

Abstract
Background
Primitive neuroectodermal tumor (PNET) is extremely rare and highly aggressive with poor prognosis. Few studies concerning PNET’s the imaging features have been published.
Methods
Six cases of PNET, all confirmed by pathology and immunohistochemical (IHC) examinations, were treated during January 2012 to December 2016. These cases’ clinical course and imaging findings were retrospectively analyzed.
Results
Among six PNET cases, one located in left superior abdomen, one case at posterior abdominal wall, one case in right orbit, one case in left frontal temporal lobe, one case in pelvic cavity, and one case located in left supraclavicular fossa. The tumor’s density was uniform for small tumor and heterogeneous for large tumors on CT images, while the size of tumors differed during presentation depending on the location of the tumor. Marked enhancement was visualized after injection of contrast media. The demarcation between the lesion and adjacent tissues or organs tended to be unclear. On magnetic resonance imaging (MRI) images, the mass mainly showed heterogeneously long T1 and long T2 signal intensity, mixed high signal intensity on fluid-attenuated inversion recovery (FLAIR) image. In two cases maximum intensity projection image reconstruction demonstrates tortuous blood vessels within the tumor on enhanced CT images. Five cases were treated by surgical resection with two cases received adjuvant radiotherapy and three cases received adjuvant chemotherapy. Six patients were followed up with a mean period of 34.5 months (ranging from 6 to 55 months). Five patients survived and one died. Among the five patients undergoing surgeries, one patient presented pelvic and abdominal recurrence/metastasis 2 months after abdominal PNET resection. One patient had a recurrent lesion in the right orbit involving the right ethmoid sinus 6 months after right orbital PNET resection. One patient’s pelvic tumor recurred 7 months after PNET operation, and this patient died after 1 year and 10 months of follow-up. During the follow-up period, the remaining three cases did not show obvious recurrence and/or metastasis.
Conclusions
Overall, the imaging appearances of PNET lack characteristics. PNETs generally do not have clear boundary, or partially so, with its adjacent organs or tissues suggesting their invasive nature. Upon further validation, maximum intensity projection image reconstruction demonstrates tortuous blood vessels within the tumor on enhanced CT images may be valuable information for the diagnosis of PNET.
Keywords: Primitive neuroectodermal tumors (PNET), computed tomography (CT), magnetic resonance imaging (MRI), surgery, chemoradiotherapy
Introduction
Primitive neuroectodermal tumor (PNET) is a kind of extremely rare and highly aggressive, small round cell tumor, with poor prognosis. It develops from the primitive nerve cells of the nervous system (1). It is a member of Ewing’s sarcoma family, mainly occurring in adolescents and in children (2). According to the differential classification, it can be divided into peripheral PNET (pPNET) and central PNET (cPNET). The cPNETs arise from a precursor cell of the subependymal matrix of the central nervous system (CNS) or external granular layer of the cerebellum, pinealocytes, and subependymal cells of the ventricles whereas pPNETs derive from the neural crest located outside the CNS (1). pPNET is the most common, it occurs most in the chest wall, followed by the pelvis, retroperitoneum, abdomen and neck in turn (3). PNET has characteristics of low incidence, high degree of malignancy, rapid progression, high rate of recurrence and metastasis with a poor prognosis (4). Although the clinical and pathological characteristics of PNET have been reported, few studies concerning the imaging features have been published (5). We report six patients with PNET in this paper.
Methods
This retrospective study was approved by the Institutional Ethics Committee of the First Affiliated Hospital of China Medical University. Informed consent was waived due to the retrospective and non-interventional nature of this analysis. Six patients with pathologically confirmed PNET were treated in the First Affiliated Hospital of China Medical University, Shenyang, China, during Jan 2012 to Dec 2016. The patients included three males and three females, ranging in age from 20 to 48 years old. The main clinical manifestation was symptoms caused by rapid-growing mass. Pathology and immunohistochemistry confirmed the tumor final diagnosis were PNETs. Pathological findings presented that heterogeneous cells were distributed in flaky nest-like shape, arranged densely, with hyperchromatic nuclei, bare nucleus and significantly atypia (Figure 1A). By immunohistochemistry, the high expression of CD99 in tumor tissues further supported the final diagnosis of PNETs (Figure 1B). All the six cases had tumor cells positive for CD99. The general information of these six cases is listed in Table 1. One case (case 4) had undergone only radiotherapy and not surgery because of her physical condition.
