

Abstract
In recent years, anthrax toxin has been reengineered to act as a highly specific antiangiogenic cancer therapeutic, shown to kill tumors in animal models. This has been achieved by modifying protective antigen (PA) so that its activation and toxicity require the presence of two proteases, matrix metalloproteinase (MMP) and urokinase plasminogen activator (uPA), which are upregulated in tumor microenvironments. These therapeutics consist of intercomplementing PA variants, which are individually nontoxic, but form functional toxins upon complementary oligomerization. Here, we have created a dual‐protease requiring PA targeting system which utilizes bismaleimide cross‐linked PA (CLPA) rather than the intercomplementing PA variants. Three different CLPA agents were tested and, as expected, found to exclusively form octamers. Two of the CLPA agents have in vitro toxicities equal to those of previous intercomplementing agents, while the third CLPA agent had compromised in vitro cleavage and was significantly less cytotoxic. We hypothesize this difference was due to steric hindrance caused by cross‐linking two PA monomers in close proximity to the PA cleavage site. Overall, this work advances the development and use of the PA and LF tumor‐targeting system as a practical cancer therapeutic, as it provides a way to reduce the drug components of the anthrax toxin drug delivery system from three to two, which may lower the cost and simplify testing in clinical trials.
HIGHLIGHT
Previously, anthrax toxin has been reengineered to act as a highly specific antiangiogenic cancer therapeutic. Here, we present a version, which utilizes bismaleimide cross‐linked protective antigen (PA) rather than intercomplementing PA variants. This advances the development of anthrax toxin as a practical cancer therapeutic as it reduces the components of the drug delivery system to two, which may lower the cost and simplify testing in clinical trials.
Keywords: anthrax toxin, protective antigen, matrix metalloprotease, urokinase plasminogen activator, oligomerization, cancer therapy, bismaleimide cross‐linker
Introduction
The anthrax lethal toxin (LT), secreted by Bacillus anthracis, consists of the cellular binding moiety protective antigen (PA, 83 kDa), and the enzymatic moiety lethal factor (LF, 90 kDa) (for review see Ref. 1). PA binds to cell surface capillary morphogenesis protein 2 and tumor endothelial marker 8 receptors,2, 3 and is cleaved by the cell‐surface furin protease.4, 5 Furin cleavage releases the amino‐terminal 20‐kDa (PA20) fragment, which allows for oligomerization of the receptor‐bound 63‐kDa fragment (PA63) to heptamers or octamers.6, 7 Oligomerization of PA63 creates binding sites for three or four molecules of LF8 and induces endocytosis of the PA63‐LF complexes. Subsequent acidification of the endosome triggers a conformational change in PA, creating a transmembrane pore that translocates LF to the cytosol. LF is a zinc metalloprotease that cleaves and inactivates mitogen‐activated protein kinase kinases (MEKs)9, 10 and NLRP1,11 leading to disruption of cellular signaling and death in experimental animals.12, 13
LT has been engineered to act as a targeted antiangiogenic cancer therapeutic by utilizing its inherent cellular targeting and cytoplasmic delivery system, combined with the consequences of MEK cleavage. This has been achieved by exchanging the native PA furin cleavage site, required for oligomer formation and thus binding and endocytosis of LF, to cleavage sites for proteases that are overexpressed in solid tumors. Singly activated anthrax toxin variants, PA‐L1, activated by matrix metalloproteinases (MMPs)14, 15, 16, 17, 18, 19 and PA‐U2, activated by urokinase plasminogen activator (uPA),20, 21, 22, 23 have been shown to be preferentially activated in tumors, and demonstrate significant tumor‐targeting potential.24, 25
LF binding sites span two adjacent monomers within the PA oligomer.8 This characteristic was utilized to design individually nontoxic PA‐L1 and PA‐U2 variants, which upon complementary oligomerization form a functional toxin.24, 26, 27 These PA oligomers require the combined activities of both cell surface MMP and uPA for activation and cytotoxicity, thereby creating (with appropriate effector moieties) tumor‐targeting agents with decreased off‐target effects and improved therapeutic indices.24, 25 The most studied of these is the intercomplementing second‐generation PA agent IC2PA, consisting of the PA‐L1 I207R and PA‐U2 R200A variants.26 The I207R and R200A substitutions disrupt different LF binding subsites such that native LF binding sites form only upon combining the two PA variants. Another intercomplementing system is composed of the PA‐L1 GN and PA‐U2 D512K variants having complementing substitutions on the PA dimer interface. These PA variants cannot form homo‐oligomers, and are active only when they combine to form heterodimers, which then assemble into functional octamers.27
In this work, we set out to create a PA oligomer which requires dual‐protease activation, as in the agents described previously, but which does not depend on specific PA substitutions like those described above. We achieved this by chemically conjugating two PA monomers, PA‐L1 and PA‐U2, to create a cross‐linked dimer, which can only assemble into an octameric complex. This agent retains the advantage of delivering up to four LF molecules per PA oligomer (like the octameric agent described above) while also reducing the number of drug components from three (PA‐L1, PA‐U2, and LF) to two (PA‐L1 + PA‐U2, LF). Having fewer components in this candidate therapeutic may reduce the time and expense of the preclinical analytical studies needed to qualify this agent for clinical trials.
Results
Cross‐linked PA formation
We set out to chemically conjugate PA‐L1 and PA‐U2 variants to create a cross‐linked PA (CLPA) dimer, which could only assemble into even‐numbered oligomers, of which the octamer is known to be the favored species. This design requires that the cross‐link does not prevent oligomerization of the dimers into an octamer but does prevent homo‐oligomerization of one member of the dimer [Fig. 1(a)]. We chose to accomplish conjugation via sulfhydryl chemistry using homo‐bifunctional bismaleimide cross‐linkers. Wild‐type PA (WTPA) has no native cysteine residues, allowing us to selectively choose the location of PA dimerization. Prior work systematically replaced every residue in PA with cysteine, thereby identifying locations where this substitution is tolerated.29 When engineering sites for PA cross‐linking, we preferentially selected amino acids that were solvent exposed and spatially adjacent within the PA octamer crystal structure (PDB ID: 3HVD).7, 8 Sites for single cysteine substitutions were identified on adjacent monomers in the PA octamer crystal structure using O software,28 and the distances between the single cysteine substitutions were estimated using the PyMol distance measurement tool. Three pairs of PA‐L1 and PA‐U2 variants having single cysteine substitutions were created (Fig. 1, Table S1). Fig. 1(b) lists the cysteine substitutions of each of the three CLPA agents tested, their estimated distances apart, the chosen bismaleimide cross‐linker, and linker length. The location of the conjugation sites within adjacent PA monomers and the PA octamer are illustrated in Figure 1(c). The CLPA1 and CLPA3 cysteine substitutions are located internally within domain 3 and 2 of PA, respectively. The CLPA2 cysteine substitutions are located at the top of domain 2 close to the protease cleavage site (residues 167/168), and therefore should have the greatest flexibility but also the potential to interfere with PA protease activation.
Figure 1.
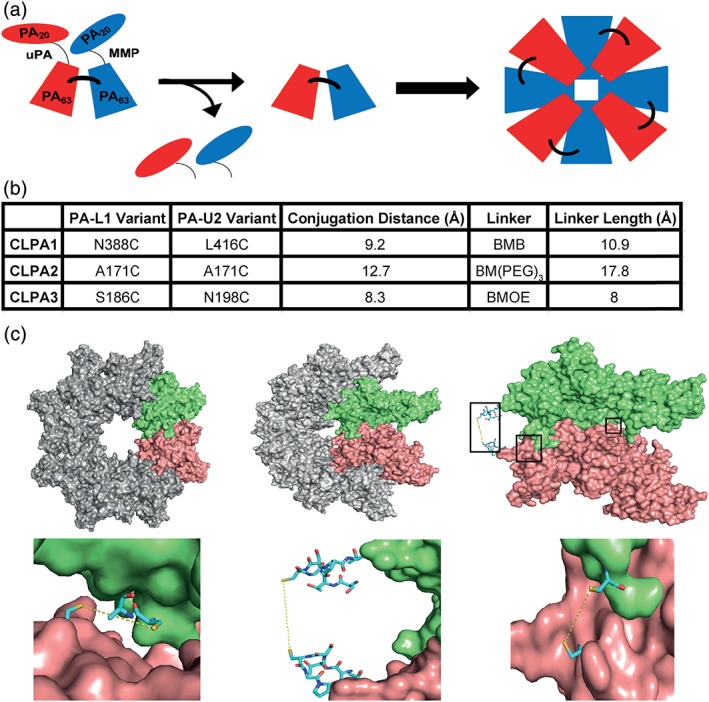
Cross‐linked PA tumor‐targeting agent design. (a) Schematic diagram of the CLPA tumor‐targeting system, which when activated by MMP and uPA proteases forms an octamer with cross‐linked PA components. (b) Table summarizing the three CLPA variants created and tested. The estimated distances between cysteine substitutions within each CLPA variant were calculated using the PyMol distance measurement tool. BMB = 1,4‐bismaleimidobutane, BMOE = bismaleimidoethane, and BM(PEG)3 = 1,11‐bismaleimido‐triethyleneglycol. (c) Illustrations of the PA cross‐linking interface, made using the crystal structure of the PA octamer (PDB ID: 3HVD) in PyMol. Each PA molecule is rendered as a surface, whereas important residues are rendered as stick models. Using O software,28 in adjacent PA monomers of the PA octamer crystal structure, single residues in PA‐L1 and PA‐U2 were substituted with a cysteine. Top left: PA octamer crystal structure with two adjacent PA monomers highlighted in lime and salmon. Top middle: PA octamer crystal structure rotated 45°. Top right: Two adjacent PA monomers within the rotated PA octamer are highlighted. The locations of the three PA cross‐linking sites are highlighted by black boxes to identify the areas expanded in the bottom three images. Bottom left: CLPA1 conjugation. Bottom middle: CLPA2. Bottom right: CLPA3.
