

Abstract
Osteoarthritis (OA) is a common multifactorial degenerative articular disease among the aging population. The current investigation aimed to elucidate the function of microRNA-495 (miR-495) in the development of OA. We found that miR-495 was upregulated in the cartilage of OA patients. Transfection of a miR-495 mimic into rat primary chondrocytes, human chondrocytes (HC) and SW1353 chondrosarcoma cells inhibited AKT1 expression, proliferation and scratch wound closure and induced apoptosis. Transfection of a miR-495 inhibitor produced an opposite effect. Furthermore, the production of cartilage degeneration-related substances was modified by miR-495. Luciferase reporter gene assay revealed that AKT1 is directly repressed by miR-495. Moreover, the levels of AKT1, p-S6 and p-mTOR diminished in chondrocytes overexpressing miR-495. AKT1 overexpression amplified p-S6 and p-mTOR levels as well as abolished miR-495 mimic-induced apoptosis and inhibition of proliferation. In the surgically induced rat OA model, apoptosis of chondrocytes and cartilage degeneration were remedied by the administration of a miR-495 antagomir. Moreover, there was an increased expression of AKT1. These findings indicate that miR-495 induces OA by targeting AKT1 and regulating the AKT/mTOR pathway. Therefore, miR-495 may be a prospective target for OA treatment.
Keywords: MicroRNA-495, apoptosis, senescence, osteoarthritis, AKT1
Introduction
Osteoarthritis (OA) is a common degenerative articular disease characterized by an imbalance between the repair and degradation of articular cartilage [1,2]. Its symptoms include joint ache, swelling, restricted movement and joint deformities; whereas the risk factors are aging, heredity, obesity, anomalous anatomy, bearing extreme weight and joint damage [3,4]. Currently, there are no effective approaches to prevent and treat OA [5,6]. In the early stages of the disease, analgesics are prescribed, and in the later stages, joint replacement surgery is frequently recommended [7,8]. Chondrocytes are the bone cells present in cartilage and are crucial for the preservation of cartilage homeostasis and the integrity of its extracellular matrix (ECM); OA involves an alteration in cartilage homeostasis toward ECM deterioration [9,10]. Several research groups have established the function of chondrocyte apoptosis in the development of OA, in which several signaling pathways and cytokines such as NO, Fas, p38 MAPK, IL-1β and TNF-α are involved [11-15]. Consequently, elucidation of the molecular mechanisms fundamental to the production and deterioration of articular cartilage has the potential to aid in the identification of new therapeutic targets for OA.
MicroRNAs (miRNAs) are short non-coding RNAs that hinder the translation of their target mRNAs [16]. It has been established that dysregulated miRNAs are involved in OA. However, the function of specific miRNAs remains unclear [17,18]. In addition, a growing number of reports have demonstrated that miRNAs also regulate bone metabolism, osteoblasts differentiation and bone formation. Previously study showed that miR-214 inhibits osteogenesis by targeting baculoviral IAP repeat-containing 7 (BIRC7) [19]. In osteoporosis models, miR-221 suppresses bone formation and osteoblasts differentiation by targeting RUNX2 directly [20]. MiR-467g regulates osteogenesis negatively by regulating Indian hedgehog (Ihh)/Runx-2 signaling pathway [21]. Previously study identified miR-495 is an important miRNA regulating tumor cell function [22]. Moreover, recently research indicated that miR-495 functions as a tumor suppressor in a variety of cancer types [23-25]. In healthy cells, miR-495 takes part in numerous developmental, apoptotic, immune and inflammatory mechanisms, but it is also associated with the proliferation, invasiveness, metastasis and drug resistance of malignant cells [22,26-28]. In addition, it has a potential use as a prognostic marker as well as a pharmacological target in the treatment of several diseases [29-31]. Previously study indicated that miR-495 regulates new bone regeneration [32]. However, the role of miR-495 in regulation of OA are still not well-understood.
In the present study, we explored the involvement of miR-495 in OA. Our results suggest that miR-495 enhances chondrocyte apoptosis and senescence and promotes OA development through AKT1 and the AKT/mTOR signaling pathway.
Materials and methods
Patient samples
Cartilage from the femur and knee was obtained from OA patients (hip OA: 61-82 years old, 3 females and 5 males; knee OA: 53-78 years old, 5 females and 3 males). Cartilage from patients (46-77 years old, 5 females and 4 males) with no joint disease but who had experienced a femoral neck fracture undergone total joint replacement was employed as a control. OA was diagnosed through medical history, examination, and X-rays. A gross pathological assessment was carried out during joint replacement. This study was authorized by the Ethical Committee of the First Hospital of JiLin University, and all patients gave written approval.
Isolation of chondrocytes
Chondrocytes were obtained from 5 neonatal Sprague-Dawley rats. The rats were sacrificed, and the articular cartilages was excised. After exclusion of muscles and connective tissue, the cartilages were cut into small pieces, washed three times with PBS and treated with 2.5 mg/mL trypsin (Gibco, Cat: 25200056) for 15 min at 37°C. Subsequently, the cartilage was rinsed with PBS and treated with 2 mg/mL type II collagenase solution (Gibco, Cat: 17101015) at 37°C for 4 h, followed by filtration to remove undigested cartilage. The cells were then rinsed with PBS three times and suspended in DMEM/F12 (Gibco, Cat: 12634-028) containing 10% FBS (Sigma, Cat: F2442), 100 U/mL ampicillin and streptomycin (Gibco, Cat: 15140122). Cells (1 × 105 cells/mL) were grown and incubated at 37°C with 5% CO2.
Cell cultures
Chondrosarcoma cells (SW1353) were acquired from the American Type Culture Collection (ATCC, Cat: HTB-94) and grown in DMEM containing of 10% FBS, 2 mM glutamine, and 100 U/mL penicillin and streptomycin. Human chondrocytes (HC; Cell Applications, USA), derived from human articular cartilage, were cultured in chondrocyte growth medium and incubated under humidified conditions at 37°C with 5% CO2.
Transient transfection
Primary chondrocytes, HC cells and SW1353 cells were seeded in 6-well plates (2.5 × 105 cells/well) and incubated for 24 h. Subsequently, they were transfected for 6 h in DMEM with Lipofectamine 2000 (Invitrogen, USA, Cat: 11668027), miR-495 mimic at 30 nM (Genepharma, Shanghai, China), miR-495 inhibitor at 50 nM (Genepharma, Shanghai, China) or the corresponding control (Genepharma, Shanghai, China). After 6 h, the medium was replaced and the cells were cultured for an additional 48 h prior to analysis. For AKT1 overexpression, AKT1 was expressed in vitro using the pLenti6.2/V5-DEST vector (Invitrogen, Cat: V36820). Transfection was carried out using Lipofectamine 2000 according to the manufacturer’s instructions.
