cells (RAS wild type) (Figure 1A). Similarly, erastin dose-dependently increased MDA levels in HL-60/NRASQ61L cells, but not in HL-60, U937, KG-1, NB4 and THP-1 cells (RAS wild type) (Figure 1A), suggesting that erastin selectively induces growth inhibition and MDA increase in HL-60/NRASQ61L cells.
Figure 1.
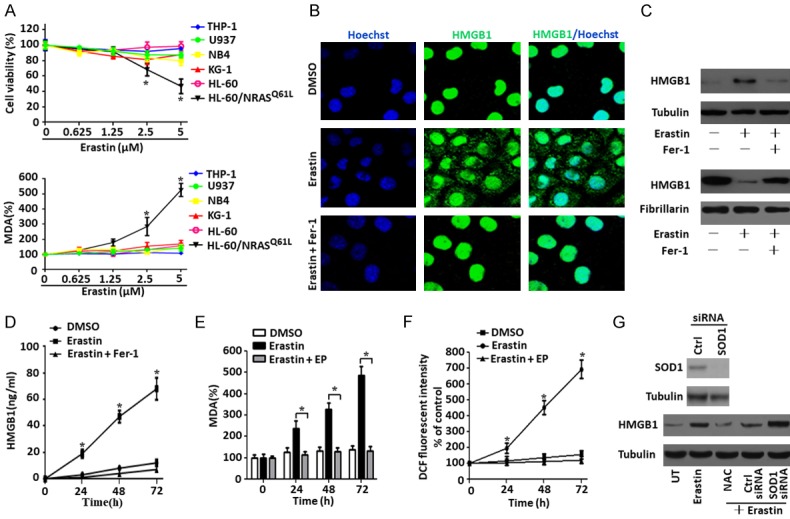
Erastin promotes ROS-dependent extranuclear HMGB1 translocation. A. Different types of leukemia cells were treated with erastin at the indicated doses for 48 h, intracellular MDA levels and cell viability were assayed (n = 3, *P < 0.05 versus untreated group or other cell lines). B and C. HL-60/NRASQ61L cells were treated with erastin (5 μM) with or without Fer-1 (1 μM) pretreatment for 48 h, and then the nuclear/cytosolic HMGB1 expression was assayed by immunofluorescence and western blot (Green, HMGB1; blue, nucleus). D. HL-60/NRASQ61L cells were treated with erastin (5 μM) for 24-72 h with or without Fer-1 (1 μM) pretreatment. The release of HMGB1 was analyzed by ELISA (n = 3, *P < 0.05 versus the erastin plus Fer-1 treatment group). E. Intracellular MDA levels were assayed in HL-60/NRASQ61L cells after treatment with erastin in the absence or presence of EP (5 mM) pretreatment for 24-72 h (n = 3, *P < 0.05). F. HL-60/NRASQ61L cells were treated with erastin (5 μM) for 24-72 h with or without (EP, 5 mM) pretreatment. ROS production was assessed by measuring the fluorescent intensity of DCF on a fluorescence plate reader. The incremental production of ROS was expressed as a percentage of the control (n = 3, *P < 0.05 versus the erastin plus EP treatment group). G. HL-60/NRASQ61L cells were transfected with control siRNA vector and SOD1 siRNA, the SOD1 expression was verified by western blot. HL-60/NRASQ61L cells were treated with erastin (5 μM) for 48 h with or without NAC (25 mM) or SOD1 RNAi or control RNAi pretreatment. Cytosolic HMGB1 expression was assayed by western blot. All experiments were conducted in triplicate, and the data are presented as the mean ± SD. Ctrl, control; UT, untreated; Fer-1, ferrostatin-1; EP, ethyl pyruvate; NAC, N-acetylcysteine.
To investigate the effect of erastin on HMGB1 translocation, we first analyzed HMGB1 expression and location by immunofluorescence and western blot. Erastin caused HMGB1 translocation from the nucleus to the cytosol in HL-60/NRASQ61L cells (Figure 1B and 1C). Treatment with ferrostatin-1 (Fer-1), a potent inhibitor of ferroptosis, prevented erastin-induced HMGB1 translocation (Figure 1B and 1C). Concurrently, in HL-60/NRASQ61L cells treated for 24-72 h with erastin, the concentration of HMGB1 in the supernatants was significantly elevated compared with untreated cells, and Fer-1 inhibited erastin-induced HMGB1 release (Figure 1D). suggesting that the ferroptosis inducer erastin is an activator of HMGB1 cytoplasmic translocation and release.
ROS function as signaling molecules in various pathway regulating both cell survival and cell death. ROS accumulation is a hallmarks of ferroptosis [3]. Given that ferroptosis is characterized by lipid peroxidation [18], we next investigated whether erastin affected MDA, an end product of lipid peroxidation, as well as ROS production in HL-60/NRASQ61L cells. It was found that erastin increased MDA and ROS levels after 24-72 h of treatment compared to DMSO-treated cells (Figure 1E and 1F). Furthermore, pretreatment of the HL-60/NRASQ61L cells with ethyl pyruvate (EP), an inhibitor of HMGB1 cytoplasmic translocation [19], blocked erastin-induced MDA and ROS levels (Figure 1E and 1F). Under physiological conditions, ROS are removed from cells by the action of SOD family members, catalase, or glutathione peroxidase [20]. As expected, knockdown of SOD1 increased erastin-induced HMGB1 cytoplasmic translocation, whereas the ROS quencher N-acetyl cysteine (NAC) significantly inhibited HMGB1 cytoplasmic translocation in HL-60/NRASQ61L cells (Figure 1G). Together, these results are consistent with the notion that lipid ROS are required for HMGB1 translocation from the nucleus to the cytosol.