The cysteine‐substituted PA variants were expressed with Tobacco Etch Virus (TEV) protease‐cleavable C‐terminal His8 or FLAG tags, that is, PA‐L1‐TEV‐His8 and PA‐U2‐TEV‐Flag (Fig. 1, Table S1). The proteins were successfully conjugated using bismaleimide cross‐linkers [Fig. 2(a)]. For an overview of the PA conjugation protocol, see Figure S1. PA dimerization by bismaleimide linkers was optimized such that the yield of each CLPA agent was approximately 50% of the starting PA monomer, as determined by size‐exclusion chromatography (Fig. S2). CLPA agents could be formed from any combination of the six single cysteine PA variants and the three bismaleimide linkers, and the efficiency of conjugation did not vary significantly between CLPA variants (Fig. S2). This was unsurprising considering all PA cysteines were created at solvent‐exposed sites. For each CLPA agent, a homodimer control reaction was performed where the linker was reacted to the free cysteine on the primary reactant (e.g., PA‐L1‐TEV‐His8), but after removal of excess linker, no secondary reactant (e.g., PA‐U2‐TEV‐Flag) was added. In such reactions, no larger cross‐linked species were found [Fig. 2(a), lanes 3]. This suggests that the protocol prevented homodimer formation. Furthermore, no extraneous nonspecific cross‐linking occurred if WTPA, lacking native cysteines, was used in the conjugation process [Fig. 2(b)]. The post‐conjugation results for each CLPA agent suggested that two major species were present: residual monomer (84 kDa) and a single cross‐linked species, which ran larger than 148 kDa [Fig. 2(a), lanes 4]. All CLPA agents were purified by size‐exclusion chromatography, which yielded distinct PA monomer and dimer peaks (Fig. S2). Subsequent sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS‐PAGE) analysis of the purified CLPA agents indicated that no significant amounts of PA monomer remained [Fig. 2(a), lanes 5].
Figure 2.
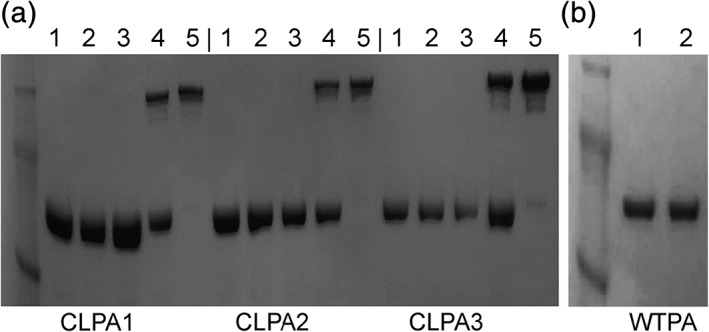
Bismaleimide cross‐linked PA formation and purification. Analysis of cross‐linked PA formation was conducted via SDS‐PAGE. (a) CLPA1‐CLPA3: (1) starting PA‐L1‐TEV‐His8 monomer, (2) starting PA‐U2‐TEV‐Flag monomer, (3) homodimer control reaction where the linker was reacted to the free cysteine on the primary reactant (PA‐L1‐TEV‐His8), but after removal of excess linker, no secondary reactant (PA‐U2‐TEV‐Flag) was added, (4) post‐conjugation CLPA (PA‐L1‐TEV‐His8 + PA‐U2‐TEV‐Flag cross‐linked by a bismaleimide linker and residual PA monomer), (5) size‐exclusion purified CLPA. CLPA1‐3 formation and purification were repeated at least 10 times with no significant difference in conjugation efficiency seen between variants. (b) (1) Initial WTPA, (2) WTPA after cross‐linking protocol. Repeated twice with similar results. A SeeBlue Plus2 prestained protein ladder was used.
Sedimentation velocity experiments were then conducted on purified CLPA1 and its constituent PA monomers (PA‐L1 N388C and PA‐U2 L416C). The input PA proteins each presented as a single homogenous peak (red line) that consisted of 95% of the absorbance signal, with a sedimentation coefficient of 4.84 S and an estimated mass of 83 kDa [Fig. 3(a)]. Purified CLPA1 exhibited the presence of a major 7.64 S species (blue line), which accounted for 89% of the absorbance signal. The sedimentation coefficient and estimated molar mass of 168 kDa indicated that this was the cross‐linked dimer. Even though there was evidence for both faster and slower sedimenting species, no prevalent monomeric species were present. Thus, the bismaleimide conjugation of monomeric PA variants consistently produced a single homogenous cross‐linked species with a mass twice that of monomeric PA. Furthermore, dynamic light scattering (DLS) experiments [Fig. 3(b)] also indicated that purified CLPA1 (blue line) was monodispersed and had a larger radius than its monomeric PA components (red line). In DLS, PA monomer had a calculated radius of 4.3 nm while purified CLPA1 had a calculated radius of 7.3 nm. These values were similar to the hydrodynamic radii calculated by sedimentation velocity for the monomer PA and CLPA species in Figure 3(a), which had radii of 4.1 and 5.6 nm, respectively. It is unsurprising that radii from sedimentation analyses were slightly smaller, as DLS reports on all the species present in the solution with a strong bias toward the larger species that scatter more strongly. Similar results were obtained from analyses of the CLPA2 and CLPA3 agents by sedimentation velocity and DLS (data not shown).
Figure 3.
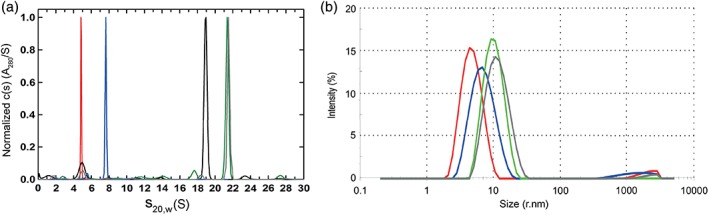
Characterization of CLPA1 and its LF complex by sedimentation velocity and dynamic light scattering. (a) Characterization of PA variant species by analytical ultracentrifugation sedimentation velocity. Normalized absorbance c(s) distributions obtained in SEDFIT. Similar profiles were observed using interference signal. Samples were run in separate cells and profiles overlaid in this single panel, for: PA monomer (red), purified CLPA1 (blue), PA63 + LF heptamer oligomer (black), furin‐cleaved PA‐GN/D512K + LF octamer oligomer (green), and MMP‐ and uPA‐cleaved CLPA1 + LF oligomer (gray). Each sample showed the presence of a single homogenous species. (b) Dynamic light scattering of purified PA variants. As in A, samples were run separately and profiles overlaid for comparison: PA monomer (red), purified CLPA1 (blue), furin‐cleaved PA‐GN and PA‐D512K + LF octamer oligomer (green), and MMP‐ and uPA‐cleaved CLPA1 + LF oligomer (gray). Each sample produced a single homogenous peak, corresponding to radii of 4.9, 7.3, 10.3, and 11.95 nm, respectively. Measurements were made for both experiments at the following concentrations: PA monomer at 0.65 mg/mL, purified CLPA‐1 at 0.61 mg/mL, furin‐cleaved PA‐GN and PA‐D512K + LF octamer oligomer at 0.76 mg/mL, and MMP‐ and uPA‐cleaved CLPA1 + LF oligomer at 0.4 mg/mL.
