

Abstract
With advancements in high‐throughput generation of phenotypic data on mutant proteins, it has become important to individually characterize different proteins or their variants rapidly and with minimal sample consumption. We have made use of a nano differential scanning fluorimetric device, from NanoTemper technologies, to rapidly carry out isothermal chemical denaturation and measure folding/unfolding kinetics of proteins and compared these to corresponding data obtained from conventional spectrofluorimetry. We show that using sample volumes 10‐50‐fold lower than with conventional fluorimetric techniques, one can rapidly and accurately measure thermodynamic and kinetic stability, as well as folding/unfolding kinetics. This method also facilitates characterization of proteins that are difficult to express and purify.
Keywords: nano differential scanning fluorimeter, isothermal denaturation, thermodynamic stability, refolding unfolding kinetics
Introduction
A protein typically adopts a folded conformation to carry out biological function. Under physiological conditions, the folded state is usually more stable than its corresponding unfolded, inactive state. Protein stability is either thermodynamically or kinetically controlled.1, 2, 3, 4 Widely used techniques for deciphering thermodynamic and kinetic stability of proteins involve measurements of intrinsic fluorescence either at equilibrium or as a function of time.5, 6, 7, 8 Such measurements are sample and labor intensive. This is a significant limitation for unstable proteins with low purification yields. Here we describe the use of a nano differential scanning fluorimeter (nanoDSF) instrument, the Prometheus NT.48 from NanoTemper technologies to overcome these limitations.
nanoDSF is an advanced differential scanning fluorimetry technology that detects small changes in the fluorescence of tryptophan upon folding or unfolding as a function of temperature or other solution conditions. The fluorescence of tryptophan in a protein is strongly dependent on its surroundings.9 By following changes in fluorescence, chemical and thermal stability10, 11 can be assessed in a truly label‐free fashion.
We describe equilibrium, isothermal, unfolding studies for two proteins, homodimeric Escherichia coli CcdB and monomeric hen egg white lysozyme (HEWL) using both a conventional fluorimeter and nanoDSF. We also measure unfolding and refolding kinetics of CcdB using nanoDSF and compare with previously published kinetic data.7, 8 The data demonstrate that nanoDSF can be used to accurately and rapidly measure protein stability and folding/unfolding kinetics with minimal sample consumption.
Results
Biophysical characterization of the proteins
CcdB protein was purified by affinity purification against immobilized ligand CcdA. The purified CcdB and HEWL [Fig. 1(a)] were subjected to thermal denaturation, monitored by intrinsic fluorescence of tryptophan and tyrosine residues as a function of temperature, and the apparent thermal melting temperature (Tm) was calculated [Fig. 1(b)]. The fluorescence of the tryptophan and tyrosine residues in a protein is strongly dependent on the surrounding environment.9 Changes in the structure of the protein have an effect on both the intensity and the emission wavelength, especially of tryptophan fluorescence. The nanoDSF (Prometheus NT.48) is equipped with fluorescence detectors that measure the fluorescence intensity at two different wavelengths, 330 nm and 350 nm, thus being sensitive for both, the change in fluorescence intensity and the shift of the fluorescence maximum upon unfolding. The apparent Tm can be calculated from the changes in tryptophan fluorescence intensity, or from the ratio of tryptophan emission at 330 nm and 350 nm, as a function of temperature. CcdB and HEWL are homodimeric and monomeric proteins, respectively. The oligomeric states of the purified CcdB and HEWL were determined by subjecting ∼120 μg of protein to size exclusion gel chromatography (SEC) using a Superdex‐200 100/300GL [Fig. 1(c)]. The expected and inferred molecular weight from SEC for CcdB is 23.4 kDa and 24 kDa, respectively and for HEWL it is 14.4 kDa and 15 kDa, respectively.
Figure 1.
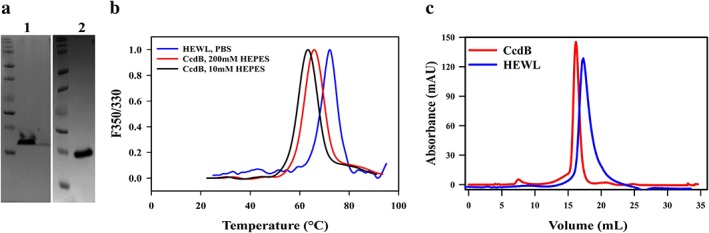
Biophysical characterization of the proteins used in the study. (a) SDS PAGE profile of CcdB (1) and HEWL (2) are shown. (b) Thermal unfolding traces of CcdB in 10 mM HEPES, CcdB in 200 mM HEPES, and HEWL are shown in black, red, and blue respectively. (c) Confirmation of oligomeric state of CcdB and HEWL using size exclusion chromatography (SEC) in PBS at 4 °C are shown in red and blue, respectively.
Isothermal denaturation
Equilibrium unfolding experiments for both CcdB and HEWL were carried out by conventional fluorimetry and with a nanoDSF (Prometheus NT.48). The data were fitted to N2 → 2D and N → D unfolding models for homodimeric CcdB and monomeric HEWL, respectively (Figs. 2, 3, Table 1). The raw data of CcdB (10 mM HEPES, pH 8.4) and HEWL (PBS, pH 7.4) in presence of GdnCl and urea is also plotted in terms of 350/330 ratio as a function of denaturant concentration (Fig. 4). A repeat experiment with a different set of capillaries demonstrated reproducibility [Fig. 5(a)]. We confirmed that the minimum concentration of protein that can be reliably used for nanoDSF is 15 μg/mL, as suggested by the manufacturer [Fig. 5(b)]. We choose these proteins because they represent different unfolding models and both have been extensively characterized previously.7, 12, 13, 14, 15, 16, 17 The midpoint of chemical denaturation (Cm), ΔG0, and m‐values measured is similar in both the instruments. The Cm, ΔG0, and m‐values of HEWL measured is similar to that reported earlier.18
Figure 2.