DAPI staining
For observation of nuclear morphology, the cells were rinsed twice with PBS, fixed with 4% paraformaldehyde (Sigma, Cat: P6148) and stained with 1 mg/mL DAPI (Beyotime, China, Cat: C1002) according to the manufacturer’s protocol. Subsequently, the cells were rinsed three times with PBS and viewed under a fluorescence microscope. Condensed or fragmented nuclei were considered an indication of apoptosis.
RNA isolation and qRT-PCR
Total cellular RNA was obtained using a TRIzol (Invitrogen, Cat: 15596026) extraction method and was reverse-transcribed into cDNA using random hexamer primers with the TaqMan MicroRNA Reverse Transcription Kit (Applied Biosystems, Cat: 4366596). qRT-PCR was carried out using SYBR® Green PCR Master Mix (Invitrogen, Cat: 4472903). β-actin (for mRNA) and U6 (for miRNA) were used as controls, and the relative change in expression was determined with the 2-ΔΔCt method. The primers used are as follows: β-actin Forward: 5’-AGAGCTACGAGCTGCCTGAC-3’; Reverse: 5’-AGCACTGTGTTGGCGTACAG-3’; U6 Forward: 5’-CTCGCTTCGGCAGCACA-3’; Reverse: 5’-AACGCTTCACGAATTTGCGT-3’; AKT1 Forward: 5’-GAAAACTATCCTGCGGGTTT-3’; Reverse: 5’-GAGGTCATGGAGCACAGGTT-3’; miR-495 Forward: 5’-GCGGAAACAAACATGGTGCA-3’; Reverse: 5’-GTTGGCTCTGGTGCAGGGTCCGAGGTATTCGCACCAGAGCCAACAAGAAG-3’.
Western blot analysis
The cartilage was ground in liquid N2, rinsed with precooled PBS and lysed in cold RIPA buffer. The total amount of protein was estimated with a BCA protein assay kit (Thermo Scirntific, Cat: 23225). The proteins were purified with 10% SDS-PAGE, transferred to PVDF membranes (Millipore, Cat: IPVH00010) and blocked for 4 h in 5% powdered milk. They were then incubated with the primary antibodies (1:1000 dilutions) at 4°C overnight, followed by rinsing and incubation for 1 h with the respective peroxidase-labeled secondary antibodies. The protein bands were visualized with an ECL kit (Pierce Chemical, Rockford, IL, USA, Cat: 34095). The primary antibodies used were: AKT1 (Cat: 75692), MMP-1 (Cat: 54376), MMP-13 (Cat: 94808), p-S6 (Cat: 4858), S6 (Cat: 2317), p-mTOR (Cat: 5536), mTOR (Cat: 2983), (Cell signaling technology, Danvers, MA, USA), p16 (Cat: ab108349), thrombospondin 5 (COMP fragment) (Cat: ab74524), COMP (Cat: ab74524), COL X (Cat: ab49945), COL II (Cat: ab34712), ADAMTS-5 (Cat: ab41037), ADAMTS-7 (Cat: ab203027), (Abcam, Cambridge, MA, USA), cleaved caspase 3 (Cat: sc-7272), Bcl-2 (Cat: sc-509), β-actin (Cat: sc-70319), Bax (Cat: sc-20067) and aggrecan (Cat: sc-70333) (Santa Cruz Biotechnology, Dallas, TX, USA). All uncropped western blot images are shown in Figures S5 and S6.
Flow cytometry
Apoptosis was evaluated with flow cytometry using the Annexin V-FITC Apoptosis Detection Kit (Sigma, USA, Cat: APOAF). Cells were rinsed two times in PBS, resuspended in binding buffer and treated with Annexin V-FITC for 15 min in the dark. Subsequently, propidium iodide (1 mg/L., Invitrogen, Cat: P1304MP) was added, and the number of stained cells was estimated via FACSCalibur (BD, USA).
MTT assay
Cell viability was estimated with MTT assay (Invitrogen, Cat: M6494). Cells were seeded in 96-well plates (2 × 105 cells/cm2), MTT (0.5 mg/mL) was introduced and the cells were cultured for 4 h at 37°C. Absorbance at 570 nm was calculated using a Bio-Rad microplate reader (Bio-Rad, USA).
Wound-healing assay
After formation of a monolayer in 6-well plates, wounds were created using a 200-μL sterile pipette tip followed by culturing in serum-free media for 24 h. Sections of the cell monolayers with wounds of equal width were photographed before and after incubation using an inverted microscope. The degree of wound repair was estimated by calculating the wound area with ImageJ (NIH).
Luciferase reporter assay
The wild-type (WT) and mutated (MUT) 3’ UTR AKT1 sequences were cloned into a pGL3-luciferase vector (control) to obtain the recombinants pGL3-AKT1 3’ UTR-WT and pGL3-AKT1 3’ UTR-MUT, respectively. Cells were grown in 12-well plates until reaching 70% confluence, after which they were transfected with the recombinant plasmids (200 ng) and miR-495 mimics (200 ng) using Lipofectamine 2000 according to the manufacturer’s instruction. Experiments were carried out in quadruplicate and repeated three times.
ELISA assay
The levels of collagenases and collagen were estimated using the following ELISA kits: Rat MMP-13 kit (Cusabio, China Cat: CSB-E07412r), Rat COL II kit (LifeSpan BioSciences, USA, Cat: LS-F11156), Rat COL X kit (Abbexa Ltd, UK, Cat: abx155379), Human MMP-13 kit (Abcam, Cat: ab100605), MMP-1 kit (Abcam, Cat: 52631), Human COL X (lifespan BioSciences, Cat: LS-F13131) and COL II kits (LifeSpan BioSciences, Cat: LS-F5640).
Establishment of the rat OA model
The animal experiments were authorized by the Animal Care and Use Committee of First Hospital of JiLin University. Thirty SD male rats (10-week-old) were separated into Sham surgery (Sham), OA-negative control (OA/NC) or OA miR-495 antagomir-treatment (OA/antagomir) groups. Six hours prior to surgery, the joints of the OA/NC and OA/antagomir rats were injected with 800 pmol of NC-antagomir or miR-495-antagomir, respectively. From each group, 9 rats were randomly selected to be sacrificed 8 weeks following the surgery. The OA rat model of was produced via anterior cruciate ligament transection (ACLT) in the right knee. In brief, the right knees were sterilized with polyvinyl iodine and a medial parapatellar skin incision was made on the joint. Then, an incision was made on the medial surface of the patellar tendon, the patella was dislocated laterally, and the ACL was transected. An anterior drawer test was performed to validate total transection, and then a medial retinaculum was repaired and the skin was sutured separately. The rats in the sham group were anesthetized and a cut was made in the articular capsule with no ACLT.
