

Abstract
The therapeutic options for pulmonary fibrosis (PF), a progressive interstitial disease of the lung, are extremely limited. Studies have shown that transforming growth factor-β1 (TGF-β1)-induced epithelial-mesenchymal transition (EMT) functions as a central mediating process that contributes to PF. Also, low-molecular-weight fucoidan (LMWF), a sulfated polysaccharide extracted from brown seaweed, has been reported to have antifibrotic characteristics that can help to alleviate kidney fibrosis by inhibiting TGF-β1-mediated EMT. Thus we hypothesized that LMWF might be an attractive candidate for alleviating PF. Eighty C57BL/6 mice and A549 cells were respectively involved in our vivo and vitro experiments. The lung fibrosis was primarily assessed by hematoxylin and eosin (H&E), Masson’s trichrome stain, lung wet-to-dry weight ratio and hydroxyproline content. TGF-β1 levels were determined by enzyme-linked immunosorbent assay (ELISA) and immunofluorescence, and the expression of EMT markers and extracellular signal-regulated kinase (ERK) signaling were mainly based on immunostaining, real-time PCR and Western blot. As expected, our vivo models showed that LMWF was associated with improved lung fibrotic histopathology and significantly reduced lung hydroxyproline content. Levels of TGF-β1 expression in bronchoalveolar lavage fluid (BALF) and lung tissue decreased than it had been before treatment. Immunostaining, real-time PCR, and Western blot demonstrated that the lung EMT phenotype was attenuated and ERK signaling downregulated after LMWF administration. The vitro experiments resulted in a similar pharmacologic inhibitory effect of TGF-β1-induced EMT with downregulated ERK signaling. Collectively, our results preliminary suggested that LMWF could attenuate bleomycin-induced PF by inhibiting TGF-β1-induced EMT through ERK signaling.
Keywords: Low-molecular-weight fucoidan, pulmonary fibrosis, epithelial-mesenchymal transition, TGF-β1/ERK
Introduction
Pulmonary fibrosis (PF) is a chronic, progressive, irreversible sequela of lung injury of various known and unknown etiologies. It causes a permanent fibrotic “scar” mainly involving the lung parenchyma (alveolar and interstitial parenchyma as well as bronchioles) and is characterized by airway remodeling, inflammation, alveolar destruction, and the deposition of an abnormal extracellular matrix (ECM) [1].
Studies have shown that many factors contribute to the pathogenesis of PF. These could include, for example, a serious connective tissue disease (CTD), granulomatous diseases, or other rare conditions such as eosinophilic pulmonary disease, lymphangioleiomyomatosis, or Langerhans histiocytosis of the lung. PF is also an environmental and aging-associated pulmonary lesion. A number of experimental and clinical trials have demonstrated that certain environmental exposures, such as cigarette smoke and suspended particulate matters, can target the pulmonary epithelium and increase the risk of PF. An increased risk has also been linked with occupational exposures, as may occur in agricultural work, farming, working with livestock, and in persons exposed to wood dust, metal dust, stone dust, or silica [2]. However, the vast majority of patients with PF are diagnosed with idiopathic interstitial pneumonia (IIP), a particularly severe form of interstitial lung disease for which the treatment options are extremely limited [3]. Therefore PF has emerged as an important public health problem that carries significant morbidity and mortality, especially since IIP may have a more dismal prognosis than many cancers [4].
Currently dysfunction of alveolar epithelial cells (AECs) is thought to be the major factor leading to honeycomb lungs. The activated epithelium, either directly or indirectly, releases a plethora of mediators through its activated neutrophils and macrophages, the abundant inducers that drive the activation, migration, proliferation, and differentiation of fibroblasts and myofibroblasts, which eventually contribute to abnormal tissue repair, the persistent production of extracellular matrix (ECM) components, and destruction of the pulmonary architecture [5]. Transforming growth factor-β1 (TGF-β1) is considered to be the central mediator in the pathogenesis of fibrotic disorders; it not only plays an important role in the activation, proliferation, and differentiation of fibroblasts and myofibroblasts but also functions as the main inducer of EMT in normal pulmonary epithelial cells, thus contributing to pulmonary fibrogenesis [6].
Experimental studies have examined a vast array of natural drugs in search of a therapeutic agent that could counteract the profibrotic activity of TGF-β1. For example, Wang et al. [7] demonstrated that corilagin, a member of the tannin family found in medicinal plants such as Phyllanthus amarusare and Geranium carolinianum, could attenuate lung fibrosis induced experimentally by aerosolized bleomycin (BLM) and TGF-β1 signaling. Wei ZF and his coworkers [8] found that paeoniflorin, a pinane monoterpene glucoside found in the root of Paeonia lactiflora Pall, could play a protective role in the lung and alleviate BLM-induced pulmonary fibrosis through a Smad-dependent pathway. In our study, we aimed to investigate the potential role of low-molecular-weight fucoidan (LMWF) in ameliorating PF. LMWF, molecular weight at 8 to 10 kDa, is a sulfated polysaccharide extracted from Laminaria japonica and degraded by the free radical [9]. Previous studies have shown that LMWF has a variety of therapeutic properties, including anti-inflammatory, antioxidant, anti-angiogenesis and immune regulating effects [10]. In the past, LMWF has been used in the treatment of kidney diseases, such as chronic nephritis and chronic kidney injury. Most importantly, accumulating evidence indicates that this monomer has a strong inhibitory effect on renal fibrosis by alleviating the TGF-β1-induced EMT phenotype in renal tubular epithelial cells [11]. These researches indicate that LMWF may also be a potential choice for improving PF on account of the common mechanisms or targets of different types of organ fibrosis. Therefore we increased our efforts to establish a well-defined and standard BLM-induced PF mouse model and in vitro experiment to determine whether LMWF could ameliorate pulmonary fibrosis by inhibition of TGF-β1-induced EMT. We hoped that our results would provide new perspectives on promising options for the treatment of PF.