GdnCl denaturation of CcdB under two different buffer conditions. Equilibrium denaturation profiles of CcdB in 10 mM HEPES, (•) pH 8.4 and 200 mM HEPES (▾), pH 8.4 at 25 °C using both fluorimeter and nanoDSF. Isothermal melts were carried out using 47 μg/mL (2 μM) protein in both cases. The experimental data are shown in dots or inverted triangles, while the fits are shown as smooth lines. The theoretical curves were obtained by fitting all the melts with two‐state unfolding models (N2↔2D) as described previously.7
Figure 3.
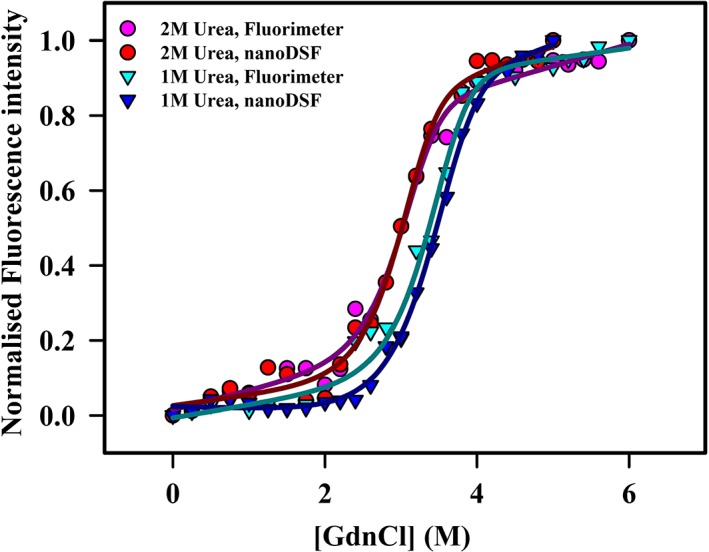
GdnCl denaturation of HEWL in the presence of two different urea concentrations. Equilibrium denaturation profiles of HEWL at PBS, pH 7.5 in the presence of 1 (▾) and 2 M (•) urea at 25 °C using both fluorimeter and nanoDSF. The protein concentration used for isothermal melts was 15 μg/mL (1.05 μM). The experimental data are shown in dots or inverted triangles, while the fits are shown in smooth lines. The theoretical curves were obtained by fitting all the melts with a two‐state unfolding model (N↔D) as described previously.13
Table 1.
Thermodynamic Parameters of CcdB and Lysozyme Measured under Different Buffer Conditionsa
| Proteins | Buffer | Fluorimeter | nanoDSF | ||||
|---|---|---|---|---|---|---|---|
| ΔG0 (kcal.mol−1) | m (kcal.mol−1 M−1) | Cm (M) | ΔG0 (kcal.mol−1) | m (kcal.mol−1 M−1) | Cm (M) | ||
| CcdB | 10 mM HEPES, pH 8.4 | 22.2 ± 0.08 | 4.89 ± 0.04 | 2.49 ± 0.05 | 22.4 ± 0.09 | 4.94 ± 0.06 | 2.51 ± 0.02 |
| CcdB | 200 mM HEPES, pH 8.4 | 23.7 ± 0.07 | 4.60 ± 0.05 | 3.03 ± 0.05 | 23.9 ± 0.05 | 4.48 ± 0.06 | 3.2 ± 0.1 |
| Lysozyme | 1 M Urea, PBS, pH 7.5 | 7.7 ± 0.07 | 2.80 ± 0.05 | 3.33 ± 0.05 | 7.5 ± 0.05 | 2.67 ± 0.06 | 3.35 ± 0.1 |
| Lysozyme | 2 M Urea, PBS, pH 7.5 | 6.9 ± 0.05 | 2.10 ± 0.05 | 2.94 ± 0.05 | 7.1 ± 0.08 | 2.3 ± 0.06 | 2.93 ± 0.02 |
Reported standard errors are derived from two independent experiments.
Figure 4.