Safranin O/fast green staining
The complete articulation of knees was maintained in 4% paraformaldehyde for 24 h fixing. Subsequent to traditional paraffin embedding, the articulation was cut into pieces with 5 μm depth. Safranin O stain kit (ICH World, Cat: IW-3011) was applied to staining.
Statistical analysis
All data are presented as the means ± standard deviation (SD). One-way or two-way ANOVA were conducted as appropriate with SPSS13.0 software. P < 0.05 was deemed statistically significant. All assays were carried out in triplicate.
Results
miR-495 promoted apoptosis and senescence of rat primary chondrocytes, HC cells and chondrosarcoma cells
To substantiate that miR-495 was associated with the pathological development of OA, we evaluated its expression in the cartilage of patients with and without OA; the expression of miR-495 in OA patients was significantly elevated compared to those without OA (Figure 1A), suggesting its involvement in the progression of OA.
Figure 1.
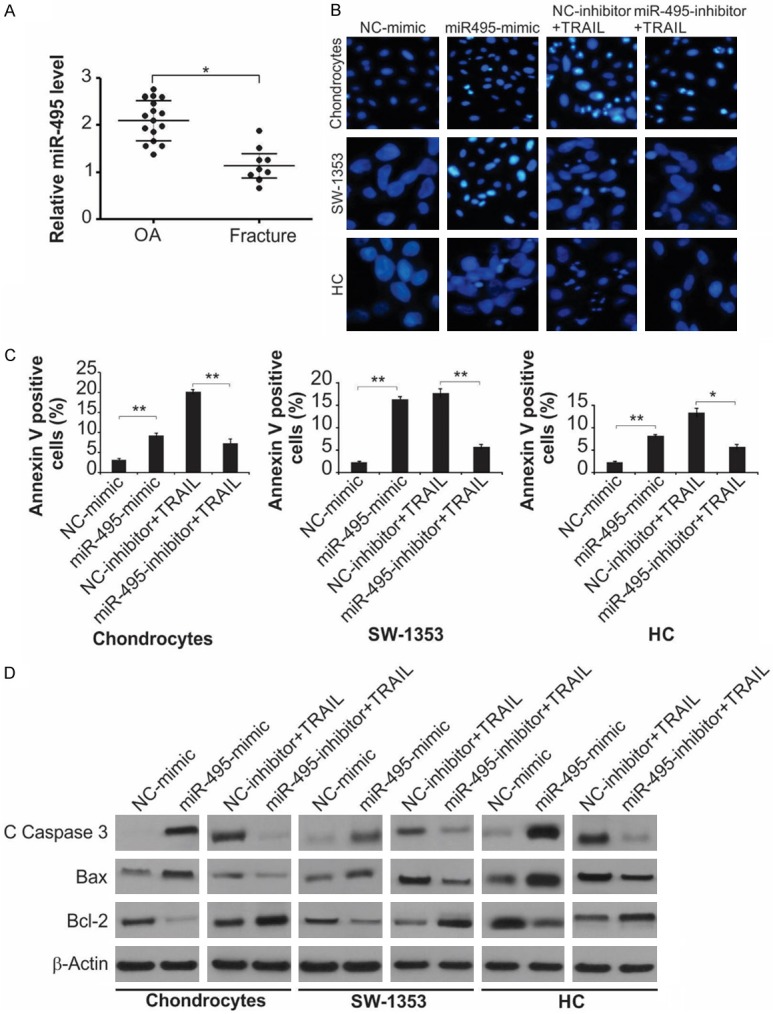
miR-495 promotes apoptosis of primary cultured rat chondrocytes, HC cells and SW1353 cells. A. Relative miR-495 level in clinical specimens (OA = 16, Fracture = 9) was detected by Real-time RT-PCR. *, P < 0.05. B. Indicated cells were treated with NC-mimic, miR-495-mimic, TRAIL + NC-inhibitor or TRAIL + miR-495-inhibitor as indicated. Photomicrographs of the nucleus the cells stained with DAPI. C. Indicated cells were treated with NC-mimic, miR-495-mimic, TRAIL + NC-inhibitor or TRAIL + miR-495-inhibitor as indicated. Cell apoptosis was analyzed by flow cytometry. Data represent the mean ± SD of three independent experiments. **, P < 0.01; *, P < 0.05 (one-way ANOVA with Tukey’s post hoc test). D. Indicated cells were treated with NC-mimic, miR-495-mimic, TRAIL + NC-inhibitor or TRAIL + miR-495-inhibitor as indicated. The protein level of cleaved caspase-3, Bax and Bcl-2 were measured by western blot assay.
In addition, the impact of miR-495 on apoptosis and senescence was explored in rat primary chondrocytes, HC cells and SW1353 cells. Subsequent to transfection with miR-495 mimic or inhibitor, DAPI staining, flow cytometry and western blot were used to evaluate apoptosis from the levels of cleaved caspase-3, Bax and Bcl-2. Furthermore, the senescence was estimated by SA-β-gal staining and p16 levels, a biomarker of cellular senescence. As shown in Figure 1B, the fraction of apoptotic cells in the miR-495-mimic-treated cells were greater compared to the NC-mimic group, and the miR-495-inhibitor markedly inhibited TRAIL-induced apoptosis. These observations were supported by flow cytometry, where the fractions of early and late apoptotic cells in the mimic-treated fraction were considerably elevated relative to the NC-inhibitor group; miR-495-inhibitor decreased the proportion of apoptotic cells (Figures 1C and S1A). Transfection with miR-495 mimic noticeably amplified cleaved caspase-3 and the relative amount of Bcl-2/Bax in the cells, and transfection with the inhibitor efficiently blocked the augmentation of these markers (Figure 1D and 1E).
A greater proportion of SA-β-gal-positive cells was detected in chondrocytes transfected with miR-495 mimic, relative to those transfected with miR-495-inhibitor (Figure S1B). The expression level of p16 was detected through western blot, consistent with SA-β-gal staining results, the p16 level was increased in miR-495-mimic-transfected chondrocytes and decreased in the inhibitor-transfected cells (Figure S1C).
miR-495 inhibited cell proliferation and repair of scratch wound closure
The effect of miR-495 on cell proliferation was explored with an MTT study and flow cytometry. Transfection with miR-495-mimic considerably suppressed proliferation (Figures 2A and S2A), whereas transfection with miR-495-inhibitor enhanced proliferation (Figures 2A and S2A). Additionally, the wound-healing assay was performed to explore the impact of miR-495 on the ability of cells to repair injury. miR-495-mimic hindered the repair of scratches, whereas the inhibitor facilitated the repair (Figures 2B and S2B). Taken together, miR-495 suppressed cell proliferation and suppressed wound closure, signifying that miR-495 inhibition might aid cartilage repair in OA.