CLPA in vitro cleavage and characterization of CLPA oligomers
CLPA agents contain both an uPA‐activation site on the PA‐U2 component and a MMP‐activation site on the PA‐L1 component. Therefore, cleavage by one of these proteases should yield a single amino‐terminal 20‐kDa PA fragment (PA20) and a half‐cleaved CLPA species of 148 kDa. Cleavage by both proteases is expected to produce two PA20 fragments and a fully cleaved CLPA species of 128 kDa. Each of the three CLPA agents was treated with MMP9, uPA, or both proteases and the digests were analyzed by SDS‐PAGE (Fig. 4). As expected, all CLPA agents resulted in half‐cleaved CLPA when treated with only one of the two proteases, while a fully cleaved CLPA species was seen when both proteases were utilized. The absence of fully cleaved CLPA upon treatment with one protease further supports the view that no significant amount of homodimer was present. For CLPA1 and CLPA3, time‐dependent cleavage was observed upon treatment with both proteases, with complete cleavage seen by 4 h. CLPA2, however, was only partially cleaved at 4 h. Doubling the concentration of each protease over an 8‐h incubation increased the level of fully cleaved CLPA2 to approximately one‐third, but complete CLPA2 cleavage was not achieved. This difference in proteolytic cleavage efficiency for CLPA2 as compared to CLPA1 and CLPA3 can be attributed to the proximity of the cross‐linking site of CLPA2 to the cleavage site. Because the cross‐linking site is only three residues away from the cleavage site, steric restrictions may limit access by MMP and uPA. Furthermore, when similar in vitro cleavage reactions were carried out with the monomeric PA‐L1‐TEV‐His8 and PA‐U2‐TEV‐Flag components, it was found that less than 2 h was required to obtain full cleavage (Fig. S3). As 4 h was required to obtain full cleavage of CLPA1 and CLPA3, we hypothesize that cleavage of the CLPA agents is less efficient, this effect being most pronounced in CLPA2, where the cross‐link is close to the protease cleavage site. We also noticed that spontaneous cleavage of the CLPA variants can occur in basal conditions, likely caused by the multistep conjugation and cleavage processes where contaminating proteases can be introduced.
Figure 4.
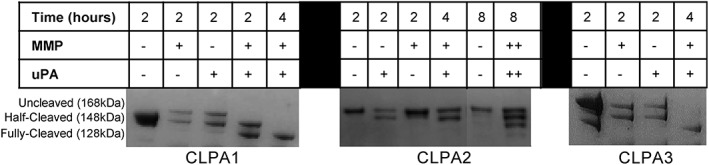
In vitro cleavage of CLPA variants by MMP and uPA. CLPA variants were incubated with MMP9 and/or uPA proteases at 37°C for various time points, and then analyzed by SDS‐PAGE and visualized with Coomassie stain. Cleavage assays were repeated two times with the same CLPA samples and a further two times with CLPAs from different CLPA conjugation preps. Cleavage results were similar in all samples. However, the amount of spontaneous PA cleavage present in basal conditions did change but with no noticeable impact on cleavage results.
The ability of fully cleaved CLPA1 to form octamers and to then bind LF in solution was then investigated by sedimentation velocity, DLS, and native gel electrophoresis. Two previously described PA oligomer species were used as controls in these experiments. WTPA when cleaved into PA63 by furin and run on Q‐Sepharose anion‐exchange resin forms a heptameric oligomer that binds three LF molecules (PA7LF3), to produce a species of approximately 715 kDa.6, 30 Alternatively, an octameric species bound to four LF molecules (PA8LF4) with a molar mass of approximately 868 kDa is produced by interaction of furin‐cleaved PA‐GN and PA‐D512K.27 To characterize the CLPA1 oligomer, the MMP‐ and uPA‐cleaved CLPA1 containing a mixture of partially and fully cleaved CLPA1 was incubated with twofold molar excess of LF and the predominant complex was purified by size‐exclusion chromatography. Native gel electrophoresis (Fig. 5) confirmed that the purified CLPA1 + LF complex formed a single oligomeric species (lane 5) that was the same size as the PA‐GN+ PA‐D512K + LF octamer oligomer (lane 4), and larger than PA63 + LF heptameric PA oligomer (lane 6). A control (lane 7) made by mixing the samples in lanes 5 and 6 showed that the oligomers do not rapidly reassort. These data indicated that the cross‐link within the CLPA1 agent does not prevent PA oligomer formation, and that as expected, CLPA1 forms an octameric species that is able to bind LF. Results were similar for CLPA2 and CLPA3 (Fig. S4).
Figure 5.

Native PhastGel electrophoresis of variant PA oligomer complexes with LF. Native gel electrophoresis of post size‐exclusion PA oligomer complexes was performed. (1) uncleaved CLPA1, (2) uncleaved WTPA, (3) LF, (4) furin‐cleaved PA‐GN/D512K + LF, (5) MMP‐ and uPA‐cleaved CLPA1 + LF, (6) cleaved WTPA + LF, and (7) samples 5 and 6 mixed immediately before electrophoresis.
Sedimentation velocity experiments were also used to characterize the oligomer formed by cleaved CLPA1 in complex with LF [Fig. 3(a)]. The control of PA63 + LF oligomer resulted in a major species, which accounted for 71% of the absorbance signal, at 18.91 S (black line), with a molar mass of 660 ± 50 kDa. This complex was consistent with the expected stoichiometry of seven PA63 units carrying three LF molecules (715 kDa). The complex of cleaved PA‐D512K and PA‐GN + LF showed a major species, which accounted for 71% of the absorbance signal, at 21.40 S (green line), with an estimated molar mass of 790 ± 150 kDa. This corresponds to the expected stoichiometry of eight PA63 units carrying four LF molecules (868 kDa). These results for heptameric PA + LF, and octameric PA + LF, are consistent with previous sedimentation data.27 Sedimentation velocity experiments for purified cleaved CLPA1 + LF verified the presence of a single homogenous species, at 21.53 S (gray line), with an estimated molar mass of 840 ± 170 kDa. Furthermore, DLS measurements of the same CLPA1 + LF complex, as well as the octameric PA‐GN and PA‐D512K in complex with LF, revealed that each sample was monodisperse, and of comparable size [Fig. 3(b), gray and green lines, respectively]. Thus, the results from native gel electrophoresis, sedimentation velocity, and DLS, all confirm that fully cleaved CLPA1 in complex with LF forms a single homogenous oligomeric species that is predominately the octameric species.
CLPA toxicity for cultured tumor cells requires dual‐protease activation
Each of the single cysteine PA‐L1 and PA‐U2 variants, after tag‐removal by TEV protease, was tested for toxicity against Lewis lung carcinoma (LLC) cells [Fig. 6(a)], which express both MMP and uPA.31 FP59, a fusion protein of the N‐terminal 254 amino acids of LF, which is the PA‐binding domain, fused to the catalytic domain of Pseudomonas exotoxin A, was used as the cytotoxic cargo. Exotoxin A ADP‐ribosylates eukaryotic elongation factor 2, inhibiting protein synthesis and leading to cell death.32 All six proteins were highly toxic, having EC50 values of approximately 0.01 nM, as did the parental PA‐L1 and PA‐U2 proteins.
Figure 6.