Materials and methods
Animal protocols
Eighty male C57BL/6 mice weighing 26 to 30 g were purchased from the Institute of Laboratory Animal Sciences (CAMS and PUMC, Beijing, China) and housed in specific pathogen-free (SPF) conditions with free access to food and water. The animals were randomly placed into 4 groups of 20 mice each: (1) Sham-operated group, (2) BLM group, (3) BLM+LMWF group, and (4) LMWF group. Briefly, mice in BLM+LMWF groups and BLM group were respectively received 100 mg/kg per day of LWMF by intraperitoneal injection and the same vehicle controls from the 7th, followed by a single slow intratracheal instillation of 3.5 mg/kg of bleomycin sulfate (S1214, Selleck, Japan) [11,12]. The LMWF group received the same volume of LMWF as the BLM+LMWF group, and sham controls only underwent the same intubation procedures as the BLM group except that saline was administered instead of bleomycin. On day 28, all the mice were sacrificed with an overdose of anesthesia. The bronchoalveolar lavage fluid (BALF) was collected with 3 separated washings. The left lung tissues were weighed, dried in an oven (60°C for 72 h), and weighted again to get the lung wet-to-dry weight ratio (W/D) [13]. The remaining tissues were isolated for histologic evaluation, hydroxyproline quantification, and analysis of gene and protein expression, respectively. The study was approved and guided by the Animal Care Review Committee of the China-Japan Friendship Hospital.
Lung histopathology
After the remaining blood was rinsed off, the left lung was fixed in 10% neutral formalin for at least 72 h, dehydrated, embedded in paraffin, and processed for hematoxylin and eosin (H&E) and Masson’s trichrome stain. The following semiquantitative grades of lung fibrosis were counted double blindly by two collaborators according to the scoring system described by Ashcroft et al. [14], and the collagen deposition areas were calculated by Image-Pro Plus 6.0 software (Media Cybernetics, Silver Spring, MD, US). For immunohistochemistry (IHC), the fixed, embedded and sliced sections were inmmunostained with antibodies against E-cadherin (1:100 diluted, ab76055, Abcam, Cambridge, UK), vimentin (1:50 diluted, 10366-1-AP, Proteintech, Chicago, IL, US) and α-SMA (1:100 diluted, A2547, Sigma-Aldrich, St. Louis, MO, US). All immunostained images were also analyzed using Image Pro-Plus software in a method of the mean density of the positive areas in the sections.
Measurement of hydroxyproline content
Total lung hydroxyproline content was measured with a standard hydroxyproline assay kit (A030-2, Nanjing Jiancheng Bioengineering Institute, Nan Jing, China). Briefly, samples were homogenized in NaOH solution with 400 rpm mechanical shaking for 20 min at 99°C. Following neutralization in HCl, the diluted samples were centrifuged at 3000 rpm for 20 min and the supernatants were detected in the Synergy4 plate reader (BioTek, Winooski, VT, US) at 550 nm absorbance. The data were expressed as micrograms of hydroxyproline per gram of wet lung tissue.
TGF-β1 measurement
The concentrations of TGF-β1 in the serum, BAL cell-free supernatant were measured via an enzyme-linked immunosorbent assay (ELISA) kit (R&D Systems, Minneapolis, MN, US) according to the manufacturer’s instructions.
Cell culture and treatment
A549 cells (CCL-185, ATCC, Rockville, MD, US) were cultured in low glucose Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (Gibco, Rockville, MD, US) and 1% antibiotics (Sigma-Aldrich) in a humidified atmosphere with 5% CO2. In our vitro experiment, cells were seeded into 6-well plates at a density of 6 × 104 cells per well and allowed to grow for 2 to 3 days until they reached 70% to 80% confluence; they were then treated with TGF-β1 (10 ng/mL, 96-100-21-10, PeproTech, Rocky Hill, NJ, US) [15] or co-treated with a predefined concentration of LMWF for 48 or 72 h for morphologic analysis (Nikon Eclipse Ti, Tokyo, Japan) and subsequent gene and protein analyses [9].
Cell viability assay
Cell viability and cytotoxicity were determined using the Cell Counting Kit-8 assay (CCK-8, Dojindo Laboratories, Kumamoto, Japan). Briefly, cells were seeded into 96-well plates at a concentration of 3 × 103 cells per well; after being starved overnight in serum-free medium, the cells were treated with 10 ng/mL TGF-β1 and/or indicated concentrations of LMWF (6.25, 12.5, 25, 50, 100, or 200 μg/mL) for 72 h [16]. Then 10 µL of CCK-8 solution was added and the cells were cultured for another hour. Optical density (OD) value of the culture medium was read at 450 nm by using the Synergy4 plate reader.
Immunofluorescence (IF)
For immunofluorescence, fresh tissue slides and cells were fixed in 4% paraformaldehyde for 30 min and then blocked, permeated, and incubated with specific antibodies. The primary antibodies used were as follows: rabbit anti-TGF-β1 (1:100 diluted, ab92486, Abcam), mouse anti-E-cadherin (1:100 diluted, Abcam), rabbit anti-vimentin (1:50 diluted, Proteintech), rabbit anti-phosphospecific ERK1/2 (1:100 diluted, 4370, CST, Boston, MA, US), rabbit anti-ERK1/2 (1:400 diluted, 4695, CST). Images were taken by the confocal laser scanning microscope (Leica Microsystems, Wetzlar, Germany).