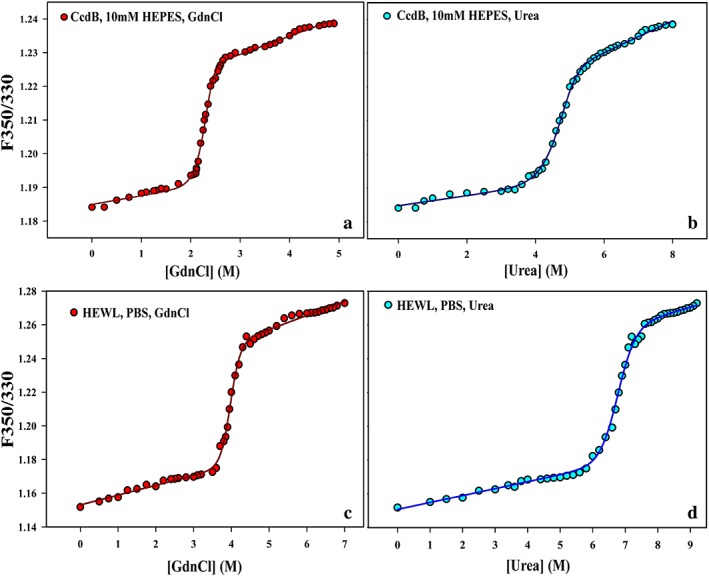
GdnCl and urea denaturation of CcdB and HEWL using 48 different denaturant concentrations. Raw equilibrium denaturation profiles of CcdB in 10 mM HEPES, pH 8.4 (a‐b) and HEWL (c‐d) at PBS, pH 7.5 using GdnCl ( ) and urea (
) and urea ( ), at 25 °C using nanoDSF. Data is plotted in terms of ratio of fluorescence intensity at 350/330 nm as a function of denaturant concentration. Isothermal melts were carried out using 47 μg/mL (2 μM) for CcdB and 15 μg/mL (1.05 μM) for HEWL. The experimental data are shown in dots, while the fits are shown as smooth lines. The theoretical curves were obtained by fitting all the melts with two‐state unfolding models (N↔D for HEWL and N2↔2D for CcdB) as described previously.7, 13
), at 25 °C using nanoDSF. Data is plotted in terms of ratio of fluorescence intensity at 350/330 nm as a function of denaturant concentration. Isothermal melts were carried out using 47 μg/mL (2 μM) for CcdB and 15 μg/mL (1.05 μM) for HEWL. The experimental data are shown in dots, while the fits are shown as smooth lines. The theoretical curves were obtained by fitting all the melts with two‐state unfolding models (N↔D for HEWL and N2↔2D for CcdB) as described previously.7, 13
Figure 5.

Measurement reproducibility and determination of sample sensitivity of nanoDSF using two different sets of capillaries for 48 different denaturant concentrations. Equilibrium GdnCl denaturation profiles of two different concentration of HEWL (a) 15 μg/mL and (b) 7.5 μg/mL in PBS, pH 7.5 at 25 °C using nanoDSF. Two set of capillaries [capillary 1 () and capillary 2 ( )] were used to examine the reproducibility of the data. The data points in denaturation profiles using 7.5 μg/mL of HEWL show increased scatter as compared to data for 15 μg/mL.
)] were used to examine the reproducibility of the data. The data points in denaturation profiles using 7.5 μg/mL of HEWL show increased scatter as compared to data for 15 μg/mL.
Isothermal refolding and unfolding kinetics study
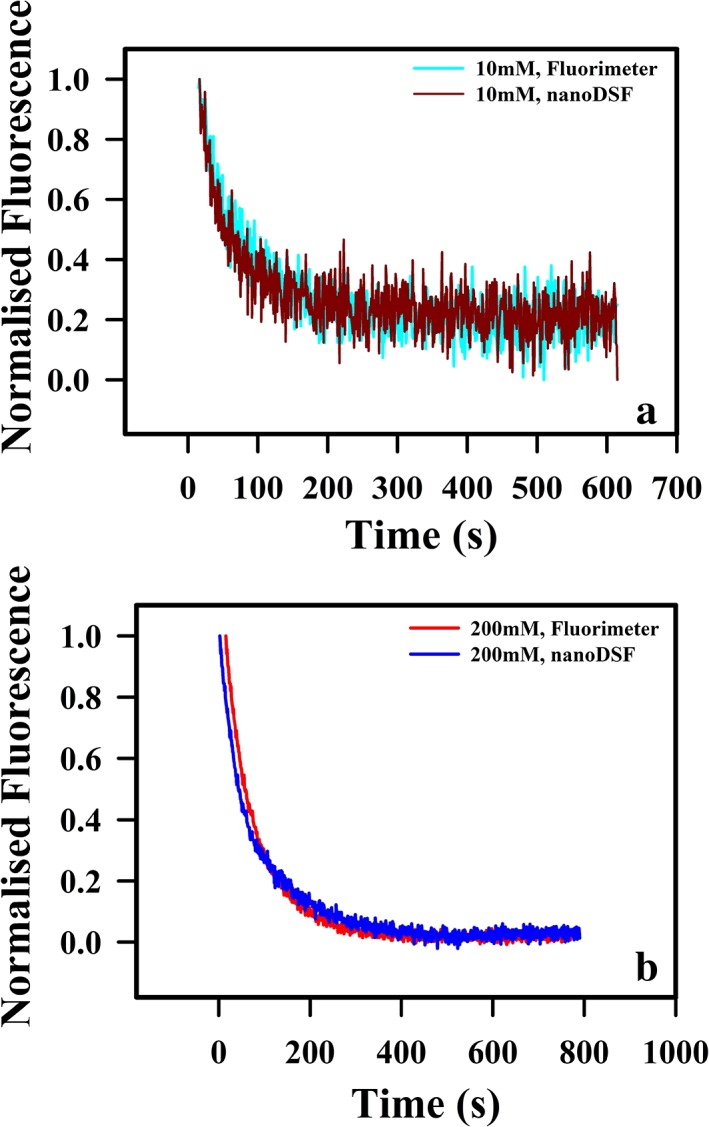
Refolding and unfolding kinetics following manual mixing for CcdB was monitored by time‐course fluorescence spectroscopy as a function of time at 25 °C. Refolding was carried out with 2 μM of purified proteins in either 10 or 200 mM HEPES, at pH 8.4 at a final GdnCl of 1.5 M. Refolding for CcdB occurs with a significant burst phase and a slow phase as observed earlier8 with measured rates similar in both the instruments in both buffer conditions (Fig. 7, Table 2). The unfolding trace of CcdB when fitted to the three‐parameter unfolding equation, gives similar fitted unfolding rates in all conditions (Fig. 6, Table 2). These experiments indicate that the capillary‐based nanoDSF can also be used to make isothermal, time‐dependent, kinetic measurements.