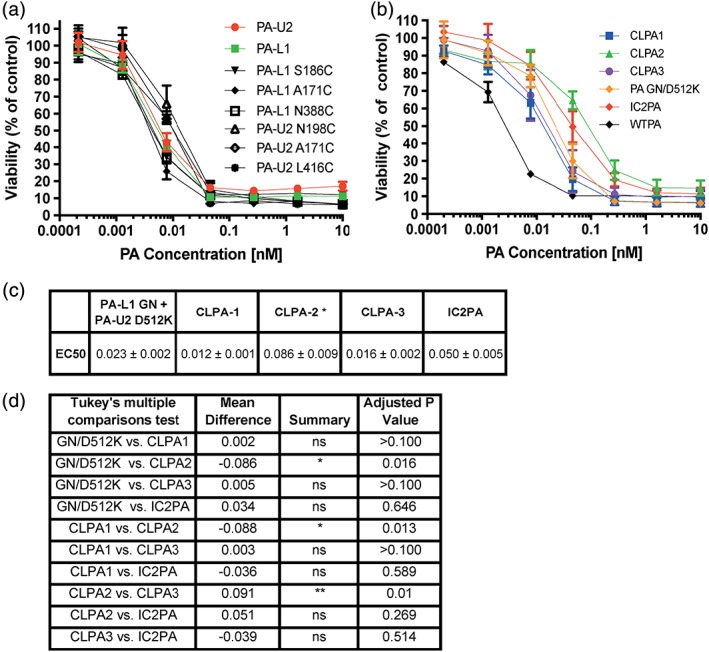
CLPA toxicity on LLC cells. (a, b) LLC cells exhibiting 30% confluence were incubated with serial dilutions of PA variants (0–10 nM) and FP59 (1.9 nM). In (a), cytotoxicities of the monomeric, unconjugated PA‐L1 and PA‐U2 single cysteine variants were assessed. In (b), cytotoxicity of the CLPA variants was compared with WTPA and the two intercomplementing models, IC2PA and PA‐L1 GN + PA‐U2 D512K. In all cases cell viability was measured using an MTT assay after 48 h. (c) Table outlining EC50 (nM) values for PA variant titrations on LLC cells. Errors are displayed as ± error of the mean of at least nine biological replicates. *represents a significant difference in toxicity for the CLPA2 variant as compared to PA‐L1 GN + PA‐U2 D512K, CLPA1 and CLPA3, but no significant difference found as compared to IC2PA. (d) Using GraphPad a one‐way Anova Tukey's multiple comparisons test was completed to compare cytotoxicity of the PA variants to each other in LLC assays. Six biological replicates of calculated EC50 values from experiments containing at least three replicates were used. The **represents a P value of ≤0.0012 and *represents a P value of ≤0.03.
The three CLPA agents were tested for toxicity to LLC cells [Fig. 6(b)], human colorectal adenocarcinoma (HT29) cells, and RAW264.7 macrophages [Fig. S5(A,B)], again in combination with FP59. Like LLC cells, HT29 and RAW264.7 cells also express both MMP and uPA.31 We verified by SDS‐PAGE that the CLPA samples used for these in vitro studies and the subsequent in vivo toxicity studies were free of spontaneously cleaved PA. The CLPA agents were compared to WTPA, as well as to the previously mentioned IC2PA26 and PA‐L1 GN + PA‐U2 D512K27 intercomplementing agents. All three CLPA agents were found to be toxic to all cell lines. The EC50 values of each PA construct's cytotoxicity to LLC cells [Fig. 6(c)] and statistical comparisons using a one‐way ANOVA multiple comparisons analysis [Fig. 6(d)] indicated that no significant differences exist between any of the PA agents except for CLPA2. CLPA2 was significantly less toxic than CLPA1 and PA‐L1 GN + PA‐U2 D512K (P values of ≤0.03) and also CLPA3 (P value ≤0.0012). These findings are consistent with the in vitro cleavage results discussed previously, which found that CLPA2 cleavage was less efficient than for CLPA1 and CLPA3. Similar results were found for RAW264.7 cells but the differences in cytotoxicity between each PA variant is less evident in HT29 cells where no significant differences were found between any of the PA variants.
Because the CLPA agents have both a PA‐L1 and PA‐U2 component, their toxicity for cells should require that both proteases be present. Consistent with this requirement, it was found that HeLa cells, which do not express uPA,21 were not sensitive to CLPA variants, even at a very high 10 nM PA concentration [Fig. 7(a)]. Additionally, the MMP inhibitors GM6001 and TIMP‐2 blocked the activation of all CLPA variants and significantly decreased killing of LLC cells [Fig. 7(b)]. These findings support requirements for activation by both proteases.
Figure 7.
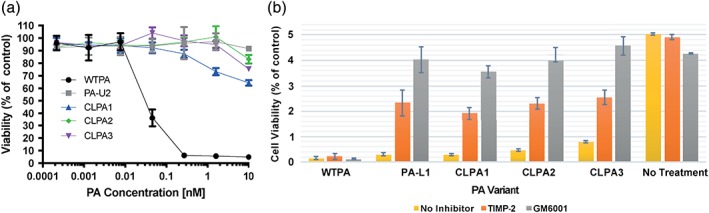
CLPA toxicity requires dual‐protease activation. (a) HeLa cells, which do not express uPA, exhibiting 50% confluence were incubated for 6 h with serial dilutions of PA variants (0–10 nM) and FP59 at 1.9 nM. Following incubation, the medium was replaced and cell viability was measured using an MTT assay after 48 h. (b) PA variant toxicity to LLC cells was assessed in the presence or absence of MMP inhibitors, GM6001 and TIMP‐2. Both inhibitors, GM6001 at 40 μM and tissue inhibitor of metalloproteinases TIMP‐2 at 20 μM, significantly reduced toxicity of CLPA variants as compared to the untreated control. In (a) and (b) results were averaged across three experiments containing four replicates of each sample.
CLPA has potent antitumor activity in vivo
To evaluate the therapeutic potential of the most promising CLPA candidate, CLPA1, we inoculated immunocompetent C57BL/6J mice intradermally with syngeneic LLC, which forms rapidly disseminating tumors in mice. When tumors had reached volumes of approximately 150 mm3 (0.75% of mouse body volume), the mice were treated five times with either PBS or with varying amounts of CLPA1 with LF (Fig. 8). As expected, tumors in mice treated with PBS grew rapidly and reached a volume of approximately 5% of mouse body volume within 9 days. Interestingly, however, CLPA1/LF‐treated mice displayed a dose‐dependent reduction in tumor growth, with tumors in mice treated with the highest dose of CLPA1 displaying only marginal growth within the 11‐day treatment period [Fig. 8(a)]. Furthermore, CLPA1 with LF was well tolerated at all doses administered, as determined by outward appearance and body weight measurements [Fig. 8(b)]. Treatment with LF or CLPA alone was not included because many previous studies have shown that neither protein affects mouse health or tumor weight.
Figure 8.
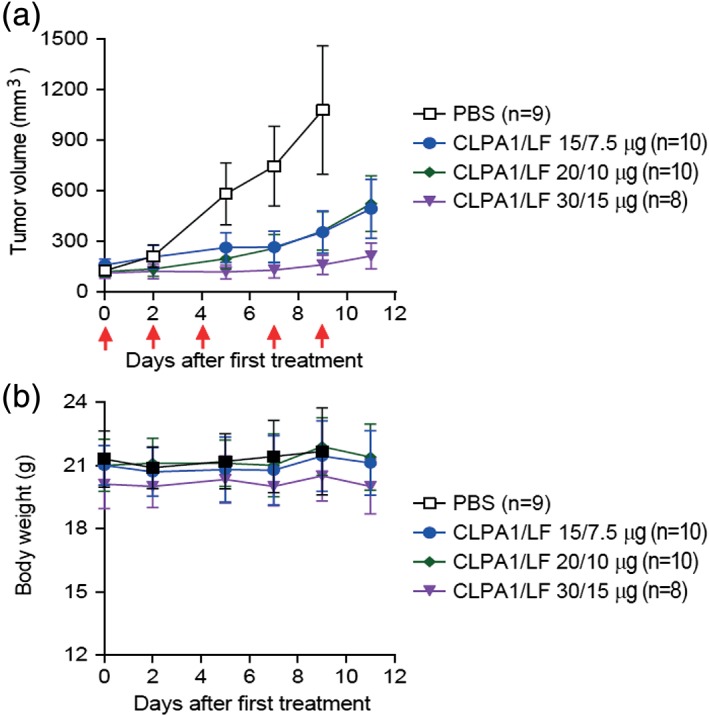
CLPA has potent antitumor activity when administered systemically to mice. C57BL/6J mice were inoculated intradermally with syngeneic LLC Lewis lung carcinoma cells. When tumors had formed, the mice were randomized into four groups (each of 8–10 mice, as indicated) that were treated with five systemic administrations (red arrows) of either PBS (open black boxes) or CLPA1 + LF at the doses shown. (a) Tumor volume and (b) body weight, as determined prior to each injection. Tumor volumes, mean ± SE; body weights, mean ± SD. One‐way ANOVA analysis (two‐tailed) for tumor size differences: PBS versus all other groups, P < 0.01; CLPA1/LF (15 μg/7.5 μg) and CLPA1/LF (20 μg/10 μg) versus CLPA1/LF (30 μg/15 μg), P < 0.05. At Day 9, the mice in the PBS group were terminated, whereas no mice were lost in any of the treated groups.