Real-time quantitative PCR
Total RNA of the lung tissue and cells were isolated by using TrIzol reagent (15596026, Invitrogen, Carlsbad, CA, US) and reversed to cDNA by using a standard Kit (High-Capacity cDNA Reverse Transcription Kit, 4368814, Invitrogen). Finally, the PCR reaction was performed in a Bio-Rad system for 44 cycles. Each cycle consisted of denaturation at 95°C for 45 s, annealing at 60°C for 60 s, and an extension at 72°C for 1 min. The quantification of each gene was normalized to that of 18S mRNA, and the fold change was calculated as 2-ΔΔCT. Primers are as follows, human α-SMA 5’-GAAGAAGAGGACAGCACTG-3’ (F) and 5’-TCCCATTCCCACCATGAC-3’ (R); human fibronectin 5’-GCTGAAGAGACTTGCTTTGACAAG-3’ (F) and 5’-GGTGTCACCAATCTTGTAGGACTG-3’ (R); human 18S: 5’-GAGCAATAACAGGTCTGTGATGC-3’ (F) and 5’-CACTTACTGGGAATTCCTCGTTC-3’ (R); mouse α-SMA 5’-GCTGGTGATGATGCTCCCA-3’ (F) and 5’-GCCCATTCCAACCATTACTCC-3’; mouse fibronectin 5’-GAGACCAGTGCATCGTTGATGAC-3’ (F) and 5’-G GTCTCTGAATCTTGGCACTGGTC-3’ (R); mouse 18S: 5’-CACGGACAGGATTGACAGATTG-3’ (F) and 5’-GCGTAACTAGTTAGCATGCCAGAG-3’ (R).
Western blot
Fresh tissues and cells were homogenized in Radio-Immunoprecipitation Assay (RIPA) lysis buffer supplemented with 25 × protease inhibitor cocktail (04693132001, Roche, Ct. Bern, Switzerland) and 10 × phosphatase inhibitor cocktail (4906845001, Roche) with subsequent protein quantification by a bicinchoninic acid (BCA) protein assay (23227, Thermo Scientific, Rockford, IL, US). After 4 × loading buffer was added, the proteins were boiled at 99°C for 5 min and then resolved on 12% SDS-PAGE gels and electrotransferred to polyvinylidene difluoride (PVDF) membranes. After blocking, the membranes were incubated with primary antibodies as follows: mouse anti-E-cadherin (1:1000 diluted, Abcam), rabbit anti-vimentin (1:2000 diluted, Proteintech), rabbit anti-fibronectin (1:2000 diluted, Proteintech), mouse anti-α-SMA (1:3000 diluted, Sigma-Aldrich), rabbit anti-TGF-β1 (1:500 diluted, ab92486, Abcam), rabbit anti-phosphospecific ERK1/2 (1:500 diluted, CST), rabbit anti-ERK1/2 (1:1000 diluted, CST), mouse anti-GAPDH (1:5000, 60004-1-Ig, Proteintech) and mouse anti-β-actin (1:5000, 60008-1-Ig, Proteintech) at 4°C overnight. Species-specific secondary antibodies were added the next day; then protein intensities were detected with the ECL Advanced Western Blot Detection Kit (Amersham Biosciences, Waltham, UK) and subsequently quantitated with Image Lab 6.0 software (Bio-Rad Laboratories, Hercules, CA, US).
Statistical analysis
All data are expressed as mean ± standard deviation (SD). Comparisons between 2 groups were made using the student’s t-test, and those among multiple groups were made using one-way analysis of variance (ANOVA). Statistical analysis was performed and is presented by using commercial statistical software Graph Pad Prism 7.0 (GraphPad, San Diego, CA, US). P value < 0.05 was considered significant.
Results
LMWF alleviated lung fibrogenesis and TGF-β1 expression in the BLM-induced PF mouse model
Intratracheal instillation of bleomycin is the classic method for establishing a well-defined mouse model of PF. H&E stain and Masson’s trichrome stain were respectively used for model identification and pharmacodynamic evaluation of LMWF. In animals treated with bleomycin alone, we observed extensive lung injury with alveolar and interstitial edema, hemorrhage, and inflammatory cellular infiltration on the 3rd day, which gradually resolved after 1 week with early fibroproliferation. By the 14th day, tissue sections showed a significant decline in pulmonary inflammatory response; on the other hand, serious fibrotic lesions developed, mainly characterized by considerable cell proliferation and obvious thickened alveolar septum. By the 28th day, fibrous foci and bullae were diffusely distributed in the lung interstitium, with significant cell proliferation and disruption of the lung architecture. Based on these preliminary experimental results, we extended our treatment from the 7th day after administration of bleomycin to 28 days in order to investigate the role of LMWF in PF. As shown in Figure 1A, the pathologic changes in the lung had improved after LMWF treatment. There was less cell proliferation, a less destroyed alveolar septum and lung structure. Masson staining clearly indicated that collagen deposition had decreased in mice subjected to LMWF compared with that in the BLM group (Figure 1B). Semiquantitative grading of fibrosis score, as well as collagen deposition areas (%) were significantly declined after treatment of LMWF administration (Figure 1C and 1D). As expected, treatment with LMWF also rendered the level of lung hydroxyproline and BALF TGF-β1 lower than in the BLM group (Figure 1E and 1F). However, there were no obvious differences in serum TGF-β1 level (Figure 1G) or lung W/D (Figure 1H) between the BLM+LMWF and BLM groups.
Figure 1.

LMWF alleviated lung fibrogenesis and TGF-β1 expression in a BLM-induced PF mouse model. (A) Representative micrographs of hematoxylin and eosin (H&E) stain for each group on day 28. (B) Masson’s trichrome staining for each group. The blue regions indicated the collagen deposition, which widely distributed in lung interstitial space. Outer original images are × 200 magnification, scale bar = 100 μm; inner original images are × 40 magnification, scale bar = 50 μm. (C and D) Ashcroft score and the collagen deposition area (%) for each group. Data were analyzed using one-way analysis of variance (ANOVA) and 5-7 animals were quantified in each group. (E) Lung hydroxyproline content. (F) Level of expression of TGF-β1 in bronchoalveolar lavage fluid (BALF). (G) Level of expression of TGF-β1 in the serum. (H) Lung wet-to-dry weight ratio (W/D) in each group. The data of (E-H) were analyzed by using ANOVA and are here presented as the mean value ± SD for 8-12 individuals in each group. Significance level was labeled as *P < 0.05, **P < 0.01, ***P < 0.001.