Depletion of HMGB1 inhibits erastin-induced cell death and anticancer activity
Our previous study showed that erastin selectively enhanced the sensitivity of HL-60 cells to chemotherapeutic agents in the ferroptosis and necroptosis processes of programmed cell death [5]. To explore the potential role for HMGB1 in the regulation of ferroptosis in leukemia cells, a target-specific shRNA against HMGB1 was transfected into HL-60/NRASQ61L cells. Depletion of HMGB1 expression inhibited erastin-induced cell death at 2.5 μM and 5 μM concentrations for 24 h compared with control groups (Figures 2A, S2A). To further characterize the role of HMGB1 in erastin-induced growth inhibition, HL-60/NRASQ61L cells were treated with erastin in the absence or presence of several potential cell death inhibitors. Treatment with iron-chelating agent DFO, inhibitor of ferroptosis Fer-1 and inhibitor of necroptosis NEC-1 significantly prevented erastin-induced growth inhibition compared with caspase inhibitor ZVAD-FMK and inhibitor of autophagy 3-MA in HL-60/NRASQ61L cells (Figures 2B, S2F). Concurrent knockdown of HMGB1 expression in HL-60/NRASQ61L cells with or without cell death inhibitor treatments almost completely prevented erastin-induced growth inhibition (Figures 2B, S2F), suggesting that HMGB1 was required for erastin-induced HL-60/NRASQ61L cells death regardless of ferroptosis and necroptosis.
Figure 2.
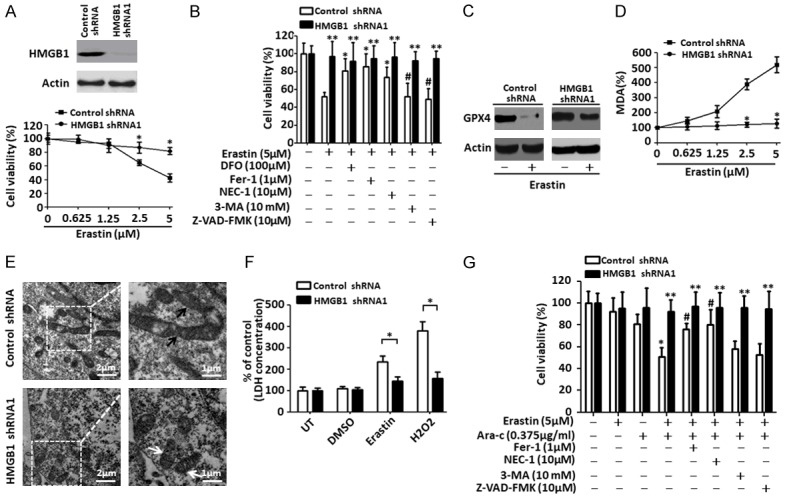
Depletion of HMGB1 inhibits erastin-induced cell death and anticancer activity. A. HL-60/NRASQ61L cells were transfected with HMGB1 shRNA1 or control shRNA vector, HMGB1 expression was verified by western blot. Then, two groups of cells were stimulated with erastin at the indicated doses for 48 h. Cell viability was assayed. (n = 3, *P < 0.05 versus the control group). B. HL-60/NRASQ61L cells were transfected with HMGB1 shRNA1 or control shRNA vector and then stimulated with erastin (5 μM) with or without the indicated inhibitors for 48 h. Cell viability was assayed. (n = 3, *P < 0.05 versus the erastin treatment only control shRNA group; #P > 0.05 versus the erastin treatment only control shRNA group; **P > 0.05 versus the untreated HMGB1 shRNA group). C and D. HL-60/NRASQ61L cells were transfected with HMGB1 shRNA1 and control shRNA vector and then stimulated with erastin (5 μM) for 48 h. GPX4 and intracellular MDA levels were assayed (n = 3, *P < 0.05 versus the control group). E. Ultrastructural features of HL-60/NRASQ61L cells with HMGB1 shRNA1 and control shRNA vector transfection plus erastin (5 μM) treatment (white arrow, normal mitochondria; black arrow, shrunken mitochondria). F. HL-60/NRASQ61L cells were transfected with HMGB1 shRNA1 and control shRNA vector and then stimulated with DMSO, erastin (5 μM), and H2O2 (50 mM) for 48 h. The LDH level in the culture medium was assayed. H2O2 was used as a positive control. UT, untreated. (n = 3, *P < 0.05). G. HL-60/NRASQ61L cells were transfected with HMGB1 shRNA1 and control shRNA vector and then stimulated with erastin (1.25 μm) combined with cytarabine (Ara-C, 0.375 μg/mL) in the absence or presence of the indicated inhibitors for 48 h. Cell viability was assayed. (n = 3, *P < 0.05 versus the erastin treatment only control shRNA group; #P < 0.05 versus the erastin plus cytarabine treatment control shRNA group; **P > 0.05 versus the untreated HMGB1 shRNA1 group). All experiments were conducted in triplicate, and the data are presented as the mean ± SD. DFO, deferoxamine; Fer-1, ferrostatin-1; NEC-1, necrostatin-1; 3-MA, 3-methyladenine; Ara-C, cytarabine.
To investigate whether HMGB1 regulated ferroptosis and necroptosis process death, we assayed different markers for ferroptosis (GPX4, MDA and signs visible via electron microscopy) and necroptosis (LDH). GPX4 is a negative regulator of ferroptosis [21]. Erastin inhibited the expression of GPX4 in HL-60/NRASQ61L cells. Depletion of HMGB1 prohibited the degradation of GPX4 compared with the control group (Figures 2C, S2B). Knockdown of HMGB1 also inhibited erastin-induced MDA production in HL-60/NRASQ61L cells, whereas erastin dose-dependently increased MDA production in the control group (Figures 2D, S2C). Our ultrastructural analysis gave more supportive evidence of ferroptosis; we observed that HL-60/NRASQ61L cells treated with HMGB1 shRNA gene transfection did not reveal changes in mitochondrial morphology, such as loss of structural integrity or increased membrane density, consistent with the previous report [17] (Figures 2E, S2D). Moreover, similar to H2O2 treatment, erastin treatment did not promote LDH release in culture supernatants of HMGB1-knockdown HL-60/NRASQ61L cells compared with the control group (Figures 2F, S2E). These findings indicate that HMGB1-mediated ferroptosis and necroptosis contribute to erastin-induced growth inhibition in HL-60/NRASQ61L cells.