Discussion
The inherent cellular targeting and cytoplasmic delivery system of anthrax toxin has become one of the best‐characterized mechanisms for delivery of proteins into cells.33, 34, 35, 36 In recent years, anthrax toxin has been reengineered to act as a targeted antiangiogenic cancer therapeutic shown to kill tumors in animal models and to display excellent therapeutic indices.25, 26, 31, 37, 38 Two conceptually distinct features were added to PA to accomplish high specificity in tumor targeting. First, the native furin cleavage site on PA, which is required for toxin activation, was exchanged to protease‐specific sequences cleaved by MMP and uPA, proteases that are upregulated in the tumor microenvironment. Second, additional changes were added into PA that introduced the requirement that individually nontoxic PA variants must be combined to generate an active oligomer, either a heptamer or octamer. These “intercomplementing” mixtures of PA variants that require both MMP and uPA activation produce highly specific tumor‐targeting agents.
In this article, we have expanded upon this approach and established a PA and LF tumor‐targeting system, which requires dual‐protease activation, but does not contain the intercomplementing PA substitutions. This was achieved by chemically conjugating single cysteine PA‐L1 and PA‐U2 variants using a bismaleimide cross‐linker. Three cross‐linked PA variants (CLPA1‐3) were designed and investigated. The PA dimerization reaction was optimized to near 50% efficiency, offering a bismaleimide cross‐linking yield sufficient for the creation of heterobifunctional protein dimers. Using size‐exclusion chromatography, pure homogenous samples of each CLPA variant were obtained. Their ability to form functional oligomers toxic to tumors in vitro and in vivo was investigated in this article.
As expected, in vitro cleavage studies using MMP and uPA showed that each CLPA formed a half‐cleaved species when one protease was added. Treatment with both proteases was required to produce a fully cleaved form that was able to oligomerize. However, as compared to noncross‐linked PA, cleavage appeared to be less efficient, and this effect was more prominent in CLPA2 compared to CLPA1 and CLPA3. CLPA + LF oligomeric complexes were then characterized by several biophysical methods and results suggest that each CLPA variant could form functional oligomers of predominately the octameric type.
We then compared the cytotoxicity of the three CLPA variants to each other and to the previously studied IC2PA and PA‐L1 GN + PA‐U2 D512K intercomplementing PA agents. We found that CLPA1 and CLPA3 had cellular and in vivo toxicities comparable to those of the previous intercomplementing agents. Interestingly, the CLPA2 variant comprises two adjacent PA monomers cross‐linked via residues immediately C‐terminal to each protease cleavage site, which had impaired in vitro cleavage, was also found to be significantly less toxic in vitro to LLC cells. This result provides insight into potential steric hindrance issues when linking two PA molecules. However, the fact that cross‐linking at two other sites does not seem to affect PA oligomer functionality or levels of toxicity is intriguing and offers interesting insights into the dynamics of PA pore maturation and LF translocation mechanisms. Future studies could investigate how conjugation at the CLPA2 site affects PA cleavage and toxicity and other PA cross‐linking sites could be designed and analyzed to further enhance our understanding of toxin assembly, maturation, and function. Finally, antitumor studies in mice revealed that CLPA1 treatment could nearly halt tumor growth with no visible dose‐toxicity affecting mouse health, suggesting that CLPA1 could provide excellent therapeutic potential.
Overall, this work further establishes anthrax toxin as a powerful tool for the delivery of proteins and drugs into cells. The ability of PA to be modified in numerous ways and the growing number of new LF drug‐conjugates makes this system a highly versatile tool not just for antitumor therapies but also other areas of biomedicine. Importantly, the CLPA agent provides a crucial step in the development and use of the PA and LF tumor‐targeting system as a practical cancer therapeutic. During all steps of preclinical and clinical drug development, drugs are tested and approved as a monotherapy and in combination. Because no components of the anthrax toxin delivery system are previously approved therapeutics, reducing the drug components from three, as is in the IC2PA and PA‐GN/NS targeting systems, to two components in the CLPA targeting system presented here, may serve to lower the cost and simplify testing in preclinical and clinical trials.
Experimental Procedures
Toxins
The Q5 site‐directed mutagenesis kit (New England Biolabs, Ipswich, MA, E0554S) was used to create single cysteine PA‐L1 and PA‐U2 variants. The Q5 mutagenesis primers used are outlined in Table S1. A His8‐tag was added to PA‐L1 and Flag‐tag was added to PA‐U2, to aid in purification and conjugation. PA‐L1‐His8 variants created were A171C, N388C, and S186C. PA‐U2‐Flag variants created were A171C, N198C, and L416C. All PA gene sequences were confirmed by sequencing (Macrogen, Rockville, MD).
PA variants, LF, and FP59 were expressed and purified as previously described.32, 39, 40 PA‐L1 and PA‐U2 monomers with cysteine substitutions were run through an additional size‐exclusion step, using a HiLoad 16/600 Superdex 200 pg column (GE Healthcare Life Sciences, Pittsburgh, PA), in a buffer of 10 mM Tris–HCl, 1 mM EDTA, 150 mM NaCl, and 2 mM 2‐mercaptoethanol (BME). The LF and the FP59 used in this article contained the native AGG N‐terminal sequence. Expected molecular weights were confirmed by mass spectrometry.
PA variants with intact C‐terminal His8 and FLAG tags were used in cytotoxicity measurements (Figs. 6 and 7), where preliminary data showed they had no effect, and in the AUC experiment [Fig. 3(a)]. All other experiments used protein from which the tags were removed by cleavage with TEV protease. The His6‐TEV(S219V)‐Arg5 variant protease was produced in this lab from plasmid pRK793,41 a gift of David Waugh, obtained from Addgene (Cambridge, MA) as plasmid #8827. Portions of 100 μg of the PA proteins were incubated with 1 μg of TEV protease at 4°C overnight in the CLPA size‐exclusion buffer. Removal of the tags was verified by His and Flag immunoblotting.
Linkers
The three bismaleimide linkers used in this article, obtained from Thermo Fisher Scientific (Waltham, MA), were bismaleimidoethane (BMOE, 22323), 1,4‐bismaleimidobutane (BMB, 22331), and 1,11‐bismaleimido‐triethyleneglycol (BM(PEG)3, 22337). Linkers were prepared as 20 mM stocks in 100% dimethyl sulfoxide.
CLPA conjugation and purification
The PA‐L1 and PA‐U2 single cysteine variants at 50 μM were reduced separately with 5 mM dithiothreitol (DTT) for 1 h at 4°C. Sephadex G‐25 PD‐10 desalting columns (GE Healthcare Life Sciences, 17085101), pre‐equilibrated with 25 mL of conjugation buffer (10 mM Tris–HCl, 1 mM EDTA, 300 mM NaCl), were used to remove excess DTT and buffer exchange proteins into the conjugation buffer. Then a 100‐fold molar excess of the bismaleimide linker, from a 20‐mM stock, was added to the primary reactant (PA‐L1‐His8) and incubated at 25°C on a shaker. After a 20‐min reaction period, this sample was then added to another pre‐equilibrated PD10 column to remove excess linker. The elutate containing PA‐L1 with its single cysteine conjugated at one end to the linker was combined with the desalted and reduced PA‐U2 single cysteine protein in an Amicon Ultra‐4 Centrifugal Filter Unit with a 30‐kDa cutoff membrane (EMD Millipore, UFC803096), concentrated to approximately 100 μM, and left on ice for 1 h. This final concentration step was found to be crucial to obtaining high dimer yield. The final reaction was quenched with 1 mM free cysteine. CLPA yield was assessed by SDS‐PAGE. A HiLoad 16/600 Superdex 200 Prep grade column (GE Healthcare Life Sciences) was used to purify CLPA from unreacted PA monomer at room temperature in a buffer of 10 mM Tris–HCl, 300 mM NaCl, 1 mM EDTA, and 0.5 mM BME.
In vitro cytotoxicity
LLC,42 human colon carcinoma cell line (HT29),43 murine macrophage cell line RAW264.7, and cervical carcinoma cell line HeLa44 were used to assess toxicity of PA variants. Cells were grown in 5% CO2 using Dulbecco's modified Eagle's medium (DMEM) with Glutamax (Thermo Fisher, 10566) supplemented with 10% fetal bovine serum, 10 mM HEPES buffer, pH 7.3, and 10 μg/mL gentamycin sulfate.