Figure 2.
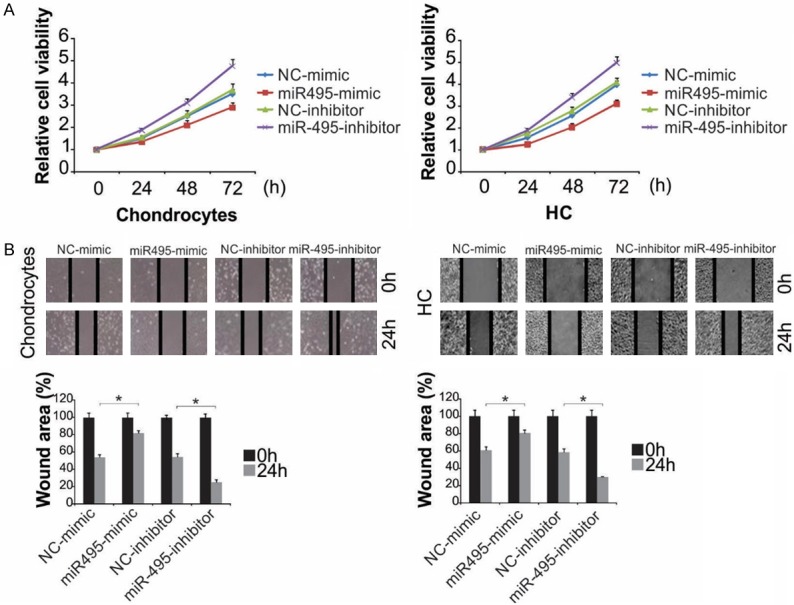
miR-495 inhibited the progressions of primary rat chondrocytes and HC cells. A. Chondrocytes and HC cells were treated with miR-495-mimic or miR-495-inhibitor. Cell proliferation was analyzed by MTT at indicated time point. B. Chondrocytes and HC cells were treated with miR-495-mimic or miR-495-inhibitor for 24 h. The wound healing assay was expressed as relative wound width. The results were expressed as the means ± SD of three independent experiments. *, P < 0.05 (Two-way ANOVA with Tukey’s post hoc test).
miR-495 modified the production of cartilage degeneration-related substances
The imbalance between ECM production and degradation of chondrocytes is a key factor for the degeneration of cartilage [10,33]. Collagenases MMP-1 and MMP-13 can breakdown COL II, the most abundant collagen in ECM [34]. COL X, produced by hypertrophic chondrocytes, is a biomarker for advanced OA [35]. The miR-495-mimic-transfected cells displayed augmented synthesis of MMP-1, MMP-13 and COL X, along with diminished COL II transcription and release. However, transfection with miR-495 inhibitor resulted in completely opposite effects on the production and secretion of collagenases and collagen (Figures 3A-D and S3A-D).
Figure 3.

miR-495 regulated cartilaginous degeneration-related molecules, including MMP-1, MMP-13, COL II and COL X. Chondrocytes and HC cells were treated with miR-495-mimic or miR-495-inhibitor for 24 h. Concentrations of secreted MMP-1 (A), MMP-13 (B), COL II (C) and COL X (D) in the media of cells were measured by ELISA assay. Data represent the mean ± SD of three independent experiments. ***, P < 0.001; **, P < 0.01; *, P < 0.05 (one-way ANOVA with Tukey’s post hoc test).
AKT1 was negatively modulated by miR-495 in rat primary chondrocytes, HC cells and chondrosarcoma cells
We used bioinformatics to investigate genes that could be targeted by miR-495 and identified AKT1 as a potential target (Figure 4A). To explore the nature (direct or indirect) of the interaction between miR-495 and AKT1 3’-UTR, the miR-495 binding site in AKT1 3’-UTR was mutated (Figure 4A). After cotransfection of the luciferase vector with mutated AKT1 3’-UTR and miR-495-mimic, the latter could not inhibit luciferase expression (Figure 4B), signifying that a direct interaction with AKT1 3’-UTR modulates luciferase expression. The influence of miR-495 on AKT1 expression and release was evaluated by transfecting cells with miR-495-mimic or miR-495-inhibitor. AKT1 was negatively modulated by miR-495 in all three cell types (Figure 4C and 4D). Taken together, our results indicate that miR-495 interacts directly with the binding site in 3’-UTR of AKT1 to regulate its expression.
Figure 4.
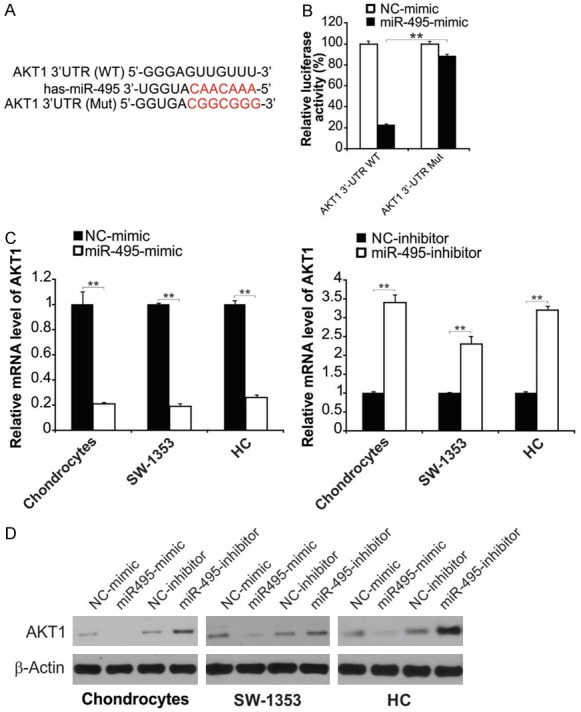
miR-495 targeted at 3’ UTR of AKT1. A. Schematic representation of miR-495 predicted binding site in the 3’-UTR of AKT1 mRNAs. B. The AKT1 3’-UTR reporter vector was transfected into NC-mimic or miR-495-mimic transfected SW1353 human chondrosarcoma cells and the reporter activities were compared. Data represent the mean ± SD of three independent experiments. **, P < 0.01 (two-way ANOVA with Tukey’s post hoc test). C. The cells were treated as indicated, mRNA level of AKT1 were analyzed by real-time RT-PCR. Data represent the mean ± SD of three independent experiments. **, P < 0.01 (one-way ANOVA with Tukey’s post hoc test). D. The indicated cells were treated with miR-495-mimic or miR-495-inhibitor for 24 h. Protein level of AKT1 were analyzed by western blot assay.