Figure 7.
Refolding kinetic studies of CcdB under different buffer conditions. CcdB exhibits biphasic refolding kinetics with a fast and a slow phase. Kinetic traces at 2 μM protein concentration of (a) CcdB in 10 mM HEPES, pH 8.4 and (b) CcdB in 200 mM HEPES, pH 8.4 using both fluorimeter and nanoDSF are shown. All studies were carried out at 25 °C.
Table 2.
Kinetic Parameters for Refolding and Unfolding of CcdB Measured under Different Buffer Conditions
| Buffer/Instrument | Refolding_1.25 M GdnCl | |||||||
|---|---|---|---|---|---|---|---|---|
| Fast phase | Slow phase | Unfolding_3M GdnCl | ||||||
| a0 | a1 | k1 (s−1) | a2 | k2 (s−1) | a0 | a1 | K1 (s−1) | |
| 10 mM HEPES, pH 8.4/Fluorimeter | 0.02 ± 0.02 | 0.7 ± 0.01 | 0.06 ± 0.005 | 0.28 ± 0.02 | 0.001 ± 0.0003 | 0.85 ± 0.01 | 0.15 ± 0.02 | 0.015 ± 0.0001 |
| 10 mM HEPES, pH 8.4/nanoDSF | 0.02 ± 0.01 | 0.8 ± 0.01 | 0.04 ± 0.005 | 0.18 ± 0.02 | 0.003 ± 0.0003 | 0.87 ± 0.04 | 0.13 ± 0.01 | 0.020 ± 0.0003 |
| 200 mM HEPES, pH 8.4/Fluorimeter | 0.02 ± 0.03 | 0.59 ± 0.02 | 0.031 ± 0.0005 | 0.41 ± 0.02 | 0.001 ± 0.003 | 0.68 ± 0.02 | 0.32 ± 0.03 | 0.006 ± 0.00005 |
| 200 mM HEPES, pH 8.4/nanoDSF | 0.02 ± 0.02 | 0.55 ± 0.03 | 0.027 ± 0.0008 | 0.43 ± 0.03 | 0.002 ± 0.001 | 0.65 ± 0.03 | 0.35 ± 0.01 | 0.005 ± 0.00005 |
Figure 6.
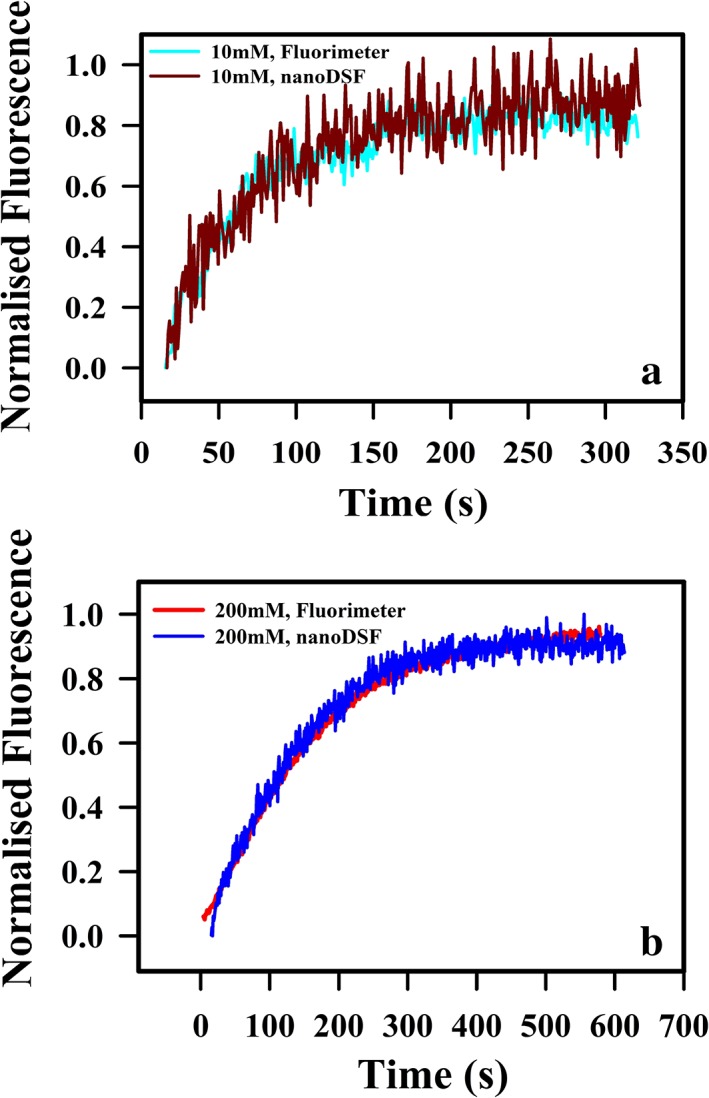
Unfolding kinetic studies of CcdB under different buffer conditions. Unfolding of CcdB follows single exponential kinetics. Kinetic traces at 2 μM protein concentration of (a) CcdB in 10 mM HEPES, pH 8.4 and (b) CcdB in 200 mM HEPES, pH 8.4 using both fluorimeter and nanoDSF are shown. All studies were carried out at 25 °C.