Low-dose erastin treatment (1.25 μm) was previously shown to enhance the anticancer activity of cytarabine and doxorubicin [5]. To characterize the role of HMGB1 in the chemosensitivity of leukemia cells, HL-60/NRASQ61L cells were transfected with HMGB1 shRNA vector and then treated with cytarabine combined with erastin for 48 h. In control shRNA groups, this weakly cytotoxic dose of erastin remarkably enhanced the anticancer activity of cytarabine, and treatment with Fer-1 and necrostatin-1 prevented combination therapy-induced growth inhibition in HL-60/NRASQ61L cells (Figures 2G, S2F). Knockdown of HMGB1 expression significantly prevented combination therapy-induced growth inhibition in HL-60/NRASQ61L cells with or without cell death inhibitor treatments (Figures 2G, S2F). These data suggest that HMGB1 is involved in erastin-enhanced anticancer activity of cytarabine in HL-60/NRASQ61L cells.
Lack of HMGB1 limits iron-mediated ROS generation and cell death
ROS, including superoxide anions (O-2), hydroxyl radicals (OH), hydrogen peroxide (H2O2), and singlet oxygen (1O2), are primarily generated by mitochondria and NADPH oxidases (NOXs) [22]. To investigate whether ROS are generated by mitochondria, intact mitochondria and mitochondrial membrane potential were quantified by flow cytometry using MitoTracker and DIOC6 dye. When HL-60/NRASQ61L cells were treated with erastin for 6-48 h, they showed no changes compared with the DMSO group (Figure 3A and 3B). To further demonstrate whether ROS are generated from NADPH, we treated cells with erastin plus the NADPH inhibitors DPI and Neopterin. ROS levels were not influenced by the inhibitors following erastin treatment after 48 h (Figure 3C). Ferroptosis is an iron-dependent type of programmed necrosis, and cellular iron is required for ROS accumulation [3]. In addition to mitochondria and NADPH, lysosomes are other sources of ROS owing to the low pH and high iron content within lysosomes [23]. DFO were endocytosed and transferred into the lysosomal compartment resulted in a major decrease of cytosolic labile iron [23]. We observed that treatment of cells with DFO prevented erastin-induced ROS generation (Figure 3C), indicating that ROS generation is caused in part by a lysosomal iron-mediated pathway in HL-60/NRASQ61L cells.
Figure 3.
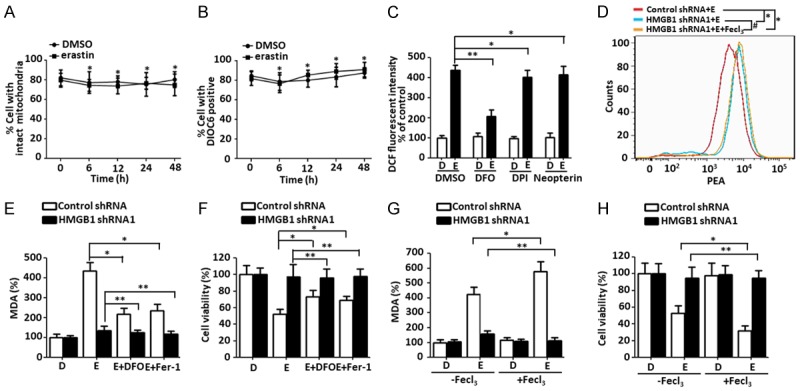
Lack of HMGB1 limits iron-mediated ROS generation and cell death. A and B. HL-60/NRASQ61L cells were treated with DMSO or erastin (5 μM) for 0-48 h, respectively. Intact mitochondria and mitochondrial membrane potential were quantified by flow cytometry with MitoTracker Red (50 nM) and DIOC6 (1 μM) as described in the Materials and Methods section, respectively (n = 3, *P > 0.05). C. HL-60/NRASQ61L cells were treated with erastin (5 μM) combined with DFO (0.1 mM), DPI (5 μM), and neopterin (50 nM) for 48 h. ROS production was assessed by measuring the fluorescent intensity of DCF on a fluorescence plate reader. The incremental production of ROS was expressed as a percentage of the control. D, DMSO; E, erastin. (n = 3, *P > 0.05, **P < 0.05). D. HL-60/NRASQ61L cells were transfected with HMGB1 shRNA1 and control shRNA vector and then stimulated with erastin (5 μM) with or without FeCl3 (30 μM, pretreated for 3 h) for 48 h. Then, cells were stained with PGSK, and the fluorescence profile of the stained cells was analyzed by flow cytometry. E, erastin. E and F. HL-60/NRASQ61L cells were transfected with HMGB1 shRNA1 and control shRNA vector and then stimulated with erastin (5 μM) combined with DFO (0.1 mM) and Fer-1 (1 μM) for 48 h. Intracellular MDA levels and cell viability were assayed. D, DMSO; E, erastin. (n = 3, *P < 0.05, **P > 0.05). G and H. HL-60/NRASQ61L cells were transfected with HMGB1 shRNA1 and control shRNA vector, and then stimulated with erastin (5 μM) with or without FeCl3 (30 μM, pretreated for 3 hours) for 48 h. Intracellular MDA levels and cell viability were then assayed. (n = 3, *P < 0.05, **P > 0.05). Necrostatin-1 pretreatment was used in all experiments. All experiments were conducted in triplicate, and the data are presented as the mean ± SD. D, DMSO; E, erastin; DFO, deferoxamine; DPI, diphenyleneiodonium chloride.