The cytotoxicity of each CLPA variant was assessed and compared to IC2PA,26 PA‐L1 GN + PA‐U2 D512K,27 and WTPA, using a colorimetric 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT) viability assay as previously described. LLC and RAW264.7 cells exhibiting 30–35% confluence and HT29 and HeLa cells exhibiting 45% confluence were treated with serial dilutions of purified PA variants starting in the presence of a constant concentration of FP59 (1.9 nM). Cell viability was measured after 48 h at 37°C. PA variant toxicity was also assessed in the presence or absence of MMP inhibitors. Either GM6001 (EMD Millipore, Billerica, MA, CC1010) or tissue inhibitor of metalloproteinases 2 (TIMP‐2, EMD Millipore, PF021) was added to cells in serum‐free DMEM at 40 or 20 μM, respectively, for 30 min prior to toxin addition (PA 300 ng/mL + FP59 50 ng/mL). After 6 h the cells were washed in serum‐free DMEM three times and incubated for 48 h prior to measuring viability by MTT assay.
In vitro PA cleavage
Digestion of WTPA and PA‐GN + PA‐D512K with furin (New England BioLabs, P8077L) was carried out as previously described.21 Monomeric PA‐L1 and PA‐U2 single cysteine variants and CLPA variants (100 μg/mL) were dialyzed into 10 mM Tris–HCl and 300 mM NaCl and then incubated with 10 μg/mL of one or both matrix metalloproteinase 9 (MMP9) (BioVision, Milpitas, CA, 7867), or uPA (EMD Millipore, CC4000), at 37°C. Aliquots were analyzed by SDS‐PAGE at 2, 4, and 8 h time points. MMP9 and uPA cleavages were conducted in a cleavage buffer of 50 mM HEPES, pH 7.5, 10 mM CaCl2, 200 mM NaCl, 0.05% Brij35, and 50 μM ZnSO4.
PA oligomer formation and purification
Furin‐cleaved WTPA was oligomerized on a MonoQ anion‐exchange column as previously described,45 and the resulting heptameric PA63 was then mixed with stoichiometric LF. CLPA cleaved by MMP and uPA (containing a mixture of fully cleaved, partially cleaved and uncleaved CLPA), and furin‐cleaved PA‐GN + PA‐D512K were combined in the presence of LF at twofold molar excess for 30 min and then purified with a Superdex‐200 Increase 10/300 GL column (GE Healthcare Life Sciences, 28990994). The column was run at 0.4 mL/min at room temperature using a buffer of 10 mM Bis‐Tris propane buffer, pH 9.0, and 300 mM NaCl. Fractions containing oligomer (as analyzed by SDS‐PAGE and native gel electrophoresis) were pooled and concentrated for further analysis. Native gel electrophoresis of PA complexes was performed as previously described.45, 46
Sedimentation velocity
Sedimentation velocity experiments were carried out at 20°C in a Beckman Optima XL‐A or Beckman Coulter ProteomeLab XI‐I analytical ultracentrifuge following standard protocols.47 A stoichiometric mixture of CLPA1 monomeric components (PA‐L1 N388C and PA‐U2 L416C) in 10 mM Tris–HCl, 300 mM NaCl, 2 mM BME, 2 mM EDTA, and pH 8.0 was studied at 0.65 mg/mL (0.325 mg/mL of each). Size‐exclusion chromatography purified CLPA1 in 10 mM Tris–HCl, pH 8.0, 400 mM NaCl, 0.25 mM BME, 2 mM EDTA was studied at 0.61 mg/mL. Cleaved PA variants in complex with LF were obtained in 10 mM Bis‐Tris propane, pH 9.0, and 350 mM NaCl. The heptameric form of cleaved WTPA + LF (PA7LF3) was analyzed at 0.43 mg/mL, whereas the octameric constructs of PA, cleaved PA‐GN and PA‐D512K + LF (PA8LF4) were analyzed at 0.72 mg/mL. Cleaved CLPA1 + LF (CLPA4LF4) was studied at 0.35 mg/mL. Monomeric PA, CLPA1, and CLPA oligomer were analyzed at 50,000 rpm, while the WTPA heptamer and PA‐GN/DK octamer were analyzed at 25,000 rpm. Samples were loaded in 2‐channel centerpiece cells and data collected using the absorbance (280 nm) and interference (655 nm, when available) optical detection systems. Sedimentation data were time‐corrected,48 and analyzed in SEDFIT 15.01c49 in terms of a continuous c(s) distribution of Lamm equation solutions with a maximum entropy regularization confidence level of 0.68. Excellent data fits were observed with r.m.s.d. values of 0.0031–0.0071 absorbance units and 0.0038–0.0086 fringes. Solution densities and viscosities were measured experimentally at 20°C on an Anton Paar DMA 5000 density meter and Anton Paar AMVn automated rolling ball viscometer, or determined based on the composition in SEDNTERP.50 Protein partial specific volumes were calculated based on the amino acid composition in SEDNTERP, and sedimentation coefficients, s, were corrected to s 20,w values under standard conditions.
Dynamic light scattering
The homogeneity of purified PA variant protein preparations was assessed by DLS. DLS measurements were made with a 10‐mm path length cuvette at 25°C using a NanoS Zetasizer (Malvern Instruments, Malvern, UK). All samples were centrifuged at 13,000 rpm for 10 min directly before measurement and screened in a disposable polystyrene cuvette (Malvern, ZEN0040) with a 40 μL sample volume. Data were processed using Malvern Zetasizer software. Measurements were made at the following concentrations: PA monomer at 0.65 mg/mL, CLPA1 at 0.61 mg/mL, furin‐cleaved PA‐GN and PA‐D512K + LF at 0.76 mg/mL, and MMP‐ and uPA‐cleaved CLPA1 + LF at 0.4 mg/mL.
Animal studies
All animal studies were carried out in accordance with protocols approved by the National Institute of Allergy and Infectious Diseases Animal Care and Use Committee. Female 10–14‐week‐old C57BL/6J mice were injected with 5 × 105 LLC cells in the mid‐scapular dermis. Tumors were measured at intervals with digital calipers (FV Fowler Company, Inc., Newton, MA) and tumor volumes were estimated from the length, width, and height of tumors using the formula: tumor volume (mm3) = ½ (length × width × height in mm). When tumors reached about 150 mm3, mice were randomized into groups and injected intraperitoneally following schedules indicated in Figure 8, with either PBS or the engineered toxins. Mice were weighed, and the tumors were measured before each injection.
Statistics
EC50 values for viability curves were calculated using GraphPad Prism 7. Curves of PA variants versus cell viability were fit using the nonlinear regression function “[inhibitor] versus normalized response.” At least nine biological replicates were used. Statistical significance between EC50 values was determined using a one‐way ANOVA Tukey's multiple comparisons test in GraphPad Prism 7. Six biological replicates of calculated EC50 values from experiments containing at least three replicates were used. One‐way ANOVA analysis (two‐tailed) was used for analysis of tumor size differences.
Conflict of Interests
The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Supporting information
Table S1 Primers used in creation of CLPA variants.
Figure S1: Flow chart outlining CLPA formation from monomeric PA.
Figure S2: Purification of CLPA by size‐exclusion chromatography. A) Two distinct peaks with nearly no overlap, a cross‐linked PA peak at 58‐70 mL and a PA monomer peak at 70‐80 mL, are seen when a mixture of monomeric PA and CLPA dimer are purified by size‐exclusion. Evidence of larger species is seen on the front edge of the dimer peak. B) SDS‐PAGE analysis of all fractions confirms the presence of larger species on the edge of the dimer peak, and small amount of residual monomer present in each dimer peak fraction. C) SDS‐PAGE analysis when only the dimer peak fractions were pooled, excluding the fractions containing larger species. Sedimentation velocity confirmed this sample did not contain a significant amount of larger species and less than 5% of the total amount is PA monomer. It is suggested that PA monomer seen by SDS‐PAGE results from break down of the bismaleimide linker when boiling samples in samples buffer containing 10 mM Tris–HCl buffer and SDS.
Figure S3: In vitro cleavage of CLPA1 monomeric components by MMP9 and uPA. A mixture of CLPA1 monomeric components (PA‐L1 N388C and PA‐U2 L416C), prior to dimerization, were incubated with none, one, or both of MMP9 and uPA for 2 h at 37°C, and then analyzed by SDS‐PAGE. The SeeBlue Plus2 pre‐stained protein ladder was used (left lane). Complete cleavage of both monomeric components is seen after 2 h only when both proteases are added. Results were similar for CLPA2 and CLPA3 monomeric components (data not shown).