Photobleaching of samples during measurement
In order to examine if visible photobleaching of intrinsic fluorophores present in the protein samples occurs during the measurement time interval, the intensity of five different concentrations of HEWL (7.5, 15, 50, 100, and 500 μg/mL) was monitored for a period of 20 min at 100% excitation power at three different temperatures (20, 40, 60 °C) and at two different emission wavelengths (330 and 350 nm). There was no detectable photobleaching observed during the measurement time course, except for one condition of HEWL (500 μg/mL) at 60 °C (Fig. 8, Fig. S2). Further photobleaching was also monitored at 330 and 350 nm for five different concentration of HEWL (7.5, 15, 50, 100, and 500 μg/mL) and CcdB (7.5, 15, 50, 100, and 500 μg/mL) at two different excitation power (100% and 80%) for 60 and 150 min, respectively (Fig. S1).
Figure 8.
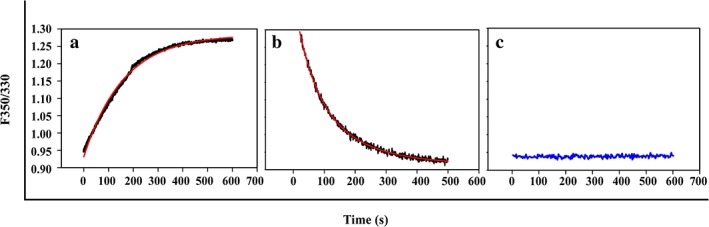
Absence of photobleaching of samples during measurement time scale. The intensity of 50 μg/mL of CcdB using two different capillaries was monitored for a period of 700 s at 100% excitation power at 20 °C (c) to detect any visible photobleaching of intrinsic fluorophores present in the protein samples, during the time course of measurement. The unfolding (a) and refolding (b) of the same sample is also plotted in terms of ratio of fluorescence at 350/330 nm in the same time window.
Discussion
The stability of a protein is governed by various factors. The folding process is both thermodynamically and kinetically controlled.1, 2, 3, 4 Thermodynamic stability is quantitated in terms of the standard state free energy difference between the folded and unfolded states 3. Kinetic stability on the other hand is related to the activation free energy barrier separating the folded and nonnative states.3 Fluorescence spectroscopy employing intrinsic protein fluorescence is a widely used tool to determine both thermodynamic and kinetic stability parameters.19, 20, 21 However, fluorescence measurements in conventional fluorimeters require relatively large sample volumes of about 300–800 μL. This can be a substantial limitation for studies that aim to characterize large numbers of mutants or for proteins that are difficult to express and purify. nanoDSF enables fluorescence intensity measurements in relatively small sample volumes of 15 μL. Earlier used for studying only relative thermal stabilities, the instrument has now added new software that can be used to measure both thermodynamic (PR.ChemControl) and kinetic stability (PR.TimeControl), using low concentrations (15 μg/mL) of proteins. Further, the capillary‐based measurements allow screening of 48 capillaries in parallel. This makes it possible to perform chemical denaturation experiments for two independent samples, exploring 24 different denaturant concentrations for each sample. In the present work, comparative isothermal denaturation studies have been carried out with CcdB and HEWL using both fluorimeter and nanoDSF. Additionally, isothermal refolding and unfolding kinetics for CcdB were also measured and compared with results from previous manual mixing experiments. The data indicate that nanoDSF is a usable alternative to conventional fluorimetry for measuring protein folding thermodynamics and kinetics with approximately 50‐fold lower sample requirements and over a 20‐fold decrease in measurement time/protein. Previously, a capillary‐based chemical denaturation carried out by Microscale Thermophoresis (MST)22 has been reported, but for kinetic experiments no such study has been done so far. Further in the above study, capillaries were loaded into the instrument as three sets of 16 point titrations to yield a final titration with 48 denaturant concentrations, thereby increasing the time for measurement per sample. In case of nanoDSF, 48 capillaries can be measured simultaneously, thereby further reducing the time of measurement per sample. The primary drawback for such studies is capillary cost that is currently half a euro per capillary for conventional capillaries and two euros per capillary for special sensitive capillaries that are used for proteins with few of tryptophan residues. Another drawback for the kinetic studies is the dead time for manual filling and insertion of the capillaries, which is about 15 s. This makes it difficult to simultaneously measure refolding/unfolding kinetics for multiple samples. However, these are both relatively minor issues when compared to the savings in sample amount and measurement time. An alternative to the nanoDSF is use of a fluorescence plate reader. In such cases, fluorescently labeled proteins can be used for improved sensitivity. However, labeling can affect the stability of the protein or produce experimental anomalies.23, 24, 25, 26 Further use of plastic microtiter plates can lead to sample adsorption issues that may lead to incorrect data interpretations, 26 and variations in the meniscus position between wells can also affect the measurement.
Materials and Methods
Reagents
Hen egg white lysozyme (HEWL) was obtained from USB corporation (Cleveland, USA) and was used without any further purification. HEWL was dissolved in PBS, pH 7.5. Ultrapure MB grade GdnCl was purchased from USB corporation. Extrapure AR grade urea was purchased from SRL Pvt. Ltd. (Maharashtra, India).