Figure 1.

Histopathology of case 6. (A) Pathological image (×200) shows heterogeneous cells arranged densely with deep staining nuclei in line with PNET; (B) immunohistochemistry CD99 (×200) demonstrates positive staining with CD99.
Table 1. General clinical information for the six patients.
| Case | Age/sex | Location | Treatment | Postoperative recurrence and metastasis | Follow-up |
|---|---|---|---|---|---|
| Case 1 (10232606) | 23/male | Periorbital area | Surgical resection | Six months post-operation recurrence of right side of the Ethmoidal sinus. | 4 years and 7 months |
| Case 2 (12199664) | 17/male | Superior abdomen | Surgical resection, chemotherapy | Two months post-operation recurrence in the pelvis and abdominal cavity. | 3 years and 7 months |
| Case 3 (13142551) | 45/female | Superior abdomen | Surgical resection | Seven months post-operation recurrence in the pelvis cavity. | 1 year and 10 months |
| Case 4 (10522313) | 40/female | Left supraclavicular | Irradiation | No | 4 years and 2 months |
| Case 5 (14338103) | 22/male | Frontal temporal lobe | Surgical resection, chemotherapy and irradiation therapy | No | 6 months |
| Case 6 (14434966) | 34/female | Pelvis | Surgical resection, chemotherapy and irradiation therapy | No | 2 years and 7 months |
Six patients were followed up, with a mean follow-up period of 34.5 months (ranging from 6 to 55 months). Five patients survived and one died. Among the five patients undergoing surgeries, one patient presented pelvic and abdominal recurrence/metastasis 2 months after abdominal PNET resection (Figure 2). One patient had a recurrent lesion in the right orbit involving the right ethmoid sinus 6 months after right orbital PNET resection. One patient’s pelvic tumor recurred 7 months after PNET operation, and this patient died after 1 year and 10 months of follow-up. During the follow-up period, the remaining three cases did not show obvious recurrence and/or metastasis.
Figure 2.

CT of case 2. (A) Nonenhanced CT image shows a giant mixed-density mass with soft tissue component measured CT value of 42 HU; (B) arterial phase image demonstrates the soft tissue portions of mass enhanced heterogeneously with CT value of 58–72 HU; (C,D) venous phase and delayed phase images demonstrate soft tissue portions further enhanced with CT value 78–79 HU; (E) maximum intensity projection image presents tortuous blood vessels in soft tissue density area; (F) pelvic effusion is noted as well as one small mural nodule attached to left pelvic wall (white arrow); (G,H) post-operation follow-up CT examinations 40 days (G) and 2 months (H) after surgery. Pelvic metastasis lesions enlarged rapidly (white arrows) with massive peritoneal effusion emerging.
Computed tomography (CT) examinations (cases 2–6) used Toshiba CT scanning system (Aquilion ONE, Japan). Scan parameters: spiral scan mode, thickness 0.5 mm, matrix 1,024×1,024. magnetic resonance (MR) examinations (cases 1 and 5) used 3.0T MR equipment (GE Signa Advantage Hdxt, USA).
Results
PNET cases’ imaging findings are presented in Table 2 and Figures 2-5. The tumor’s density was uniform for small tumor and heterogeneous for large tumors on CT images, while the size of tumors differed during presentation depending on the location of the tumor. Marked enhancement was visualized after injection of contrast media. The demarcation between the lesion and adjacent tissues or organs tended to be unclear (Figures 3,5). Tortuous blood vessels within the masses could be observed after enhancement (Figures 2E,3E). On magnetic resonance imaging (MRI) images, the mass mainly showed heterogeneously long T1 and long T2 signal intensity, mixed high signal intensity on fluid-attenuated inversion recovery (FLAIR) image (Figure 4B,C,D).