Ferroptosis is characterized by iron-mediated lipid peroxidation, so we next investigated whether HMGB1 regulated iron levels and affected MDA production and cell death. Intracellular chelatable iron was determined using the fluorescent indicator PGSK, the fluorescence of which is quenched by iron [24]. Depletion of HMGB1 expression with or without exogenous Fe (FeCl3) treatment showed a similar profile, whereas the control group cells showed decreased fluorescence (Figures 3D, S3A). Meanwhile, knockdown of HMGB1 almost completely prevented erastin-induced MDA production and growth inhibition with or without DFO and Fer-1 treatment (Figures 3E, 3F, S3B and S3C). This indicates that HMGB1 is essential for intracellular iron levels and ferroptotic cell death in erastin-treated HL-60/NRASQ61L cells. To further confirm whether HMGB1 regulated iron-dependent cell death, FeCl3 was added, and the amount of MDA production and cell death was further increased in control groups (Figures 3G, 3H, S3D and S3E). Under knockdown of HMGB1 expression in HL-60/NRASQ61L cells, FeCl3 did not increased MDA production and cell death (Figures 3G, 3H, S3D and S3E). These data suggest that HMGB1 playes a major role in maintaining iron levels and regulating erastin-induced ferroptosis.
HMGB1 regulates TfR1 expression in a RAS-JNK/p38-dependent pathway
TfR1 is a membrane protein that binds to the transferrin-iron complex and is internalized to release iron within the cytoplasm [25]. In this study, we detected an increase of TfR1 level after erastin treatment, which could be inhibited by Fer-1 (Figure 4A). Moreover, knockdown of TfR1 expression by a specific siRNA significantly decreased the cell death caused by erastin (Figure 4B), indicating that TfR1 is involved in erastin-induced cell death. Oncogenic RAS and MAPK signaling have been proposed to enrich the cellular iron pool mainly by upregulation of TfR1 [26-28]. Our prevous findings indicated that JNK and p38, but not the extracellular signal-regulated kinase (ERK)/mitogen-activated protein kinase (MAPK) pathway, were responsible for erastin-induced HL-60/NRASQ61L cells death [5]. Whether oncogenic RAS or the JNK/p38 pathway exists in leukemia cells and whether HMGB1 regulates TfR1 levels and ferroptosis through this pathway is unclear. Here, we found that co-treatment with JNK inhibitor (SP600125) or p38 inhibitor (SB202190) and erastin led to a significant decrease in TfR1 expression and JNK or p38 phosphorylation, respectively (Figure 4C), suggesting that the JNK/p38 pathway may be upstream of TfR1 protein expression. To further explore the role of RAS in erastin-induced ferroptosis via the JNK/p38 pathway, we used an RNA interference approach. Knockdown of NRAS significantly reduced TfR1 expression and JNK or p38 phosphorylation (Figure 4D), suggesting that the RAS-JNK/p38 pathway may be involved in erastin-induced cellular iron uptake.
Figure 4.

HMGB1 regulates TfR1 expression in the RAS-JNK/p38-dependent pathway. A. HL-60/NRASQ61L cells were lysed after treatment with erastin (5 μM) and/or Fer-1 (1 μM), then TfR1 expression was verified by western blot. B. HL-60/NRASQ61L cells were transfected with TfR1 siRNA and control siRNA, and TfR1 expression was verified by western blot. Then the two groups of cells were stimulated with erastin at the indicated doses for 48 h. Cell viability was assayed. Ctrl, control. (n = 3, *P < 0.05 versus the control group). C. HL-60/NRASQ61L cells were treated with erastin (5 μM) combined with SP600125 (10 μM) and SB202190 (10 μM) for 48 h. TfR1 expression and the phosphorylation of p38 (p-P38) and JNK1/2 (p-JNK1/2) were assayed by western blot. D. HL-60/NRASQ61L cells were transfected with NRAS siRNA and control siRNA vector, and NRAS expression was verified by western blot. Then two groups of cells were stimulated with erastin (5 μM) for 48 h. TfR1, p-P38 and p-JNK1/2 were assayed by Western blot. Ctrl, control. E. HL-60/NRASQ61L cells were transfected with HMGB1 shRNA (HMGB1 shRNA1 and HMGB1 shRNA2) and control shRNA vector and then stimulated with erastin (5 μM) for 48 h. TfR1, p-P38 and p-JNK1/2 expression levels were assayed by western blot. F. HL-60/NRASQ61L cells were transfected with HMGB1 plasmid and empty vector, and HMGB1 expression was verified by western blot. Then, two groups of cells were stimulated with erastin (5 μM) combined with SP600125 (10 μM) and SB202190 (10 μM) for 48 h. TfR1 expression was assayed by western blot. Necrostatin-1 pretreatment was used in all experiments. All experiments were conducted in triplicate, and the data are presented as the mean ± SD. Fer-1, ferrostatin-1.
To further characterize the role of HMGB1 in regulating the RAS-JNK/p38 pathway and TfR1 expression, HL-60/NRASQ61L cells were transfected with the HMGB1 shRNA vector (including HMGB1 shRNA1 and HMGB1 shRNA2) and HMGB1 plasmid. Following HMGB1 shRNA transfection, there was an obvious decrease in TfR1 levels and phosphorylation of JNK and p38 compared with the control group (Figure 4E). In contrast, up-regulated HMGB1 expression significantly increased the level of TfR1, and its expression was inhibited by SP600125 and SB202190 in erastin-treated HL-60/NRASQ61L cells (Figure 4F). Collectively, these data indicate that HMGB1 regulates iron metabolism-related proteins in the RAS-JNK/p38-dependent pathway.