CcdB expression
CcdB was expressed under control of the arabinose promoter PBAD in the pBAD24 vector using the CcdB resistant Top10Gyrase strain of E. coli, and purified using affinity chromatography as described previously.27 A single colony was inoculated into LB medium (HiMedia) and grown at 37 °C overnight. Five hundred milliliters of LB medium was inoculated with 1% of the primary inoculum and grown at 37 °C until the OD600 reached 0.6. Cells were then induced with 0.2% (w/v) arabinose and grown at 37 °C for 5 h. Cells were harvested by centrifugation (1800g, 20 min, 4 °C). The pellet was resuspended in HEG resuspension buffer pH 7.4 (10 mM HEPES, 50 mM EDTA, 10% glycerol containing 10 mM PMSF) and sonicated, followed by centrifugation at 25,000 g, 30 min, 4 °C. The supernatant was incubated with Affi‐Gel15 (Biorad) coupled to CcdA peptide (residues 46–72) and incubated overnight at 4 °C as described previously.27 The unbound fraction was removed and washed with five times the bed volume of coupling buffer pH 8.3 (0.05 M sodium bicarbonate, 0.5 M sodium chloride). Bound CcdB was eluted with 0.2 M Glycine, pH 2.5 into a tube containing an equal volume of 400 mM HEPES, pH 8.5, 4 °C. The eluted fractions were subjected to 15% Tricine SDS‐PAGE and the protein concentration was determined using an ε280 of 16,500 M−1.cm‐1 and 39000 M−1.cm‐1 for CcdB and HEWL, respectively.12, 28 Fractions containing pure protein were pooled to a final volume of 2 mL and were dialyzed overnight three times against 6 L of 10 mM HEPES, pH 8.4 and 200 mM HEPES, pH 8.4 using Tube‐O‐Dialyser (4 kDa MWCO, G‐Biosciences).
nanoDSF studies
Thermal unfolding experiments of the purified CcdB and HEWL were carried out by nanoDSF [Instrument weblink: (https://nanotempertech.com/prometheus/)]. nanoDSF is an advanced differential scanning fluorimetry technology which detects small changes in the fluorescence of tryptophan upon folding or unfolding in a truly label‐free manner as a function of temperature10, 11 or denaturant. The fluorescence is excited at 280 nm and is detected at 330 nm and 350 nm to derive F350/330 for data analysis. The bandwidth of measurement is 5 nm. The Prometheus NT.48 can accommodate 48 capillaries. The sample is filled into the capillaries by capillary force action and then placed into the instrument. The capillaries are single‐use and therefore no equilibration or cleaning is required. The capillaries were heated from 20 to 90 °C with a heating rate of 1 °C /min and the changes in the fluorescence ratio (F350/F330) was monitored to determine the apparent Tm. Thermal denaturation of CcdB in 10 or 200 mM HEPES, pH 8.4 was carried out at a fixed protein concentration of 47 μg/mL (2 μM) and HEWL in PBS, pH 7.5 was used at 15 μg/mL (1.05 μM). For isothermal denaturation and kinetic measurements, blanks were also subtracted. For each experiment, capillaries containing five different GdnCl concentration were used to calculate blank values. A linear fit to the data was used to estimate the fluorescence of buffer alone as a function of GdnCl concentration.
Isothermal denaturation
Equilibrium unfolding experiments of CcdB and HEWL18 were carried out by conventional fluorimetry using a JASCO FP‐6300 spectrofluorometer and nanoDSF. The changes in either the fluorescence (in case of the fluorimeter) or the fluorescence ratio (F350/F330, in case of nanoDSF using PR.ChemControl software, Prometheus NT.48) were monitored, after overnight incubation at 25 °C in different buffer conditions (10 or 200 mM HEPES, pH 8.4 for CcdB and PBS, pH 7.5 for HEWL) containing various concentrations of the denaturant, guanidium chloride (GdnCl), to determine the stability parameters for chemical denaturation (Cm). GdnCl concentrations were calculated from measurements of the refractive index using a refractometer. The data was analyzed using SigmaPlot™ version 12.5 for Windows™ scientific graphing software, from Systat Software, Inc., San Jose California USA, (www.systatsoftware.com), and the plots were fitted to a two‐state unfolding model (N↔D for HEWL and N2↔2D for CcdB). Isothermal denaturation of CcdB in 10 or 200 mM HEPES, pH 8.4 was carried out at a fixed protein concentration of 47 μg/mL (2 μM) and HEWL in PBS, pH 7.5 was used at 15 μg/mL (1.05 μM). CcdB is a homodimer29 and HEWL is a monomer in the native state. The fraction unfolded for both CcdB and HEWL that is calculated in a similar way to the earlier studies7 is given below.
The spectroscopic signal (Y) of a protein solution (in our case, fluorescence intensity at 340 nm by fluorimeter or F350/F330 ratio by nanoDSF) is related to the fraction of unfolded protein (f u) by:
| (1) |
where Y f and Y u are the values of Y for the folded and unfolded protein, respectively. These change linearly with the denaturant concentration ([D]), as follows:
| (2) |
| (3) |
where y f and y u are the folded and unfolded parameters at zero denaturant concentration. d f and d u are the denaturation dependence of Y for the folded and unfolded state, respectively.