LMWF attenuated lung EMT phenotype in the BLM-induced PF mouse model
Consistent with our histologic findings in Figure 1, a marked EMT phenotype was observed in the bleomycin administrated alveolar cells. As shown in Figure 2A, the epithelial marker of E-cadherin in BLM group was significantly reduced as opposed to the dramatically elevated mesenchymal phenotype markers of vimentin and α-SMA, compared to the controls. Mice given LMWF continued to show higher expression levels of E-cadherin and lower expression levels of vimentin and α-SMA than models. The results of real-time PCR suggested that the mice subjected to both bleomycin and LMWF had lower expression of α-SMA and fibronectin than those treated only with bleomycin (Figure 2B). Western blot also demonstrated similar expression trends of E-cadherin, α-SMA, and fibronectin in mice of BLM groups, which could partly be reversed with LMWF treatment (Figures 2C, S1). Collectively, these findings strongly indicate that LMWF plays a role in inhibiting EMT in the lungs during lung fibrogenesis in the mouse model of BLM-induced PF.
Figure 2.

LMWF attenuated lung EMT phenotype in a BLM-induced PF mouse model. A. Lung immunocytochemistry (IHC) staining of E-cadherin, vimentin, and α-SMA that performed on paraffin-embedded sections (upper panel, brown identify regions of positive staining). Original images are × 200 magnification. n = 5; scale bar = 100 μm. The mean density of E-cadherin, vimentin and α-SMA for each group (lower panel); values were analyzed by using ANOVA and expressed by fold-changes of sham. B. Real-time PCR for analysis the expression of α-SMA and fibronectin on the transcriptional level on the 28th day, 18S served as an internal control. Values were analyzed by using student’s t-test and expressed by fold-changes of sham. C. Western blot analysis for E-cadherin, α-SMA and fibronectin with each experimental group (upper panel), and the ratio of corresponding protein in lung homogenate (lower panel). Data are determined using the student’s t-test and are presented as mean value ± SD. Significance was set at *P < 0.05, **P < 0.01, and ***P < 0.001.
LMWF inhibited the cell morphologic alterations and proliferation in TGF-β1-induced A549 cells
To further explore the potential mechanism of LMWF in TGF-β1-induced EMT, A549 cells were incubated with LMWF in the absence or presence of TGF-β1 (10 ng/mL). Morphologic alterations were observed under a phase-contrast microscope (Figure 3A). In short, we clearly found that TGF-β1-driven A549 cells exhibited a signature morphologic lesion of fibroblastic appearance with an elongated spindle-like shape and markedly decreased cell-cell contacts, whereas untreated cells still maintained their normal epithelial-like appearance with close cell-to-cell contacts. Low concentrations of 6.25, 12.5, or 25 μg/mL LMWF were not associated with any obvious morphologic alterations, and relatively higher doses of LMWF (50, 100, or 200 μg/mL) produced slight changes; however, changes were especially pronounced with 200 μg/mL. The cytotoxicity of LMWF was observed by the means of a CCK-8 assay. Figure 3B showed the growth curve of cells that were cultured with different concentrations of LMWF (50, 100, and 200 μg /mL) up to 72 h. It indicated that cells treated with either low or high doses of LMWF displayed similar cell viability to that of the untreated normal cells over the indicated time spans (12, 24, 48, and 72 h), suggesting that there was no concentration- or time-dependent cytotoxic effect of LMWF on A549 cells [17]. Figure 3C reflected the cell viability of groups of cells that were cultured with TGF-β1 and/or LMWF for 72 h. We found that at 48 h cell viability was enhanced by co-culture with TGF-β1 compared to with normal cells, and that these values could return to normal levels when cells were cocultured with LMWF at relative higher dose of 100 or 200 μg/mL. Like the cells treated with TGF-β1 only, the cells in TGF-β1 and 200 μg/mL LMWF co-administrated group also showed decreased cell viability at 72 h; however, under other conditions, no detectable differences in the cell viability of each group were found.
Figure 3.
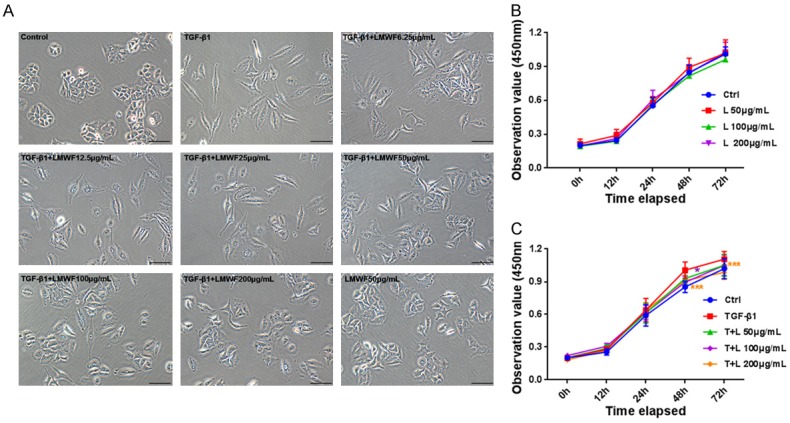
Cell morphology and cell viability of A549 up to 72 h. A. Representative microscopic images of A549 cells incubated with different concentrations of LMWF (6.25, 12.5, 25, 50, 100, and 200 μg/mL) in the presence and absence of TGF-β1 (10 ng/mL) for up to 72 h. Original images are × 200 magnification; scale bar = 100 μm. B and C. Cell cytotoxic assay and cell proliferation test. A549 cells were cultured with different concentrations of LMWF (50, 100, and 200 μg/mL) and/or TGF-β1 up to 72 h. This shows there to be no concentration- or time-dependent cytotoxic effect of LMWF on A549 cells, and LMWF at relatively high dose of 100 or 200 μg/mL could inhibited the proliferation of A549 cells caused by TGF-β1 at 48 and 72 h effectively. Data were analyzed using the student’s t-test and are presented as mean value ± SD of three independent experiments. Significance was set at *P < 0.05 and ***P < 0.001 relative to the control at a same time.