Table 2. Radiological findings and immunohistochemistry results for the six patients.
| Case | Image methods | Imaging findings | Immunohistochemistry |
|---|---|---|---|
| 1 | MRI | A right inferior orbital mass with long T1 & long T2 signal intensity in right periorbita area, and mass measured 2.5 cm × 3.2 cm in size. Marked enhancement. The mass extended to the sinus which deformed the globe due to compression | CK(+), CD3(−), CD20(−) Pax-5(−), CD30(−), CD21(−), CD68(+), Bcl-2(+), CD56(+), synaptophysin(−), CD99(+), Ki-67(+), NSE(+), CD57(+), vimentin(+), CD4(+), CD5(−) |
| 2 | CT | Abdominal CT shows a heterogeneous density mass measured 15.4 cm × 8.6 cm in size in left superior abdominal part. Marked enhancement and enhanced blood vessels can be visualized within the tumor. The surrounding fat gap disappeared, and the adjacent intestinal wall thickened with abnormal enhancement (Figure 2) | CK(−), vimentin(−), CD56(+), MPO(−), TdT(−), CD3(−), CD20(−), Pax-5(−), desmin(−), MyoD1(−), MDM2(+), S-100(+), INI-1(+), CD99(+), CD38(−), Muml(−), NSE(−), Ki-67(+) |
| 3 | CT | Abdominal CT shows a retroperitoneal olive-shaped heterogeneous mass with measured size of 7.5 cm × 6.3 cm. no distinguished boundary between tumor and adjacent psoas major muscle. Marked enhancement and tortuous enhanced blood vessel can be observed after injection of contrast media (Figure 3) | CD20(−), CD3(−), CD56(+), CD99(+), CK(PAN)(−), chromogranin A(−), ki-67(−), NSE(+), Pax-5(−), synaptophysin(+), TIF-1(−), vimentin(+) |
| 4 | CT | Supraclavicular fossa ultrasonography examination shows a left superior clavicle mass measured 4.0 cm × 2.4 cm in size. The CT value was 36 HU for the mass. Enhanced scan showing marked enhancement with arterial phase CT value of 56 HU and venous phase CT value of 71 HU | CK(−), CD3(−), CD20(−), Pax-5(−), CD30(−), Bcl-6(−), CD10(−), MUM1(−), Bcl-2(+), synaptophysin(+), CD56(−), CD99(+), S-100(−), CK-7(−), CK5/6(−), P63(−), vimentin(+), CgA(−), NSE(−), Ki-67(+),EBV(−) |
| 5 | CT, MRI | Head CT shows a left temporal lobe patchy low-density mass measured 4.81 cm × 4.33 cm, CT value 21 HU. MR images show the lesion with heterogeneously long T1 and long T2 signal intensity (Figure 4) | CK(+), CD3(−), CD20(−), EMA(−), vimentin(+), CD68(+), GFAP(+), S-100(−), NeuN(−), synaptophysin(+), NF(−), Olig2(+), P53(+++), Ki-67 (80%), IDH1(−), PR(−), CD99(+), CD56(+), Bcl-6(−), CD10(−), MUM1(−), EBV(−), INI-1(+), desmin(−), MyoD1(−), myogenin(−), myoglobin(−), HMB-45(+), MelanA(+), MiTF(−), CD30(−), Inhibin-a(−), PLAP(−), SALL4(−) |
| 6 | CT | Pelvis CT shows an olive-shaped mass with soft tissue density in the right superior vagina fornix wall. Measured mass size of 3.6 cm × 2.1 cm. Homogeneous enhancement (Figure 5) | CK(−), Vim(+), P40(−), P63(−), Pax-8(−), ER(−), PR(+), P16(−), S-100(−), Ki-67(+), EMA(−), HMB45(−), MelanA(−), desmin(−), CD31(−), CD10(−), CD99(+) |
CT, computed tomography; MRI, magnetic resonance imaging.
Figure 3.