Knockdown of HMGB1 expression inhibits anticancer activity of erastin in vivo
To investigate if suppression of HMGB1 expression affected tumor sensitivity to erastin in vivo, stable HMGB1-knockdown HL-60/NRASQ61L cells were subcutaneously introduced into the right flank of NOD/SCID mice. Beginning at day seven, these mice were treated with erastin or DMSO (vehicle). Compared with the control shRNA group, erastin treatment did not effectively reduce the size of tumors formed by HMGB1-knockdown cells (Figures 5A, S4A). Meanwhile, it should be noted that mice body weight did not significantly differ between groups with or without HMGB1 depletion (Figures 5B, S4B). At the end of the experiments, we found that the weights of xenografts formed by HMGB1-knockdown cells were not reduced compared with the control shRNA mice (Figures 5C, S4C), indicating that these HMGB1-knockdown cells formed tumors that well-tolerated to the erastin regimen. Based on qPCR analysis the expression of PTGS2, a marker for assessment of ferroptosis in vivo [21], indicated that knockdown of HMGB1 decreased erastin-induced PTGS2 expression and ferroptosis (Figures 5D, S4D). Moreover, qPCR and immunohistochemistry analysis revealed that TfR1 was also weakly expressed in tumors formed by HMGB1-knockdown cells (Figures 5E, 5F, S4E and S4F). Together, these findings demonstrated that HMGB1 was required for the anticancer activity of erastin-mediated ferroptosis in vivo.
Figure 5.
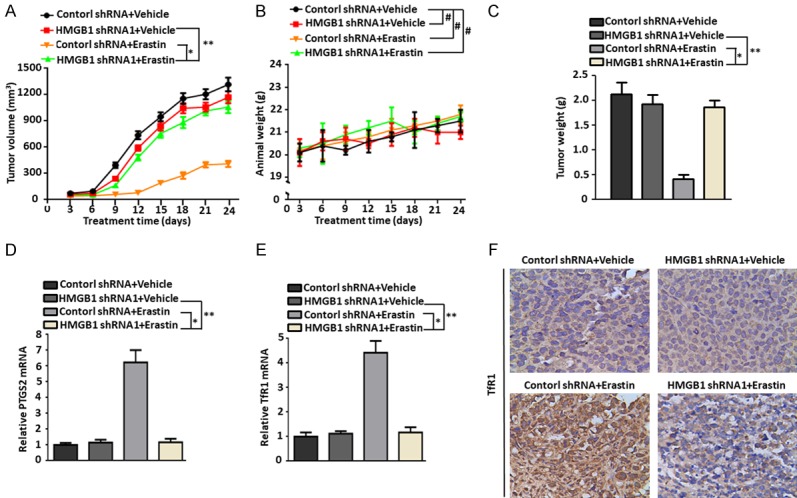
Knockdown of HMGB1 expression inhibited anticancer activity of erastin in vivo. A-C. NOD/SCID mice were injected subcutaneously with HMGB1 shRNA1 HL-60/NRASQ61L cells (1 × 106 cells/mouse) and treated with erastin (20 mg/kg i.v., twice every other day) starting at day seven for two weeks. Tumor volumes and animal weight were measured twice a week. At the termination of the experiments, all xenografts were removed and weighted (n = 4 mice/group, *P < 0.05, **P > 0.05, #P > 0.05). D and E. qPCR analysis of PTGS2 and TfR1 gene expressions in isolated tumors at the termination of experiments (*P < 0.05, **P > 0.05). F. Immunohistochemical staining of TfR1 was performed with an isolated tumor at the termination of the experiments. All experiments were conducted in triplicate, and the data are presented as the mean ± SD.
Discussion
Ferroptosis is emerging as a potential mechanism for targeting tumor growth because it has been shown to potentiate cell death in some malignancies. Classical ferroptotic stimuli, such as erastin, sulfasalazine or RSL3, have been the most often investigated [3]. In this study, we demonstrated that a novel function of HMGB1 was directly involved in the positive regulation and maintenance of ferroptosis in erastin-treated HL-60/NRASQ61L cells, possibly through regulating iron-mediated lipid ROS production. Depletion of HMGB1 inhibited lipid peroxidation and decreased cellular iron pools and the anticancer activity of erastin in vitro and in vivo through a RAS-JNK/p38-dependent pathway. Our findings may therefore provide novel treatment options for patients with AML.
HMGB1 protein is both a nuclear DNA binding factor and a secreted protein. Its activities are determined by its intracellular localization and posttranslational modifications [6]. Chemotherapeutics and cellular stress promoted the translocation of HMGB1 from the nucleus into the cytosol [8]. We have previously demonstrated that HMGB1 plays an important role in leukemia pathogenesis and chemotherapy resistance. HMGB1 acts as a negative regulator of apoptosis or a positive regulator of autophagy, rendering leukemia cells resistant to chemotherapeutic drugs [10,14]. Here, we showe that the ferroptosis-inducer erastin promoted HMGB1 translocation from nucleus into cytosol. Conversely, inhibition of ferroptosis limited HMGB1 translocation, suggesting that HMGB1 was central to the regulation of ferroptosis. Moreover, we have demonstrated that HMGB1 translocation in ferroptosis was ROS dependent. Erastin induced ROS and MDA production in a time-dependent manner, which could be decreased by the HMGB1 cytoplasmic translocation inhibitor ethyl pyruvate (EP). Increasing ROS levels by SOD1 depletion or decreasing ROS levels by NAC treatment significantly affected HMGB1 translocation and levels in cytosol. These findings suggest that HMGB1 is directly involved in the positive regulation and maintenance of ferroptosis in erastin-treated cells through ROS-dependent signals.