LMWF attenuated TGF-β1-induced EMT in A549 cells
In the TGF-β1-induced cells, a significant EMT phenotype was observed. The result of dual IF of E-cadherin and vimentin suggested an intermediate transitional stage of EMT (Figure 4A). In the cells cocultured with both TGF-β1 and LMWF, mainly the cells treated with 200 μg/mL LMWF, the expression of vimentin was dramatically decreased with the higher expression of E-cadherin. The results of real-time PCR indicated that cells treated with TGF-β1 had significantly less expression of α-SMA and fibronectin than controls. However, the cells incubated with both TGF-β1 and LMWF showed less expression of α-SMA and fibronectin mRNA compared to those only treated with TGF-β1 (Figure 4B). As determined by Western blot analysis, results showed that TGF-β1 administration also resulted in reduced expression of E-cadherin and raised expression levels of vimentin and fibronectin, whereas the expression of corresponding proteins was reversed with the treatment of LMWF. As expected, the changes were most pronounced when LMWF was administered at a dose of 200 μg/mL (Figures 4C, S2).
Figure 4.

LMWF attenuated TGF-β1-induced EMT in A549 cells. A. Dual-immunofluorescence of E-cadherin and vimentin in A549 cells incubated with TGF-β1 and/or LMWF at 72 h; Ctrl: cells treated with vehicle; T: cells treated with 10 ng/mL TGF-β1; T+L: cells treated with 10 ng/mL TGF-β1 plus 200 μg/mL LMWF. Original images are × 400 magnification. B. Real time PCR for analysis of the relative expression levels of α-SMA and fibronectin in each group. C. Western blot analysis for E-cadherin, fibronectin and vimentin in A549 cells that cultured with TGF-β1 and/or LMWF (50, 100, and 200 μg/mL) at 72 h. Data were analyzed using the student’s t-test and are presented as mean value ± SD of three independent experiments. Significance was set to *P < 0.05, **P < 0.01, and ***P < 0.001.
LMWF downregulated TGF-β1/ERK signaling both in vivo and in vitro
ERK signaling is an important mechanism implicated in the process of TGF-β1-induced EMT during the lung fibrogenesis. As shown in Figure 5A and 5B, we determined the relative expression of TGF-β1 and p-ERK1/2 by IF and IHC, respectively. Results showed that LMWF treatment could inhibit the expression of TGF-β1 and p-ERK1/2 in mice given bleomycin. In our Western blot analysis, we also found the level of expression of TGF-β1 was increased in lung homogenates, accompanied with higher expression levels of p-ERK1/2. As predicted, expression of both agents was downregulated after treatment of LMWF (Figures 5C, S1). We also performed in vitro experiments. As shown in Figure 5D, we found the relative expression level of p-ERK1/2 to be significantly higher in cells with exogenous TGF-β1, which could be effectively inhibited with the treatment of LMWF. Accordingly, Western blot (Figures 5E, S2) results also suggested that the cells treated with LMWF, mainly at doses of 100 and 200 μg/mL LMWF, were associated with the downregulation of ERK1/2 signaling.
Figure 5.
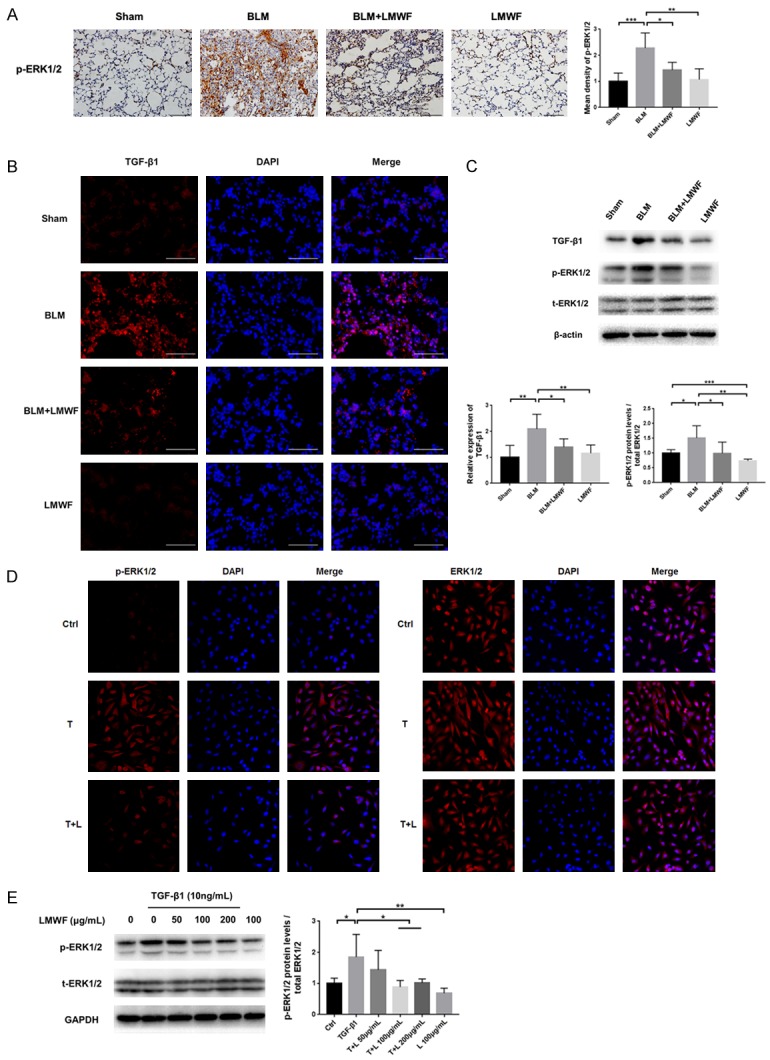
LMWF downregulated TGF-β1/ERKs pathway both in vivo and in vitro. A. IHC staining for p-ERK1/2 for each animal group at 28th day, Original images are × 200 magnification. n = 4-5; scale bar = 100 μm. The mean density of p-ERK1/2 was analyzed by using ANOVA and expressed by fold-changes of sham. B. Immunofluorescence (IF) staining of TGF-β1 in lung tissue sections. Original images are × 400 magnification. scale bar = 50 μm. C. Representative blots of TGF-β1, ERK1/2, p-ERK1/2 on day 28 (upper panel) and the ratio of specific protein in lung homogenate (lower panel). D. IF staining for p-ERK1/2 and ERK1/2 in A549 cells that incubated with TGF-β1 and/or LMWF up to 72 h. Original images are × 400 magnification. E. Western blot analysis for ERK1/2, p-ERK1/2 at 72 h (left panel) and the densitometry values of p-ERK1/2 normalized to ERK1/2 (right panel). Data are determined using the student’s t-test and are presented as mean ± SD of three independent experiments. Significance level was set at *P < 0.05, **P < 0.01, and ***P < 0.001.