CT of case3. (A) CT plain image shows a quasi-circular tumor with a capsule and mixed density located on the right side of the aorta with CT value of 40 HU for solid area. There is no distinguishable boundary between tumor and adjacent psoas major muscle; (B) arterial phase image shows solid portions and capsule of the tumor markedly enhanced with CT value increase by 28 HU. The white arrow within solid area indicates an enhanced blood vessel; (C) the enhancement for solid tumor portions slightly decreased with CT value of 65 HU on venous phase; (D) on delay phase, enhancement for solid portions of tumor further decreased and the right ureter is compressed and shifted to the right side (white arrow); (E) MIP image demonstrated tortuous blood vessels within the tumor (white arrows). MIP, maximum intensity projection.
Figure 4.
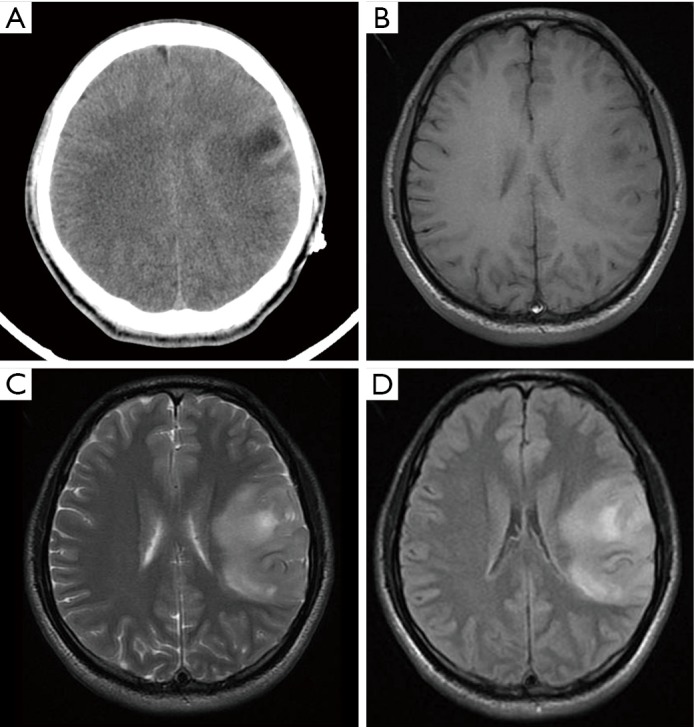
CT/MRI of case5. (A) Axial CT image show a low-density lesion at the left temporal lobe with slight edema surrounding the lesion; (B,C) the lesion in the left temporal lobe presented as a heterogeneously long T1 and long T2 signal intensity on T1 (B) and T2 (C) weighted images; (D) FLAIR image shows similar lesion signal intensity to T2WI. FLAIR, fluid-attenuated inversion recovery; CT, computed tomography; MRI, magnetic resonance imaging.
Figure 5.

CT of case 6. (A) Axial plain image shows a mass located between bladder and vaginal anterior wall with an obscure margin and CT value of 39 HU; (B,C) on arterial phase (B) and venous phase (C), the mass enhanced markedly with CT reached 66 and 76 HU, respectively. The mass adhered to bladder, vaginal anterior wall and pelvic lateral wall; (D) delayed phase image, mass’s enhancement decreased. The bladder shows compression and deformation due to the external mass. CT, computed tomography.
Discussion
PNET is a rare and highly malignant tumor arising from the neural crest that was. It is a member of Ewing’s sarcoma family (1), often occur in youth and children. PNETs can be divided into cPNET, that develops from the CNS, most commonly the cerebellum, and pPNET arising from the neural crest. Both are aggressive tumors and have similar survival rates (2). Both types have characteristics of a high degree of malignancy, a common distant metastasis, and local recurrence. The incidence ratio of male to female is around 3:1 (5,6). PNET tends to recur locally and to metastasize early to regional lymph nodes, lungs, liver, bone, and bone marrow within 2 to 3 years after surgery, with a resulting of very poor prognosis.