Refolding and unfolding kinetics
Refolding and unfolding kinetics for CcdB were monitored by both fluorimeter8 (fluorescence intensity at 340 nm) and nanoDSF (F350/F330 using PR. TimeControl software, Prometheus NT.48) at 25 °C. To measure the rates of refolding, 4 μM of the dialyzed protein in either 10 or 200 mM HEPES, pH 8.4 was denatured in 3 M GdnCl and subsequently diluted to a final concentration of 1.5 M GdnCl. The changes in signal were monitored as a function of time. To measure the unfolding kinetics, protein in native buffer (10 or 200 mM HEPES, pH 8.4) was diluted into the same buffer containing 6 M GdnCl to a final concentration of 3 M GdnCl and the changes in either the fluorescence intensity at 340 nm (in case of the fluorimeter) or the fluorescence ratio (F350/F330, in case of nanoDSF) were monitored as a function of time. Refolding kinetic traces of fluorescence intensity in 1.5 M GdnCl as a function of time for CcdB in 10 mM or 200 mM HEPES, pH 8.4 were normalized from 0 to 1 between native and denatured baseline at 1.5 M GdnCl. Unfolding kinetic traces of fluorescence intensity in 3 M GdnCl as a function of time for CcdB in 10 mM or 200 mM HEPES, pH 8.4 were normalized from 0 to 1 between native and denatured baseline at 3 M GdnCl. The data was analyzed using SigmaPlot™ version 12.5 for Windows™ scientific graphing software, from Systat Software, Inc., San Jose, CA, USA, (www.systatsoftware.com), and plots were fitted to a 5 parameter equation for exponential decay for refolding (y = y0 + a*exp(−bx) + c*exp(−dx)), yielding slow and fast phase rate constants and a 3 parameter exponential rise for unfolding (y = y0 + a*exp[b*x]) as described previously, where x is the time (in seconds) of refolding/unfolding.8, 27
Conflict of Interest
The authors claim no conflict of interest.
Supporting information
Supplementary Figure S1 Detection of photobleaching of samples during measurement time scale using two different laser powers. The intensity of five different concentration of HEWL (7.5, 15, 50, 100 and 500 μg/mL, increasing concentration from bottom to top, A‐B and E‐F) and CcdB (7.5, 15, 50, 100 and 500 μg/mL, increasing concentration from bottom to top, C‐D and G‐H) using two different capillaries for each set, was monitored at two different laser powers (100% and 80%) for 60 and 150 minutes respectively at two different emission wavelengths (330 and 350 nm) to detect any visible photobleaching of intrinsic fluorophores present in the protein samples, during the time course of measurement.
Supplementary Figure S2. Detection of photobleaching of samples during measurement time scale at three different temperature. The intensity of five different concentration of HEWL (7.5, 15, 50, 100 and 500 μg/mL, increasing concentration from bottom to top) using two different capillaries for each set, at 100% laser power was monitored at three different temperatures (20 (A, D), 40 (B, E) and 60 (C, F) °C) for 20 minutes at two different emission wavelengths (330 and 350 nm) to detect any visible photobleaching of intrinsic fluorophores present in the protein samples, during the time course of measurement.
Acknowledgments
This work was funded in part by a grant to RV from the Department of Biotechnology, grant number‐BT/COE/34/SP15219/2015, November 20, 2015), Government of India and also supported by National Institutes of Health Grant R01A1118366‐01. We also acknowledge funding for infrastructural support from the following programs of the Government of India: DST FIST, UGC Centre for Advanced study, Ministry of Human Resource Development (MHRD), and the DBT IISc Partnership Program. The funders had no role in study design, data collection, and interpretation, or the decision to submit the work for publication. We are thankful to Dr. Sivaramaiah Nallapeta, Head, Business Operations, NanoTemper Technologies; Dr. Saji Menon, Application Specialist, NanoTemper Technologies, and Dr. Andreas Langer, Application Specialist, NanoTemper Technologies for their valuable inputs and for the nanoDSF facility. We also thank all the members of the RV lab for their valuable suggestions.
Broader Impact Statement: Protein thermodynamic stability and kinetic measurements are important to understand different aspects of folding properties but are sample intensive. Here, we validate the use of a capillary‐based nano differential scanning fluorimeter to make rapid, accurate measurements with minimal sample volumes.
References
- 1. Baker D, Agard DA (1994) Kinetics versus thermodynamics in protein folding. Biochemistry 33:7505–7509. [DOI] [PubMed] [Google Scholar]
- 2. Demetrius L (2002) Thermodynamics and kinetics of protein folding: an evolutionary perspective. J Theor Biol 217:397–411. [DOI] [PubMed] [Google Scholar]
- 3. Sanchez‐Ruiz JM (2010) Protein kinetic stability. Biophys Chem 148:1–15. [DOI] [PubMed] [Google Scholar]
- 4. Brissos V, Gonçalves N, Melo EP, Martins LO (2014) Improving kinetic or thermodynamic stability of an azoreductase by directed evolution. PLoS One 9:e87209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shortle D (1986) Guanidine hydrochloride denaturation studies of mutant forms of staphylococcal nuclease. J Cell Biochem 30:281–289. [DOI] [PubMed] [Google Scholar]
- 6. Ganesh C, Shah AN, Swaminathan CP, Surolia A, Varadarajan R (1997) Thermodynamic characterization of the reversible, two‐state unfolding of maltose binding protein, a large two‐domain protein. Biochemistry 36:5020–5028. [DOI] [PubMed] [Google Scholar]
- 7. Bajaj K, Chakshusmathi G, Bachhawat‐Sikder K, Surolia A, Varadarajan R (2004) Thermodynamic characterization of monomeric and dimeric forms of CcdB. Biochem J 380:409–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Baliga C, Varadarajan R, Aghera N (2016) Homodimeric Escherichia coli toxin CcdB (controller of cell division or death B protein) folds via parallel pathways. Biochemistry 55:6019–6031. [DOI] [PubMed] [Google Scholar]