Discussion
Fucoidan comprises an intriguing group of natural fucose-enriched sulfated polysaccharides that are extracted from brown seeds. LMWF is obtained through the radical degradation of fucoidan [17]. Previous studies showed that LMWF has a range of bioactivities, having anti-inflammatory and antioxidant properties and promoting wound healing, as well as a significant role in regulating the circulation and the immune system [18,19]. It has therefore offered a supplementary option for the clinical management of patients with cardiovascular and kidney disease. In our study we described a novel role for LMWF in that it could effectively alleviate experimental BLM-induced PF as compared with controls. By inhibiting the process of TGF-β1-induced EMT, we also demonstrated the downregulation of ERK signaling behind the LMWF-attenuated fibrotic response.
Fibroblasts and myofibroblasts are the effector cells responsible for the production of lung matrix components. They can be derived from 3 potential sources, as follows: the expansion and differentiation of local mesenchymal cells, the recruitment and differentiation of circulating bone marrow progenitors known as fibrocytes, or via a process called EMT [20,21]. The overall importance of each fibroblast population remains unclear; however, EMT is well recognized to be the most important factor related to the dysfunction of AECs. Specifically, EMT refer to the transdifferentiation of epithelial cells into fibroblast-like cells, including morphologic transformation, phenotype marker transition, cytoskeletal rearrangements, and the activation of cell signaling pathways, which would eventually lead to excessive collagen secretion and ECM deposition [22,23]. It is an important process that is involved not only in embryonic development, tumor metastasis but also significantly participates in the pathogenesis of PF. In general, the epithelial markers include surface active protein (SP-A, SP-B), protein (ZO-1, E-cad), cytokeratins, and β-catenin; whereas N-cadherin, vimentin, fibronectin, α-smooth muscle actin (α-SMA), connective tissue growth factor (CTGF) and matrix metalloproteinases (MMPs) are the classic mesenchymal markers [15,24]. In our experiment, we found strong evidence for a possible intermediate transitional stage of EMT in BLM-induced mice and TGF-β1-stimulated cells. In the samples treated with LMWF, these alterations were partly alleviated or even completely abrogated in some areas.
It is well known that a variety of cytokines, chemokines, and growth factors are significantly involved in the process of EMT, such as CXC chemokines CXCL1 (KC), platelet-derived growth factor (PDGF), basic fibroblast growth factor (bFGF), TGF-β1, and others [25,26]. TGF-β1 is considered to be the most central mediator in the pathogenesis of PF and for the activation, proliferation, and differentiation of epithelial cells and collagen-producing myofibroblasts [21]. Studies have shown, as an upstream factor, that once overexpressed TGF-β1 is bound to TGF-β ligands, type II TGF-β receptors (TβRII) form heterodimers with TβRI and excessively activate regulatory Smad2/3 signaling, thus initiating abnormal ECM accumulation and tissue repair. An important transcriptional pathway is the Smad-independent pathway [27,28]. Apart from this canonical signaling, the non-Smad pathway is also significantly involved in the development of PF, as has been reported with a synergistic reaction in regulating TGF-β1 signaling with MAPK, EGFR, p53, and Notch signaling [29,30]. ERK, a member of the MAPK family, has been established as major participants in the regulation of cell growth and differentiation. When improperly activated, however, it contributes to malignant transformation. ERK1 and 2 are central components of the MAPK cascade. Once activated, ERK can translocate to the nucleus to phosphorylate and activate transcription factors while other pools of activated ERK phosphorylate a number of cytoplasmic targets. Upon phosphorylation, nuclear translocation of ERK1 and ERK2 is critical for both gene expression and DNA replication induced by growth factors [31]. Studies have shown that the ERK1/2 signaling pathways are essential for EMT, as well as fibroblast-to-myofibroblast differentiation induced by TGF-β1, which eventually contributes to the pathobiology of fibrosis [32,33]. Here, we preliminarily explored the role of LMWF in regulation of ERK signaling during the process of TGF-β1-induced EMT. As expected, we found that the expression levels of TGF-β1 were raised in BLM-treated mice and decreased in mice treated with LMWF. We also found that the protein levels of p-ERK1/2 were increased in BLM-induced mice and decreased after LMWF treatment.