Figure S4: Native PhastGel electrophoresis of CLPA1‐3 oligomers. LF (lane 1), untreated CLPA1‐3 (lanes 2‐4), MMP & uPA cleaved CLPA1‐3 in complex with LF (lanes 5‐7), furin cleaved PA‐GN + PA‐D512K + LF (lane 8). All CLPA1‐3 oligomers run at the same size as the PA‐GN + PA‐D512K + LF oligomer, previously shown to be an octamer. These results show that all CLPA variants assemble as octamers.
Figure S5: CLPA toxicity on RAW264.7 cells (A) and HT29 cells (B). (A) RAW264.7 cells exhibiting 65% confluence were incubated with serial dilutions of PA variants (0–7 nM) and LF (10 nM) for 5 h (B) HT29 cells exhibiting 45% confluence were incubated with serial dilutions of PA variants (0‐10 nM) and FP59 (1.9 nM) for 48 h. Cell viability was then measured using an MTT assay.
Acknowledgments
We thank Apostolos Gittis and David Garboczi for performing DLS experiments and helping with figure modeling. Mahtab Moayeri and Jonathan Renn provided experimental advice and troubleshooting. This research was supported by funds from the Divisions of Intramural Research of the National Institute of Dental and Craniofacial Research, the National Institute of Allergy and Infectious Diseases, and the National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health.
References
- 1. Liu S, Moayeri M, Leppla SH (2014) Anthrax lethal and edema toxins in anthrax pathogenesis. Trends Microbiol 22:317–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bradley KA, Mogridge J, Mourez M, Collier RJ, Young JA (2001) Identification of the cellular receptor for anthrax toxin. Nature 414:225–229. [DOI] [PubMed] [Google Scholar]
- 3. Scobie HM, Rainey GJ, Bradley KA, Young JA (2003) Human capillary morphogenesis protein 2 functions as an anthrax toxin receptor. Proc Natl Acad Sci U S A 100:5170–5174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Klimpel KR, Molloy SS, Thomas G, Leppla SH (1992) Anthrax toxin protective antigen is activated by a cell surface protease with the sequence specificity and catalytic properties of furin. Proc Natl Acad Sci U S A 89:10277–10281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Molloy SS, Bresnahan PA, Leppla SH, Klimpel KR, Thomas G (1992) Human furin is a calcium‐dependent serine endoprotease that recognizes the sequence Arg‐X‐X‐Arg and efficiently cleaves anthrax toxin protective antigen. J Biol Chem 267:16396–16402. [PubMed] [Google Scholar]
- 6. Milne JC, Furlong D, Hanna PC, Wall JS, Collier RJ (1994) Anthrax protective antigen forms oligomers during intoxication of mammalian cells. J Biol Chem 269:20607–20612. [PubMed] [Google Scholar]
- 7. Kintzer AF, Thoren KL, Sterling HJ, Dong KC, Feld GK, Tang II, Zhang TT, Williams ER, Berger JM, Krantz BA (2009) The protective antigen component of anthrax toxin forms functional octameric complexes. J Mol Biol 392:614–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cunningham K, Lacy DB, Mogridge J, Collier RJ (2002) Mapping the lethal factor and edema factor binding sites on oligomeric anthrax protective antigen. Proc Natl Acad Sci U S A 99:7049–7053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Duesbery NS, Webb CP, Leppla SH, Gordon VM, Klimpel KR, Copeland TD, Ahn NG, Oskarsson MK, Fukasawa K, Paull KD, Vande Woude GF (1998) Proteolytic inactivation of MAP‐kinase‐kinase by anthrax lethal factor. Science 280:734–737. [DOI] [PubMed] [Google Scholar]
- 10. Vitale G, Pellizzari R, Recchi C, Napolitani G, Mock M, Montecucco C (1998) Anthrax lethal factor cleaves the N‐terminus of MAPKKs and induces tyrosine/threonine phosphorylation of MAPKs in cultured macrophages. Biochem Biophys Res Commun 248:706–711. [DOI] [PubMed] [Google Scholar]
- 11. Levinsohn JL, Newman ZL, Hellmich KA, Fattah R, Getz MA, Liu S, Sastalla I, Leppla SH, Moayeri M (2012) Anthrax lethal factor cleavage of Nlrp1 is required for activation of the inflammasome. PLoS Pathog 8:e1002638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liu S, Zhang Y, Moayeri M, Liu J, Crown D, Fattah RJ, Wein AN, Yu ZX, Finkel T, Leppla SH (2013) Key tissue targets responsible for anthrax‐toxin‐induced lethality. Nature 501:63–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Moayeri M, Haines D, Young HA, Leppla SH (2003) Bacillus anthracis lethal toxin induces TNF‐á‐independent hypoxia‐mediated toxicity in mice. J Clin Invest 112:670–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Liu S, Netzel‐Arnett S, Birkedal‐Hansen H, Leppla SH (2000) Tumor cell‐selective cytotoxicity of matrix metalloproteinase‐activated anthrax toxin. Cancer Res 60:6061–6067. [PubMed] [Google Scholar]
- 15. Liu S, Wang H, Currie BM, Molinolo A, Leung HJ, Moayeri M, Basile JR, Alfano RW, Gutkind JS, Frankel AE, Bugge TH, Leppla SH (2008) Matrix metalloproteinase‐activated anthrax lethal toxin demonstrates high potency in targeting tumor vasculature. J Biol Chem 283:529–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Alfano RW, Leppla SH, Liu S, Bugge TH, Ortiz JM, Lairmore TC, Duesbery NS, Mitchell IC, Nwariaku F, Frankel AE (2010) Inhibition of tumor angiogenesis by the matrix metalloproteinase‐activated anthrax lethal toxin in an orthotopic model of anaplastic thyroid carcinoma. Mol Cancer Ther 9:190–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Alfano RW, Leppla SH, Liu S, Bugge TH, Meininger CJ, Lairmore TC, Mulne AF, Davis SH, Duesbery NS, Frankel AE (2009) Matrix metalloproteinase‐activated anthrax lethal toxin inhibits endothelial invasion and neovasculature formation during in vitro morphogenesis. Mol Cancer Res 7:452–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Alfano RW, Leppla SH, Liu S, Bugge TH, Herlyn M, Smalley KS, Bromberg‐White JL, Duesbery NS, Frankel AE (2008) Cytotoxicity of the matrix metalloproteinase‐activated anthrax lethal toxin is dependent on gelatinase expression and B‐RAF status in human melanoma cells. Mol Cancer Ther 7:1218–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Alfano RW, Leppla SH, Liu S, Bugge TH, Duesbery NS, Frankel AE (2008) Potent inhibition of tumor angiogenesis by the matrix metalloproteinase‐activated anthrax lethal toxin: implications for broad anti‐tumor efficacy. Cell Cycle 7:745–749. [DOI] [PubMed] [Google Scholar]
- 20. Liu S, Aaronson H, Mitola DJ, Leppla SH, Bugge TH (2003) Potent antitumor activity of a urokinase‐activated engineered anthrax toxin. Proc Natl Acad Sci U S A 100:657–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liu S, Bugge TH, Leppla SH (2001) Targeting of tumor cells by cell surface urokinase plasminogen activator‐dependent anthrax toxin. J Biol Chem 276:17976–17984. [DOI] [PubMed] [Google Scholar]
- 22. Su Y, Ortiz J, Liu S, Bugge TH, Singh R, Leppla SH, Frankel AE (2007) Systematic urokinase‐activated anthrax toxin therapy produces regressions of subcutaneous human non‐small cell lung tumor in athymic nude mice. Cancer Res 67:3329–3336. [DOI] [PubMed] [Google Scholar]
- 23. Abi‐Habib RJ, Singh R, Liu S, Bugge TH, Leppla SH, Frankel AE (2006) A urokinase‐activated recombinant anthrax toxin is selectively cytotoxic to many human tumor cell types. Mol Cancer Ther 5:2556–2562. [DOI] [PubMed] [Google Scholar]
- 24. Liu S, Redeye V, Kuremsky JG, Kuhnen M, Molinolo A, Bugge TH, Leppla SH (2005) Intermolecular complementation achieves high‐specificity tumor targeting by anthrax toxin. Nat Biotechnol 23:725–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Peters DE, Hoover B, Cloud LG, Liu S, Molinolo AA, Leppla SH, Bugge TH (2014) Comparative toxicity and efficacy of engineered anthrax lethal toxin variants with broad anti‐tumor activities. Toxicol Appl Pharmacol 279:220–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wein AN, Peters DE, Valivullah Z, Hoover BJ, Tatineni A, Ma Q, Fattah R, Bugge TH, Leppla SH, Liu S (2015) An anthrax toxin variant with an improved activity in tumor targeting. Sci Rep 5:16267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Phillips DD, Fattah RJ, Crown D, Zhang Y, Liu SH, Moayeri M, Fischer ER, Hansen BT, Ghirlando R, Nestorovich EM, Wein AN, Simons L, Leppla SH, Leysath CE (2013) Engineering anthrax toxin variants that exclusively form octamers and their application to targeting tumors. J Biol Chem 288:9058–9065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jones TA (2004) Interactive electron‐density map interpretation: from INTER to O . Acta Crystallogr D60:2115–2125. [DOI] [PubMed] [Google Scholar]