Proteins in the Ras family (K-Ras4A, K-Ras4B, H-Ras and N-Ras) are members of the superfamily of small G proteins. RAS genes are mutated in 33% of cancers, stimulating an intensive effort to develop RAS inhibitors for cancer therapy [29]. In addition to their role in solid cancers, RAS mutations have been linked to leukemic progression in myelodysplastic syndrome (MDS) and commonly occur in patients with leukemia [30,31]. Indeed, RAS mutations have been shown to occur in 25% of patients with AML and enhanced sensitivity to drugs [32]. Overexpression of HMGB1 has been demonstrated in numerous types of cancer, including breast cancer, colorectal cancer, lung cancer, hepatocellular carcinoma and leukemia [13,33]. Our findings indicate that HMGB1 is required for erastin-induced ferroptosis in HL-60/NRASQ61L cells. Knockdown of HMGB1 expression inhibited erastin-induced cell death, MDA production, GPX4 protein degradation, and typical mitochondrial morphology changes associated with ferroptosis. In addition, knockdown of HMGB1 expression prevented the erastin-promoted the anticancer activity of cytarabine, a first-line chemotherapy drug for inducing remission of non-acute promyelocytic leukemia. These observations may provide insights that enable the development of novel therapies to overcome resistance in AML patients.
One of the keys in regulating ferroptosis is the generation of ROS [3]. NADPH, the mitochondrial electron transport chain, and lysosomes are the main sources of ROS [22,23]. NAPDH is produced by several cellular reactions, but the pentose phosphate pathway is particularly important for ferroptosis in RAS-mutant cancer cells [3,34]. Lysosomal degradation of ferritin is important to maintain normal iron metabolism. High intracellular iron concentrations can trigger ferroptosis by enhancing the generation of lipid peroxides, and this can be reverted using iron chelators such as DFO [23]. However, ROS generated from the mitochondrial electron transport chain, which was originally found not to contribute to ferroptosis, may contribute under certain circumstances [3,35,36]. Our findings indicate that erastin-induced ROS generation in HL-60/NRASQ61L cells came in part from lysosomes, but not NADPH or mitochondria. Ferroptosis is an iron-dependent PCD, and cellular iron is required for ROS accumulation [3]. Moreover, we showed that HMGB1 was a positive regulator of intracellular iron levels, MDA production, and cell death in erastin-treated HL-60/NRASQ61L cells. The lower iron content in HMGB1-depleted HL-60/NRASQ61L cells, together with the iron-dependent action of erastin, led us to the hypothesis that HMGB1 maintaining iron levels within cancer cells might be a target for inducing cancer-cell-specific lethality.
TfR1 is an important iron regulatory protein that mediates most of the cellular iron uptake by binding iron transferrin at the cell surface, which is internalized by receptor-mediated endocytosis, permitting the release and reduction of the iron in endosomes and transport of the released iron into the cytosol [37,38]. As the number of TfR1 molecules at the cell surface is the rate-limiting factor for iron entry into cells and is essential for iron overload, TfR1 expression is precisely controlled at multiple levels, and TfR1 imbalance has a significant effect on tumor cell death [39,40]. Here, we demonstrate that erastin treatment increased TfR1 expression in HL-60/NRASQ61L cells. Knockdown of TfR1 significantly decreased cell death caused by erastin. This suggests that erastin enriches the cellular iron levels and that the induction of ferroptosis may be dependent on TfR1-dependent iron transport in HL-60/NRASQ61L cells.
MAPKs are activated by a variety of environmental stressors and regulate multiple cell processes ranging from cell survival to cell death [41,42]. The ERK-dependent signaling pathway is required for RAS-dependent ferroptosis in solid cancer cells [17]. However, our previous findings indicated that JNK and p38, but not the ERK/MAPK pathway, were responsible for erastin-induced HL-60/NRASQ61L cells death [5]. Previously, oncogenic RAS and MAPK signaling have been proposed to enrich the cellular iron pool mainly by upregulation of TfR1 [26-28]. Here, we showe that oncogenic RAS and JNK and p38 MAPK signaling were involved in erastin-induced TfR1 expression. Moreover, we provide evidence that knockdown of HMGB1 inhibited erastin-induced TfR1 expression and JNK or p38 phosphorylation, whereas overexpression of HMGB1 increased erastin-induced TfR1 expression. Furthermore, our findings suggeste that knockdown of HMGB1 significantly inhibited HL-60/NRASQ61L xenograft growth and expression of PTGS2 and TfR1 in NOD/SCID mice. Importantly, the in vivo erastin regimens and HMGB1 expression did not affect mice weights, nor did they induce any significant toxicities to the experimental animals. These preclinical results suggeste that HMGB1-mediated ferroptosis could be a potential target for therapeutic interventions in leukemia.