In order to better clarify the mechanisms involved, a further test on the cellular level was performed. Recombinant TGF-β1 at a concentration of 10 ng/mL was used to stimulate A549 cells. As a result, the cells’ morphology and ability to proliferate were altered to make them more like mesenchymal cells; the associated EMT markers were also dysregulated at the transcriptional and protein level. Most importantly, in the TGF-β1-overexpressed cells, we observed a significantly higher expression of p-ERK1/2, consistently with the results of our in vivo research. All these alterations were alleviated in the cells treated with LMWF, pointing to the important role of LMWF in ameliorating the TGF-β1-induced EMT phenotype via inhibition of ERK signaling during the lung fibrogenesis.
This study has several limitations. For instance, even if the mouse model used for BLM-induced PF is validated, it is not fully representative of the complexity of conditions seen in humans. Additionally, the A549 cell was a lung adenocarcinoma epithelial cell. It may differ from the primary epithelial cells, thus bias may have been created. Our study marks only the preliminary stage of pharmacodynamic and pharmacologic research on this issue; further studies at multiple levels may be needed in future.
In conclusion, our study preliminarily demonstrated that LMWF can ameliorate TGF-β1-induced EMT in both vivo and in vitro models of PF by downregulating ERK signaling. This suggests that it may be a promising therapeutic option for the treatment of PF and other fibrotic disorders in the future.
Acknowledgements
This work was supported by the fund of National Natural Science Foundation of China (Project No. 81470270) and The National Key Research and Development Program of China (2016YFC1304300).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Sgalla G, Iovene B, Calvello M, Ori M, Varone F, Richeldi L. Idiopathic pulmonary fibrosis: pathogenesis and management. Respir Res. 2018;19:32. doi: 10.1186/s12931-018-0730-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Martinez FJ, Collard HR, Pardo A, Raghu G, Richeldi L, Selman M, Swigris JJ, Taniguchi H, Wells AU. Idiopathic pulmonary fibrosis. Nat Rev Dis Primers. 2017;3:17074. doi: 10.1038/nrdp.2017.74. [DOI] [PubMed] [Google Scholar]
- 3.Canestaro WJ, Forrester SH, Raghu G, Ho L, Devine BE. Drug treatment of idiopathic pulmonary fibrosis: systematic review and network meta-analysis. Chest. 2016;149:756–766. doi: 10.1016/j.chest.2015.11.013. [DOI] [PubMed] [Google Scholar]
- 4.Raimundo K, Chang E, Broder MS, Alexander K, Zazzali J, Swigris JJ. Clinical and economic burden of idiopathic pulmonary fibrosis: a retrospective cohort study. BMC Pulm Med. 2016;16:1–8. doi: 10.1186/s12890-015-0165-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.King TJ, Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet. 2011;378:1949–1961. doi: 10.1016/S0140-6736(11)60052-4. [DOI] [PubMed] [Google Scholar]
- 6.Wei Y, Kim TJ, Peng DH, Duan D, Gibbons DL, Yamauchi M, Jackson JR. Fibroblast-specific inhibition of TGF-β1 signaling attenuates lung and tumor fibrosis. J Clin Invest. 2017;127:3675–3688. doi: 10.1172/JCI94624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang Z, Guo QY, Zhang XJ, Li X, Li WT, Ma XT, Ma LJ. Corilagin attenuates aerosol bleomycin-induced experimental lung injury. Int J Mol Sci. 2014;15:9762–9779. doi: 10.3390/ijms15069762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ji Y, Dou Y, Zhao Q, Zhang J, Yang Y, Wang T, Xia YF, Dai Y, Wei ZF. Paeoniflorin suppresses TGF-β mediated epithelial-mesenchymal transition in pulmonary fibrosis through a smad-dependent pathway. Acta Pharmacol Sin. 2016;37:794–804. doi: 10.1038/aps.2016.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li X, Li X, Zhang Q, Zhao T. Low molecular weight fucoidan and its fractions inhibit renalepithelial mesenchymal transition induced by TGF-β1 or FGF-2. Int J Biol Macromol. 2017;105:1482–1490. doi: 10.1016/j.ijbiomac.2017.06.058. [DOI] [PubMed] [Google Scholar]
- 10.Chen MC, Hsu WL, Hwang PA, Chou TC. Low molecular weight fucoidan inhibits tumor angiogenesis through downregulation of HIF-1/VEGF signaling under hypoxia. Mar Drugs. 2015;13:4436–4451. doi: 10.3390/md13074436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen J, Cui W, Zhang Q, Jia Y, Sun Y, Weng L, Luo D, Zhou H, Yang B. Low molecular weight fucoidan ameliorates diabetic nephropathy via inhibiting epithelial-mesenchymal transition and fibrotic processes. Am J Transl Res. 2015;7:1553–1563. [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Q, Tong M, Ou B, Liu C, Hu C, Yang Y. Isorhamnetin protects against bleomycin-induced pulmonary fibrosis by inhibiting endoplasmic reticulum stress and epithelial-mesenchymal transition. Int J Mol Med. 2019;43:117–126. doi: 10.3892/ijmm.2018.3965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.González-López A, Astudillo A, García-Prieto E, Fernández-García MS, López-Vázquez A, Batalla-Solís E, Taboada F, Fueyo A, Albaiceta GM. Inflammation and matrix remodeling during repair of ventilator-induced lung injury. Am J Physiol Lung Cell Mol Physiol. 2011;301:500–509. doi: 10.1152/ajplung.00010.2011. [DOI] [PubMed] [Google Scholar]