PNET patients usually have no specific symptoms/signs on physical examination or no special presentations on imaging exams and serum biochemical tests. The clinical presentation of PNET is variable, and related to tumor location, size, and invasiveness. The tumors in our study are located in the orbit, temporal region, pelvic, and abdomen, which is consistent with previous reports (7). The diagnosis of PNET is dependent on pathological examination and immunohistochemistry. Microscopically, PNET is mainly composed of small round cells, which are diffusively distributed or form several lobulated structures. In addition, tumor cells may be positive for other proteins related to neural differentiation, including neuron-specific enolase (NSE), S-100 protein, neurofilament, synaptophysin, and chromogranin A. NSE, CD99 and vimentin positive can help confirm the diagnosis. In addition, chromosome ectopic (11; 22) (q24; q12) is highly specific and it can have additional diagnostic standards (7-10).
There is no current consensus regarding the optimal treatment protocol for PNET (11). The cornerstone of therapy has been surgical resection of the tumor (12,13). Due to the scarcity of cases of this disease, the standard first-line adjuvant treatment remains unclear. There is no consensus on the chemotherapy regimens in the patients with PNET; however, there are reports recommending cyclophosphamide or ifosfamide, cisplatin or carboplatin and VP-16 (etoposide) (14). In recent years, some literature reported that chemotherapy using sunitinib, sorafenib, and bevacizumab might be valuable in the treating PNET (8). Radiotherapy is believed to be an important adjuvant treatment option for PNET. Our study tentatively supports the effectiveness of radiotherapy as an adjuvant treatment option for PNET (Table 1).
Previous studies (15) reported that PNET mostly shows mixed isointense to hypointense signals on T1-weighted imaging, and isointense to hyperintense signals on T2-weighted imaging. PNET may have heterogeneous enhancement on CT scan and significant enhancement on the MRI (16). It can be seen from our cases that the tumor density may appear uniform when it is small and the tumor may appear heterogeneous when it is large in size. PNETs generally do not have clear boundary with its adjacent organs or tissues suggesting their invasive nature. It may be a diagnostic valuable sign when maximum intensity projection reconstruction image demonstrates tortuous blood vessels within the tumor on enhanced CT images.
In conclusion, the clinics and imaging manifestations of six cases of PNET are presented in this report. Generally, the imaging appearances of PNET lack characteristics. When the tumor is small its density may appear uniform and it tends to become heterogeneous when the tumor is large. PNETs generally do not have clear boundary, or partially so, with its adjacent organs or tissues suggesting their invasive nature. Upon further validation, maximum intensity projection image reconstruction demonstrates tortuous blood vessels within the tumor on enhanced CT images may be valuable information for diagnosis of PNET.
Acknowledgements
None.
Ethical Statement: This retrospective study was approved by the Institutional Ethics Committee of the First Affiliated Hospital of China Medical University. Informed consent was waivered due to the retrospective and non-interventional nature of this analysis.
Footnotes