AKT1 participates in miR-495 mimic-induced cell death
We used western blot to test whether miR-495 could regulate the AKT/mTOR pathway involved in OA development and progression. The results showed that p-mTOR and p-S6 levels were diminished in the miR-495-transfected group (Figure 5A). Furthermore, we found that AKT1 overexpression blocked miR-495-mediated mTOR and p-S6 downregulation (Figure 5B). Since miR-495 could inhibit the synthesis of AKT1, we hypothesized that the impact of miR-495-mimic on the growth and apoptosis of primary rat chondrocytes, HC cells and chondrosarcoma cells was related to AKT1 deficiency. We thus transfected cells with miR-495-mimic during AKT1 overexpression and evaluated apoptosis, proliferation and wound-healing. Overexpression of AKT1 diminished the upregulation of cleaved caspase-3 induced by miR-495 mimic and may consequently have inhibited apoptosis (Figures 5C and S4A). Overexpression of AKT1 inhibited the effect of proliferation by the mimic (Figure 5D) and hastened wound repair relative to cells treated solely with the mimic (Figure 5E), suggesting that miR-495 affects the chondrosarcoma cells via modulation of AKT1 expression.
Figure 5.
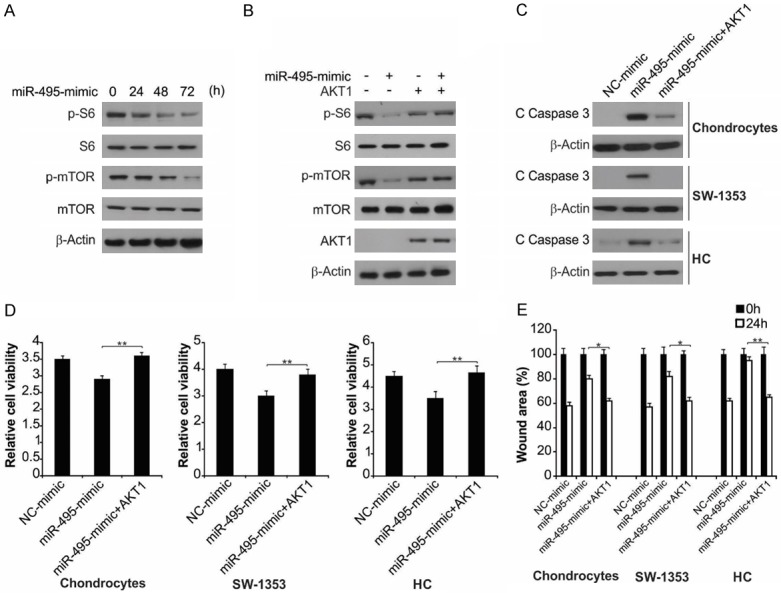
AKT1 overexpression affected miR-495-mimic-induced cell apoptosis and growth inhibition. A. SW1353 cells were treated with miR-495-mimic at indicated time points. Indicated protein level was analyzed by western blot assay. B. SW1353 cells transfected with AKT1 were treated with miR-495-mimic. Indicated protein level was analyzed by western blot assay. C. The indicated cells with or without AKT1 transfection were treated with miR-495-mimic for 24 h. The protein level of cleaved caspase-3 was measured by western blot assay. D. The indicated cells with or without AKT1 transfection were treated with miR-495-mimic for 72 h. Cell proliferation measured with MTT kit. Data represent the mean ± SD of three independent experiments. **, P < 0.01 (one-way ANOVA with Tukey’s post hoc test). E. The indicated cells with or without AKT1 transfection were treated with miR-495-mimic for 24 h. Percentage of wound closure measured on the basis of scratch wound assay. The wound healing assay was expressed as relative wound width. The results were expressed as the means ± SD of three independent experiments. **, P < 0.01; *, P < 0.05 (Two-way ANOVA with Tukey’s post hoc test).
Effect of miR-495 on OA progression in vivo
To investigate the role of miR-495 in OA in vivo, an OA model was generated through ACLT. A significantly higher miR-495 expression was observed in the cartilage of OA rats relative to that in the sham surgery (SS) group (Figure 6A). To study the curative effect of miR-495 in OA, the rats were injected intra-articularly with miR-495 antagomir before the induction of OA through surgery, and chondrocytes apoptosis was subsequently evaluated. miR-495 antagomir significantly inhibited OA-induced upregulation of miR-495 (Figure 6A). The effect of miR-495 antagomir on cartilage loss in OA was then confirmed by safranin O-fast green staining. The sham-operated rats showed intact knee-joint cartilage structure and uniformly stained Safranin O (Figure 6B). However, rats in the OA/NC group exhibited symptoms of knee-joint OA, including the formation of surface fissures, a sharp decrease in Safranin O staining of the surface layer and polycrystalline layer, and the appearance of deep hypertrophic chondrocytes layers, which was reversed by miR-495 antagomir injection (Figure 6B). AKT1 expression in the cartilage of the OA/NC group was greater relative to the sham group, which was significantly upregulated by the antagomir pretreatment (Figure 6C). Moreover, the level of cleaved caspase-3 has considerably elevated, and pretreatment with the miR-495 antagomir inhibited the augmentation of apoptosis (Figure 6D). The major pathological characteristic of OA is the imbalance in the production and breakdown of ECM, resulting in the loss of cartilage matrix. After establishing that miR-495 antagomir has an anti-apoptosis role in OA, we evaluated the impact on cartilage degradation. Subsequent to the induction of OA, the expression levels of matrix-degrading enzymes were upregulated, leading to the degradation of articular cartilage, characterized by the increased degeneration of COMP, COL II and Aggrecan. In this context, miR-495 antagomir could decrease the synthesis of these enzymes (Figure 6E). Therefore, these results indicate that downregulation of miR-495 significantly inhibits OA progression in vivo.
Figure 6.