In conclusion, this study shows that HMGB1 is translocated from the nucleus to the cytosol in HL-60/NRASQ61L cell lines in response to erastin and functions as a positive regulator of ferroptosis, which may enhance resistance to anticancer therapies. Our discovery of HMGB1 as a critical regulator of ferroptotic cancer cell death has provided new insights into the molecular mechanism of ferroptosis. This research confirms not only the involvement of iron uptake and lipid oxidization in ferroptosis, but also provides a basis for possible combinational cancer therapy targeting both HMGB1 and ferroptosis. Furthermore, this study points to new therapeutic strategies that can be developed to overcome chemotherapy resistance in AML.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (Grant Nos. 81770178 and 81601528) and the Natural Science Foundation of Hunan Province of China (Grant No. 2015J-J6110).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Ross DD. Novel mechanisms of drug resistance in leukemia. Leukemia. 2000;14:467–473. doi: 10.1038/sj.leu.2401694. [DOI] [PubMed] [Google Scholar]
- 2.Metzig M, Gdynia G, Roth W. Mechanisms of cell death. Novel insights and implications for tumor pathology. Pathologe. 2012;33(Suppl 2):241–245. doi: 10.1007/s00292-012-1678-5. [DOI] [PubMed] [Google Scholar]
- 3.Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, Morrison B 3rd, Stockwell BR. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fatokun AA, Dawson VL, Dawson TM. Parthanatos: mitochondrial-linked mechanisms and therapeutic opportunities. Br J Pharmacol. 2014;171:2000–2016. doi: 10.1111/bph.12416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yu Y, Xie Y, Cao L, Yang L, Yang M, Lotze MT, Zeh HJ, Kang R, Tang D. The ferroptosis inducer erastin enhances sensitivity of acute myeloid leukemia cells to chemotherapeutic agents. Mol Cell Oncol. 2015;2:e1054549. doi: 10.1080/23723556.2015.1054549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lotze MT, Tracey KJ. High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol. 2005;5:331–342. doi: 10.1038/nri1594. [DOI] [PubMed] [Google Scholar]
- 7.Tang D, Kang R, Zeh HJ 3rd, Lotze MT. High-mobility group box 1 and cancer. Biochim Biophys Acta. 2010;1799:131–140. doi: 10.1016/j.bbagrm.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tang D, Kang R, Livesey KM, Cheh CW, Farkas A, Loughran P, Hoppe G, Bianchi ME, Tracey KJ, Zeh HJ 3rd, Lotze MT. Endogenous HMGB1 regulates autophagy. J Cell Biol. 2010;190:881–892. doi: 10.1083/jcb.200911078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang J, Ni J, Liu K, Yu Y, Xie M, Kang R, Vernon P, Cao L, Tang D. HMGB1 promotes drug resistance in osteosarcoma. Cancer Res. 2012;72:230–238. doi: 10.1158/0008-5472.CAN-11-2001. [DOI] [PubMed] [Google Scholar]
- 10.Yu Y, Xie M, He YL, Xu WQ, Zhu S, Cao LZ. Role of high mobility group box 1 in adriamycin-induced apoptosis in leukemia K562 cells. Ai Zheng. 2008;27:929–933. [PubMed] [Google Scholar]
- 11.Kang R, Tang DL, Cao LZ, Yu Y, Zhang GY, Xiao XZ. High mobility group box 1 is increased in children with acute lymphocytic leukemia and stimulates the release of tumor necrosis factor-alpha in leukemic cell. Zhonghua Er Ke Za Zhi. 2007;45:329–333. [PubMed] [Google Scholar]
- 12.Xie M, Kang R, Yu Y, Zhu S, He YL, Xu WQ, Tang DL, Cao LZ. Enhancive effect of HMGB1 gene silence on adriamycin-induced apoptosis in K562/A02 drug resistance leukemia cells. Zhonghua Xue Ye Xue Za Zhi. 2008;29:549–552. [PubMed] [Google Scholar]
- 13.Yang L, Yu Y, Kang R, Yang M, Xie M, Wang Z, Tang D, Zhao M, Liu L, Zhang H, Cao L. Up-regulated autophagy by endogenous high mobility group box-1 promotes chemoresistance in leukemia cells. Leuk Lymphoma. 2012;53:315–322. doi: 10.3109/10428194.2011.616962. [DOI] [PubMed] [Google Scholar]
- 14.Liu L, Yang M, Kang R, Wang Z, Zhao Y, Yu Y, Xie M, Yin X, Livesey KM, Lotze MT, Tang D, Cao L. HMGB1-induced autophagy promotes chemotherapy resistance in leukemia cells. Leukemia. 2011;25:23–31. doi: 10.1038/leu.2010.225. [DOI] [PubMed] [Google Scholar]
- 15.Zhang Z, Wu B, Chai W, Cao L, Wang Y, Yu Y, Yang L. Knockdown of WAVE1 enhances apoptosis of leukemia cells by downregulating autophagy. Int J Oncol. 2016;48:2647–2656. doi: 10.3892/ijo.2016.3446. [DOI] [PubMed] [Google Scholar]
- 16.Yang L, Chai W, Wang Y, Cao L, Xie M, Yang M, Kang R, Yu Y. Reactive oxygen species regulate the differentiation of acute promyelocytic leukemia cells through HMGB1-mediated autophagy. Am J Cancer Res. 2015;5:714–725. [PMC free article] [PubMed] [Google Scholar]
- 17.Yagoda N, von Rechenberg M, Zaganjor E, Bauer AJ, Yang WS, Fridman DJ, Wolpaw AJ, Smukste I, Peltier JM, Boniface JJ, Smith R, Lessnick SL, Sahasrabudhe S, Stockwell BR. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature. 2007;447:864–868. doi: 10.1038/nature05859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang WS, Stockwell BR. Ferroptosis: death by lipid peroxidation. Trends Cell Biol. 2016;26:165–176. doi: 10.1016/j.tcb.2015.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ulloa L, Ochani M, Yang H, Tanovic M, Halperin D, Yang R, Czura CJ, Fink MP, Tracey KJ. Ethyl pyruvate prevents lethality in mice with established lethal sepsis and systemic inflammation. Proc Natl Acad Sci U S A. 2002;99:12351–12356. doi: 10.1073/pnas.192222999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scherz-Shouval R, Elazar Z. ROS, mitochondria and the regulation of autophagy. Trends Cell Biol. 2007;17:422–427. doi: 10.1016/j.tcb.2007.07.009. [DOI] [PubMed] [Google Scholar]