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- 1.Dai J, He HC, Huang X, Sun FK, Zhu Y, Xu DF. Long-term survival of a patient with a large adrenal primitive neuroectodermal tumor: A case report. World J Clin Cases 2019;7:340-6. 10.12998/wjcc.v7.i3.340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nery B, Pereira LCT, Costa RAF, Queiroz RM, Abud LG, Quaggio E, Coronatto LH, Prado IST, Miyake CH, Filho FB. Cervicomedullary primitive neuroectodermal tumor of the spine: Case report. Surg Neurol Int 2018;9:241. 10.4103/sni.sni_41_18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rahbar M, Rahbar M, Bahoush G. Peripheral primitive neuroectodermal tumor associated with paraneoplastic Cushing's syndrome: The rare case. Ann Med Surg (Lond) 2018;37:21-4. 10.1016/j.amsu.2018.11.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ma J, Ma S, Yang J, Jia G, Jia W. Primary spinal primitive neuroectodermal tumor: A single center series with literature review. J Spinal Cord Med 2018:1-9. [Epub ahead of print]. doi: . 10.1080/10790268.2018.1547862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aguiar TF, Barbosa-Teixeira AC, Costa SS, Ezquina S, Gimenez TM, Novak E, Cristofani LM, Rosenberg C, Odone Filho V, Krepischi ACV. Atypical presentation of a germline APC mutation in a child with supratentorial primitive neuroectodermal tumor. Pediatr Blood Cancer 2019;66:e27566. 10.1002/pbc.27566 [DOI] [PubMed] [Google Scholar]
- 6.Abolhasani M, Salarinejad S, Moslemi MK. Ewing sarcoma/primitive neuroectodermal tumor of the kidney: A report of three cases. Int J Surg Case Rep 2016;28:330-4. 10.1016/j.ijscr.2016.10.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen J, Jiang Q, Zhang Y, Yu Y, Zheng Y, Chen J, Zhao Y, Miao Z, Fan F, Wang Y. Clinical Features and Long-Term Outcome of Primary Intracranial Ewing Sarcoma/Peripheral Primitive Neuroectodermal Tumors: 14 Cases From a Single Institution. World Neurosurg 2019;122:e1606-14. 10.1016/j.wneu.2018.11.151 [DOI] [PubMed] [Google Scholar]
- 8.Thomas AC, Rajashekharan R. A Rare Case of Dumbbell-shaped Primary Intraspinal Peripheral Primitive Neuroectodermal Tumor Involving Thoracic Spinal Epidural Space. Asian J Neurosurg 2018;13:1216-8. 10.4103/ajns.AJNS_91_17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen J, Yuan T, Liu X, Hua B, Dong C, Liu Y, Quan G. Ewing's Sarcoma/Peripheral Primitive Neuroectodermal Tumors in Bronchus. Am J Med Sci 2019;357:75-80. 10.1016/j.amjms.2018.08.009 [DOI] [PubMed] [Google Scholar]
- 10.Zhang C, Zhang J, Wang G, Xu J, Li Y, Guo Q, Zheng T, Zhang Y. Benefit of Sunitinib in the treatment of pulmonary primitive neuroectodermal tumors: a case report and literature review. Oncotarget 2016;7:87543-51. 10.18632/oncotarget.13896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jaramillo-Huff A, Bakkar R, McKee JQ, Sokkary N. Primary Primitive Neuroectodermal Tumor Arising from an Ovarian Mature Cystic Teratoma in a 12-Year-Old Girl: A Case Report. J Pediatr Adolesc Gynecol 2017;30:511-2. 10.1016/j.jpag.2016.11.007 [DOI] [PubMed] [Google Scholar]
- 12.Soles BS, Wilson A, Lucas DR, Heider A. Melanotic Neuroectodermal Tumor of Infancy. Arch Pathol Lab Med 2018;142:1358-63. 10.5858/arpa.2018-0241-RA [DOI] [PubMed] [Google Scholar]
- 13.Liu Z, Xu YH, Ge CL, Long J, Du RX, Guo KJ. Huge peripheral primitive neuroectodermal tumor of the small bowel mesentery at nonage: A case report and review of the literature. World J Clin Cases 2016;4:306-9. 10.12998/wjcc.v4.i9.306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sublett JM, Davenport C, Eisenbrock H, Dalal S, Jaffar Kazmi SA, Kershenovich A. Pediatric Primary Diffuse Leptomeningeal Primitive Neuroectodermal Tumor: A Case Report and Literature Review. Pediatr Neurosurg 2017;52:114-21. 10.1159/000452807 [DOI] [PubMed] [Google Scholar]
- 15.Cherif El Asri A, Benzagmout M, Chakour K, Chaoui MF, Laaguili J, Chahdi H, Gazzaz M, El Mostarchid B. Primary Intracranial pPNET/Ewing Sarcoma: Diagnosis, Management, and Prognostic Factors Dilemma-A Systematic Review of the Literature. World Neurosurg 2018;115:346-56. 10.1016/j.wneu.2018.04.164 [DOI] [PubMed] [Google Scholar]
- 16.Baleato-González S, Tirapu-de-Sagrario MG, Pintos-Martínez E, García-Figueiras R. Scrotal Peripheral Primitive Neuroectodermal Tumor. Curr Urol 2018;12:50-3. 10.1159/000447231 [DOI] [PMC free article] [PubMed] [Google Scholar]