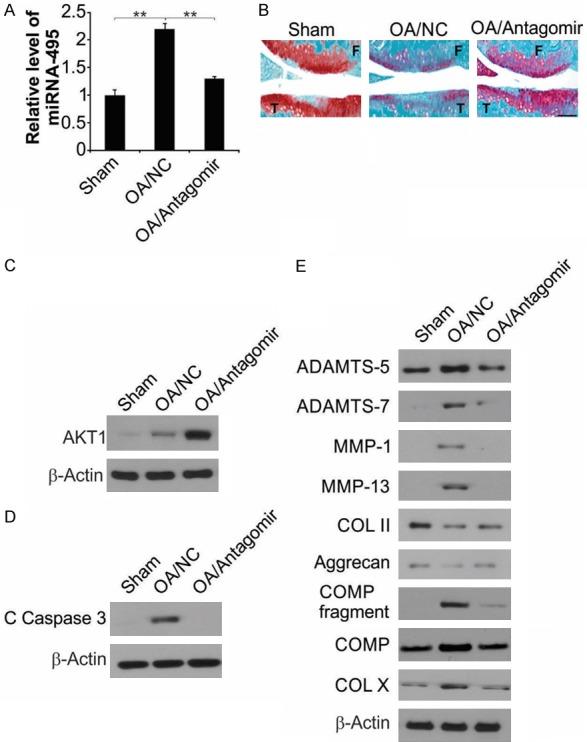
Downregulation of miR-495 resulted in decreased cell apoptosis of articular chondrocytes of osteoarthritic rats. A. Eight weeks after surgery, miR-495 level in the tibial cartilage was detected by qRT-PCR. The results were expressed as the means ± SD of three independent experiments. **, P < 0.01 (Two-way ANOVA with Tukey’s post hoc test). B. Representative safranin O-fast green staining of the knee joint from each group. F: femur; T: tibia. Scale bars: 100 μm. C. AKT1 in the tibial cartilage was measured by western blot assay. D. Cleavage of caspase-3 in the cartilage was measured by western blot assay. E. Protein levels of catabolic biomarkers (ADAMTS-5, -7, MMP-1, -13 and COMP fragment), anabolic biomarkers (COL II and Aggrecan) and the hypertrophic chondrocyte marker (COL X) were analyzed by western blot assay.
Discussion
OA is a multifactorial inflammatory disease affecting articular cartilage, ligaments, synovial membrane and the adjacent muscles [36]. The causative factors of OA are heredity, obesity, aging, cartilage abnormalities, metabolic imbalance, chondrocyte apoptosis, inflammation and oxidative stress [37,38]. MicroRNAs are a group of noncoding, 22-23 nucleotide long RNAs, and extensively occur among all living organisms [39]. Growing evidence shows that greater than 25 miRNAs are associated with the incidence and progression of OA, and several are functionally associated with its pathogenesis [40,41]. It has been revealed that miR-21 is upregulated in OA, where it attenuates the process of chondrogenesis [42]. In addition, earlier studies have demonstrated that miR-210 might be linked to OA [43]. Furthermore, miR-22 suppressed the expression of BMP-7 and PPARA, resulting in augmented expression of IL-1 and MMP-13 [44], leading to the breakdown of chondrocytes and the cartilage matrix.
In this context, we initially evaluated the consequence of miR-495 overexpression or knockdown on apoptosis, growth and wound repair in articular cells in vitro. Overexpression of miR-495 considerably facilitated apoptosis and suppressed proliferation, while knockdown led to the opposite effects. Additionally, transfection of miR-495 mimic considerably hindered the repair of scratch wound closure, signifying reduced damage repair ability in these cells. Furthermore, we observed that miR-495 mimic promoted the activation of caspase-3, which may promote apoptosis. Interestingly, it was established that miR-495 could directly modulate AKT1 expression. miR-495 mimic considerably diminished luciferase expression through mutations in the 3’ UTR of AKT1, the binding site for miR-495. Additionally, miR-495 mimic notably downregulated AKT1 expression, while miR-495 inhibitor enhanced AKT1 expression. Therefore, miR-495 might induce chondrocyte apoptosis by targeting AKT1, which was supported by the fact that AKT1 overexpression reversed the impact of miR-495 mimic on apoptosis, growth and wound repair of chondrocytes.
The correlation between chondrocyte apoptosis and OA is still uncertain. Some researchers have reported it as an early occurrence in OA development, while others suggest that it occurs at later stages in OA [45-47]. Nevertheless, numerous reports in the literature have indicated that apoptosis plays a key function in the degeneration of articular cartilage [48,49]. Therefore, the pathways of chondrocyte apoptosis might act as therapeutic targets for OA. In the present work, an OA rat model was established and the degeneration of cartilage brought about by chondrocyte apoptosis was appreciably suppressed by miR-495 knockdown in vivo. Moreover, overexpression of miR-495 stimulated ECM degradation-related enzymes, whereas downregulation miR-495 in vivo suppressed ADAMTS-5, ADAMTS-5, MMP-1 and MMP-13 production. along with increasing the levels of COL II, and Aggrecan.
In summary, we established that miR-495 induced chondrocyte apoptosis and senescence via directly targeting AKT1 and regulating the AKT/mTOR signaling pathway. Knockdown of miR-495 hindered apoptosis and loss of articular cartilage in the surgically-induced OA rat model. Altogether, these outcomes imply that miR-495 and AKT1 could be exploited as a pharmacological target for OA treatment.
Acknowledgements
This work was supported by Youth Funding Project of JiLin Provincial Technology office. Funding Number: 20140520021JH.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Sinusas K. Osteoarthritis: diagnosis and treatment. Am Fam Physician. 2012;85:49–56. [PubMed] [Google Scholar]
- 2.Whitney KE, Liebowitz A, Bolia IK, Chahla J, Ravuri S, Evans TA, Philippon MJ, Huard J. Current perspectives on biological approaches for osteoarthritis. Ann N Y Acad Sci. 2017;1410:26–43. doi: 10.1111/nyas.13554. [DOI] [PubMed] [Google Scholar]
- 3.Timmins KA, Leech RD, Batt ME, Edwards KL. Running and knee osteoarthritis: a systematic review and meta-analysis. Am J Sports Med. 2017;45:1447–1457. doi: 10.1177/0363546516657531. [DOI] [PubMed] [Google Scholar]
- 4.Bian Q, Wang YJ, Liu SF, Li YP. Osteoarthritis: genetic factors, animal models, mechanisms, and therapies. Front Biosci (Elite Ed) 2012;4:74–100. doi: 10.2741/361. [DOI] [PubMed] [Google Scholar]
- 5.Tools for earlier diagnosis and treatment of osteoarthritis. Earlier detection via magnetic resonance imaging and optical coherence tomography, and treatment with exercise and weight loss emerge as a new paradigm. Duke Med Health News. 2012;18:3–4. [PubMed] [Google Scholar]