- 29. Mourez M, Yan M, Lacy DB, Dillon L, Bentsen L, Marpoe A, Maurin C, Hotze E, Wigelsworth D, Pimental RA, Ballard JD, Collier RJ, Tweten RK (2003) Mapping dominant‐negative mutations of anthrax protective antigen by scanning mutagenesis. Proc Natl Acad Sci U S A 100:13803–13808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Blaustein RO, Koehler TM, Collier RJ, Finkelstein A (1989) Anthrax toxin: channel‐forming activity of protective antigen in planar phospholipid bilayers. Proc Natl Acad Sci U S A 86:2209–2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schafer JM, Peters DE, Morley T, Liu S, Molinolo AA, Leppla SH, Bugge TH (2011) Efficient targeting of head and neck squamous cell carcinoma by systemic administration of a dual uPA and MMP‐activated engineered anthrax toxin. PLoS One 6:e20532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Arora N, Leppla SH (1993) Residues 1‐254 of anthrax toxin lethal factor are sufficient to cause cellular uptake of fused polypeptides. J Biol Chem 268:3334–3341. [PubMed] [Google Scholar]
- 33. Liu S, Schubert RL, Bugge TH, Leppla SH (2003) Anthrax toxin: Structures, functions and tumour targeting. Expert Opin Biol Ther 3:843–853. [DOI] [PubMed] [Google Scholar]
- 34. Young JA, Collier RJ (2007) Anthrax toxin: Receptor‐binding, internalization, pore formation, and translocation. Annu Rev Biochem 76:243–265. [DOI] [PubMed] [Google Scholar]
- 35. Rabideau AE, Pentelute BL (2016) Delivery of non‐native cargo into mammalian cells using anthrax lethal toxin. ACS Chem Biol 11:1490–1501. [DOI] [PubMed] [Google Scholar]
- 36. Verdurmen WP, Luginbuhl M, Honegger A, Pluckthun A (2015) Efficient cell‐specific uptake of binding proteins into the cytoplasm through engineered modular transport systems. J Control Release 200:13–22. [DOI] [PubMed] [Google Scholar]
- 37. Liu S, Ma Q, Fattah R, Bugge TH, Leppla SH (2017) Anti‐tumor activity of anthrax toxin variants that form a functional translocation pore by intermolecular complementation. Oncotarget 8:65123–65131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Liu S, Liu J, Ma Q, Cao L, Fattah RJ, Yu Z, Bugge TH, Finkel T, Leppla SH (2016) Solid tumor therapy by selectively targeting stromal endothelial cells. Proc Natl Acad Sci U S A 113:E4079–E4087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ramirez DM, Leppla SH, Schneerson R, Shiloach J (2002) Production, recovery and immunogenicity of the protective antigen from a recombinant strain of Bacillus anthracis . J Ind Microbiol Biotechnol 28:232–238. [DOI] [PubMed] [Google Scholar]
- 40. Leppla SH. The anthrax toxin complex In: Alouf JE, Freer JH, Eds. (1991) Sourcebook of bacterial protein toxins. London: Academic Press; p. 277–302. [Google Scholar]
- 41. Kapust RB, Tozser J, Fox JD, Anderson DE, Cherry S, Copeland TD, Waugh DS (2001) Tobacco etch virus protease: mechanism of autolysis and rational design of stable mutants with wild‐type catalytic proficiency. Protein Eng 14:993–1000. [DOI] [PubMed] [Google Scholar]
- 42. O'Reilly MS, Holmgren L, Shing Y, Chen C, Rosenthal RA, Moses M, Lane WS, Cao Y, Sage EH, Folkman J (1994) Angiostatin: a novel angiogenesis inhibitor that mediates the suppression of metastases by a Lewis lung carcinoma. Cell 79:315–328. [DOI] [PubMed] [Google Scholar]
- 43. von Kleist S, Chany E, Burtin P, King M, Fogh J (1975) Immunohistology of the antigenic pattern of a continuous cell line from a human colon tumor. J Natl Cancer Inst 55:555–560. [DOI] [PubMed] [Google Scholar]
- 44. Scherer WF, Syverton JT, Gey GO (1953) Studies on the propagation in vitro of poliomyelitis viruses. IV. Viral multiplication in a stable strain of human malignant epithelial cells (strain HeLa) derived from an epidermoid carcinoma of the cervix. J Exp Med 97:695–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Singh Y, Klimpel KR, Goel S, Swain PK, Leppla SH (1999) Oligomerization of anthrax toxin protective antigen and binding of lethal factor during endocytic uptake into mammalian cells. Infect Immun 67:1853–1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Leysath CE, Phillips DD, Crown D, Fattah RJ, Moayeri M, Leppla SH (2013) Anthrax edema factor toxicity is strongly mediated by the N‐end rule. PLoS One 8:e74474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhao H, Brautigam CA, Ghirlando R, Schuck P (2013) Overview of current methods in sedimentation velocity and sedimentation equilibrium analytical ultracentrifugation. Curr Protoc Protein Sci 71:20.12.1–20.12.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ghirlando R, Balbo A, Piszczek G, Brown PH, Lewis MS, Brautigam CA, Schuck P, Zhao H (2013) Improving the thermal, radial, and temporal accuracy of the analytical ultracentrifuge through external references. Anal Biochem 440:81–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Schuck P (2000) Size distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophysical Journal 78:1606–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cole JL, Lary JW, T PM, Laue TM (2008) Analytical ultracentrifugation: sedimentation velocity and sedimentation equilibrium. Methods Cell Biol 84:143–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Primers used in creation of CLPA variants.
Figure S1: Flow chart outlining CLPA formation from monomeric PA.
Figure S2: Purification of CLPA by size‐exclusion chromatography. A) Two distinct peaks with nearly no overlap, a cross‐linked PA peak at 58‐70 mL and a PA monomer peak at 70‐80 mL, are seen when a mixture of monomeric PA and CLPA dimer are purified by size‐exclusion. Evidence of larger species is seen on the front edge of the dimer peak. B) SDS‐PAGE analysis of all fractions confirms the presence of larger species on the edge of the dimer peak, and small amount of residual monomer present in each dimer peak fraction. C) SDS‐PAGE analysis when only the dimer peak fractions were pooled, excluding the fractions containing larger species. Sedimentation velocity confirmed this sample did not contain a significant amount of larger species and less than 5% of the total amount is PA monomer. It is suggested that PA monomer seen by SDS‐PAGE results from break down of the bismaleimide linker when boiling samples in samples buffer containing 10 mM Tris–HCl buffer and SDS.
Figure S3: In vitro cleavage of CLPA1 monomeric components by MMP9 and uPA. A mixture of CLPA1 monomeric components (PA‐L1 N388C and PA‐U2 L416C), prior to dimerization, were incubated with none, one, or both of MMP9 and uPA for 2 h at 37°C, and then analyzed by SDS‐PAGE. The SeeBlue Plus2 pre‐stained protein ladder was used (left lane). Complete cleavage of both monomeric components is seen after 2 h only when both proteases are added. Results were similar for CLPA2 and CLPA3 monomeric components (data not shown).
Figure S4: Native PhastGel electrophoresis of CLPA1‐3 oligomers. LF (lane 1), untreated CLPA1‐3 (lanes 2‐4), MMP & uPA cleaved CLPA1‐3 in complex with LF (lanes 5‐7), furin cleaved PA‐GN + PA‐D512K + LF (lane 8). All CLPA1‐3 oligomers run at the same size as the PA‐GN + PA‐D512K + LF oligomer, previously shown to be an octamer. These results show that all CLPA variants assemble as octamers.
Figure S5: CLPA toxicity on RAW264.7 cells (A) and HT29 cells (B). (A) RAW264.7 cells exhibiting 65% confluence were incubated with serial dilutions of PA variants (0–7 nM) and LF (10 nM) for 5 h (B) HT29 cells exhibiting 45% confluence were incubated with serial dilutions of PA variants (0‐10 nM) and FP59 (1.9 nM) for 48 h. Cell viability was then measured using an MTT assay.