- 9. Teale FWJ, Weber G (1957) Ultraviolet fluorescence of the aromatic amino acids. Biochem J 65:476–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Magnusson AO, Szekrenyi A, Joosten HJ, Finnigan J, Charnock S, Fessner WD (2019) nanoDSF as screening tool for enzyme libraries and biotechnology development. FEBS J 286:184–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bruce D, Cardew E, Freitag‐Pohl S, Pohl E (2019) How to stabilize protein: stability screens for thermal shift assays and nano differential scanning fluorimetry in the virus‐X project. J Vis Exp (144):e58666. [DOI] [PubMed] [Google Scholar]
- 12. Steyaert J, Van Melderen L, Bernard P, Thi MHD, Loris R, Wyns L, Couturier M (1993) Purification, circular dichroism analysis, crystallization and preliminary X‐ray diffraction analysis of the F plasmid CcdB killer protein. J Mol Biol 231:513–515. [DOI] [PubMed] [Google Scholar]
- 13. Laurents DV, Baldwin RL (1997) Characterization of the unfolding pathway of hen egg white lysozyme. Biochemistry 36:1496–1504. [DOI] [PubMed] [Google Scholar]
- 14. Brinkmann C, Weiss MS, Weckert E (2006) The structure of the hexagonal crystal form of hen egg‐white lysozyme. Acta Cryst D62:349–355. [DOI] [PubMed] [Google Scholar]
- 15. Tankrathok A, Daduang S, Patramanon R, Araki T, Thammasirirak S (2009) Purification process for the preparation and characterizations of hen egg white ovalbumin, lysozyme, ovotransferrin, and ovomucoid. Prep Biochem Biotechnol 39:380–399. [DOI] [PubMed] [Google Scholar]
- 16. Ansari M, Zubair S, Atif S, Kashif M, Khan N, Rehan M, Anwar T, Iqbal A, Owais M (2010) Identification and characterization of molten globule‐like state of hen egg‐white lysozyme in presence of salts under alkaline conditions. Protein Pept Lett 17:11–17. [DOI] [PubMed] [Google Scholar]
- 17. Šimić M, Vesnaver G, Lah J (2009) Thermodynamic stability of the dimeric toxin CcdB. Acta Chim Slov 56:139–144. [Google Scholar]
- 18. Schön A, Freire E (2016) Three easy pieces. Biochim Biophys Acta Gen Subj 1860:975–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ladokhin AS, Jayasinghe S, White SH (2000) How to measure and analyze tryptophan fluorescence in membranes properly, and why bother? Anal Biochem 285:235–245. [DOI] [PubMed] [Google Scholar]
- 20. Lakowicz JR. Principles of fluorescence spectroscopy. U.S.A: Springer US, 2006. [Google Scholar]
- 21. Sauer M, Hofkens J, Enderlein J. Handbook of fluorescence spectroscopy. U.S.A: Wiley‐VCH Verlag GmbH & Co. KGaA, 2011. [Google Scholar]
- 22. Alexander CG, Wanner R, Johnson CM, Breitsprecher D, Winter G, Duhr S, Baaske P, Ferguson N (2014) Novel microscale approaches for easy, rapid determination of protein stability in academic and commercial settings. Biochim Biophys Acta Prot Proteom 1844:2241–2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sato S, Ferguson N, Schartau PJ, Fersht AR, Sharpe TD (2004) One‐state downhill versus conventional protein folding. J Mol Biol 344:295–301. [DOI] [PubMed] [Google Scholar]
- 24. Allen MD, Johnson CM, Sato S, Ferguson N, Rutherford TJ, Schartau PJ, Fersht AR, Sharpe TD (2005) Ultra‐fast barrier‐limited folding in the peripheral subunit‐binding domain family. J Mol Biol 353:427–446. [DOI] [PubMed] [Google Scholar]
- 25. Schartau PJ, Ferguson N, Johnson CM, Sharpe TD, Fersht AR (2007) Analysis of “downhill” protein folding. Nature 445:E14–E15. [DOI] [PubMed] [Google Scholar]
- 26. Murray AN, Palhano FL, Bieschke J, Kelly JW (2013) Surface adsorption considerations when working with amyloid fibrils in multiwell plates and Eppendorf tubes. Protein Sci 22:1531–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tripathi A, Gupta K, Khare S, Jain PC, Patel S, Kumar P, Pulianmackal AJ, Aghera N, Varadarajan R (2016) Molecular determinants of mutant phenotypes, inferred from saturation mutagenesis data. Mol Biol Evol 33:2960–2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hamaguchi K, Kurono A (1963) Structure of muramidase (lysozyme) I: effect of guanidine hydrochloride on muramidase. J Biochem 54:111–122. [PubMed] [Google Scholar]
- 29. Loris R, Bahassi EM, Van Melderen L, Poortmans F, Liddington R (1999) Crystal structure of CcdB, a topoisomerase poison from E. coli . J Mol Biol 285:1667–1677. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1 Detection of photobleaching of samples during measurement time scale using two different laser powers. The intensity of five different concentration of HEWL (7.5, 15, 50, 100 and 500 μg/mL, increasing concentration from bottom to top, A‐B and E‐F) and CcdB (7.5, 15, 50, 100 and 500 μg/mL, increasing concentration from bottom to top, C‐D and G‐H) using two different capillaries for each set, was monitored at two different laser powers (100% and 80%) for 60 and 150 minutes respectively at two different emission wavelengths (330 and 350 nm) to detect any visible photobleaching of intrinsic fluorophores present in the protein samples, during the time course of measurement.
Supplementary Figure S2. Detection of photobleaching of samples during measurement time scale at three different temperature. The intensity of five different concentration of HEWL (7.5, 15, 50, 100 and 500 μg/mL, increasing concentration from bottom to top) using two different capillaries for each set, at 100% laser power was monitored at three different temperatures (20 (A, D), 40 (B, E) and 60 (C, F) °C) for 20 minutes at two different emission wavelengths (330 and 350 nm) to detect any visible photobleaching of intrinsic fluorophores present in the protein samples, during the time course of measurement.