- 6.Wood AM, Brock TM, Heil K, Holmes R, Weusten A. A review on the management of hip and knee osteoarthritis. Int J Chronic Dis. 2013;2013:845015. doi: 10.1155/2013/845015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Newberry SJ, FitzGerald J, SooHoo NF, Booth M, Marks J, Motala A, Apaydin E, Chen C, Raaen L, Shanman R, Shekelle PG. Treatment of osteoarthritis of the knee: an update review. Rockville (MD): Agency for Healthcare Research and Quality (US); 2017. [PubMed] [Google Scholar]
- 8.Tieppo Francio V, Dima RS, Towery C, Davani S. Prolotherapy and low level laser therapy: a synergistic approach to pain management in chronic osteoarthritis. Anesth Pain Med. 2017;7:e14470. doi: 10.5812/aapm.14470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen JL, Duan L, Zhu W, Xiong J, Wang D. Extracellular matrix production in vitro in cartilage tissue engineering. J Transl Med. 2014;12:88. doi: 10.1186/1479-5876-12-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gao Y, Liu S, Huang J, Guo W, Chen J, Zhang L, Zhao B, Peng J, Wang A, Wang Y, Xu W, Lu S, Yuan M, Guo Q. The ECM-cell interaction of cartilage extracellular matrix on chondrocytes. Biomed Res Int. 2014;2014:648459. doi: 10.1155/2014/648459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zamli Z, Sharif M. Chondrocyte apoptosis: a cause or consequence of osteoarthritis? Int J Rheum Dis. 2011;14:159–166. doi: 10.1111/j.1756-185X.2011.01618.x. [DOI] [PubMed] [Google Scholar]
- 12.Zhou Y, Ming J, Li Y, Du X, Deng M, He B, Zhou J, Wang G, Liu S. Surfactant protein D attenuates nitric oxide-stimulated apoptosis in rat chondrocyte by suppressing p38 MAPK signaling. Biochem Biophys Res Commun. 2018;495:526–532. doi: 10.1016/j.bbrc.2017.10.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tu Y, Xue H, Francis W, Davies AP, Pallister I, Kanamarlapudi V, Xia Z. Lactoferrin inhibits dexamethasone-induced chondrocyte impairment from osteoarthritic cartilage through up-regulation of extracellular signal-regulated kinase 1/2 and suppression of FASL, FAS, and Caspase 3. Biochem Biophys Res Commun. 2013;441:249–255. doi: 10.1016/j.bbrc.2013.10.047. [DOI] [PubMed] [Google Scholar]
- 14.Chen J, Gu YT, Xie JJ, Wu CC, Xuan J, Guo WJ, Yan YZ, Chen L, Wu YS, Zhang XL, Xiao J, Wang XY. Gastrodin reduces IL-1beta-induced apoptosis, inflammation, and matrix catabolism in osteoarthritis chondrocytes and attenuates rat cartilage degeneration in vivo. Biomed Pharmacother. 2018;97:642–651. doi: 10.1016/j.biopha.2017.10.067. [DOI] [PubMed] [Google Scholar]
- 15.Lee SW, Rho JH, Lee SY, Kim JH, Cheong JH, Kim HY, Jeong NY, Chung WT, Yoo YH. Leptin protects rat articular chondrocytes from cytotoxicity induced by TNF-alpha in the presence of cyclohexamide. Osteoarthritis Cartilage. 2015;23:2269–2278. doi: 10.1016/j.joca.2015.06.005. [DOI] [PubMed] [Google Scholar]
- 16.Hayes J, Peruzzi PP, Lawler S. MicroRNAs in cancer: biomarkers, functions and therapy. Trends Mol Med. 2014;20:460–469. doi: 10.1016/j.molmed.2014.06.005. [DOI] [PubMed] [Google Scholar]
- 17.Yu C, Chen WP, Wang XH. MicroRNA in osteoarthritis. J Int Med Res. 2011;39:1–9. doi: 10.1177/147323001103900101. [DOI] [PubMed] [Google Scholar]
- 18.Zhang M, Lygrisse K, Wang J. Role of MicroRNA in osteoarthritis. J Arthritis. 2017;6 doi: 10.4172/2167-7921.1000239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu J, Li Y, Luo M, Yuan Z, Liu J. MicroRNA-214 inhibits the osteogenic differentiation of human osteoblasts through the direct regulation of baculoviral IAP repeat-containing 7. Exp Cell Res. 2017;351:157–162. doi: 10.1016/j.yexcr.2017.01.006. [DOI] [PubMed] [Google Scholar]
- 20.Zhang Y, Gao Y, Cai L, Li F, Lou Y, Xu N, Kang Y, Yang H. MicroRNA-221 is involved in the regulation of osteoporosis through regulates RUNX2 protein expression and osteoblast differentiation. Am J Transl Res. 2017;9:126–135. [PMC free article] [PubMed] [Google Scholar]
- 21.Kureel J, John AA, Dixit M, Singh D. MicroRNA-467g inhibits new bone regeneration by targeting Ihh/Runx-2 signaling. Int J Biochem Cell Biol. 2017;85:35–43. doi: 10.1016/j.biocel.2017.01.018. [DOI] [PubMed] [Google Scholar]
- 22.Chen G, Xie Y. miR-495 inhibits proliferation, migration, and invasion and induces apoptosis via inhibiting PBX3 in melanoma cells. Onco Targets Ther. 2018;11:1909–1920. doi: 10.2147/OTT.S152362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu C, Jian M, Qi H, Mao WZ. MicroRNA-495 inhibits proliferation, metastasis and promotes apoptosis by targeting Twist1 in gastric cancer cells. Oncol Res. 2019;27:389–397. doi: 10.3727/096504018X15223159811838. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 24.Bai Z, Wang J, Wang T, Li Y, Zhao X, Wu G, Yang Y, Deng W, Zhang Z. The MiR-495/annexin A3/P53 axis inhibits the invasion and EMT of colorectal cancer cells. Cell Physiol Biochem. 2017;44:1882–1895. doi: 10.1159/000485877. [DOI] [PubMed] [Google Scholar]
- 25.Yan L, Yao J, Qiu J. miRNA-495 suppresses proliferation and migration of colorectal cancer cells by targeting FAM83D. Biomed Pharmacother. 2017;96:974–981. doi: 10.1016/j.biopha.2017.11.138. [DOI] [PubMed] [Google Scholar]
- 26.Chen H, Wang X, Bai J, He A. Expression, regulation and function of miR-495 in healthy and tumor tissues. Oncol Lett. 2017;13:2021–2026. doi: 10.3892/ol.2017.5727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen Y, Luo D, Tian W, Li Z, Zhang X. Demethylation of miR-495 inhibits cell proliferation, migration and promotes apoptosis by targeting STAT-3 in breast cancer. Oncol Rep. 2017;37:3581–3589. doi: 10.3892/or.2017.5621. [DOI] [PubMed] [Google Scholar]