- 21.Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji AF, Clish CB, Brown LM, Girotti AW, Cornish VW, Schreiber SL, Stockwell BR. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156:317–331. doi: 10.1016/j.cell.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dixon SJ, Stockwell BR. The role of iron and reactive oxygen species in cell death. Nat Chem Biol. 2014;10:9–17. doi: 10.1038/nchembio.1416. [DOI] [PubMed] [Google Scholar]
- 23.Kurz T, Eaton JW, Brunk UT. The role of lysosomes in iron metabolism and recycling. Int J Biochem Cell Biol. 2011;43:1686–1697. doi: 10.1016/j.biocel.2011.08.016. [DOI] [PubMed] [Google Scholar]
- 24.Shingles R, North M, McCarty RE. Direct measurement of ferrous ion transport across membranes using a sensitive fluorometric assay. Anal Biochem. 2001;296:106–113. doi: 10.1006/abio.2001.5209. [DOI] [PubMed] [Google Scholar]
- 25.Cheng Y, Zak O, Aisen P, Harrison SC, Walz T. Structure of the human transferrin receptor-transferrin complex. Cell. 2004;116:565–576. doi: 10.1016/s0092-8674(04)00130-8. [DOI] [PubMed] [Google Scholar]
- 26.Ha YM, Park MK, Kim HJ, Seo HG, Lee JH, Chang KC. High concentrations of ascorbic acid induces apoptosis of human gastric cancer cell by p38-MAP kinase-dependent up-regulation of transferrin receptor. Cancer Lett. 2009;277:48–54. doi: 10.1016/j.canlet.2008.11.020. [DOI] [PubMed] [Google Scholar]
- 27.Yang WS, Stockwell BR. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem Biol. 2008;15:234–245. doi: 10.1016/j.chembiol.2008.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ouyang Q, Bommakanti M, Miskimins WK. A mitogen-responsive promoter region that is synergistically activated through multiple signalling pathways. Mol Cell Biol. 1993;13:1796–1804. doi: 10.1128/mcb.13.3.1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Drugging the undruggable RAS: mission possible? Nat Rev Drug Discov. 2014;13:828–851. doi: 10.1038/nrd4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hirai H, Okada M, Mizoguchi H, Mano H, Kobayashi Y, Nishida J, Takaku F. Relationship between an activated N-ras oncogene and chromosomal abnormality during leukemic progression from myelodysplastic syndrome. Blood. 1988;71:256–258. [PubMed] [Google Scholar]
- 31.Reuter CW, Morgan MA, Bergmann L. Targeting the Ras signaling pathway: a rational, mechanism-based treatment for hematologic malignancies? Blood. 2000;96:1655–1669. [PubMed] [Google Scholar]
- 32.Neubauer A, Maharry K, Mrozek K, Thiede C, Marcucci G, Paschka P, Mayer RJ, Larson RA, Liu ET, Bloomfield CD. Patients with acute myeloid leukemia and RAS mutations benefit most from postremission high-dose cytarabine: a Cancer and Leukemia Group B study. J. Clin. Oncol. 2008;26:4603–4609. doi: 10.1200/JCO.2007.14.0418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ellerman JE, Brown CK, de Vera M, Zeh HJ, Billiar T, Rubartelli A, Lotze MT. Masquerader: high mobility group box-1 and cancer. Clin Cancer Res. 2007;13:2836–2848. doi: 10.1158/1078-0432.CCR-06-1953. [DOI] [PubMed] [Google Scholar]
- 34.Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, Kalyanaraman B, Mutlu GM, Budinger GR, Chandel NS. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci U S A. 2010;107:8788–8793. doi: 10.1073/pnas.1003428107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ma S, Henson ES, Chen Y, Gibson SB. Ferroptosis is induced following siramesine and lapatinib treatment of breast cancer cells. Cell Death Dis. 2016;7:e2307. doi: 10.1038/cddis.2016.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Louandre C, Marcq I, Bouhlal H, Lachaier E, Godin C, Saidak Z, Francois C, Chatelain D, Debuysscher V, Barbare JC, Chauffert B, Galmiche A. The retinoblastoma (Rb) protein regulates ferroptosis induced by sorafenib in human hepatocellular carcinoma cells. Cancer Lett. 2015;356:971–977. doi: 10.1016/j.canlet.2014.11.014. [DOI] [PubMed] [Google Scholar]
- 37.Chitambar CR, Wereley JP. Transferrin receptor-dependent and -independent iron transport in gallium-resistant human lymphoid leukemic cells. Blood. 1998;91:4686–4693. [PubMed] [Google Scholar]
- 38.Yamanishi H, Iyama S, Yamaguchi Y, Kanakura Y, Iwatani Y. Total iron-binding capacity calculated from serum transferrin concentration or serum iron concentration and unsaturated iron-binding capacity. Clin Chem. 2003;49:175–178. doi: 10.1373/49.1.175. [DOI] [PubMed] [Google Scholar]
- 39.Gammella E, Buratti P, Cairo G, Recalcati S. The transferrin receptor: the cellular iron gate. Metallomics. 2017;9:1367–1375. doi: 10.1039/c7mt00143f. [DOI] [PubMed] [Google Scholar]
- 40.Basuli D, Tesfay L, Deng Z, Paul B, Yamamoto Y, Ning G, Xian W, McKeon F, Lynch M, Crum CP, Hegde P, Brewer M, Wang X, Miller LD, Dyment N, Torti FM, Torti SV. Iron addiction: a novel therapeutic target in ovarian cancer. Oncogene. 2017;36:4089–4099. doi: 10.1038/onc.2017.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wada T, Penninger JM. Mitogen-activated protein kinases in apoptosis regulation. Oncogene. 2004;23:2838–2849. doi: 10.1038/sj.onc.1207556. [DOI] [PubMed] [Google Scholar]
- 42.Arthur JS, Ley SC. Mitogen-activated protein kinases in innate immunity. Nat Rev Immunol. 2013;13:679–692. doi: 10.1038/nri3495. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.