- 14.Ashcroft T, Simpson JM, Timbrell V. Simple method of estimating severity of pulmonary fibrosis on a numerical scale. J Clin Pathol. 1988;41:467–470. doi: 10.1136/jcp.41.4.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kasai H, Allen JT, Mason RM, Kamimura T, Zhang Z. TGF-beta1 induces human alveolar epithelial to mesenchymal cell transition (EMT) Respir Res. 2005;6:1–15. doi: 10.1186/1465-9921-6-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boo HJ, Hyun JH, Kim SC, Kang JJ, Kim MK, Kim SY, Cho H, Yoo ES, Kang HK. Fucoidan from undaria pinnatifida, induces apoptosis in a549 human lung carcinoma cells. Phytother Res. 2011;25:1082–1086. doi: 10.1002/ptr.3489. [DOI] [PubMed] [Google Scholar]
- 17.Hwang PA, Yan MD, Lin HT, Li KL, Lin YC. Toxicological evaluation of low molecular weight fucoidan in vitro and in vivo. Mar Drugs. 2016;14:E121. doi: 10.3390/md14070121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hwang PA, Phan NN, Lu WJ, Ngoc Hieu BT, Lin YC. Low-molecular-weight fucoidan and high-stability fucoxanthin from brown seaweed exert prebiotics and anti-inflammatory activities in caco-2 cells. Food Nutr Res. 2016;60:32033. doi: 10.3402/fnr.v60.32033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Park JH, Choi SH, Park SJ, Lee YJ, Park JH, Song PH, Cho CM, Ku SK, Song CH. Promoting wound healing using low molecular weight fucoidan in a full-thickness dermal excision rat model. Mar Drugs. 2017;15:112. doi: 10.3390/md15040112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Andersson-Sjöland A, de Alba CG, Nihlberg K, Becerril C, Ramírez R, Pardo A, Westergren-Thorsson G, Selman M. Fibrocytes are a potential source of lung fibroblasts in idiopathic pulmonary fibrosis. Int J Biochem Cell Biol. 2008;40:2129–2140. doi: 10.1016/j.biocel.2008.02.012. [DOI] [PubMed] [Google Scholar]
- 21.Wynn TA. Integrating mechanisms of pulmonary fibrosis. J Exp Med. 2011;208:1339–1350. doi: 10.1084/jem.20110551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu T, Liu Y, Miller M, Cao L, Zhao J, Wu J, Wang J, Liu L, Li S, Zou M, Xu J, Broide DH, Dong L. Autophagy plays a role in fstl1-induced emt and airway remodeling in asthma. Am J Physiol Lung Cell Mol Physiol. 2017;313:L27–L40. doi: 10.1152/ajplung.00510.2016. [DOI] [PubMed] [Google Scholar]
- 23.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tian R, Zhu Y, Yao J, Meng X, Wang J, Xie H, Wang R. NLRP3 participates in the regulation of EMT in bleomycin-induced pulmonary fibrosis. Exp Cell Res. 2017;357:328–334. doi: 10.1016/j.yexcr.2017.05.028. [DOI] [PubMed] [Google Scholar]
- 25.Guan S, Zhou J. Cxcr7 attenuates the TGF-β-induced endothelial-to-mesenchymal transition and pulmonary fibrosis. Mol Biosyst. 2017;13:2116–2124. doi: 10.1039/c7mb00247e. [DOI] [PubMed] [Google Scholar]
- 26.Thuan DTB, Zayed H, Eid AH, Abou-Saleh H, Nasrallah GK, Mangoni AA, Pintus G. A potential link between oxidative stress and endothelial-to-mesenchymal transition in systemic sclerosis. Front Immunol. 2018;9:1985. doi: 10.3389/fimmu.2018.01985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li J, Chen K, Li S, Feng J, Liu T, Wang F, Zhang R, Xu S, Zhou Y, Zhou S, Xia Y, Lu J, Zhou Y, Guo C. Protective effect of fucoidan fromfucus vesiculosuson liver fibrosis via the TGF-β1/smad pathway-mediated inhibition of extracellular matrix and autophagy. Drug Des Devel Ther. 2016;10:619–630. doi: 10.2147/DDDT.S98740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhou XM, Wang GL, Wang XB, Liu L, Zhang Q, Yin Y, Wang QY, Kang J, Hou G. Ghk peptide inhibits bleomycin-induced pulmonary fibrosis in mice by suppressing TGF-β1/smad-mediated epithelial-to-mesenchymal transition. Front Pharmacol. 2017;8:904. doi: 10.3389/fphar.2017.00904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xi Y, Tan K, Brumwell AN, Chen SC, Kim YH, Kim TJ, Wei Y, Chapman HA. Inhibition of epithelial-to-mesenchymal transition and pulmonary fibrosis by methacycline. Am J Respir Cell Mol Biol. 2014;50:51–60. doi: 10.1165/rcmb.2013-0099OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zheng X, Qi C, Zhang S, Fang Y, Ning W. TGF-β1 induces fstl1 via the smad3-c-jun pathway in lung fibroblasts. Am J Physiol Lung Cell Mol Physiol. 2017;313:L240–L251. doi: 10.1152/ajplung.00523.2016. [DOI] [PubMed] [Google Scholar]
- 31.Islam MS, Ciavattini A, Petraglia F, Castellucci M, Ciarmela P. Extracellular matrix in uterine leiomyoma pathogenesis: a potential target for future therapeutics. Hum Reprod Update. 2017;24:1–27. doi: 10.1093/humupd/dmx032. [DOI] [PubMed] [Google Scholar]
- 32.Lu J, Zhong Y, Lin Z, Lin X, Chen Z, Wu X, Wang N, Zhang H, Huang S, Zhu Y, Wang Y, Lin S. Baicalin alleviates radiation-induced epithelial-mesenchymal transition of primary type ii alveolar epithelial cells via TGF-β and ERK/GSK3β signaling pathways. Biomed Pharmacother. 2017;95:1219–1224. doi: 10.1016/j.biopha.2017.09.037. [DOI] [PubMed] [Google Scholar]
- 33.Huang X, He Y, Chen Y, Wu P, Gui D, Cai H, Chen A, Chen M, Dai C, Yao D, Wang L. Baicalin attenuates bleomycin-induced pulmonary fibrosis via adenosine a2a receptor related TGF-β1-induced ERK1/2 signaling pathway. BMC Pulm Med. 2016;16:132. doi: 10.1186/s12890-016-0294-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.