- 28.Wei T, Zhu W, Fang S, Zeng X, Huang J, Yang J, Zhang J, Guo L. miR-495 promotes the chemoresistance of SCLC through the epithelial-mesenchymal transition via Etk/BMX. Am J Cancer Res. 2017;7:628–646. [PMC free article] [PubMed] [Google Scholar]
- 29.Ye M, Wei T, Wang Q, Sun Y, Tang R, Guo L, Zhu W. TSPAN12 promotes chemoresistance and proliferation of SCLC under the regulation of miR-495. Biochem Biophys Res Commun. 2017;486:349–356. doi: 10.1016/j.bbrc.2017.03.044. [DOI] [PubMed] [Google Scholar]
- 30.Li W, Yang Y, Hou X, Zhuang H, Wu Z, Li Z, Guo R, Chen H, Lin C, Zhong W, Chen Y, Wu K, Zhang L, Feng D. MicroRNA-495 regulates starvation-induced autophagy by targeting ATG3. FEBS Lett. 2016;590:726–738. doi: 10.1002/1873-3468.12108. [DOI] [PubMed] [Google Scholar]
- 31.Nie S, Li K, Huang Y, Hu Q, Gao X, Jie S. miR-495 mediates metabolic shift in glioma cells via targeting Glut1. J Craniofac Surg. 2015;26:e155–158. doi: 10.1097/SCS.0000000000001385. [DOI] [PubMed] [Google Scholar]
- 32.Tian Z, Zhou H, Xu Y, Bai J. MicroRNA-495 inhibits new bone regeneration via targeting high mobility group AT-Hook 2 (HMGA2) Med Sci Monit. 2017;23:4689–4698. doi: 10.12659/MSM.904404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sanchez C, Bay-Jensen AC, Pap T, Dvir-Ginzberg M, Quasnichka H, Barrett-Jolley R, Mobasheri A, Henrotin Y. Chondrocyte secretome: a source of novel insights and exploratory biomarkers of osteoarthritis. Osteoarthritis Cartilage. 2017;25:1199–1209. doi: 10.1016/j.joca.2017.02.797. [DOI] [PubMed] [Google Scholar]
- 34.Vincenti MP, Brinckerhoff CE. Transcriptional regulation of collagenase (MMP-1, MMP-13) genes in arthritis: integration of complex signaling pathways for the recruitment of gene-specific transcription factors. Arthritis Res. 2002;4:157–164. doi: 10.1186/ar401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang YJ, Shen M, Wang S, Wen X, Han XR, Zhang ZF, Li H, Wang F, Wu DM, Lu J, Zheng YL. Inhibition of the TGF-beta1/Smad signaling pathway protects against cartilage injury and osteoarthritis in a rat model. Life Sci. 2017;189:106–113. doi: 10.1016/j.lfs.2017.09.022. [DOI] [PubMed] [Google Scholar]
- 36.Al-Khazraji BK, Appleton CT, Beier F, Birmingham TB, Shoemaker JK. Osteoarthritis, cerebrovascular dysfunction and the common denominator of inflammation: a narrative review. Osteoarthritis Cartilage. 2018;26:462–470. doi: 10.1016/j.joca.2018.01.011. [DOI] [PubMed] [Google Scholar]
- 37.Komori T. Cell death in chondrocytes, osteoblasts, and osteocytes. Int J Mol Sci. 2016;17 doi: 10.3390/ijms17122045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hwang HS, Kim HA. Chondrocyte apoptosis in the pathogenesis of osteoarthritis. Int J Mol Sci. 2015;16:26035–26054. doi: 10.3390/ijms161125943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chandra S, Vimal D, Sharma D, Rai V, Gupta SC, Chowdhuri DK. Role of miRNAs in development and disease: lessons learnt from small organisms. Life Sci. 2017;185:8–14. doi: 10.1016/j.lfs.2017.07.017. [DOI] [PubMed] [Google Scholar]
- 40.Cong L, Zhu Y, Tu G. A bioinformatic analysis of microRNAs role in osteoarthritis. Osteoarthritis Cartilage. 2017;25:1362–1371. doi: 10.1016/j.joca.2017.03.012. [DOI] [PubMed] [Google Scholar]
- 41.Seeliger C, Balmayor ER, van Griensven M. miRNAs related to skeletal diseases. Stem Cells Dev. 2016;25:1261–1281. doi: 10.1089/scd.2016.0133. [DOI] [PubMed] [Google Scholar]
- 42.Sun C, Huang F, Liu X, Xiao X, Yang M, Hu G, Liu H, Liao L. miR-21 regulates triglyceride and cholesterol metabolism in non-alcoholic fatty liver disease by targeting HMGCR. Int J Mol Med. 2015;35:847–853. doi: 10.3892/ijmm.2015.2076. [DOI] [PubMed] [Google Scholar]
- 43.Zhang D, Cao X, Li J, Zhao G. MiR-210 inhibits NF-kappaB signaling pathway by targeting DR6 in osteoarthritis. Sci Rep. 2015;5:12775. doi: 10.1038/srep12775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang R, Zhang D, Yu K, Sun L, Yang J, Zhao C, Li X, Chen Y. Detection of miR-22, miR-140 and bone morphogenetic proteins (BMP)-2 expression levels in synovial fluid of osteoarthritis patients before and after arthroscopic debridement. Med Sci Monit. 2018;24:863–868. doi: 10.12659/MSM.908110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pan T, Chen R, Wu D, Cai N, Shi X, Li B, Pan J. Alpha-Mangostin suppresses interleukin-1beta-induced apoptosis in rat chondrocytes by inhibiting the NF-kappaB signaling pathway and delays the progression of osteoarthritis in a rat model. Int Immunopharmacol. 2017;52:156–162. doi: 10.1016/j.intimp.2017.08.021. [DOI] [PubMed] [Google Scholar]
- 46.Kapitanov GI, Ayati BP, Martin JA. Modeling the effect of blunt impact on mitochondrial function in cartilage: implications for development of osteoarthritis. PeerJ. 2017;5:e3468. doi: 10.7717/peerj.3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu Y, Zhu H, Yan X, Gu H, Gu Z, Liu F. Endoplasmic reticulum stress participates in the progress of senescence and apoptosis of osteoarthritis chondrocytes. Biochem Biophys Res Commun. 2017;491:368–373. doi: 10.1016/j.bbrc.2017.07.094. [DOI] [PubMed] [Google Scholar]
- 48.Ni Z, Shang X, Tang G, Niu L. Expression of miR-206 in human knee articular chondrocytes and effects of miR-206 on proliferation and apoptosis of articular chondrocytes. Am J Med Sci. 2018;355:240–246. doi: 10.1016/j.amjms.2017.11.003. [DOI] [PubMed] [Google Scholar]
- 49.Jia PT, Zhang XL, Zuo HN, Lu X, Li L. Articular cartilage degradation is prevented by tanshinone IIA through inhibiting apoptosis and the expression of inflammatory cytokines. Mol Med Rep. 2017;16:6285–6289. doi: 10.3892/mmr.2017.7340. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.